 S. Di Sabatino
S. Di Sabatino J. Koskelo2,3
J. Koskelo2,3 J. Prodhon
J. Prodhon P. Romaniello
P. Romaniello- 1Laboratoire de Chimie et Physique Quantiques, Université de Toulouse, CNRS, UPS, Toulouse, France
- 2Laboratoire de Physique Théorique, Université de Toulouse, CNRS, UPS, Toulouse, France
- 3European Theoretical Spectroscopy Facility (ETSF), Toulouse, France
The Extended Koopman’s Theorem (EKT) provides a straightforward way to compute charged excitations from any level of theory. In this work we make the link with the many-body effective energy theory (MEET) that we derived to calculate the spectral function, which is directly related to photoemission spectra. In particular, we show that at its lowest level of approximation the MEET removal and addition energies correspond to the so-called diagonal approximation of the EKT. Thanks to this link, the EKT and the MEET can benefit from mutual insight. In particular, one can readily extend the EKT to calculate the full spectral function, and choose a more optimal basis set for the MEET by solving the EKT secular equation. We illustrate these findings with the examples of the Hubbard dimer and bulk silicon.
1 Introduction
The Extended Koopman’s Theorem (EKT) (Morrell et al., 1975; Smith and Day, 1975) has been derived in quantum chemistry and used within various frameworks, from functional theories based on reduced quantities, such as reduced-density matrix functional theory (Gilbert, 1975) (e.g., Pernal and Cioslowski, 2005; Leiva and Piris, 2005; Piris et al., 2012; Piris et al., 2013) and many-body perturbation theory based on Green’s functions (Hedin, 1965) (e.g., Dahlen and van Leeuwen, 2005; Stan et al., 2006; Stan et al., 2009), to wavefunction-based methods (e.g., Cioslowski et al., 1997; Kent et al., 1998; Bozkaya, 2013; Zheng, 2016; Bozkaya and Ünal, 2018; Pavlyukh, 2019; Lee et al., 2021). The EKT allows one to calculate energies corresponding to charged excitations. Although it can be formulated both for ionization potentials (IPs) and electron affinities (EAs), it has been widely used only for the former, whereas for the latter applications have been limited to the calculation of the lowest EA as the first IP of the (N+1)-electron system (in case of finite systems), with N the number of electrons in the reference system. There exist hence many benchmarks for the IPs. So far, the method has been mainly used for finite systems. The EKT is known to be in principle (i.e., using exact ingredients, namely the one- and two body density matrices, as we shall see) exact for the first ionization potential (Katriel and Davidson, 1980; Sundholm and Olsen, 1993). In the solid state instead there are only a few applications which only focus on the band structure. It would be desirable to have also the spectral function, i.e., the spectrum of electron addition and removal energies weighted by the Dyson amplitudes, which measure the overlap between the eigenstates of the (N + 1) − electron ((N − 1) − electron) system and the ground state of the N-electron system where an electron has been added (removed). The spectral function is related to photoemission spectroscopy, which gives precious information about the electronic structure and excitations in a system, and, moreover, allows one to study metal-insulator transitions, of paramount importance in condensed matter.
A simple way to calculate the spectral function A(ω) is through the imaginary part of the one-body Green’s function G(ω), as
The paper is organized as follows. In Theory we give the basic equations of the EKT and the MEET and we make the link between them. The Hubbard dimer and bulk silicon are used to illustrate the difference between the EKT and the MEET (removal and addition) energies in Mutual Insights and Illustration. In Conclusions and Perspectives we draw our conclusions and perspectives.
2 Theory
In this section we briefly review the MEET and EKT methods, and we make the link between the two. We will consider an N-electron system governed by the following Hamiltonian in second quantization
where
2.1 Key Equations of the MEET
Within the MEET the time-ordered 1-body Green’s function G(ω) at zero temperature is split into removal (R) and addition (A) parts as G(ω) = GR(ω) + GA(ω). In the following we concentrate on the diagonal elements of G, which are related to photoemission spectra. Within the MEET the diagonal matrix elements of GR/A(ω) are written in terms of an effective energy
with γii the diagonal matrix element of the one-body density matrix in a given basis set. We note that a similar effective energy can be introduced also for the off-diagonal elements of GR/A. The spectral function is hence expressed as
where the symbol δ on the right-hand side indicates the Dirac delta function. In our previous works we have chosen the basis set of natural orbitals, i.e., the orbitals which diagonalize the one-body reduced density matrix. In this case γii = ni, i.e., the natural occupation numbers. This choice has been made based on our results on exactly solvable Hubbard clusters, where the MEET performs very well. (Di Sabatino et al., 2016) However this does not guarantee that it remains the best choice for more realistic systems. In fact this is not the case as we shall see.
The effective energy
where
of the
with
(and similarly for
2.2 Key Equations of the EKT
Within the EKT one starts from the following approximation for the removal energy
with
with
The eigenvalues of ΛR are the removal energies. (Morrell et al., 1975; Pernal and Cioslowski, 2005) By comparing to Eq. 4 it becomes clear that the diagonal element of ΛR are the removal energy of the MEET within the low-order approximation. The diagonal element of ΛR are also referred in literature as the energies of the EKT within the diagonal approximation (DEKT).
Similar equations hold for the addition energies. One can indeed define the addition energy
and in a similar way as for
with
Again, the diagonal elements of ΛA are the MEET addition energies within the approximation given in Eq. 5.2
3 Mutual Insights and Illustration
Now that we have established the link between the EKT and the MEET we will study how these theories can benefit from mutual insight.
3.1 Hubbard Dimer
We use a modified version of the Hubbard dimer in which the on-site Coulomb interaction is different for the two sites. Its Hamiltonian is given by
where i, j run over the sites,
3.1.1 Insights Into the EKT
Making the parallel with the MEET, one can readily define the EKT spectral function as.
with γii and 1 − γii the diagonal matrix element of the one-body density matrix in the basis which diagonalizes
which is a weighted average of the poles of
We notice that very recently Lee et al. (Lee et al., 2021), have also proposed an expression for the spectral function from the EKT.
3.1.2 Insights Into the MEET
Several choices for an optimal basis set for the MEET expressions are now possible. In previous works we considered the basis of natural orbitals as optimal basis set for the MEET based on the following findings (Di Sabatino, 2016): i) the MEET (in this basis of natural orbitals) gives the exact spectral function at all level of approximations for the symmetric Hubbard dimer using exact density matrices; ii) the MEET in its lowest level of approximation in terms of one- and two-body density matrices gives good results for the spectral function of the (symmetric) Hubbard model with more sites using approximate density matrices. Moreover, for these (symmetric) model systems the ΛR/A matrices of the EKT are diagonal in the basis of natural orbitals, therefore there is not another better option. For the asymmetric Hubbard dimer instead, and in general for realistic systems, the basis of natural orbitals does not diagonalize the ΛR/A matrices, therefore the set which diagonalizes these matrices can be a better option for the MEET. We notice that this choice of the optimal basis set can be generally applied to other methods which express G as a continued fraction, such as the Lanczos method (Balzer et al., 2011), in order to have more accurate results at a given order of truncation of the series. For example in Figure 1 we report the spectral function of the Hubbard dimer governed by the Hamiltonian in Eq. 13 for two different values of |U1 − U2|. The (D)EKT results are obtained using exact density matrices. The results show that the basis which diagonalizes the ΛR/A matrices is a much better choice than the basis of natural orbitals the more the difference |U1 − U2| is large. We also observe that the removal part is less affected by the diagonal approximation than the addition part, and we observe this trend also in more complex systems. The diagonal approximation has been addressed in literature also for realistic systems. (Piris et al., 2013; Kent et al., 1998) In particular in bulk silicon QMC results show that the DEKT slightly overestimates the EKT band gaps. Below we will address this system in more details.
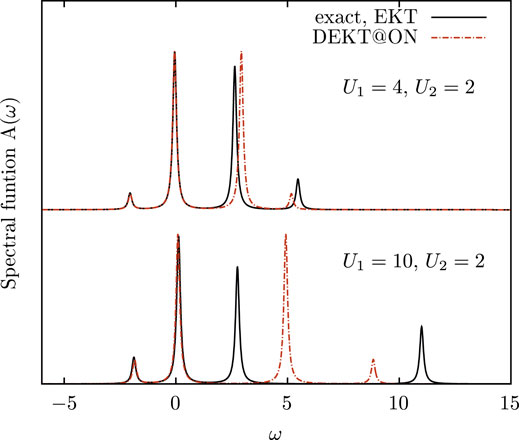
FIGURE 1. Spectral function of the Hubbard dimer with two different on-site interactions U1 and U2 for site 1 and site 2, respectively: exact solution (black curves) vs. EKT (black curves, EKT is exact in this case, see text) and DEKT/MEET in the basis of natural orbitals (red curves).
3.2 Realistic Systems: The Example of Bulk Si
As an example of realistic systems we use bulk silicon, for which results using the EKT within QMC are reported in Ref. (Kent et al., 1998). The diagonal approximation to the EKT within QMC works very well for the valence states and slightly less well for the conduction states, with a band gap at the Γ point of 4.4 eV vs. 3.8 eV from the full EKT compared to 3.4 eV in experiment. (Kent et al., 1998) We note that this discrepency is largely due to the energy of the conduction band at Γ. However, bulk silicon is a relatively weakly correlated system, with hence a predominant quasiparticle-like spectral function for which the EKT is a good approximation. Larger overestimation of the band gap can be expected for strongly correlated systems. This can be understood from the interpretation of the EKT energies as first moments of the one-body Green’s function. However, an important point to stress is that even for these systems, which are a challenge for state-of-the-art ab initio methods, such as GW, the EKT would open a gap, in accordance with experiment.
As pointed out in Ref. (Kent et al., 1998) the choice of the trial wave function for QMC calculations is of critical importance. Indeed, as a result of the fixed-node approximation, QMC calculations of the matrix elements of the density matrix and operators VR,A (see EKT Equations 8, 11) are expected to critically depend on the nodal structure of the trial wave function employed. For the weakly correlated bulk silicon the accurate QMC value of 3.8 eV reported above has been obtained by (Kent et al., 1998) using a standard Slater-Jastrow trial wave function whose nodes are those of a single determinant consisting of LDA orbitals. For more strongly correlated systems the wave function acquires a significant multi-determinant character and getting physically meaningful nodes becomes much more difficult. It is thus useful to use the EKT within alternative approaches.
In our previous works (Di Sabatino et al., 2016, 2019, 2021) we used reduced-density matrix functional theory (RDMFT) (Gilbert, 1975) to find approximations to the one- and two-body density matrices which are needed in the MEET equations. More specifically the two-body density matrix in the MEET equations is approximated using the Power functional, which is given by
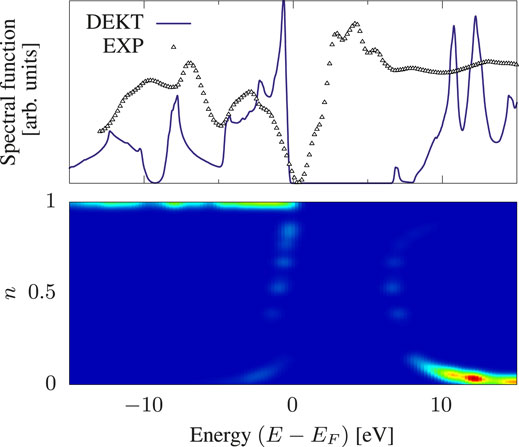
FIGURE 2. Spectral function of bulk Si within DEKT (violet solid line). The experimental photoemission spectrum (small triangles) is taken from Ref. (Chelikowsky et al., 1989). The color map illustrates the occupation numbers ni that play a role into the spectrum for the reported energy range.
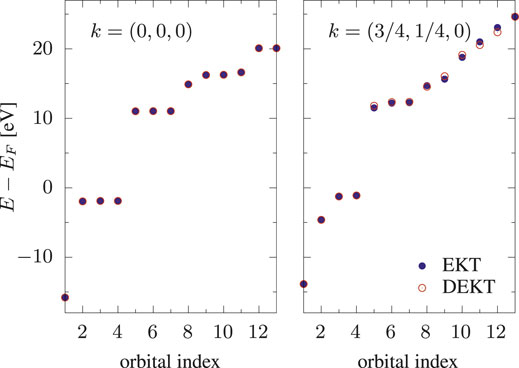
FIGURE 3. Removal/addition energies of bulk Si: EKT (filled circles) vs DEKT (empty circles) for two k-points, k = (0, 0, 0) and k = (3/4, 1/4, 0).
4 Conclusions and Perspectives
We linked our recently derived Many-Body Effective Energy Theory (MEET) for the calculation of photoemission spectra to the Extended Koopman’s Theorem (EKT). Within the lowest level of approximation in terms of one- and two-body density matrices, the MEET equations correspond to the so-called diagonal approximation to the EKT (DEKT) equations. This allowed us to readily extend the EKT to the calculation of an approximate spectral function as well as to give an alternative interpretation of the EKT in terms of moments of the one-body Green’s function. Using the test case of the Hubbard dimer with two different on-site interactions U1 and U2 for site 1 and site 2 we showed the effect of the basis set on the MEET (removal and addition) energies: in particular HOMO-LUMO gap in the basis sets which solve the EKT secular equations (one basis set for the valence part and one for the conduction part) is smaller than the HOMO-LUMO gap obtained using the natural orbital basis set. These results are in line with the EKT results reported in literature for bulk Si using QMC. We have implemented the EKT within reduced-density matrix functional theory (RDMFT), which offers a convenient computationally affordable framework to treat extended systems. However one has to rely on approximate one- and two-body density matrices. We showed that using the currently available approximations the DEKT band gap of Si largely deviates from the DEKT value obtained using QMC (12.9 eV vs 4.4 eV at the Γ point) and, moreover, there is no effect of the basis set (EKT vs DEKT) on the DEKT energies, contrary to what is observed within QMC, where, although small, there is a significant difference. These results on bulk Si indicate that, although the EKT/DEKT are expected to overestimate the band gap (even using very accurate density matrices), this overestimation can be much amplified by commonly used approximations in RDMFT. This also explains the huge overestimation of the band gap obtained by the MEET within RDMFT in strongly correlated systems such as paramagnetic NiO. We are currently working on improving approximations to correlation in RDMFT by introducing some form of screening (for example the screening due to electron-hole excitations as in GW), which is of particular importance in solids.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This study has been supported through the EUR grant NanoX ANR-17-EURE-0009 in the framework of the “Programme des Investissements d’Avenir” and by ANR (project ANR-18-CE30-0025 and ANR-19-CE30-0011).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
1Since in the basis of natural orbitals the SR (SA) matrix is a diagonal matrix with the natural occupation numbers ni (1-ni) as elements, the invertibility of this matrix is strictly related to the non-existence of so-called pinned states, i.e. states with occupation numbers equal to 1 or 0. This is an important question that has several consequences (e.g., Giesbertz and van Leeuwen, 2013; Baldsiefen et al., 2015). Here we assume that SR (SA) is invertible in a restricted space (of natural orbitals) in which the corresponding KS orbitals are occupied (unoccupied). This is a reasonable assumption.
2Note that the standard EKT equations in RDMFT (Morrell et al., 1975) present a prefactor
References
Baldsiefen, T., Cangi, A., and Gross, E. K. U. (2015). Reduced-density-matrix-functional Theory at Finite Temperature: Theoretical Foundations. Phys. Rev. A. 92, 052514. doi:10.1103/PhysRevA.92.052514
Balzer, M., Gdaniec, N., and Potthoff, M. (2011). Krylov-space Approach to the Equilibrium and Nonequilibrium Single-Particle Green's Function. J. Phys. Condens. Matter 24, 035603. doi:10.1088/0953-8984/24/3/035603
Berger, J. A., Romaniello, P., Tandetzky, F., Mendoza, B. S., Brouder, C., and Reining, L. (2014). Solution to the many-body Problem in One point. New J. Phys. 16, 113025. doi:10.1088/1367-2630/16/11/113025
Bozkaya, U. (2013). The Extended Koopmans' Theorem for Orbital-Optimized Methods: Accurate Computation of Ionization Potentials. J. Chem. Phys. 139, 154105. doi:10.1063/1.4825041
Bozkaya, U., and Ünal, A. (2018). State-of-the-Art Computations of Vertical Ionization Potentials with the Extended Koopmans' Theorem Integrated with the CCSD(T) Method. J. Phys. Chem. A. 122, 4375–4380. doi:10.1021/acs.jpca.8b01851
Chelikowsky, J. R., Wagener, T. J., Weaver, J. H., and Jin, A. (1989). Valence-and Conduction-Band Densities of States for Tetrahedral Semiconductors: Theory and Experiment. Phys. Rev. 40, 9644–9651. doi:10.1103/PhysRevB.40.9644
Cioslowski, J., Piskorz, P., and Liu, G. (1997). Ionization Potentials and Electron Affinities from the Extended Koopmans' Theorem Applied to Energy-Derivative Density Matrices: The EKTMPn and EKTQCISD Methods. J. Chem. Phys. 107, 6804–6811. doi:10.1063/1.474921
Dahlen, N. E., and van Leeuwen, R. (2005). Self-consistent Solution of the Dyson Equation for Atoms and Molecules within a Conserving Approximation. J. Chem. Phys. 122, 164102. doi:10.1063/1.1884965
Di Sabatino, S., Berger, J. A., Reining, L., and Romaniello, P. (2016). Photoemission Spectra from Reduced Density Matrices: The Band gap in Strongly Correlated Systems. Phys. Rev. B 94, 155141. doi:10.1103/PhysRevB.94.155141
Di Sabatino, S., Berger, J. A., Reining, L., and Romaniello, P. (2015). Reduced Density-Matrix Functional Theory: Correlation and Spectroscopy. J. Chem. Phys. 143, 024108. doi:10.1063/1.4926327
Di Sabatino, S., Berger, J. A., and Romaniello, P. (2019). Many-body Effective Energy Theory: Photoemission at strong Correlation. J. Chem. Theor. Comput. 15, 5080–5086. doi:10.1021/acs.jctc.9b00427
Di Sabatino, S., Koskelo, J., Berger, J. A., and Romaniello, P. (2021). Photoemission Spectrum in Paramagnetic Feo under Pressure: Towards an Ab Initio Description. Phys. Rev. Res. 3, 013172. doi:10.1103/PhysRevResearch.3.013172
Elk (2004). Elk. Available at: http://elk.sourceforge.net.
Giesbertz, K. J. H., and van Leeuwen, R. (2013). Natural Occupation Numbers: When Do They Vanish? J. Chem. Phys. 139, 104109. doi:10.1063/1.4820419
Gilbert, T. L. (1975). Hohenberg-kohn Theorem for Nonlocal External Potentials. Phys. Rev. B 12, 2111–2120. doi:10.1103/PhysRevB.12.2111
Guzzo, M., Lani, G., Sottile, F., Romaniello, P., Gatti, M., Kas, J. J., et al. (2011). Valence Electron Photoemission Spectrum of Semiconductors:Ab InitioDescription of Multiple Satellites. Phys. Rev. Lett. 107, 166401. doi:10.1103/physrevlett.107.166401
Hedin, L. (1965). New Method for Calculating the One-Particle Green’s Function with Application to the Electron-Gas Problem. Phys. Rev. 139, A796–A823. doi:10.1103/physrev.139.a796
Katriel, J., and Davidson, E. R. (1980). Asymptotic Behavior of Atomic and Molecular Wave Functions. Proc. Natl. Acad. Sci. 77, 4403–4406. doi:10.1073/pnas.77.8.4403
Kent, P. R. C., Hood, R. Q., Towler, M. D., Needs, R. J., and Rajagopal, G. (1998). Quantum Monte Carlo Calculations of the One-Body Density Matrix and Excitation Energies of Silicon. Phys. Rev. B 57, 15293–15302. doi:10.1103/PhysRevB.57.15293
Kuneš, J., Anisimov, V. I., Skornyakov, S. L., Lukoyanov, A. V., and Vollhardt, D. (2007). Nio: Correlated Band Structure of a Charge-Transfer Insulator. Phys. Rev. Lett. 99, 156404. doi:10.1103/PhysRevLett.99.156404
Lani, G., Romaniello, P., and Reining, L. (2012). Approximations for many-body Green's Functions: Insights from the Fundamental Equations. New J. Phys. 14, 013056. doi:10.1088/1367-2630/14/1/013056
Lee, J., Malone, F. D., Morales, M. A., and Reichman, D. R. (2021). Spectral Functions from Auxiliary-Field Quantum Monte Carlo without Analytic Continuation: The Extended Koopmans' Theorem Approach. J. Chem. Theor. Comput. 17, 3372–3387. PMID: 33983735. doi:10.1021/acs.jctc.1c00100
Leiva, P., and Piris, M. (2005). Assessment of a New Approach for the Two-Electron Cumulant in Natural-Orbital-Functional Theory. J. Chem. Phys. 123, 214102. doi:10.1063/1.2135289
Lischner, J., Vigil-Fowler, D., and Louie, S. G. (2013). Physical Origin of Satellites in Photoemission of Doped Graphene: AnAb InitioGWPlus Cumulant Study. Phys. Rev. Lett. 110, 146801. doi:10.1103/PhysRevLett.110.146801
Morrell, M. M., Parr, R. G., and Levy, M. (1975). Calculation of Ionization Potentials from Density Matrices and Natural Functions, and the Long-Range Behavior of Natural Orbitals and Electron Density. J. Chem. Phys. 62, 549–554. doi:10.1063/1.430509
Pavlyukh, Y. (2019). The Ubiquitous Extended Koopmans' Theorem. Phys. Status Solidi B 256, 1800591. doi:10.1002/pssb.201800591
Pernal, K., and Cioslowski, J. (2005). Ionization Potentials from the Extended Koopmans' Theorem Applied to Density Matrix Functional Theory. Chem. Phys. Lett. 412, 71–75. doi:10.1016/j.cplett.2005.06.103
Piris, M., Matxain, J. M., Lopez, X., and Ugalde, J. M. (2012). The Extended Koopmans’ Theorem: Vertical Ionization Potentials from Natural Orbital Functional Theory. J. Chem. Phys. 136, 174116. doi:10.1063/1.4709769
Piris, M., Matxain, J. M., Lopez, X., and Ugalde, J. M. (2013). The One-Electron Picture in the Piris Natural Orbital Functional 5 (Pnof5). Theor. Chem. Acc. 132, 1298. doi:10.1007/s00214-012-1298-4
Romaniello, P., Bechstedt, F., and Reining, L. (2012). Beyond theGWapproximation: Combining Correlation Channels. Phys. Rev. B 85, 155131. doi:10.1103/physrevb.85.155131
Romaniello, P., Guyot, S., and Reining, L. (2009). The Self-Energy beyond Gw: Local and Nonlocal Vertex Corrections. J. Chem. Phys. 131, 154111. doi:10.1063/1.3249965
Sharma, S., Dewhurst, J. K., Lathiotakis, N. N., and Gross, E. K. U. (2008). Reduced Density Matrix Functional for many-electron Systems. Phys. Rev. B 78, 201103. doi:10.1103/PhysRevB.78.201103
Sharma, S., Dewhurst, J. K., Shallcross, S., and Gross, E. K. U. (2013). Spectral Density and Metal-Insulator Phase Transition in mott Insulators within Reduced Density Matrix Functional Theory. Phys. Rev. Lett. 110, 116403. doi:10.1103/physrevlett.110.116403
Shishkin, M., Marsman, M., and Kresse, G. (2007). Accurate Quasiparticle Spectra from Self-ConsistentGWCalculations with Vertex Corrections. Phys. Rev. Lett. 99, 246403. doi:10.1103/PhysRevLett.99.246403
Smith, D. W., and Day, O. W. (1975). Extension of Koopmans' Theorem. I. Derivation. J. Chem. Phys. 62, 113–114. doi:10.1063/1.430253
Springer, M., Aryasetiawan, F., and Karlsson, K. (1998). First-PrinciplesT-Matrix Theory with Application to the 6 eV Satellite in Ni. Phys. Rev. Lett. 80, 2389–2392. doi:10.1103/PhysRevLett.80.2389
Stan, A., Dahlen, N. E., and Leeuwen, R. v. (2006). Fully Self-Consistent GW Calculations for Atoms and Molecules. Europhys. Lett. 76, 298–304. doi:10.1209/epl/i2006-10266-6
Stan, A., Dahlen, N. E., and van Leeuwen, R. (2009). Levels of Self-Consistency in the Gw Approximation. J. Chem. Phys. 130, 114105. doi:10.1063/1.3089567
Stefanucci, G., Pavlyukh, Y., Uimonen, A.-M., and van Leeuwen, R. (2014). Diagrammatic Expansion for Positive Spectral Functions beyondGW: Application to Vertex Corrections in the Electron Gas. Phys. Rev. B 90, 115134. doi:10.1103/PhysRevB.90.115134
Sundholm, D., and Olsen, J. (1993). The Exactness of the Extended Koopmans' Theorem: A Numerical Study. J. Chem. Phys. 98, 3999–4002. doi:10.1063/1.464028
Zheng, H. (2016). First Principles Quantum Monte Carlo Study of Correlated Electronic Systems. University of Illinois at Urbana-Champaign. Ph.D. thesis.
Keywords: extended Koopman’s theorem, strong correlation, photoemission, one-body Green’s function, RDMFT, QMC
Citation: Di Sabatino S, Koskelo J, Prodhon J, Berger JA, Caffarel M and Romaniello P (2021) Photoemission Spectra from the Extended Koopman’s Theorem, Revisited. Front. Chem. 9:746735. doi: 10.3389/fchem.2021.746735
Received: 24 July 2021; Accepted: 15 September 2021;
Published: 08 October 2021.
Edited by:
Dorothea Golze, Aalto University, FinlandReviewed by:
Thibaud Etienne, Universite de Montpellier, FranceKatarzyna Pernal, Lodz University of Technology, Poland
Copyright © 2021 Di Sabatino, Koskelo, Prodhon, Berger, Caffarel and Romaniello. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: S. Di Sabatino, ZGlzYWJhdGlub0BpcnNhbWMudXBzLXRsc2UuZnI=; P. Romaniello, cm9tYW5pZWxsb0BpcnNhbWMudXBzLXRsc2UuZnI=