 Mariela M. Nolasco1*
Mariela M. Nolasco1* Leonor C. Rodrigues1
Leonor C. Rodrigues1 Catarina F. Araújo1
Catarina F. Araújo1 Mariana M. Coimbra1
Mariana M. Coimbra1 Paulo Ribeiro-Claro1
Paulo Ribeiro-Claro1 Pedro D. Vaz2
Pedro D. Vaz2 Svemir Rudić3
Svemir Rudić3 Armando J. D. Silvestre1
Armando J. D. Silvestre1 Chaima Bouyahya1,4Mustapha Majdoub4
Chaima Bouyahya1,4Mustapha Majdoub4 Andreia F. Sousa1,5*
Andreia F. Sousa1,5*- 1CICECO, Departamento de Química, Universidade de Aveiro, Aveiro, Portugal
- 2Champalimaud Foundation, Champalimaud Centre for the Unknown, Lisboa, Portugal
- 3ISIS Neutron & Muon Source, STFC Rutherford Appleton Laboratory, Didcot, United Kingdom
- 4Laboratoire des Interfaces et Matériaux Avancés, Université de Monastir, Monastir, Tunisia
- 5Centre for Mechanical Engineering, Materials and Processes, Department of Chemical Engineering, University of Coimbra, Coimbra, Portugal
This work explores the conformational preferences and the structure-property correlations of poly(butylene 2,5-furandicarboxylate) (PBF), a longer chain analogue of the most well-known biobased polyester from the furan family, poly(ethylene 2,5-furandicarboxylate) (PEF). A thorough computational spectroscopic study–including infrared, Raman and inelastic neutron scattering spectroscopy, combined with discrete and periodic density functional theory calculations–allowed the identification of dominant structural motifs in the amorphous and crystalline regions. Discrete calculations and vibrational spectroscopy of semi-crystalline and amorphous samples strongly support the predominance of gauche, trans, gauche conformations of the butylene glycol fragment in both the crystalline and amorphous domains. In what concerns the furandicarboxylate fragment, amorphous domains are dominated by syn,syn conformations, while in the crystalline domains the anti,anti forms prevail. A possible crystalline structure–built from these conformational preferences and including a network of C-H···O hydrogen bond contacts—was optimized using periodic density functional theory. This proposed crystal structure avoids the unrealistic structural features of the previously proposed X-ray structure, provides an excellent description of the inelastic neutron scattering spectrum of the semi-crystalline form, and allows the correlation between microscopic structure and macroscopic properties of the polymer.
Introduction
Progress in renewable based polymers has accelerated since they offer the possibility to reduce the environmental impact of plastics, paving the way to the UN Sustainable Development Goals (United Nations, 2020) and towards a circular economy as set forth by the European Commission (European Commission, 2015). Despite this, biobased polymers still represent today a minor fraction of all commercial polymers produced yearly (ca. 864 tonnes in 2021), accounting for less than 1% of the global production (European Bioplastics, 2021). A complex set of factors underlie this fact, in which economic constraints (high production costs) and their typical inferior mechanical and thermal properties have limited its market production and penetration. Nevertheless, the extensive research carried out by both academia and industry have brought new prospects with the arising of the 2,5-furandicarboxylic acid (FDCA)–a key building-block for polymers development which can impart high performance to the polymers thereof (Werpy and Petersen, 2004; Bozell and Petersen, 2010; Loos et al., 2020; de Jong et al., 2022).
Poly(ethylene 2,5-furandicarboxylate) (PEF) and poly(butylene 2,5-furandicarboxylate) (PBF)—Figures 1 – are among the most interesting FDCA-based polymers because, besides having a renewable origin and a more favorable sustainable performance, they have improved properties arising from their chemical structure. The in-depth characterization carried out so far focused mostly on PEF, due to its potential to replace the fossil-based poly(ethylene terephthalate) (PET) on packaging, e.g. plastic bottles (Fei et al., 2020; de Jong et al., 2022). Our group reported a vibrational spectroscopy and computational modeling study on this polymer, shedding light on important structure-property correlations (Araujo C. F. et al., 2018). A clear picture emerged, then, on how the most favorable conformational preferences both in the amorphous and crystalline regions, as well as a and ß polymorphs, are built in. Results show that, in the amorphous domains, PEF chains prefer a winding structure based on energetically favorable gauche conformation of the ethylene-glycol fragment. Yet, in the crystalline domains, polymeric chains adopt an energetically unfavorable extended all-trans geometry, which is stabilized by a network of C-H···O bonds linking adjacent chains. Interestingly the INS spectrum, revealing distinct low-frequency vibrational profiles for PEF and PET, confirmed the furanic “ring flipping” hindrance and stiffer polymeric chains, typically associated with enhanced O2, CO2, and H2O gas barrier properties (Burgess et al., 2014a; 2014c; 2014b, 2015), as well as a higher Young’s modulus. To the best of our knowledge, a similar picture has not been drawn for PBF, besides an attempt study made by (Zhu et al., 2013). Indeed, most studies focus instead on its synthesis (Carlos Morales-Huerta et al., 2016), thermal and crystallinity properties (Ma et al., 2012; Sousa et al., 2018), mechanical behavior (Zhu et al., 2013; Robles-Hernandez et al., 2020), molecular dynamics (Soccio et al., 2017; Papamokos et al., 2019; Guidotti et al., 2020; Klonos et al., 2020; Bianchi et al., 2021; Fosse et al., 2022; Poulopoulou et al., 2022) or in some of these aspects (Matos et al., 2018).
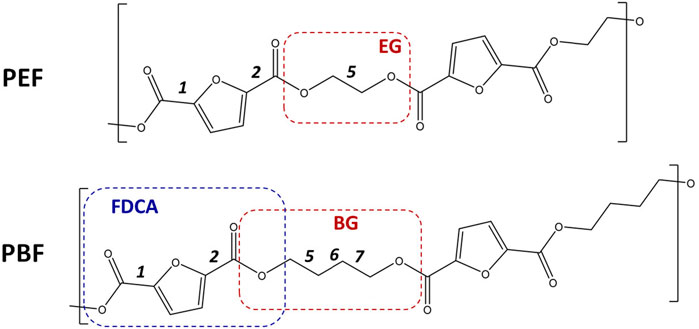
FIGURE 1. Skeletal formulas of the repeating units for poly(ethylene 2,5-furandicarboxylate) ((PEF, top) and poly(butylene 2,5-furandicarboxylate) (PBF, bottom). Dashes delimit the 1,2- and 1,4-alkyl glycol moieties—EG, BG—and the first 2,5-furandicarboxylic acid moiety–FDCA. The labels 1, 2, five to seven identify the single bonds for which rotational freedom exists. For 1 and 2, the orientation of C=O bonds relative to ring C=C bonds are described as syn or anti. For five EG and 5,6,7 BG bonds, conformations are either trans or gauche. In this way, the PBF structure shown here is described as anti,anti-trans,trans, trans, or aa-ttt for short.
Recent publications demonstrate the high-quality simulations of the vibrational spectra of crystals using periodic boundary conditions, usually described as periodic density functional theory (DFT) calculations. Inelastic neutron scattering (INS) intensities, which are a straightforward result of the eigenvectors (atomic displacements) determined for the vibrational normal modes, are particularly well-predicted from periodic DFT, allowing the confident assignment of molecular and lattice modes in the crystal. In the case of amorphous polymer samples—or in absence of a reliable crystal structure for the crystalline domains in a semi-crystalline sample—discrete (or molecular) calculations have been used with success (Araujo C. et al., 2018; Vilela et al., 2020; Druzbicki et al., 2021; Stroupe et al., 2022). In this case, the possible polymer chain structures are simulated by resorting to short polymer fragments, typically triads. Then, the experimental spectra can be matched to a combination of proposed structures whose spectral contributions are determined from calculations. Due to the high reliability of calculations, structures that fail to adequately contribute to the experimental spectra can be excluded. This approach applies to infrared (IR) and Raman spectroscopy with some caveats, since selection rules could provide different weightings for different geometries, resulting in a non-quantitative intensity/population relationship. However, the approach is particularly suitable for INS spectroscopy, because the INS spectrum is a quantitative measurement of the vibrational density of states—i.e., it is a direct sum of contributions from the different populations—and so the INS scattering profile can be decomposed into a linear combination of contributions from dominant structural motifs (see, e.g. (Harrelson et al., 2017)). Despite the potential of the combination of these approaches they have not yet been used to describe PBF.
Further, a comparison between PEF and PBF conformational preferences and related properties was missing, apart from the obvious gain in flexibility due to the extra methylene groups of PBF. All in all, what difference does the longer alkyl chain make?
In this vein, in this work, the computational spectroscopy approach was extended to biobased PBF and a comparison with PEF is provided. By combining vibrational spectroscopy techniques—including infrared absorption (IR), Raman scattering and inelastic neutron scattering (INS)—and quantum mechanical calculations at the density functional theory (DFT) level, new insights on the structure and properties of this polymer, compared to PEF were herein achieved.
The syn/anti terms refer to the orientation of C=O bonds relative to the nearest ring C=C bond. In the previous work with PEF (Araujo C. F. et al., 2018), the terms syn/anti were defined relative to the position of furanic oxygen atom; using the more adequate definition herein adopted simply reverses the anti/syn meaning.
Experimental details
Synthesis of PBF
Dimethyl 2,5-furandicarboxylate (DMFDC) was synthesized following a previously reported procedure (Matos et al., 2017). Briefly, DMFDC was prepared by refluxing FDCA (5 g, 32.0 mmol) with an excess of methanol, under acidic conditions (HCl) at 80°C, for 15 h. Then, the reaction mixture was cooled down to room temperature and the resulting insoluble product was filtered off in ca. 70% yield.
PBF was synthesized by an adapted polytransesterification reaction procedure previously reported (Matos et al., 2018). In a first stage, DMFDC (8 g, 43.5 mmol), 1,4-butanediol (6 g, 66.5 mmol) and titanium(IV) butoxide (20 mg, 0.1 wt%) were allowed to react under a nitrogen atmosphere from 190°C up to 210°C, for 7–8 h. During the second stage, a high vacuum of 10–3 mbar was gradually applied, and the reaction was carried out at 210°C for 3 h. Then, the reaction was stopped and cooled down to room temperature. The polymer was purified by dissolving in a chloroform–trifluoroacetic acid mixture (6:1) and poured in an excess of methanol, filtered and dried in a vacuum oven at 40°C. The ensuing PBF was isolated in ca. 70% yield. The amorphous and semi-crystalline samples of PBF were obtained as described for PEF (Araujo C. F. et al., 2018) and characterized from powder X-ray diffraction (XRD). The XRD pattern of the amorphous sample did not reveal the presence of a crystalline fraction. The crystallinity of the semi-crystalline sample, estimated from its XRD pattern, was ca. 60%.
X-ray diffraction
PBF samples were analyzed using X-ray diffraction studies. XRD powder patterns were collected at room temperature on a Panalytical Empyrean instrument operating with CuKα radiation at 40 kV and 50 mA. Samples were scanned in the 2θ range of 5°–70° with a step size of 0.026° and step time of 67 s.
Vibrational spectra
PBF samples were studied using optical techniques (IR, Raman) and inelastic neutron scattering (INS). FTIR-ATR spectra were measured at room temperature using a FT Bruker IFS 55 spectrometer with a Golden Gate ATR accessory with a resolution of 2 cm−1. Raman spectra were collected at room temperature on a Bruker MultiRAM FT-Raman instrument with an Nd:YAG laser and using a resolution of 2 cm−1. The INS spectra of PBF samples were collected in the scope of project RB2000214 (Nolasco et al., 2021), using the TOSCA instrument (Parker et al., 2014; Pinna et al., 2018) at the ISIS Neutron and Muon Source of STFC’s Rutherford Appleton Laboratory (Chilton, United Kingdom) (ISIS, 2022). The samples, weighing 0.5–1 g, were placed inside a flat thin-walled aluminum can, which was then mounted perpendicular to the incident beam using a regular TOSCA centered stick. Spectra were collected below 20 K and samples were “shock-frozen” by quenching in liquid nitrogen before placement in the beam path, in order to preserve the room-temperature morphology of possible amorphous and crystalline regions. The contribution of aluminum can to the final INS spectra was found to be not negligible and has been removed by subtraction.
Quantum chemical calculations
Discrete (molecular) calculations: Geometry optimizations and vibrational frequency calculations of PBF oligomers (triads, BG3FDCA3) were computed using the Gaussian 09 software, using the B3LYP density functional with the 6–311G (d,p) basis set. This method was found to provide a reliable description of the conformational preferences of molecular models and allows a direct comparison with previous results for PEF (Araujo C. F. et al., 2018; Papamokos et al., 2019). The initial structures were based on the well-known minima for these systems (see., e.g. (Papamokos et al., 2019)) described in Figure 1. Optimizations were performed without constraints and all the optimized structures were found to be real minima, with no imaginary frequencies. For calculated Raman and infrared spectra, vibrational frequencies were scaled by a factor of 0.967 (NIST Computational Chemistry Comparison and Benchmark Database, 2022). The inelastic neutron scattering simulated intensities were estimated from the calculated eigenvectors using the AbINS software (Dymkowski et al., 2018), a part of the Mantid package (Arnold et al., 2014). The energy values mentioned throughout the text refer to the electronic energy without zero-point correction.
Periodic DFT calculations: calculations were performed using the plane wave pseudopotential method as implemented in CASTEP 8.0 code (Clark et al., 2005; Refson et al., 2006). All calculations were done using the Perdew−Burke−Ernzerhof (PBE) functional based on the generalized gradient gauge (GGA) approximation (Perdew et al., 1996) supplemented with the semi-empirical dispersion correction of Tkatchenko and Scheffler (Tkatchenko and Scheffler, 2009). The plane-wave cutoff energy was set at 830 eV. Brillouin zone sampling of electronic states was performed on 2×4×5 Monkhorst−Pack grid. The initial structures were built from selected triads, considering a triclinic crystal with P1 symmetry, or obtained from (Zhu et al., 2013). Geometry optimizations were carried out with no constraints (i.e., both cell parameters and internal coordinates were relaxed) and accuracy of the optimization requested residual forces to fall below 0.005 eV A−1. Phonon frequencies were obtained by diagonalization of dynamical matrices calculated using density-functional perturbation theory (Milman et al., 2009). The calculated atomic displacements in each mode that are part of the CASTEP output enable visualization of the atomic motions and support the assignment of vibrational modes. The simulated inelastic neutron scattering intensities were predicted from the calculated eigenvectors using AbINS, and values were not scaled.
Results and discussion
Conformational landscape (from discrete molecular modelling)
The existence of a crystalline model structure is a pre-requisite to periodic DFT calculations. In the case of PBF, Zhu et al. proposed a crystal structure from fiber X-ray diffraction scans on a stretched PBF film (Zhu et al., 2013). However, the proposed structure presents several unrealistic structural features that cast serious doubts on its reliability and render it unavailing for structural characterization of the system. Among other geometrical issues, the BG alkyl chain presents C–C–C angles up to 129°, far from acceptable values for an alkyl chain. In the absence of doubtless crystallographic data, discrete (or molecular) calculations on polymer fragments offer a reliable alternative to assess conformational preferences of the polymer chain.
In this way, the conformational landscape of PBF was explored through DFT calculations on BG3FDCA3 triads. Due to the large number of possible combinations for this oligomer, calculations were performed for uniform conformations along the BG3FDCA3 chain. For instance, the lowest energy conformation for BG3FDCA3 was found to be ss-gtg, which means that all three FDCA fragments have syn-syn orientation and all three BG chains have gauche-trans-gauche conformations (see Figure 1). This “uniform chain” approach is the model expected to prompt easiest close packing of chains and, thus, to better describe the crystalline domains in the polymer. Of course, a variety of non-uniform sequences are predictable for the amorphous domains.
Figure 2 compares the structure and energies of the lower energy triads found for PEF and PBF. These triads present uniform conformations, resulting from the internal rotation around FDCA bonds labelled 1,2 and EG and BG bonds labelled 5 and 5,6,7 in Figure 1, respectively.
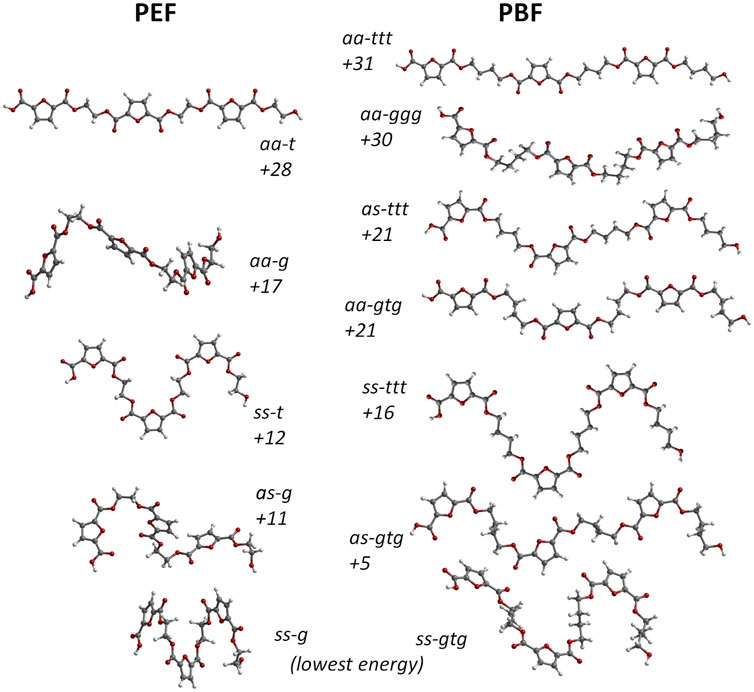
FIGURE 2. Comparison between the molecular structure of PBF triads BG3FDCA3 (right) and PEF triads EG3FDCA3 (left). The numbers indicate their relative stability expressed in terms of electronic energy, at the B3LYP/6311G (d,p) level, in kJ/mol. The aa, as and ss labels indicate the anti- and syn- orientation of the carbonyl bonds relative to nearest ring C=C bond in each FDCA fragment; for PBF, ttt and gtg indicate trans- and gauche-conformations along the BG skeleton. The as structures with alternate sequence as-sa-as for the three FDCA fragments present marginally lower energy than structures with as-as-as sequence and are used in this figure. Only the three ttt forms present a fully planar skeleton.
As in the case of PEF, in the FDCA fragment the syn orientation is clearly preferred over the anti orientation, while the alkoxy CC-CO fragments prefer the gauche conformation relative to the trans conformation. The longer alkyl chain in PBF brings an additional CC-CC torsion angle with a preference for the trans conformation. Hence, the lowest energy conformation in PBF is ss-gtg and energy raises ca. 30 kJ/mol up to the aa-ggg and aa-ttt conformations. The high energy aa-ggg structure results from the optimization of a triad extracted from the defective X-ray structure of Zhu et al., discussed above. Due to the correction of unrealistic geometrical parameters during the geometry optimization, this triad adopts the curved shape evident in Figure 2.
A noticeable feature of Figure 2 is the correlation between PBF and PEF triads and their relative energies. For instance, the lowest energy ss-g PEF form directly correlates with the lowest energy ss-gtg PBF form. In the same way, as-g (PEF) correlates with as-gtg (PBF), but the energy gap to the lowest energy form is substantially lower for PBF: 5 kJ/mol vs 11 kJ/mol. More interesting is the correlation between the “crystal-prone” (non-winding) structures in both polymers: due to the longer alkyl chain of PBF, the aa-t form of PEF correlates with both the aa-ttt and aa-gtg forms of PBF. And while aa-t and aa-ttt present similar energy values relative to the minima (28 and 31 kJ/mol, respectively), the aa-gtg form is ca. 7 kJ/mol below aa-t.
On average, the change from gtg to ttt conformation in a single BG fragment has an energy penalty of ca. 5 kJ/mol. Changes to mixed conformations, such as gtt or tgg, will require a fraction of this value. The energy penalty for a single syn-to-anti change in a single FDCA fragment falls in the range of ca. 2–3.5 kJ/mol. These values turn a large number of conformations accessible for the amorphous domain at room temperature, and, from this point of view (energetic considerations alone) also for crystalline forms. However, it is possible to discriminate the conformational preferences for crystalline and amorphous domains from a computational spectroscopy approach, as described below.
INS spectroscopic patterns
As stated above, since the INS intensities are relatively easy to simulate and predict, it is possible to generate reliable INS spectrum for each triad on Figure 2 and, thus, identify the INS patterns associated with each conformation. The conformations with dominant contribution to the crystalline and amorphous domains are selected from the patterns that best match the experimental spectrum of semi-crystalline and amorphous samples. Figure 3 shows the INS spectra predicted for the seven uniform triads considered, compared with the experimental INS spectra for amorphous and semi-crystalline samples.
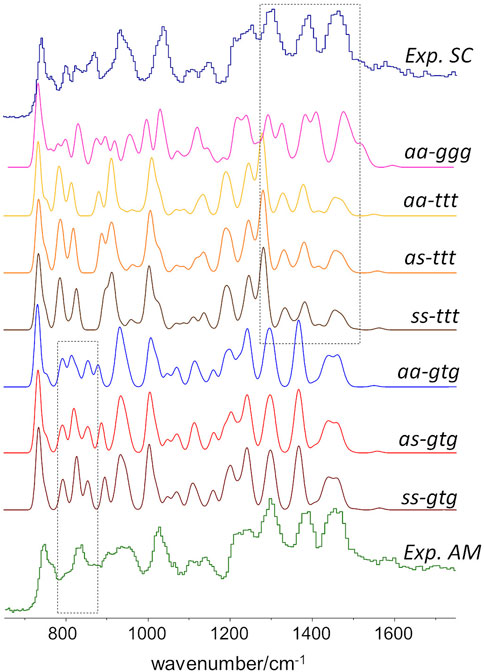
FIGURE 3. Comparison of the INS patterns in the 650–1750 cm−1 region theoretically predicted for the seven triads shown in Figure.2, with the experimental INS patterns of PBF semi-crystalline (top trace) and amorphous (bottom trace) samples. The dashed rectangles highlight two regions of clear pattern definition.
As it can be seen, the triads bearing a BG group with ttt conformation fail to reproduce the intensity pattern in the ca. 1200–1450 cm−1 region (associated with CH2 bending modes). Since the intensity pattern associated with the gtg conformation is observed for both amorphous and semi-crystalline samples, it can be assumed that this conformation dominates both crystalline and amorphous domains. It should be mentioned that the all-trans (ttt) conformation of the alkyl chain was observed for the crystalline domains of PEF and PET, and has been reported for one polymorphic form of PBT (poly(2,5-butylene terephathalate)) (Milani and Galimberti, 2014). However, the herein described results do not support a similar behavior on PBF.
A noticeable difference between the INS spectra of amorphous and semi-crystalline samples occurs in the region of ca. 800–900 cm−1 (which embraces the out-of-plane bending of the furanic C-H bonds and the stretching of the butylene O-CH2 bonds). In this case, the triads combining gtg with syn, syn or syn, anti conformations provide a better description of the INS spectrum of the amorphous sample. The anti,anti-gtg triad (Figure 3, middle blue trace) relates better with the INS spectrum of the semi-crystalline sample, suggesting the predominance of this conformation in the crystalline domains, hence shedding light into the crystalline structure puzzle of PBF.
Further details from IR and Raman spectroscopy
For PEF, it was possible to infer the conformational preferences of the crystalline and amorphous domains from optical spectroscopy, i.e., infrared and Raman spectroscopy (Araujo C. F. et al., 2018). In particular, the trans vs. gauche infrared profiles could be related with similar cases previously described (namely, from PET studies). The anti vs. syn forms of FDCA fragment were identified with support from discrete quantum mechanical frequency calculations of PEF triads.
In the case of PBF, and regarding the conformation of the butylene chain, the comparison with the PBT analogue is the most straightforward. According to Milani et al. (Milani and Galimberti, 2014), there are several infrared markers associated with gauche and trans orientations of the butylene chain in PBT. For instance, the 917 cm−1 band is an unambiguous marker of the PBT polymorph with gtg chain (α polymorph) and the one at 960 cm−1 is a marker of the crystal form possessing chains in all-trans conformation (β polymorph). Other α/β markers in PBT are observed in the 1300–1550 cm−1 region (Milani and Galimberti, 2014). Unfortunately, in the case of PBF, the infrared spectra in these regions are not unambiguous. Nevertheless, the infrared spectra of amorphous and semi-crystalline PBF samples (Figure 4), when compared with spectra of the α/β forms of PBT in the same region, evidences a few features compatible with the prevalence of gtg configuration in both the crystalline and amorphous domains.
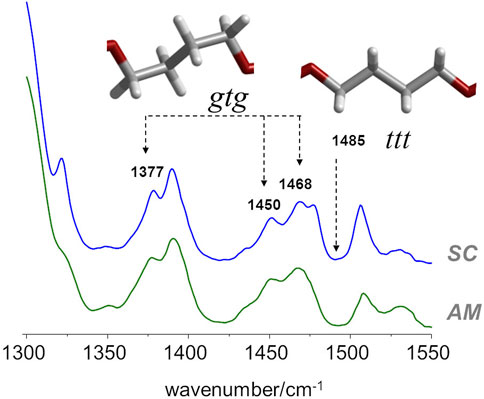
FIGURE 4. FTIR-ATR spectra of semicrystalline (top) and amorphous (bottom) samples of PBF, with identification of some bands sensitive to gauche−trans isomerism.
In particular, both samples present the same general profile of the a polymorph, with bands at 1377, 1450 and 1468 cm−1, and the absence of the ß polymorph marker at 1485 cm−1. It should be mentioned that in the case of PEF, there are large intensity changes from the amorphous to the semi-crystalline samples in this region, signaling the change from gauche to trans conformation in the BG fragment. The absence of such changes in PBF is indirect evidence of the commonness of gtg forms in both amorphous and crystalline domains.
A more definite conclusion can be drawn from the infrared bands associated with syn and anti conformations of the FDCA moiety. For PEF, the frequencies of two vibrational modes - ring out-of-plane deformation and C=C stretching—were found to be sensitive to the syn/anti conformation (Araujo C. F. et al., 2018). In PBF triads, these modes are consistently predicted to fall in the same wavenumbers and follow the same pattern, with the lowest wavenumbers associated with anti,anti conformation and the highest wavenumbers associated with the syn,syn conformation. Figure 5 compares the infrared spectra of amorphous and semi-crystalline samples in the relevant regions. The semi-crystalline sample is richer in the anti-FDCA forms, while the syn-FDCA forms are dominant in the amorphous sample.
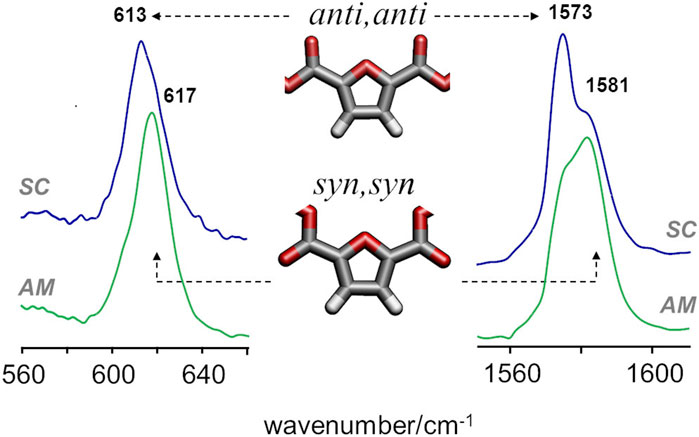
FIGURE 5. FTIR-ATR spectra of semicrystalline (top) and amorphous (bottom) samples of PBF, with identification of the bands sensitive to syn−anti isomerism.
These observations concerning the syn-anti conformations are consistent with the energetic profile described above and the expected changes upon crystalline packing. As observed for PEF, the syn,syn conformation has lower energy (and hence dominates the amorphous regions) but the anti,anti conformation is crucial for the establishment of C-H···O hydrogen bonds that stabilize the crystalline domains.
In the case of PEF, the presence of C-H···O hydrogen bond interactions was inferred from a few spectroscopic changes, namely, those observed for the in-plane deformation (δ) and the stretching (ν) of furanic C−H modes and the stretching of the carbonyl C=O mode (Araujo C. F. et al., 2018). In PBF, these vibrational modes follow the same trends reported for PEF, as shown in Figure 6: from the amorphous to semicrystalline samples, the νsym CHring mode displays a pronounced intensification in infrared intensity, along with a red-shift from 3125 to 3118 cm−1, a behaviour associated with the formation of C−H···O bonds; The blue-shift of the δCHring deformation mode, clearly observed in the Raman spectra, is also a direct consequence of the restricted motion of CHring moieties due to the formation of C−H···O bonds; And the band profile of carbonyl stretching mode shows the competing effects already discussed for PEF, with a broad profile characteristic of the amorphous sample and two sharper maxima emerging with increasing crystallinity.
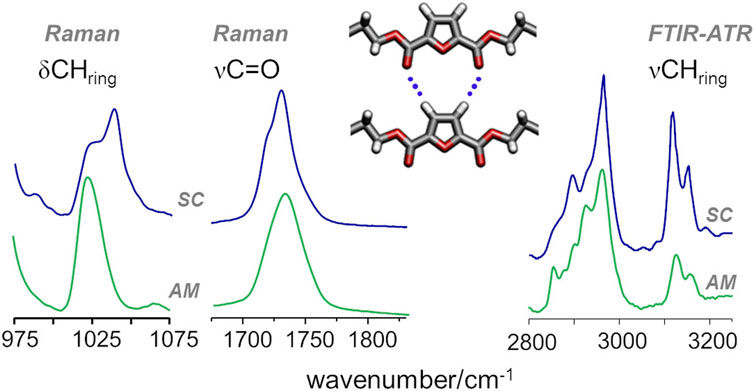
FIGURE 6. Raman and infrared spectra of amorphous (bottom) and semicrystalline (top) samples of PBF, highlighting the CH deformation (δCHring) and stretching (νCHring) and the C=O stretching (νC=O) vibrational modes of FDCA moieties.
Crystal structure: What can be learned from computational spectroscopy?
On the whole, what can be said about the amorphous and crystalline domains of PBF?
The amorphous domains are dominated by gtg-BG and ss-FDCA conformations, with contributions from aa-FDCA. “Mixed” conformations, such as gtt- or ggt-BG and as-FDCA forms are also probable, considering the energy balances within the amorphous polymer chain.
For the crystalline domains, the most reasonable structures steam from “extended” conformations, such as the ones observed for aa-ttt or aa-gtg. “Winding” conformations, such as those observed for the lower energy triads are not prone to crystal packing. In addition, the above mentioned observations strongly support the existence of hydrogen-bonded FDCA fragments (which requires anti, anti orientation of the carbonyl groups) and the prevalence of gtg BG chains. In this way, periodic DFT calculations were performed for a crystalline structure based on the aa-gtg triad. In order to have a comparison set, similar calculations were performed for two other starting structures: 1) a crystal structure built from the most linear triad (aa-ttt), that mimics the planar structure observed for PEF; 2) a crystal using the reported X-ray as a starting point for geometry optimization. In this last case, (and as already mentioned for discrete calculations, above) the required geometry optimization corrected the unrealistic structural parameters, and led to a geometry that can be described as aa-ggg.
Figure 7 compares the experimental INS spectrum of the semi-crystalline sample with the simulated INS spectra for the three crystal models. As it can be seen, the best match is provided by the periodic structure based on the aa-gtg conformation.
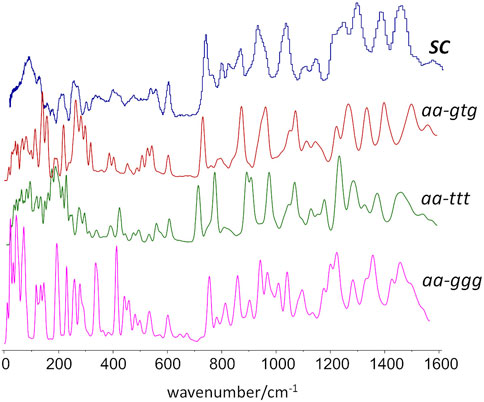
FIGURE 7. Experimental and calculated INS spectra obtained from periodic DFT calculations based on different conformational arrangements of the butylene skeleton. From top to bottom, experimental spectrum, calculated spectra for crystalline models with gtg, ttt, and ggg conformations.
For instance, the “intensity gap” at ca. 700 cm−1 was only correctly predicted for the aa-gtg. Above this region, the aa-ttt structure clearly failed to reproduce the observed intensities for the CH2 rock, twist, wag and scissor modes at ca. 1200–1450 cm−1, as already observed from discrete calculations with the molecular triad models. The aa-ggg spectrum produced a large number of bands in the region of 900–1200 cm−1, whose general profile did not match the experimental spectrum. In what concerns the 300–600 cm−1 interval, the bands of the experimental spectrum are nearly described by a one-to-one match to the aa-gtg spectrum, while both aa-ggg and aa-ttt forms deviate from this profile. Of course, below 200 cm−1, the intensities become increasingly “external” - or intermolecular - modes, and more dependent on crystal packing details. The description of external modes is generally known to be hampered by natural limitations of the periodic calculations (e.g. harmonic oscillator approximation, incomplete description of dispersion interactions, energy cut-offs, and the sum of numerical errors, which accumulate in the low wavenumber modes). Nevertheless, the simulated spectrum for the aa-gtg crystal structure provides a reasonable description of this region, allowing a reliable assignment of the low wavenumber bands, as shown in Figure 8.
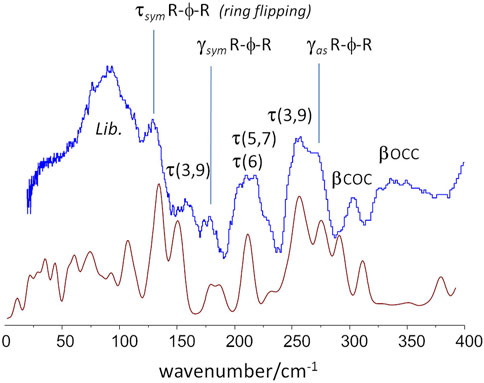
FIGURE 8. Low-wavenumber region of the INS spectra of PBF: experimental (top, blue) and calculated (bottom, red) for the aa-gtg crystal model.
This Figure 8 reveals some dynamical properties of crystalline PBF, which can be related with the longer alkyl chain of the BG fragment. For instance, the ring flipping mode, identified at ca. 160 cm−1 in PEF is observed at ca 130 cm−1 in PBF. This significant reduction of the barrier to rotation of the furanic ring can be ascribed to a larger flexibility of the longer BG chain in the gtg conformation–corroborated from the presence multiple torsional modes in the 160–260 cm−1 range. This larger flexibility is expected to affect the polymer properties, as discussed below.
An important vibrational mode in the comparison between 2,5-PEF and 2,4-PEF was the low-wavenumber mode described as “asymmetric out-of-plane bend of ring substituents” (Nolasco et al., 2020). In 2,4-PEF the mode was found to be sensitive to 2,4-FDCA orientation, becoming a broad band due to the random orientation of furanic rings. In the symmetrical 2,5-analogue the mode occurs as a sharp band at 272 cm−1, very close to the value of 270 cm−1 in PBF, indicating that this mode is not sensitive to the length or conformation of the alkyl chain.
How to derive macroscopic properties from the microscopic structure?
The in-depth understanding (or even prediction) of PBF macroscopic properties—namely, thermal and mechanical properties—using microscopic structure insights is the ultimate goal of the computational spectroscopy approach used in this study, and a highly desired exponent of polymer physics. However, it still remains a challenge, in large part due to the diversity of processes which ramp up into measurable properties, depending on such diverse factors such as the fine structure, chain regularity/symmetry and the chain flexibility, together with the intermolecular forces.
In addition, properties are dependent on factors such as average molecular weight, degree of crystallinity, and thermal history of the polymer sample (see, e.g. (Terzopoulou et al., 2020)).
The melting (Tm) and the glass transition (Tg) temperatures are thermal properties whose enthalpic component can be straightforwardly related to the intermolecular forces within the polymer bulk. While Tm is dependent on the interactions in the crystalline domains, Tg depends on cohesion of the amorphous form. Both transition temperatures are assumed to decrease with the increase of chain mobility and flexibility (Balani et al., 2015).
Crystal packing of PBF chains relies on the same intermolecular forces as PEF, namely the C-H···O contacts forming a planar layer, but the interactions between layers is hampered the gtg “ladder” motif of PBF, absent in PEF. This is in line with the Tm values of 211°C and 170°C reported for PEF and PBF, respectively (Burgess et al., 2014b; Guidotti et al., 2020). In what concerns the Tg, both intermolecular forces and chain stiffness are in the equation. The presence of some C-H···O contacts, even in the amorphous form, can be assumed for both PEF and PBF. However, in PBF the longer alkyl chain reduces the probability of C-H···O contacts in the amorphous domains, thus favouring an increase in free volume and reducing chain stiffness (due to the several gauche/trans conformations available). All these effects contribute to the huge decrease of Tg, from ca. 85°C in PEF to ca. 39°C in PBF (Burgess et al., 2014b; Guidotti et al., 2020).
The increased chain mobility of PBF relative to PEF brought by the presence of the longer alkyl chain spacer is expected to also affect properties such as gas permeation and elasticity. The gas barrier properties of PEF have been related with the restriction of the ring flipping motion (Burgess et al., 2014b; Araujo C. F. et al., 2018). As mentioned above, the ring flipping mode, identified at ca. 160 cm−1 in PEF is observed at ca. 130 cm−1 in PBF, signalling a significant reduction of the barrier to ring rotation. Hence, larger gas permeability can be predicted for PBF compared to PEF, a prediction in agreement with the recently reported values (Guidotti et al., 2020; Zhao et al., 2021).
A relevant mechanical parameter of polymeric materials due to its relevance for applications is the Young’s modulus, E, which is a measure of the elastic response to applied stress. For this property, which is more dependent on the above mentioned sample composition and processing, the experimental results are somewhat scattered. Nevertheless, a recent review (Terzopoulou et al., 2020) lists six values for PEF Young’s modulus with average value of 2.5 GPa, and twelve values for PBF Young’s modulus with average value of 1.4 GPa. Assuming a connection between the dihedral angles flexibility and polymer elasticity during the viscoelastic regime—an assumption that gets grounds on the molecular interpretation of Stirnemann (Stirnemann, 2022) for protein elasticity—the presence of a longer alkyl chain in PBF, with multiple low energy torsional modes, clearly supports the reduced stiffness of PBF relative to PEF, and, thus, in accordance with a lower PBF modulus.
Conclusion
This work explores the conformational preferences and the structure-property correlations of biobased PBF, a longer chain analogue of PEF, from a computational spectroscopy approach, i.e., combining experimental results with computational chemistry. The approach combined infrared, Raman and inelastic neutron scattering spectroscopy with discrete and periodic density functional theory calculations, aiming at the identification of dominant structural motifs in the amorphous and crystalline regions—and, from this information at the microscopic level, predict and describe the macroscopic properties of the material.
In comparison with PEF, PBF presents higher conformational flexibility due to the presence of additional torsional degrees of freedom in the alkyl chain. Discrete calculations for triad models revealed a large number of conformations energetically accessible at room temperature, for both amorphous and crystalline forms. Nevertheless, it was possible to discriminate the conformational preferences for crystalline and amorphous domains by comparing the predicted and observed INS spectroscopic patterns and analyzing the infrared and Raman profiles in regions previously known to be sensitive to structural motifs.
The results strongly support the predominance of gtg conformations of the BG fragment in both the crystalline and amorphous domains. In what concerns the furandicarboxylate fragment, amorphous domains are dominated by syn,syn conformations, while in the crystalline domains the anti,anti form prevails. In addition, Raman and infrared spectra of the semi-crystalline sample unveil the spectral signature of the C-H···O hydrogen bond contacts, as found for PEF.
A possible crystalline structure, built from these conformational preferences (aa-gtg) and including a network of C-H···O hydrogen bond contacts, was optimized using periodic density functional theory. The gtg conformation of the BG fragment leads to a “ladder-like” chain. This is a relevant difference relative to PEF, for which only the fully planar aa-t structure allows the formation of the C-H···O hydrogen bond network. In PBF, a fully planar chain is also possible from the ttt conformation of the BG fragment, but with a substantially higher energy cost (ca. 10 kJ/mol per triad, as shown in Figure 2). This energy penalty is probably determinant in crystallization of PBF. Nevertheless, the existence of a higher energy PBF polymorph based on aa-ttt conformation cannot be discarded (and the INS main features for such aa-ttt polymorph are predicted from periodic DFT calculations in Figure 7).
As an ultimate goal of the computational spectroscopy approach herein described, some correlations between microscopic structure and macroscopic properties of PBF were addressed. In comparison with PEF, differences in melting and glass transition temperatures, as well as in elastic modulus and gas permeability can generally be understood at a deeper level. Furthermore, the experimental trend on lower thermal and Young’s modulus of PBF could be predicted from the increased molecular flexibility resulting from the longer alkyl chain.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: http://www.crystallography.net/tcod/index.php, 30000103 and 30000104; https://data.isis.stfc.ac.uk/doi/INVESTIGATION/113612429/.
Author contributions
All authors contributed toward the planning, preparation and implementation of the fieldwork, data analysis and interpretation, and writing the manuscript. In addition, AFS and MMN were the principal investigators for ‘polymer science’ and ‘computational chemistry’ tasks, respectively; MMN was responsible for INS project at ISIS (DOI: 10.5286/ISIS.E.RB2000214); PDV and MMN were responsible for periodic DFT calculations; LCR was responsible for initial laboratory work and discrete DFT calculations, as a part of her graduation thesis under supervision of AFS and PR-C; PR-C, CFA, and MMC were engaged in discrete DFT calculations and sample environment, data acquisition and data processing tasks; SR, the instrument scientist at ISIS, provided the expertise on INS data collection and analysis; AFS and AJDS provided student supervision for preparation, processing, and physical characterization of polymer samples; CB and MM were engaged in the synthesis and processing of polymer samples.
Funding
This work was developed within the scope of the project CICECO-Aveiro Institute of Materials, UIDB/50011/2020, UIDP/50011/2020 and LA/P/0006/2020, financed by national funds through the FCT/MCTES (PIDDAC). This research is also sponsored by FEDER funds through the program COMPETE—Programa Operacional Factores de Competitividade—and by national funds through the FCT under the project UID/EMS/00285/2020. FCT is also acknowledged for the research contract under Scientific Employment Stimulus to AFS (CEECIND/02322/2020) and for the grant to CFA (SFRH/BD/129040/2017). The STFC Rutherford Appleton Laboratory is thanked for access to neutron beam facilities (TOSCA/RB2000214, DOI: 10.5286/ISIS.E.RB2000214). CASTEP calculations were made possible due to the computing resources provided by STFC Scientific Computing Department’s SCARF cluster. This publication was also supported by COST Action FUR4Sustain—European network of FURan based chemicals and materials FOR a Sustainable development, CA18220.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Araujo, C. F., Nolasco, M. M., Ribeiro-Claro, P. J., Rudić, S., Silvestre, A. J., Vaz, P. D., et al. (2018). Inside PEF: Chain conformation and dynamics in crystalline and amorphous domains. Macromolecules 51, 3515–3526. doi:10.1021/acs.macromol.8b00192
Araujo, C., Freire, C. S. R., Nolasco, M. M., Ribeiro-Claro, P. J. A., Rudić, S., Silvestre, A. J. D., et al. (2018). Hydrogen bond dynamics of cellulose through inelastic neutron scattering spectroscopy. Biomacromolecules 19, 1305–1313. doi:10.1021/acs.biomac.8b00110
Arnold, O., Bilheux, J. C., Borreguero, J. M., Buts, A., Campbell, S. I., Chapon, L., et al. (2014). Mantid—data analysis and visualization package for neutron scattering and μ SR experiments. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrom. Detect. Assoc. Equip. 764, 156–166. doi:10.1016/j.nima.2014.07.029
Balani, K., Verma, V., Agarwal, A., and Narayan, R. (2015). Biosurfaces: A materials science and engineering perspective. United States: Wiley.
Bianchi, E., Soccio, M., Siracusa, V., Gazzano, M., Thiyagarajan, S., and Lotti, N. (2021). Poly(butylene 2, 4-furanoate), an added member to the class of smart furan-based polyesters for sustainable packaging: Structural isomerism as a key to tune the final properties. ACS Sustain. Chem. Eng. 9, 11937–11949. doi:10.1021/acssuschemeng.1c04104
Bozell, J., and Petersen, G. R. (2010). Technology development for the production of biobased products from biorefinery carbohydrates - the US Department of Energy’s “top 10” revisited. Green Chem. 12, 539–554. doi:10.1039/b922014c
Burgess, S. K., Karvan, O., Johnson, J. R., Kriegel, R. M., and Koros, W. J. (2014a). Oxygen sorption and transport in amorphous poly(ethylene furanoate). Polymer 55, 4748–4756. doi:10.1016/j.polymer.2014.07.041
Burgess, S. K., Kriegel, R. M., and Koros, W. J. (2015). Carbon dioxide sorption and transport in amorphous poly(ethylene furanoate). Macromolecules 48, 2184–2193. doi:10.1021/acs.macromol.5b00333
Burgess, S. K., Leisen, J. E., Kraftschik, B. E., Mubarak, C. R., Kriegel, R. M., and Koros, W. J. (2014b). Chain mobility, thermal, and mechanical properties of poly(ethylene furanoate) compared to poly(ethylene terephthalate). Macromolecules 47, 1383–1391. doi:10.1021/ma5000199
Burgess, S. K., Mikkilineni, D. S., Yu, D. B., Kim, D. J., Mubarak, C. R., Kriegel, R. M., et al. (2014c). Water sorption in poly(ethylene furanoate) compared to poly(ethylene terephthalate). Part 2: Kinetic sorption. Polymer 55, 6870–6882. doi:10.1016/j.polymer.2014.10.065
Carlos Morales-Huerta, J., Martínez de Ilarduya, A., and Muñoz-Guerra, S. (2016). Poly(alkylene 2, 5-furandicarboxylate)s (PEF and PBF) by ring opening polymerization. Polymer 87, 148–158. doi:10.1016/j.polymer.2016.02.003
Clark, S. J., Segall, M. D., Pickard, C. J., Hasnip, P. J., Probert, M. I. J., Refson, K., et al. (2005). First principles methods using CASTEP. Cryst. Mat. 220, 567–570. doi:10.1524/zkri.220.5.567.65075
NIST Computational Chemistry Comparison Database, B. (2022). NIST stand. Ref. Database number 101. Available at: https://cccbdb.nist.gov/vibscalejustx.asp (Accessed September 15, 2022).
de Jong, E., Visser, H. A., Dias, A. S., Harvey, C., and Gruter, G. J. M. (2022). The road to bring FDCA and PEF to the market. Polym. (Basel) 14, 943–1032. doi:10.3390/polym14050943
Druzbicki, K., Gaboardi, M., and Fernandez-Alonso, F. (2021). Dynamics & spectroscopy with neutrons—recent developments & emerging opportunities. Polym. (Basel) 13, 1440. doi:10.3390/polym13091440
Dymkowski, K., Parker, S. F., Fernandez-Alonso, F., and Mukhopadhyay, S. (2018). AbINS: The modern software for INS interpretation. Phys. B Condens. Matter 551, 443–448. doi:10.1016/j.physb.2018.02.034
European Bioplastics (2021). European bio-plastics. Available at: https://www.european-bioplastics.org/market/.
European Commission (2015). COMMUNICATION FROM THE COMMISSION TO THE EUROPEAN PARLIAMENT, THE COUNCIL, THE EUROPEAN ECONOMIC AND SOCIAL COMMITTEE AND THE COMMITTEE OF THE REGIONS Closing the loop—An EU action plan for the Circular Economy Available at: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:52015DC0614.
Fei, X., Wang, J., Zhu, J., Wang, X., and Liu, X. (2020). Biobased poly(ethylene 2, 5-furancoate): No longer an alternative, but an irreplaceable polyester in the polymer industry. ACS Sustain. Chem. Eng. 8, 8471–8485. doi:10.1021/acssuschemeng.0c01862
Fosse, C., Esposito, A., Thiyagarajan, S., Soccio, M., Lotti, N., Dargent, E., et al. (2022). Cooperativity and fragility in furan-based polyesters with different glycolic subunits as compared to their terephthalic counterparts. J. Non-Crystalline Solids 597, 121907. doi:10.1016/j.jnoncrysol.2022.121907
Guidotti, G., Soccio, M., Cruz Garcia-Gutierrez, M., Ezquerra, T., Siracusa, V., Gutierrez-Fernandez, E., et al. (2020). Fully biobased superpolymers of 2, 5-furandicarboxylic acid with different functional properties: From rigid to flexible, high performant packaging materials. ACS Sustain. Chem. Eng. 8, 9558–9568. doi:10.1021/acssuschemeng.0c02840
Harrelson, T. F., Cheng, Y. Q., Li, J., Jacobs, I. E., Ramirez-Cuesta, A. J., Faller, R., et al. (2017). Identifying atomic scale structure in undoped/doped semicrystalline P3HT using Inelastic Neutron Scattering. Macromolecules 50, 2424–2435. doi:10.1021/acs.macromol.6b02410
ISIS (2022). ISIS facility INS/TOSCA. Available at: https://www.isis.stfc.ac.uk/Pages/tosca.aspx (Accessed September 15, 2022).
Klonos, P. A., Papadopoulos, L., Terzopoulou, Z., Papageorgiou, G. Z., Kyritsis, A., and Bikiaris, D. N. (2020). Molecular dynamics in nanocomposites based on renewable poly(butylene 2, 5-furan-dicarboxylate) in situ reinforced by montmorillonite nanoclays: Effects of clay modification, crystallization, and hydration. J. Phys. Chem. B 124, 7306–7317. doi:10.1021/acs.jpcb.0c04306
Loos, K., Zhang, R., Pereira, I., Agostinho, B., Hu, H., Maniar, D., et al. (2020). A perspective on PEF synthesis, properties, and end-life. Front. Chem. 8, 585–618. doi:10.3389/fchem.2020.00585
Ma, J., Yu, X., Xu, J., and Pang, Y. (2012). Synthesis and crystallinity of poly(butylene 2, 5-furandicarboxylate). Polym. Guildf. 53, 4145–4151. doi:10.1016/j.polymer.2012.07.022
Matos, M., Sousa, A. F., Silva, N. H. C. S., Freire, C. S. R., Andrade, M., Mendes, A., et al. (2018). Furanoate-based nanocomposites: A case study using poly(butylene 2, 5-furanoate) and poly(butylene 2, 5-furanoate)-co-(butylene diglycolate) and bacterial cellulose. Polymers 10 (1–16), 810. doi:10.3390/polym10080810
Matos, M., Sousa, A. F., and Silvestre, A. J. D. (2017). Improving the thermal properties of poly(2, 5-furandicarboxylate)s using cyclohexylene moieties: A comparative study. Macromol. Chem. Phys. 218 (1–10), 1600492. doi:10.1002/macp.201600492
Milani, A., and Galimberti, D. (2014). Polymorphism of poly(butylene terephthalate) investigated by means of periodic density functional theory calculations. Macromolecules 47, 1046–1052. doi:10.1021/ma402602f
Milman, V., Perlov, A., Refson, K., Clark, S. J., Gavartin, J., and Winkler, B. (2009). Structural, electronic and vibrational properties of tetragonal zirconia under pressure: A density functional theory study. J. Phys. Condens. Matter 21, 485404. doi:10.1088/0953-8984/21/48/485404
Nolasco, M. M., Araujo, C. F., Ribeiro-Claro, P., Rudic, S., and Vaz, P. D. (2021). Inside bio-based furan polymers: Conformation and dynamics in crystalline and amorphous domains. STFC ISIS Neutron Muon Source. doi:10.5286/ISIS.E.RB2000214
Nolasco, M. M., Araujo, C. F., Thiyagarajan, S., Rudić, S., Vaz, P. D., Silvestre, A. J. D., et al. (2020). Asymmetric monomer, amorphous polymer structure–property relationships in 2, 4-FDCA and 2, 4-PEF. Macromolecules 53, 1380–1387. doi:10.1021/acs.macromol.9b02449
Papamokos, G., Dimitriadis, T., Bikiaris, D. N., Papageorgiou, G. Z., and Floudas, G. (2019). Chain conformation, molecular dynamics, and thermal properties of poly(n-methylene 2, 5-furanoates) as a function of methylene unit sequence length. Macromolecules 52, 6533–6546. doi:10.1021/acs.macromol.9b01320
Parker, S. F., Fernandez-Alonso, F., Ramirez-Cuesta, A. J., Tomkinson, J., Rudic, S., Pinna, R. S., et al. (2014). “Recent and future developments on TOSCA at ISIS,” in J. Phys.: Conf. Ser.. Editors S. JimenezRuiz, and M. Parker 554, 012003. doi:10.1088/1742-6596/554/1/012003
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868. doi:10.1103/PhysRevLett.77.3865
Pinna, R. S., Rudić, S., Parker, S. F., Armstrong, J., Zanetti, M., Škoro, G., et al. (2018). The neutron guide upgrade of the TOSCA spectrometer. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrom. Detect. Assoc. Equip. 896, 68–74. doi:10.1016/j.nima.2018.04.009
Poulopoulou, N., Nikolaidis, G. N., Ioannidis, R. O., Efstathiadou, V. L., Terzopoulou, Z., Papageorgiou, D. G., et al. (2022). Aromatic but sustainable: Poly(butylene 2, 5-furandicarboxylate) as a crystallizing thermoplastic in the bioeconomy. Ind. Eng. Chem. Res. 61, 13461–13473. doi:10.1021/acs.iecr.2c02069
Refson, K., Tulip, P. R., and Clark, S. J. (2006). Variational density-functional perturbation theory for dielectrics and lattice dynamics. Phys. Rev. B 73, 155114. doi:10.1103/PhysRevB.73.155114
Robles-Hernandez, B., Soccio, M., Castrillo, I., Guidotti, G., Lotti, N., Alegria, A., et al. (2020). Poly(alkylene 2, 5-furanoate)s thin films: Morphology, crystallinity and nanomechanical properties. Polym. Guildf. 204, 122825. doi:10.1016/j.polymer.2020.122825
Soccio, M., Martinez-Tong, D. E., Alegria, A., Munari, A., and Lotti, N. (2017). Molecular dynamics of fully biobased poly(butylene 2, 5-furanoate) as revealed by broadband dielectric spectroscopy. Polym. Guildf. 128, 24–30. doi:10.1016/j.polymer.2017.09.007
Sousa, A. F., Guigo, N., Pożycka, M., Delgado, M., Soares, J., Mendonça, P. V., et al. (2018). Tailored design of renewable copolymers based on poly(1, 4-butylene 2, 5-furandicarboxylate) and poly(ethylene glycol) with refined thermal properties. Polym. Chem. 9, 722–731. doi:10.1039/c7py01627a
Stirnemann, G. (2022). Molecular interpretation of single-molecule force spectroscopy experiments with computational approaches. Chem. Commun. 58, 7110–7119. doi:10.1039/D2CC01350A
Stroupe, Z. D., Strange, N. A., Daemen, L. L., and Larese, J. Z. (2022). Inelastic neutron scattering from thin film biaxially oriented polyethylene terephthalate. J. Phys. Chem. A 126, 7491–7501. doi:10.1021/acs.jpca.2c05397
Terzopoulou, Z., Papadopoulos, L., Zamboulis, A., Papageorgiou, D. G., Papageorgiou, G. Z., and Bikiaris, D. N. (2020). Tuning the properties of furandicarboxylic acid-based polyesters with copolymerization: A review. Polym. (Basel) 12. doi:10.3390/polym12061209
Tkatchenko, A., and Scheffler, M. (2009). Accurate molecular van der Waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 102, 073005. doi:10.1103/PhysRevLett.102.073005
United Nations (2020). Sustainable development goals. Available at: https://unstats.un.org/sdgs/report/2020/(Accessed September 15, 2022).
Vilela, C., Freire, C. S. R., Araújo, C., Rudić, S., Silvestre, A. J. D., Vaz, P. D., et al. (2020). Understanding the structure and dynamics of nanocellulose-based composites with neutral and ionic poly(methacrylate) derivatives using inelastic neutron scattering and DFT calculations. Molecules 25, 1689. Article Number 1689. doi:10.3390/molecules25071689
Werpy, T., and Petersen, G. (2004). Top value added chemicals from biomass. Volume I—results of screening for potential candidates from sugars and synthesis gas. United States: US Department of Energy. doi:10.2172/926125
Zhao, M., Zhang, C., Yang, F., and Weng, Y. (2021). Gas barrier properties of furan-based polyester films analyzed experimentally and by molecular simulations. Polym. Guildf. 233, 124200. doi:10.1016/j.polymer.2021.124200
Keywords: computational spectroscopy, inelastic neutron scattering (INS), C-H...O hydrogen bond, molecular interpretation, physical properties, 2,5-furan dicarboxylate, butylene glycol
Citation: Nolasco MM, Rodrigues LC, Araújo CF, Coimbra MM, Ribeiro-Claro P, Vaz PD, Rudić S, Silvestre AJD, Bouyahya C, Majdoub M and Sousa AF (2022) From PEF to PBF: What difference does the longer alkyl chain make a computational spectroscopy study of poly(butylene 2,5-furandicarboxylate). Front. Chem. 10:1056286. doi: 10.3389/fchem.2022.1056286
Received: 28 September 2022; Accepted: 21 November 2022;
Published: 06 December 2022.
Edited by:
Vadim G. Kessler, Swedish University of Agricultural Sciences, SwedenReviewed by:
Rocco Di Girolamo, University of Naples Federico II, ItalyPatrizia Cinelli, University of Pisa, Italy
Copyright © 2022 Nolasco, Rodrigues, Araújo, Coimbra, Ribeiro-Claro, Vaz, Rudić, Silvestre, Bouyahya, Majdoub and Sousa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mariela M. Nolasco, bW5vbGFzY29AdWEucHQ=; Andreia F. Sousa, YW5kcmVpYWZzQHVhLnB0