 Tanuja T. Yadav
Tanuja T. Yadav Gulam Moin Shaikh
Gulam Moin Shaikh Maushmi S. Kumar
Maushmi S. Kumar Meena Chintamaneni
Meena Chintamaneni Mayur YC
Mayur YC- Department of Pharmaceutical Chemistry, SPP School of Pharmacy & Technology Management, SVKM’s NMIMS- Deemed to be University, Mumbai, India
Epidermal growth factor receptor (EGFR) belongs to the family of tyrosine kinase that is activated when a specific ligand binds to it. The EGFR plays a vital role in the cellular proliferation process, differentiation, and apoptosis. In the case of cancer, EGFR undergoes uncontrolled auto-phosphorylation that results in increased cellular proliferation and decreased apoptosis, causing cancer promotion. From the literature, it shows that pyrimidine is one of the most commonly studied heterocycles for its antiproliferative activity against EGFR inhibition. The authors have collated some interesting results in the heterocycle-fused pyrimidines that have been studied using different cell lines (sensitive and mutational) and in animal models to determine their activity and potency. It is quite clear that the fused systems are highly effective in inhibiting EGFR activity in cancer cells. Therefore, the structure–activity relationship (SAR) comes into play in determining the nature of the heterocycle and the substituents that are responsible for the increased activity and toxicity. Understanding the SAR of heterocycle-fused pyrimidines will help in getting a better overview of the molecules concerning their activity and potency profile as future EGFR inhibitors.
1 Introduction
As per the recent WHO report published in 2020, cancer is the leading cause of mortality in the world. In 2018, the most diagnosed cancer was lung cancer followed by breast cancer (World Health Organization, 2020). The epidermal growth factor receptor (EGFR) belongs to the receptor tyrosine kinase (TK) family and plays an important role in the cancer progression as v-ErbB oncogene present in the avian erythroblastosis virus that has proved to be a mutant homolog of the human EGFR. The similarities were found to exist in the transmembrane and the cytoplasmic domain of the EGFR (Downward et al., 1984; Ullrich et al., 1984).
The EGFR family consists of four members of receptors mainly ErbB1, ErbB2, ErbB3, and ErbB4, and they differ in their binding interactions with the ligand (Teramura et al., 2006). There are 28 distinct combinations of these family members of EGFR that are identified till now (Lemmon and Schlessinger, 2010). Irregularities in the signaling pathway lead to malfunctioning of the EGFR, and various studies have revealed the determining cause of cancer and most of the results correlate to the overexpression of EGFR (Raymond et al., 2000). In normal cells, EGFR expression is believed to be between 40,000 and 100,000 receptors per cell, while cancer cells overexpress EGFR to the tune of more than 1,000,000 receptors per cell (Merlino et al., 1985). The EGFR plays an important role in the cellular proliferation, cell differentiation, angiogenesis, and apoptosis. In cancer, it is observed that EGFR is overexpressed and results in increased cellular proliferation with decreased apoptosis, thereby promoting tumor growth (Liu et al., 2018).
Structurally, EGFR consists of an extracellular ligand-binding site with a dimerization arm (exons 1–16), a transmembrane hydrophobic membrane (exon 17), and an intracellular tyrosine kinase and a C-terminal tail (exons 18–28) (Roskoski, 2014). In the extracellular region, the kinase domain and the C-terminal tail are the mutational “hotspots” under which the EGFR undergoes further mutations. The ectodomain alterations result in the loss of inhibitory regulatory domains that are required for dimerization. It is also worth noting that roughly 20% of glioblastomas have EGFRvIII, which results from the loss of exons 2–7. There is no ligand-dependent signaling for EGFRvIII, although there is a low level of constitutive activity for it. Even though these receptors are not downregulated by endocytosis, the modest constitutive activity is sufficient to enhance signaling in cancer cells (Gan et al., 2013).
There is roughly 45% of EGFR point exon 19 deletion in the tyrosine kinase domain of the EGFR exon 21 L858R mutation. In the activation loop, the L858R mutation gives a 50-fold greater activity with a higher Michaelis constant (Km) compared to the wild-type EGFR (Paez et al., 2004). The EGFR exon 19 in-frame deletions are another type of activating mutation in the kinase domain that is often seen in non-small-cell lung cancer (NSCLC) (Yasuda et al., 2013). In addition to imparting resistance to tyrosine kinase inhibitors, another kinase domain mutant, T790M, is known as the “gatekeeper residue” because of its ability to increase EGFR phosphorylation levels (Yun et al., 2008). The structure of EGFR with possible mutations is shown in Figure 1A.
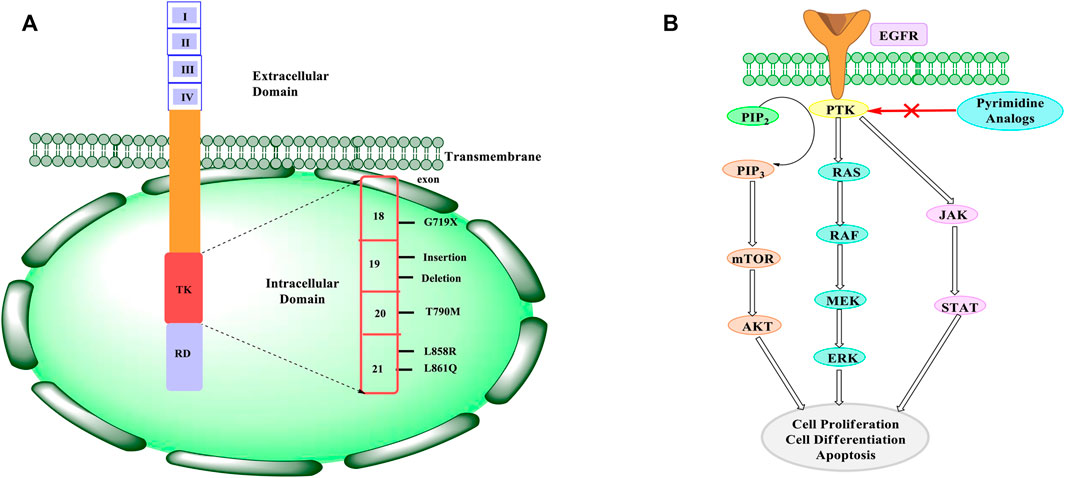
FIGURE 1. (A) EGFR structure and mutations. EGFR consists of three domains namely an extracellular domain, a transmembrane domain, and an intracellular domain. The extracellular domain consists of four subunits that are responsible for binding to the ligand and resulting in dimerization. This results in downstream signaling. The intracellular domain consists of tyrosine kinase (TK) and a regulatory domain (RD), which are responsible for regulating the signaling and physiological process of the EGFR. The TK domain consists of an exon (18–21) that shows mutation. The mutations mainly occur at T790M and L858R. (B) Mechanism of inhibiting EGFR. When an EGF ligand binds to the PTK site, it results in the activation of the downstream signalling. Two different pathways are possible. It can activate PI3K pathway or the RAS pathway. PI3K results in the conversion of PIP2 to PIP3, which is further followed by the activation of mTOR and AKT, whereas RAS activation results in the activation of RAF and MEK. Also, there can be activation of JAK-STAT pathway. Whichever pathway it follows, there’s an increase in the cell proliferation, differentiation, and apoptosis of the cell. In cancer, these downstream signaling are enhanced and hence there is uncontrolled cell growth. Therefore, pyrimidine analogues are found to inhibit the PTK site where the EGF ligand binds and results in decreasing the downstream signaling. This reduction in downstream signaling helps in reducing the cell proliferation and differentiation of cancerous cell.
The EGFR has a PTK (phosphorylated tyrosine kinase) site for attachment of the EGF (epidermal growth factor). For PI3K (phosphoionositide-3-kinase) and RAS, PTK serves as a docking location. When EGFR binds to ligands, it stimulates the PI3K, JAK-STAT, and RAS pathways, which control cell proliferation, transcription, and apoptosis (Liu et al., 2018). The phosphorylation of PIP2 (phosphatidylionositol-4,5-bisphospahte) to PIP3 (phosphatidylionositol-3,4,5-triphospahte) is carried out by PI3K. The AKT is activated because of this conversion, which then activates mTOR that oversees cell proliferation, protein synthesis, cell survival, and transcription, whereas AKT oversees glucose metabolism, cell proliferation, apoptosis, and transcription. When EGFR dimerizes, the RAS pathway is triggered. Also, RAS kinases are activated by dimerization, while RAF kinase is activated by the phosphorylation of MEK (mitogen activated protein kinase). Increased cell proliferation, differentiation, and angiogenesis are caused by the results of both PTK JAK-STAT and RAS activation pathways (Wu and Zhang, 2020). The pathways are elevated in malignant cells, resulting in increased autophosphorylation and cell proliferation, which is followed by reduced apoptosis, promoting cancer progression (Chen et al., 2014). When an inhibitor is given, it binds to EGFR and inhibits downstream signaling, as indicated in Figure 1B. Therefore, inhibiting EGFR will result in reduced cell proliferation, angiogenesis, and metastasis, as well as cell death.
The EGFR is widely located in the lung, brain, colon, and breast. It is estimated that 85% of lung cancers include non-small cell lung cancer (NSCLC) and large cell carcinoma (LCC), out of which NSCLC has been deeply understood (Chen et al., 2014). Therefore, inhibiting the EGFR expression can prove beneficial in controlling tumor progression. This results in the development of EGFR kinase inhibitors based on the identified activity from the animal models (Levitzki and Mishani, 2006). The approved list of drugs that are used to target NSCLC includes lazertinib, osimertinib, nazartinib, avitinib, erlotinib, gefitinib, afatinib, and lapatinib. Out of which avitinib is commercially available as a fused-pyrimidine derivative in the treatment of cancer (Figure 2). Lazertinib is a potent irreversible EGFR inhibitor that consists of pyrimidine as a heterocycle (Park et al., 2020; Nagasaka et al., 2021). Osimertinib is a pyrimidine containing molecule that is an oral irreversible inhibitor of wild-type EGFR and mutation T790M. It inhibits EGFR proliferation by binding to the ATP site and hence reduces the downstream signaling of EGFR (Soria et al., 2018; Lazzari et al., 2020). Nazartinib is an indole containing selective EGFR inhibitor that inhibits EGFRT790M and EGFRL858R (Murtuza et al., 2019; Tan et al., 2020). Avitinib is a pyrrolopyrimidine containing mutant-selective T790M EGFR inhibitor (Wang et al., 2020). Erlotinib, gefitinib, afatinib, and lapatinib are all quinazoline-containing EGFR inhibitors. Erlotinib is a reversible inhibitor of EGFR. It binds to ATP binding sites and inhibits EGFR signaling (Abdelgalil et al., 2020). Gefitinib shows selectivity in binding the ATP site of EGFR and inhibiting its autophosphorylation (Giaccone, 2004). Afatinib is an oral irreversible inhibitor of EGFRWT and EGFRT790M that inhibits the kinase domain of EGFR (Wind et al., 2017a; Harvey et al., 2020). Lapatinib is also an oral reversible inhibitor of EGFR and HER2 that inhibits the autophosphorylation and downstream signaling of EGFR (Collins et al., 2019) (refer Table 1).
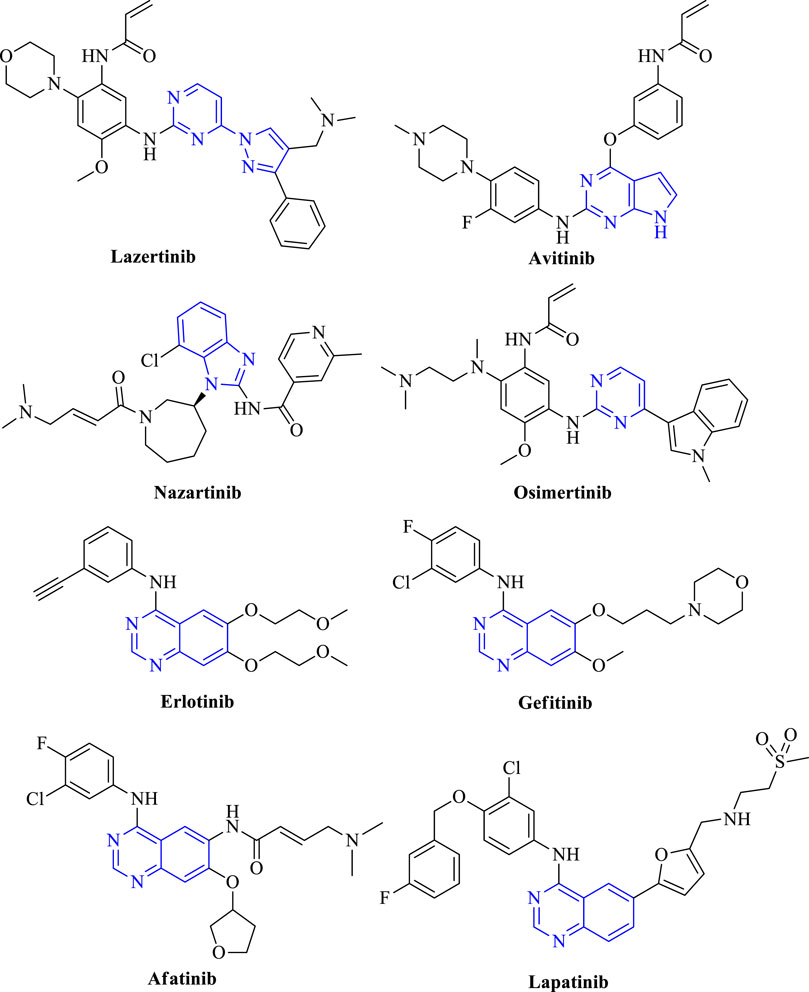
FIGURE 2. Structures of approved EGFR inhibitors.
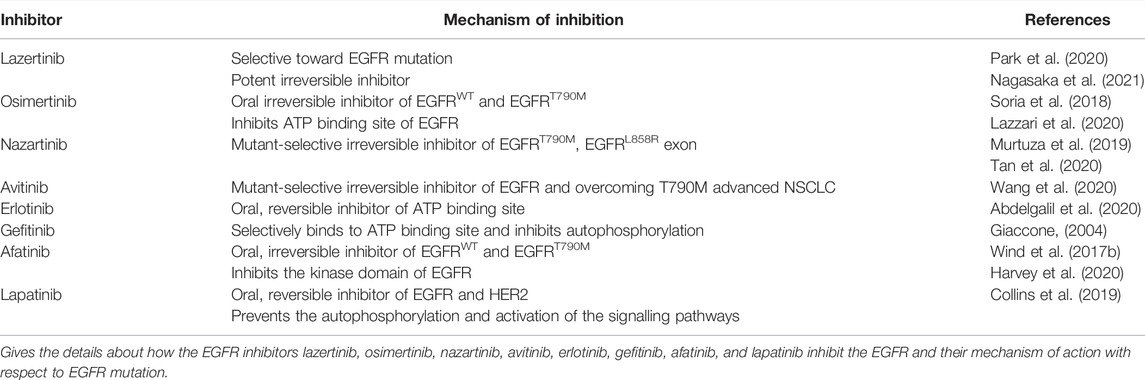
TABLE 1. Details of various EGFR inhibitors and their mechanisms.
Various heterocycles have been studied for their anticancer properties, out of which pyrimidine is found to be the most promising one. Most of the approved drugs have pyrimidine heterocycle common in them, hence making pyrimidine an important heterocycle for targeting the inhibition of EGFR. Pyrimidine is a heteroaryl system that consists of nitrogen at the first and third positions. In recent years, pyrimidine has gained focus for its use in many diseases as they have been used as antiviral (Rashad and Ali, 2006; Rashad et al., 2009; Perlíková and Hocek, 2017; Wang et al., 2018), anticancer (Galmarini et al., 2003; Song et al., 2011; Perlíková and Hocek, 2017), anti-hypertensive (El-Hamouly et al., 2006; Ismail et al., 2006), anti-mycobacterial (Garg et al., 2016; Khandazhinskaya et al., 2018), anti-microbial (Okasha et al., 2016; Beyzaei et al., 2017; Marepu et al., 2018), anti-diabetic (Spasov et al., 2017; Fang et al., 2018) and anti-inflammatory activities (Zhang et al., 2017; Abdelgawad et al., 2018).
In the past few years, researchers have synthesized heteroaryl-based pyrimidine derivatives and found that these compounds are active as EGFR inhibitors. One of the recent literatures reports that the pyrimidine system is considered as the bioisosteres to the purine analog of ATP. This results in the better uptake of the pyrimidine compound into the cell and inhibits cell functioning (Abdellatif and Bakr, 2021). Moreover, fused pyrimidines are also reported as CDK inhibitors. The CDK is responsible for the progression of cell cycle. Increase in the CDK activity results in uncontrolled tumor growth (Lee and Han, 2008). As per the reported literatures, pyrimidine fused with different heterocycles such as furan, thiophene, pyrrole, pyrimidine, indole, pyrimidine, acridone, pyrazole, thiazole, and pyridine acts as EGFR inhibitors. Out of which pyrimidine combined with pyrazole (Zhang et al., 2016; Jorda et al., 2019; Massaro et al., 2021), pyrrole (Shi et al., 2021), pyrimidine (El-Moghazy et al., 2011), and pyridine (Barvian et al., 2000; Caballero et al., 2008; Abbas et al., 2019) shows inhibitory activity not only against EGFR but also against CDK. It is suggested that pyrimidine fused with the N-containing heterocyclic system acts as a dual inhibitor. However, furopyrimidine, thienopyrimidine, pyrimidoindole, pyrimidoacridone, and thiazolopyrimidine are selective in inhibiting EGFR. This selectivity is due to the presence of a heteroatom O in furan, S in thiophene, N and S in thiazole, N in indole, and an acridone ring system. The focus of this review is to provide an insight into the substituents and structural changes that can be incorporated to find potent EGFR inhibitors.
2 Pyrimidines
Pyrimidines have been explored and are widely used heterocycles for determining their activity against various types of cancer, but most reported articles suggest the pyrimidine’s potential in inhibiting EGFR in different types of cancers. This review considers the various fused derivatives of pyrimidine that have been synthesized and are active against EGFR.
3 Fused Pyrimidine Derivatives
Pyrimidines can be fused with heterocycles like pyrrole, thiophene, furan, pyran, pyrazole, pyridine, piperidine, acridine, azepine, and even with phenyl rings to obtain a variety of different classes of molecules, which can increase the potency of these heterocycles as anticancer agents. Figure 3 exhibits various fused-pyrimidine systems that have been reported as EGFR inhibitor.
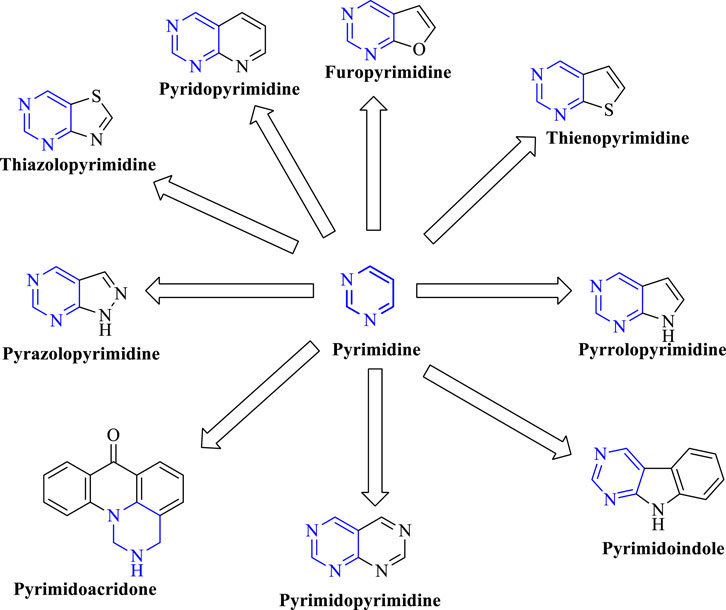
FIGURE 3. Exhibits various fused-pyrimidine systems that have been reported as EGFR inhibitors..
3.1 Pyridopyrimidines
Pyridine is a heterocycle that consists of one nitrogen atom in the system. There are a total of five positions where substitution can take place, depending on the fusion of pyridine and pyrimidine.
3.1.1 Disubstituted Pyridopyrimidines
A novel series of 4-anilino-7,8-dihydropyrido [4,3-d]pyrimidine (1–4, Figure 4A) was designed and synthesized. These molecules were further investigated for their cytotoxicity against A549, HT29, H460, and H1975 and kinase inhibitory activity using EGFR, HER2, and VEGFR. The compounds among the series 1–4 were found to be potent against the aforesaid cell lines. Compound 1 showed the highest potency and selectivity to EGFR and HER2 with an IC50 of 14.8 and 682 nM, respectively. With the increase in the size of the substituent at the meta position of the phenyl ring, a reduction in the EGFR inhibitory activity was observed (A549 IC50 values are 5.67 ± 0.08 µM for 1, 10.31 ± 0.12 µM for 2, 9.76 ± 0.08 µM for 3, and 7.22 ± 0.08 µM for 4). EGFR IC50 values are 14.8 nM for 1, 26.2 nM for 2, 21.4 nM for 3, and 28.9 nM for 4 (Zhang D. et al., 2018). This study indicates that increasing the length of the substituents and such modifications at R2 positions can lead to a decrease in the potency of the derivatives.
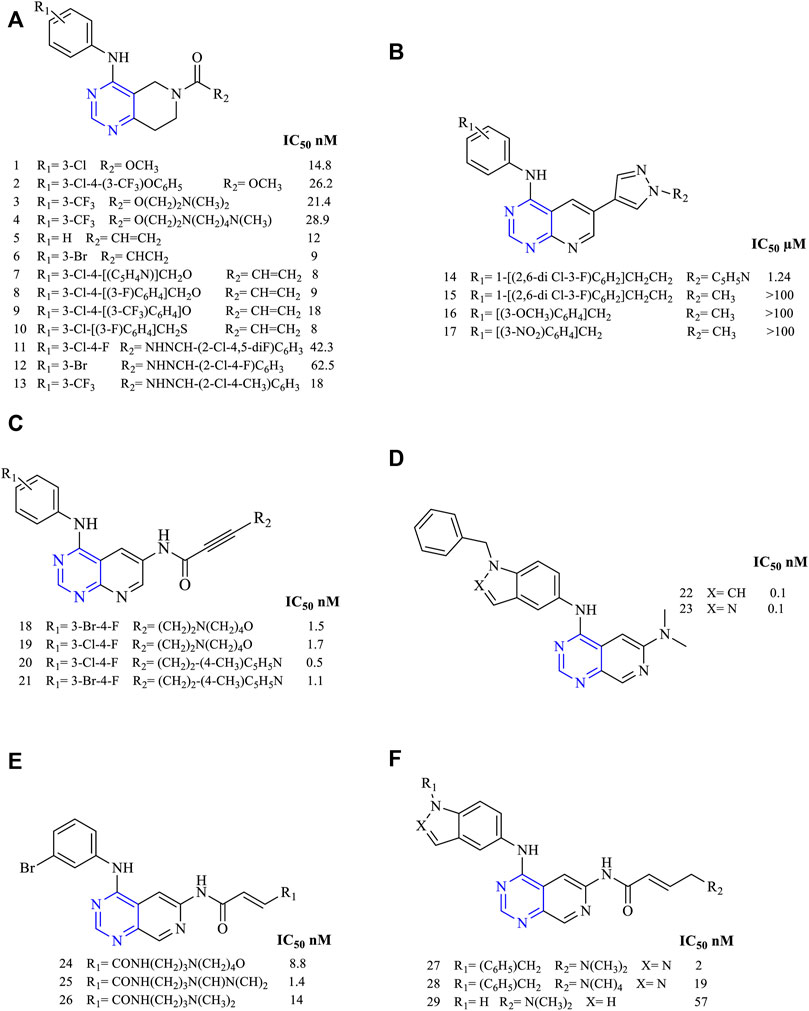
FIGURE 4. Chemical structure of (A) 4-anilino-7,8-dihydropyrido [4,3-d]pyrimidine, (B) 4,6- disubstituted [2,3-d]pyrimidine, (C) 4,6- disubstituted [2,3-d]pyrimidine, (D) 4,6- disubstituted pyrido [3,4-d]pyrimidine, (E) 4-anilinopyrido [3,4-d]pyrimidino-6-acrylamide, and (F) 4-fused anilinopyrido [3,4-d]pyrimidino-6-acrylamide.
A study regarding tetrahydropyrido [4,3-d]pyrimidine (5–10, Figure 4A) for its anticancer activity was investigated against cell lines HT29, A549, H460, and H1975. In vitro kinase inhibition studies were carried out using EGFR and HER2 (HT29 IC50 42.38 µM for 5, 24.62 µM for 6, 13.87 µM for 7, 11.69 µM for 8, 7.91 µM for 9, and 7.48 µM for 10; EGFR IC50 12 nM for 5, 9 nM for 6, 8 nM for 7, 9 nM for 8, 18 nM for 9, and 8 nM for 10). Among the series compounds, compounds 5–10 were found to be the most potent in EGFR inhibition with an IC50 range of 8–18 nM. Also, this study found that compounds 7–10 were found to inhibit HER2, which is more potent, giving rise to the dual inhibition activity (Zhang Y. et al., 2015). These heterocycles have shown good cytotoxicity in this study.
Recently, a series of tetrahydropyrido [4,3-d]pyrimidine (11–13, Figure 4A) were studied for anticancer properties using cell lines A549, H1975, MKN-45, and SGC (A549 IC50 7.55 ± 0.41 µM for 11, 1.61 ± 0.21 µM for 12, and 4.96 ± 0.34 µM for 13; EGFR IC50 42.3 nM for 11, 62.5 nM for 12, and 18 nM for 13). Further, kinase inhibition studies were carried out for its EGFR, VEFGR2, and EGFRT790M/L858R for its inhibitory potency. Compound 5 was highly potent and selective to EGFR with an IC50 of 18 nM, but it was less potent toward VEFGR2 and EGFRT790M/L858R because of the presence of the hydrophobic group trifluoromethyl, which resulted in less activity (Jing et al., 2018).
The series of molecules of 4,6-disubstituted [2,3-d]pyrimidine (14–17, Figure 4B) have been designed and studied for their anticancer activity, and 14 was found to be more effective in inhibiting MCF-7 and MDA-MB-231 human cancer cell lines (IC50 1.24 µM for 14 and that for 15–17, the values are greater than 100 µM). When studied for its EGFR inhibitory activity, compound 14 was capable of inhibiting EGFR by only 17%, showing less potency toward the enzyme. This was due to the lack of hydrogen bonding with the target, as revealed by the docking simulation studies. The docking simulation was carried using the protein with PDB ID: 4WKQ. It has gefitinib as a co-crystallized ligand. During the docking simulation, it was found that N1 of gefitinib showed a hydrogen-bonding MET793 residue and this bonding was absent in 14 and hence the EFGR inhibitory activity was found to be reduced. It shows that hydrogen bonding is important for a molecule to show the EGFR inhibitory activity (Hou et al., 2016).
A study was carried out by synthesizing 4,6-disubstituted [2,3-d]pyrimidine (18–21, Figure 4C) derivatives for investigating their anticancer activity. Compound 20 was found to be more potent against the NIH3T3 and MDA-MB-453 (EGFR IC50 of 1.5 nM for 18, 1.7 nM for 19, 0.5 nM for 20, and 1.1 nM for 21). Its activity was also determined using ErbB1 and ErbB2, which was found to be more potent toward both the receptors. Western blot analysis suggested that 20 was very selective toward EGFR and showed complete inhibition at 6 nM as compared to the positive control canertinib, which gave complete inhibition at 100 nM (Klutchko et al., 2006), clearly showing compound 20 as a future EGFR inhibitor.
A study carried out on 4,6-disubstituted pyrido [3,4-d]pyrimidine (22–23, Figure 4D) reported that the pyridopyrimidines were highly potent in inhibiting EGFR and c-ErbB-2, proving them as dual inhibitors (EGFR IC50 0.0001 µM for 22 and 23). Performing an ex vivo study on the BT474 breast tumor xenograft model revealed that the compounds were capable of inhibiting the tumor completely at a dose of 10 mg/kg BID administered for 20 days (Cockerill et al., 2001).
The studies on the class of compounds belonging to 4-anilinopyrido [3,4-d]pyrimidino-6-acrylamide (24–26, Figure 4E) were found to be highly active against EGFR and ErbB2 (EGFR IC50 8.8 nM for 24, 14 nM for 25, and 14 nM for 26), and hence these classes of compounds were found to be highly potent toward both the kinases. Their potencies could be attributed due to the presence of an acrylamide chain that was capable of interacting with the kinases (Smaill et al., 2001).
A series of 4-fused anilinopyrido [3,4-d]pyrimidino-6-acrylamide (27–29, Figure 4F) were synthesized (EGFR IC50 2 nM for 27, 19 nM for 28, and 57 nM for 29). Its antiproliferative activity was determined using kinases ErbB1, ErbB2, and ErbB3. Compound 27 was the most potent with activity ranging from 2 to 87 nM. Its pharmacokinetic profile was investigated using rats and monkeys, and it was found to be effective with 60% and 4% bioavailabilities and no mortality of animals observed (Smaill et al., 2016).
3.1.1.1 SAR of Disubstituted Pyridopyrimidine
Depending on the types of substituent and the nature of fusion, disubstituted pyridopyrimidines exhibited a wide range of activity. The following section gives the compilation of SAR:
1) On changing the fused ring from [2,3-d] to [3,4-d], it showed less inhibitory activity toward EGFR but increased activity against ErbB2 (Smaill et al., 2001) and changing to [4,3-d], which revealed that the molecules obtained were highly specific in EGFR inhibition (Zhang D. et al., 2018).
2) The presence of substituted phenyl amine is required to show inhibitory activity.
3) Substitution at R1: the presence of electron-withdrawing substituents like OCH3 and NO2 exhibited poor inhibition activity (Smaill et al., 2001). Halo substitutions at both (third and fourth) positions showed 2.5–6-fold more potent inhibitory activity toward EGFR than the existing drugs (canertinib, gefitinib, and erlotinib) (Klutchko et al., 2006), whereas mono-substitution with 3-Br gave molecules that showed dual inhibitory activity against EGFR and ErbB2 (Smaill et al., 2001). Increasing the distance between phenyl and nitrogen resulted in less active compounds (Hou et al., 2016). Changing the phenyl ring to indole or indazole gave compounds that were less effective as EGFR inhibitors (Smaill et al., 2016). The presence of benzyloxy and phenoxy increases the selectivity to EGFR rather than to HER2 (Zhang Y. et al., 2015).
4) Substitution at R2: the presence of nitrogen (N) is essential for its activity, whereas replacing it with carbon (C) showed a significant loss in its activity (Hou et al., 2016). The presence of tertiary N makes the molecules selective to EGFR (Cockerill et al., 2001), whereas substituting it with C=O retains activity (Zhang D. et al., 2018). The addition of acrylamide (Smaill et al., 2001), benzyloxy (Zhang Y. et al., 2015), phenoxy (Zhang D. et al., 2018), piperidine (Klutchko et al., 2006), and alkene enabled the molecules to exhibit selective inhibition against EGFR, whereas substituting with morpholine and imidazole imbibes a dual inhibitory activity (Smaill et al., 2001).
The summary of the SAR is depicted in Figure 5.
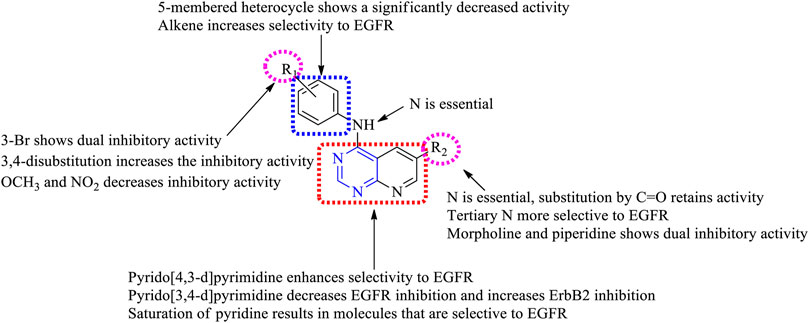
FIGURE 5. Summary SAR of disubstituted pyridopyrimidine.
3.1.2 Trisubstituted Pyridopyrimidines
Studies were conducted by synthesizing 4,6,7-trisubstituted pyrido [3,2-d]pyrimidine-based derivatives (30–32, Figure 6A) (EGFR IC50 0.95 nM for 30, 0.97 nM for 31, and 1.5 nM for 32). These derivatives’ kinase inhibition activity was determined using A431 cell lines and was found to be irreversible inhibitors of EGFR by using immune-affinity chromatography. A permeability study using the Caco-2 cell line suggested that these molecules have good absorption capacity when compared to vinblastine, but on performing a xenograft study using the nude mice model, it was found to be less effective (Smaill et al., 2000).
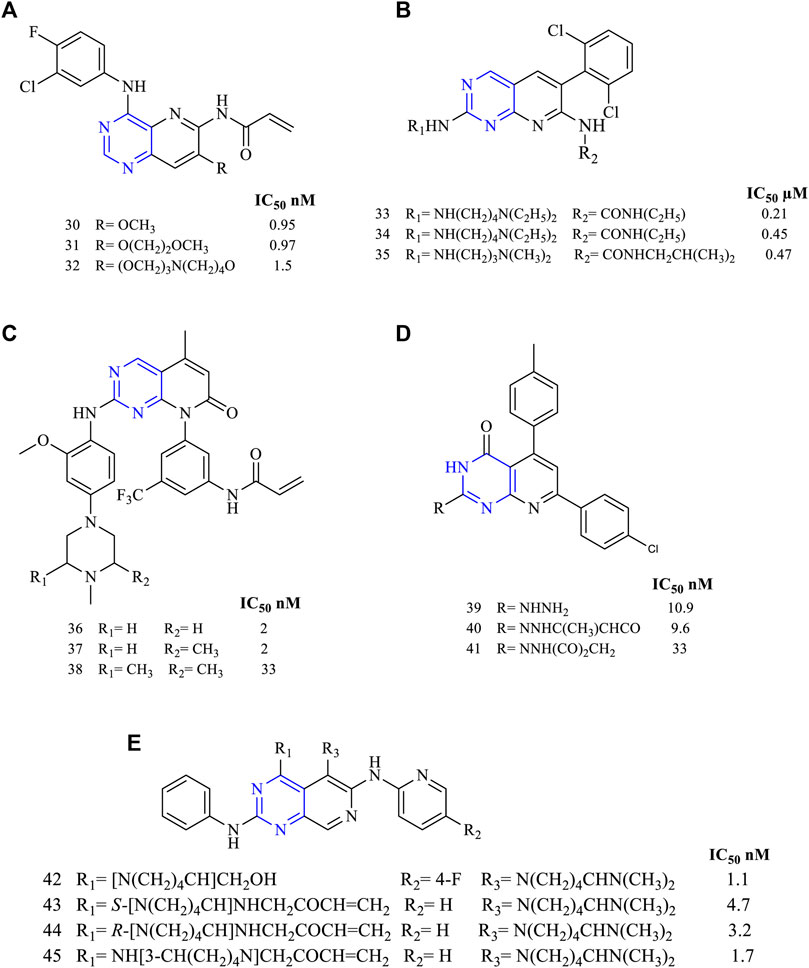
FIGURE 6. Chemical structure of (A) 4,6,7-trisubstituted pyrido [3,2-d]pyrimidine-based derivatives, (B) 6-aminopyrido [2,3-d]pyrimidino-7-urea, (C) 2,5,8-trisubstituted pyrido [2,3-d]pyrimidine, (D) trisubstituted pyrido [2,3-d]pyrimidinone, and (E) 2,4,6-trisubstituted pyrido [3,4-d]pyrimidine.
A series of 6-aminopyrido [2,3-d]pyrimidino-7-urea (33–35, Figure 6B) were synthesized to investigate its ability to inhibit receptor TK (EGFR IC50 0.21 µM for 33, 0.45 µM for 34, and 0.47 µM for 35). The studies indicated that these molecules 33–35 were used for studying their inhibitory activities. It was also reported that molecule 33 was highly potent against the MEK/ERK pathway. Also, 33 was effective in suppressing tumors in Colo 205 (Schroeder et al., 2001).
Studies found that 2,5,8-trisubstituted pyrido [2,3-d]pyrimidine (36–38, Figure 6C) were highly potent in inhibiting EGFR kinase activities with an IC50 of 2 nM for 34, 2 nM for 37, and 33 nM for 38. Compound 36 was further studied for its pharmacokinetic properties using the rat model and found to be orally active with a dose of 25 mg/kg. To predict its selectivity, the Western blot experiment revealed that compound 36 was found to be highly selective toward the cell line H1975 (Yu et al., 2017).
A novel series of trisubstituted pyrido [2,3-d]pyrimidinone (39–41, Figure 6D) were designed and studied for its anticancer activity using different cancer cell lines, and compound 40 was found to be highly potent against HepG-2, PC-3, HCT116, MCF-7, and A549 cell lines at a dose of 100 µM. An IC50 value of 40 was found to be around 9.6 µM in A549 cancer cell lines, and further, it was used to study the kinase inhibition activity. The study revealed that 40 was capable of inhibiting 81%–86% of the EGFR activity at a dose of 50 and 100 µM, respectively, suggesting that 40 was showing anticancer activity by strongly inhibiting kinases (Elzahabi et al., 2018).
A new series of 2,4,6-trisubstituted pyrido [3,4-d]pyrimidine (42, Figure 6E) were synthesized and investigated for its antiproliferative activity using HCC827, H1975, and A549 and kinase inhibitory activity using EGFRL858R, EGFRL858R/T790M, and EGFRL858R/T790M/C797S. Compound 42 was found to be more potent in inhibiting EGFRL858R, EGFRL858R/T790M, and EGFRL858R/T790M/C797S, and the IC50 values were found to be 1.1, 34, and 7.2 nM, respectively. Docking studies revealed that the hydroxyl group of 42 was interacting with Ser797, representing a stronger interaction with EGFR (Zhang et al., 2018b).
Zhang et al. carried out the studies in determining the specificity in inhibiting EGFR. They designed and synthesized 2,4,6-trisubstituted pyrido [3,4-d]pyrimidine (43–45, Figure 6E) and investigated for its antiproliferative activity against HCC827, H1975, and A549 cell lines and kinase inhibitory activity using EGFRL858R, EGFRL858R/T790M, and EGFRL858R/T790M/C797S. Compound 45 was found to be more potent against EGFRL858R and EGFRL858R/T790M with an IC50 of 1.7 and 23.3 nM, respectively, whereas for EGFRL858R/T790M/C797S, IC50 was 582.2 nM. The potency of 45 is due to the presence of the acrylamide group and the S configuration of the substituent. Also, an in vivo study using a mice model study revealed 45 was the most potent molecule to reduce the tumor growth and reported no mortality (Zhang et al., 2018c).
3.1.2.1 SAR of Trisubstituted Pyridopyrimidine
Depending on the types of substituent and the nature of fusion, trisubstituted pyridopyrimidine exhibits a wide range of activities. Following is the compilation that gives the SAR insights:
1) Maximum inhibitory activity is seen with 2,4,6-trisubstituted [3,4-d], 2,5,8-trisubstituted [2,3-d] and 4,6,7-trisubstituted [3,2-d]. Changing the rings to 2,5,7-trisubstituted [2,3-d] showed a decrease in activity.
2) At R1: N is essential for its activity. Molecules with 4-(piperazinyl)-phenyl were highly active against the triple mutant EGFR (Yu et al., 2017), whereas substituting with alkyl amino decreases the activity (Schroeder et al., 2001). The presence of pyrazole (Elzahabi et al., 2018) and phenyl resulted in the loss of inhibitory activity (Zhang et al., 2018b).
3) At R2: the presence of disubstituted phenyl is highly selective and is an irreversible inhibitor of EGFR (Smaill et al., 2000). The presence of piperidine and pyrrolidine makes the molecule active against the mutant EGFR and shows a dual inhibitory activity (Zhang et al., 2018c).
4) At R3: the presence of CH3 is active against the mutant (Yu et al., 2017). Substituting with acrylamide improves activity toward EGFR inhibition and also exhibits weak inhibitory activity toward ErbB2 (Smaill et al., 2000). Substituting with the phenyl group results in a significant loss of inhibitory activity (Elzahabi et al., 2018).
5) At R4: pyridine is highly active against the mutant form (Zhang et al., 2018b; 2018c) but disubstituted phenyl showed decreased activity (Schroeder et al., 2001).
6) At R5: saturating N and substituting with disubstituted phenyl improves activity against the mutant form (Zhang et al., 2018b). The N can be replaced by O, which showed enhanced potency. The presence of the alkoxy group revealed the irreversible inhibition of EGFR and weak activity to ErbB2 (Smaill et al., 2000). Acrylamide showed a significant loss of activity (Schroeder et al., 2001).
The summary of the SAR is depicted in Figure 7.
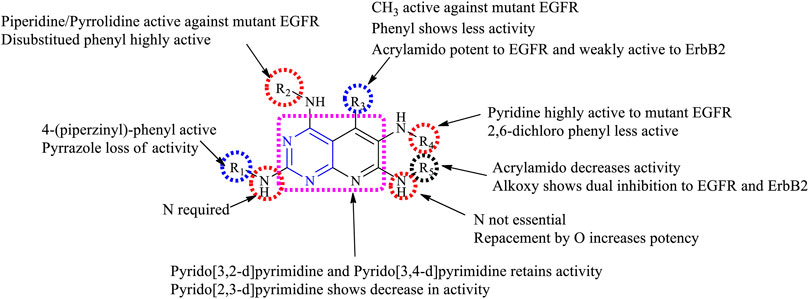
FIGURE 7. Summary SAR of trisubstituted pyridopyrimidine.
3.2 Pyrrolopyrimidine
Pyrrole is a five-membered heterocycle that consists of a nitrogen atom in the system. There are a total of five places where substitution can take place, depending on the fusion of pyrrole and pyrimidine rings with different fusion points.
3.2.1 Disubstituted Pyrrolopyrimidine
Disubstituted pyrrolo [2,3-d]pyrimidines have been widely studied for determining their activity against EGFR. A study revealed using 2,4-disubstituted pyrrolo [2,3-d]pyrimidine (46–50, Figure 8A) proved that the molecules were highly active against EGFR in nanomolar ranges (EGFR IC50 3.76 nM for 46, 5.98 nM for 47, 3.63 nM for 48, 383.7 nM for 49, and 63.29 nM for 50). The presence of halogen makes 46 a highly potent EGFR inhibitor with an IC50 of 3.76 nM. These molecules also exhibited cytotoxicity toward AURKA cell lines, enabling dual inhibitory activity (Kurup et al., 2018).
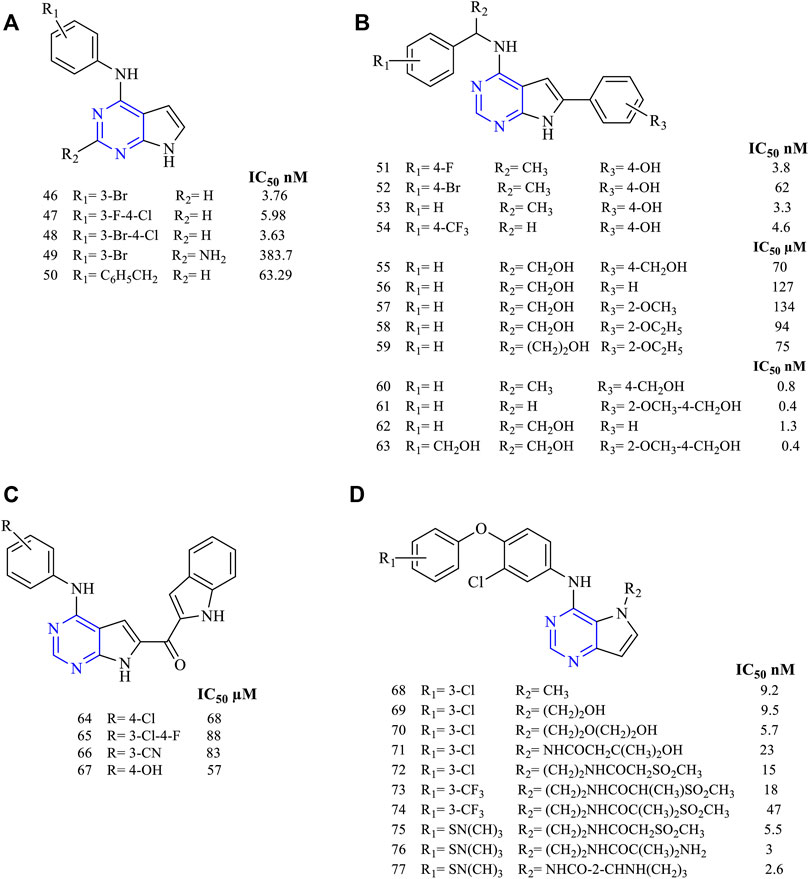
FIGURE 8. Chemical structure of (A) 2,4-disubstituted pyrrolo[2,3-d]pyrimidine, (B) 4,6-disubstituted pyrrolo[2,3-d]pyrimidine, (C) 4,6-disubstituted-7H-pyrrolo[2,3-d]pyrimidine, and (D) 4,5-disubstituted pyrrolo[3,2-d]pyrimidine.
The various studies carried out for investigating the EGFR inhibitory activity of 4,6-disubstituted pyrrolo [2,3-d]pyrimidines (Figure 8B) showed that the presence of N4-phenyl substitutions is necessary for the inhibitory activity. Compounds 51–54 (EGFR IC50 3.8 nM for 51, 62 nM for 52, 3.3 nM for 53, and 4.608 µM for 54) were found to be highly potent toward Hela cell lines that have shown IC50 in the range of 3.3–62 nM (Kaspersen et al., 2011). Substitution on the 6-phenyl in 55–59 (Sundby et al., 2015; Han et al., 2016a) (EGFR IC50 0.07 nM for 55, 127 µM for 56, 134 µM for 57, 94 µM for 58, and 75 µM for 59) revealed that they were active against a wide range of cell lines but showed a slight reduction in the activity with an IC50 range of 94–127 µM compared to erlotinib (IC50 87 µM) against the EGFR-WT (Han et al., 2016a). Another study showed that the presence of methoxy groups (60–63, Figure 8B) (EGFR IC50 0.8 nM for 60, 0.4 nM for 61, 1.3 nM for 62 and 0.4 nM for 63) increased the inhibitory potential of the molecules. Also, they were more potent against the mutant EGFR when compared to the standard erlotinib (Kaspersen et al., 2014).
Becker et al. synthesized 4,6-disubstituted-7H-pyrrolo [2,3-d]pyrimidines as methanone derivatives (64–67, Figure 8C) with a novel approach of attaching indole on the sixth position (EGFR IC50 68 µM for 64, 88 µM for 65, 83 µM for 66, and 57 µM for 67). They found that these molecules were highly active against EGFR and ErbB2, thereby showing a dual inhibitory activity. Western blot revealed that they were capable of inhibiting the receptors irreversibly (Beckers et al., 2012).
Various studies have concluded that by having substitutions in the fourth and fifth positions, they showed enhanced inhibitory activity. It was also reported that 4,5-disubstituted pyrrolo [3,2-d] pyrimidine showed potent antiproliferative activity. Compounds 68–71 (Figure 8D) (EGFR IC50 9.2 nM for 68, 9.5 nM for 69, 5.7 nM for 70, and 23 nM for 71) proved that they were highly effective in their in vitro studies against EGFR and ErbB2 with an IC50 range of 5.7–9.5 nM and 2.1–4.1 nM, respectively, thereby exhibiting dual inhibitory activity against EGFR and ErbB2 (Ishikawa et al., 2011).
Compounds 72–74 (Figure 8D) (EGFR IC50 15 nM for 72, 18 nM for 73, and 47 nM for 74) were found to be active against EGFR and ErbB2, but their affinity and selectivity were found to be less in the free state. On converting them into the tosylate salt, these compounds showed improved activity in the rat model (Kawakita et al., 2012b). Compound 71 was further taken up for its preclinical studies and is currently in phase-II clinical trial.
Compounds 75–77 (Figure 8D) (EGFR IC50 5.5 nM for 75, 3 nM for 76, and 2.6 nM for 77) have been reported with high potency and selectivity toward EGFR and ErbB2 with an IC50 in the range of 2.6–5.5 nM and 0.92–2 nM, respectively, showing greater activity toward ErbB2. Compound 76 was taken up for further preclinical studies using the xenograft mice model and was reported to be effective at a dose of 50 and 100 mg/kg with no mortality (Kawakita et al., 2012a).
3.2.1.1. SAR of Disubstituted Pyrrolopyrimidine
Depending on the types of substituent and the nature of fusion, disubstituted pyrrolopyrimidine-based molecules exhibit a wide range of activity. The following points are the outcomes, and the SAR for the same are compiled here:
1) Changing the ring from [2,3-d] to [3,2-d] exhibited dual inhibitory activity toward EGFR and ErbB2 (Ishikawa et al., 2011).
2) At R1: the presence of any group decreased its activity. Substitution with NH2 decreased its inhibitory activity (Kurup et al., 2018).
3) At R2: the presence of N is essential for activity. Increasing the distance between the phenyl and N decreased the activity (Kaspersen et al., 2011). The presence of 4-Br and 4-OH showed good inhibitory activity (Kurup et al., 2018). Substituting with phenyl and 4-CF3 resulted in a significant loss of activity (Kaspersen et al., 2014; Sundby et al., 2015; Han et al., 2016a) but substituting it with 4-Cl and 4-CN improved its potency and made the compound active toward EGFR and ErbB2 (Beckers et al., 2012). Substituting with 3-CF3 proved the compound to be selective to ErbB2 (Kawakita et al., 2012a).
4) At R3: compounds with alkoxy substituents were active (Kawakita et al., 2012b), but the addition of sulfonyl group or substituted side chains significantly reduced the activity (Kawakita et al., 2012a). Pyrrolidine and NH2 are well tolerated and showed enhanced activity (Kawakita et al., 2012a).
The summary of the SAR is depicted in Figure 9.
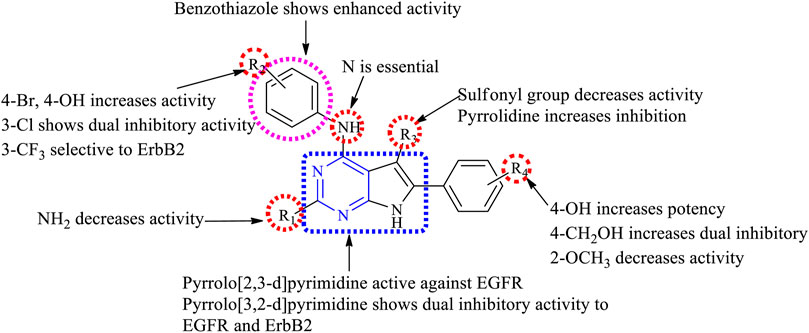
FIGURE 9. Summary SAR of disubstituted pyrrolopyrimidine.
3.2.2 Trisubstituted Pyrrolopyrimidine
A wide range of trisubstituted pyrrolopyrimidine were synthesized and studied for their antiproliferative activities against EGFR. All the trisubstituted pyrrolopyrimidine compounds showed the presence of pyrrolo [2,3-d]pyrimidine fused rings. The 2,4,6-trisubstituted pyrrolo [2,3-d]pyrimidine derivatives also showed that they were cytotoxic against the cancer cell lines. A study indicating 78–81 (Figure 10A) reported that their activity was comparable to the standard used (PD153035) in the studies (EGFR IC50 values are 0.2 µM for PD153035, 0.3 µM for 78, 2.2 µM for 79, 3.4 µM for 80, and 4.7 µM for 81) (Gangjee et al., 2008). Similar results were also reported using the A431 cell line for the identification of cytotoxic studies. Compounds 82–85 (Figure 10A) (EGFR IC50 0.32 µM for 82, 22.8 µM for 83, 122 µM for 84, and 1.32 µM for 85) exhibited good cytotoxic activity and EGFR inhibitory activity (Gangjee et al., 2010a). By varying the substitution at the R2 position with groups like methoxy and chloro, compounds 86–91 (IC50 45.7 µM for 86, 50.9 µM for 87, 197.4 µM for 88, 121 µM for 89, 5.6 µM for 90, and 8.5 µM for 91) significantly reduced its activity (Figure 10A) (Gangjee et al., 2010b, 2012).
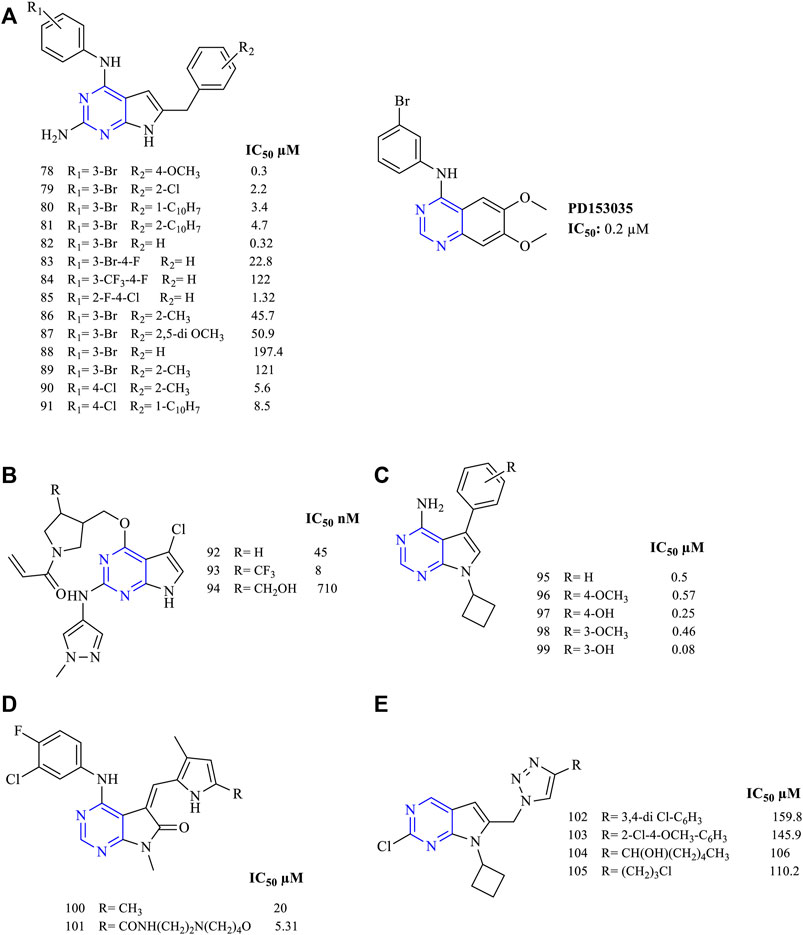
FIGURE 10. Chemical structure of (A) 2,4,6-trisubstituted pyrrolo[2,3-d]pyrimidine, (B) (2,4,6-trisubstituted pyrrolo[2,3-d] pyrimido)-prop-2-en-1-one, (C) 4,5,6-trisubstituted pyrrolo[2,3-d]pyrimidine, (D) 4,5,7-trisubstituted pyrrolo[2,3-d]pyrimidine, and (E) 2,5,6-trisubstituted pyrrolo[2,3-d]pyrimidine.
This study reported the design and synthesis of (2,4,6-trisubstituted pyrrolo [2,3-d] pyrimido)-prop-2-en-1-one for the antiproliferative action. Compounds 92–94 (Figure 10B) proved that they are highly effective against the mutant EGFR cell line with an IC50 of 4.5–710 nM. The presence of 1,2-diazole helped in improving its activity by inhibiting the EGFR (Cheng et al., 2016).
Substitution of hydroxy and methoxy substituents at R2 positions on the phenyl ring yielded compounds that exhibited high selectivity in inhibiting EGFR. Compounds 95–99 (Figure 10C) (EGFR IC50 0.5 µM for 95, 0.57 µM for 96, 0.25 µM for 97, 0.46 µM for 98, and 0.08 µM for 99) were found to be highly selective to EGFR, which showed that this type of action and this activity may be due to the presence of a cyclobutyl substituent at the seventh position. The cycloalkyl ring present at the N7 position resembles the ribose moiety of ATP, and hence the cyclobutyl ring is capable of inhibiting the ATP binding site resulting in higher EGFR inhibitory activity (Widler et al., 2001).
A study of trisubstituted pyrrolo [2,3-d]pyrimidine substituted with methyl-pyrrole (100–101, Figure 10D) (EGFR IC50 20 µM for 100, and 5.31 for 101) showed that the compounds were less active in inhibiting EGFR (Sun et al., 2002).
This study indicated the design and synthesis of 2,5,6-trisubtituted pyrrolo [2,3-d]pyrimidine compounds 102–105 (Figure 10E) (EGFR IC50 159.8 µM for 102, 145.9 µM for 103, 106 µM for 104, and 110.2 µM for 105). The study outcome was that the aryl and the alkyl functionalities can be proved to be beneficial for its anticancer cytotoxicity. Also, the presence of triazole helped in improving its anti-inhibitory activity (Thiriveedhi et al., 2019).
3.2.2.1 SAR of Trisubstituted Pyrrolopyrimidine
Depending on the type of substituents and the nature of fusion, trisubstituted pyrrolopyrimidine compounds exhibited a wide range of activity. The following section gives the compilation of the SAR:
1) Pyrrolo [2,3-d]pyrimidine is reported to show anticancer activity.
2) At R1: the presence of amino (Gangjee et al., 2008; 2010a; 2010b, 2012) and substituted amino (Cheng et al., 2016) provides good anticancer activity against the cell lines. Substituting with Cl significantly reduces the activity (Thiriveedhi et al., 2019).
3) At R2: 3-Br is required for pyrrolopyrimidine to exhibit the EGFR inhibitory activity (Gangjee et al., 2008). Substituting with ethynyl or CF3 results in a significant loss of activity (Gangjee et al., 2010a). The 4-Cl substitution is effective against EGFR (Gangjee et al., 2012); however, pyrrolidine makes the compound highly selective and effective (Cheng et al., 2016).
4) At R3: substituting phenyl with OH and OCH3 makes the compound highly selective and increases the potency (Widler et al., 2001). Substitution with methyl-pyrrole, however, reduces the activity (Sun et al., 2002).
5) At R4: the presence of phenyl moiety is responsible for its biological activity (Gangjee et al., 2008). The presence of naphthyl coupled with 3-Br at R2 showed significant improvement in the activity (Gangjee et al., 2008; 2010b). However, substituting with triazole retains the activity (Thiriveedhi et al., 2019).
6) At R5: the cyclobutyl retains the activity and also helps in improving its pharmacokinetic properties by delaying metabolism (Widler et al., 2001; Thiriveedhi et al., 2019); however, changing to CH3 reduces its activity (Sun et al., 2002).
The summary of the SAR is depicted in Figure 11.
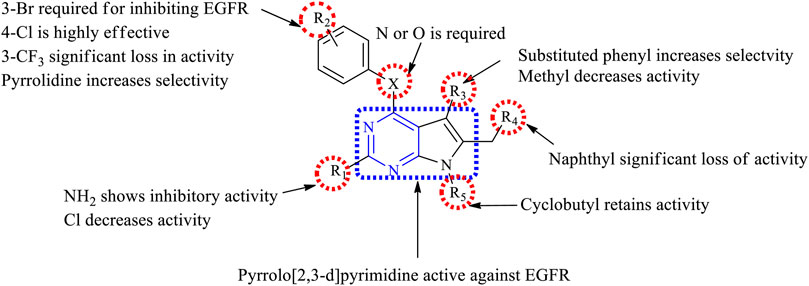
FIGURE 11. Summary SAR of trisubstituted pyrrolopyrimidine.
3.3 Thienopyrimidine
Thiophene is a five-membered heterocycle that consists of a sulfur atom in the system. The literature reported that the fourth and sixth positions have been used for substitutions and studies have been conducted for determining their anticancer potency.
3.3.1 Disubstituted Thieno[2,3-d]Pyrimidine
The compound 4,6-disubstituted thieno [2,3-d]pyrimidine has been widely studied for its anticancer activity. The literature suggests that this moiety is beneficial for exhibiting anticancer activity via inhibition of EGFR. A study on 4-substituted anilinothieno [2,3-d] pyrimidine-6-methanone (106–109, Figure 12A) (EGFR IC50 5.54 nM for 106, 18.7 nM for 107, 43 nM for 108, and 82 nM for 109)-based derivatives was found to be a dual inhibitor of EGFR and ErbB2. Further studies were carried out using a mutant and wild-type EGFR kinase and the selectivity profile was calculated. It was found that compounds 106 and 108 were found to be highly potent to EGFR and ErbB2, respectively, coupled with good inhibitory activity (Beckers et al., 2012).
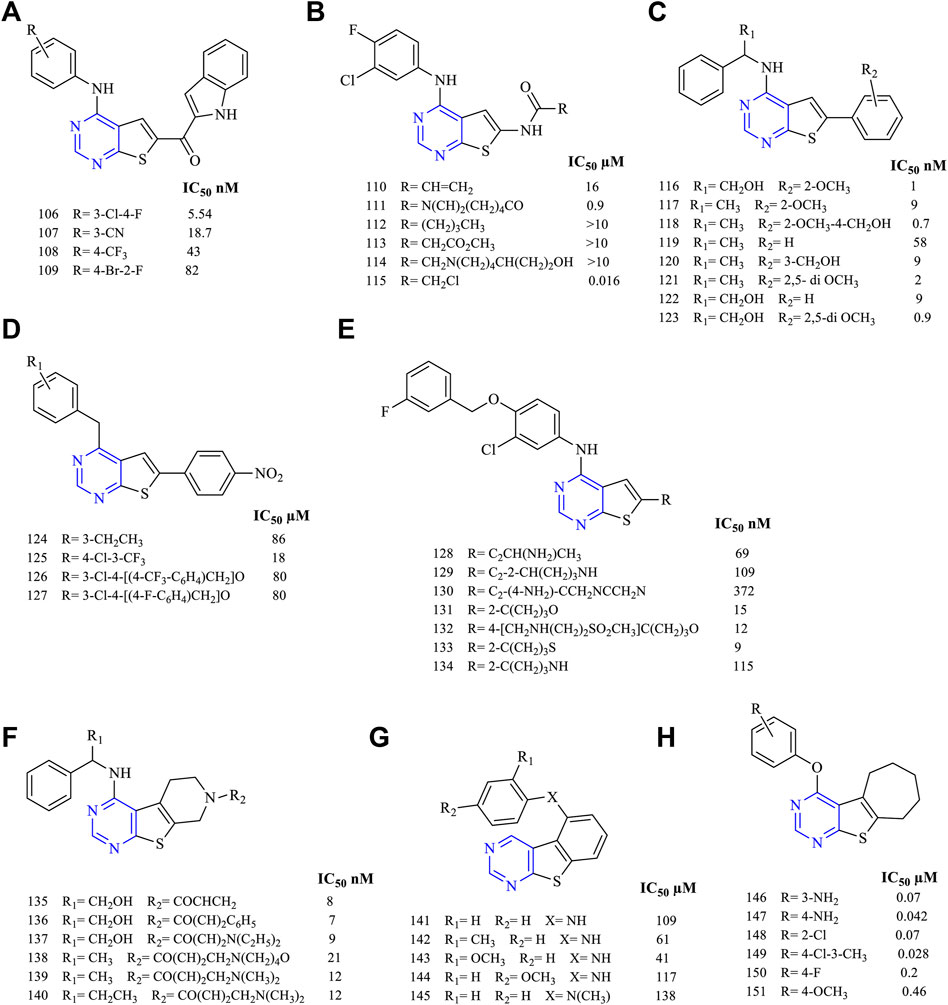
FIGURE 12. Chemical structure of (A) 4-substituted anilinothieno[2,3-d]pyrimidine-6-methanone, (B) 4-substituted anilinothieno[2,3-d]pyrimidin-6-amide, (C) 4-substituted amino-6-phenylthieno[2,3-d]pyrimidine, (D) 4-anilino-6-phenylthieno[2,3-d]pyrimidine, (E) substituted phenyl thieno[2,3-d]pyrimidine, (F) fused pyridine thieno[2,3-d]pyrimidine, (G) fused substituted phenyl thieno[2,3-d]pyrimidine, and (H) fused cycloheptane thieno[2,3-d]pyrimidine.
A series of 4-substituted anilinothieno [2,3-d] pyrimidin-6-amide (110–115, Figure 12B) (EGFR IC50 16 µM for 110, 0.9 µM for 111, >10 µM for 112–114, and 0.016 µM for 115) were synthesized and subjected to inhibitory studies. The IC50 was determined using EGFRWT and EGFRT790M/L858R. It was reported that 111 was highly potent and effective (IC50 0.9 nM for EGFRWT and IC50 4 nM for EGFRT790M/L858R). Also, compounds 110 and 111 were found to be active against ErbB2, suggesting a dual inhibitory activity (Ji et al., 2014).
It was also investigated that 4-substituted amino-6-phenylthieno [2,3-d]pyrimidine (116–123, Figure 12C) (EGFR IC50 1 nM for 116, 9 nM for 117, 0.7 nM for 118, 58 nM for 119, 9 nM for 120, 2 nM for 121, 9 nM for 122, and 0.9 nM for 123) was found to be less effective except compound 118. The compounds are erlotinib derivatives with an additional aromatic ring that makes the compounds less effective irrespective of the similar dock pose. The potency of 118 (IC50 0.7 nM) was very close to erlotinib (IC50 0.4 nM) but was found to be more cytotoxic (Bugge et al., 2015, 2016).
The studies have further reported that 4-anilino-6-phenylthieno [2,3-d]pyrimidine (124–127, Figure 12D) (EGFR IC50 86 µM for 124, 18 µM for 125, 80 µM for 126, and 86 µM for 127) was found to be effective in kinase inhibitory activity and proved compounds were exhibiting dual inhibitory characteristics against EGFR and ErbB2. Compounds 126 and 127 were found to be more cytotoxic than lapatinib (Milik et al., 2018).
To understand the inhibitory activity of thienopyrimidine, compounds with phenyl alkynyl substituent (128–130, Figure 12E) (EGFR IC50 69 nm for 128, 109 nM for 129, and 372 nM for 130) and heteroaryl substituent (131–134, Figure 12E) (EGFR IC50 15 nM for 131, 12 nM for 132, 9 nM for 133, and 115 nM for 134) were studied. Both types of substituents were capable of inhibiting EGFR and ErbB2 in the kinase study. The presence of a hetero-aromatic system yielded better inhibition activity, and 112 was found to be more effective and potent (Wood et al., 2008; Rheault et al., 2009).
Further studies suggested that when thienopyrimidine was fused with a ring (saturated ring or an aromatic ring), there was an improvement in its inhibition activity. The fusion of pyrido with thieno [2,3-d]pyrimidine resulted in compounds 135–140 (Figure 12F) (EGFR IC50 8 nM for 135, 7 nM for 136, 9 nM for 137, 21 nM for 138, 12 nM for 139–140, and 20 nM for gefitinib) that were highly potent against the double mutant EGFR cell line. Moreover, 135 was found to be more selective against EGFR from Western blotting analysis. Compound 136 was further taken up for preclinical studies using the xenograft mice model, which proved that they was a significant decrease in the tumor size (Wu et al., 2010).
It was found that fusing the substituted phenyl with thieno [2,3-d]pyrimidine resulted in compounds 141–145 (Figure 12G) (IC50 109 µM for 141, 61 µM for 142, 41 µM for 143, 117 µM for 144, and 138 µM for 145) that were highly effective against the glioblastoma. Compound 143 exhibited a strong inhibition against the cell lines U87-MG and DBTRG.05-MG. The results obtained from the Western blot analysis proved its selectivity toward glioblastoma cell line DBTRG.05-MG harboring EGFR, which showed that compound 143 has the capability of inhibiting the EGFR downstream signaling (Pédeboscq et al., 2010).
A recent study proposed that the fusion of cycloheptane with thieno [2,3-d]pyrimidine resulted in compounds 146–151 (Figure 12H) (EGFR IC50 0.07 µM for 146, 0.042 µM for 147, 0.07 µM for 148, 0.028 µM for 149, 0.2 µM for 150, and 0.46 µM for 151) that were capable of showing promising results in inhibiting EGFR and VEGFR. The IC50 of 147 (0.042 µM) obtained was found to be potent compared with erlotinib (0.03 µM), whereas 149 showed the highest apoptotic activity against the VEGFR (Mghwary et al., 2019).
3.3.2 Disubstituted Thieno[3,2-d]Pyrimidine
A series of 4,6-disubstituted thieno [3,2-d]pyrimidines were synthesized and studied for their anticancer activity. The results proved that the pyrrolidinyl-acetylenic thieno [3,2-d]pyrimidine (152–157, Figure 13A) (EGFR IC50 14 nM for 152, 90 nM for 153, 20 nM for 154, 32 nM for 155, 28 nM for 156, and 65 nM for 157) was found to have not only good inhibitory properties but also good covalent binding with EGFR. These compounds exhibited a dual inhibitory potential toward EGFR and ErbB2 (Hubbard et al., 2008). It was further reported that modifying the carbamate helps in improving the inhibitory activity and enhances the oral bioavailability of the same (Stevens et al., 2009).
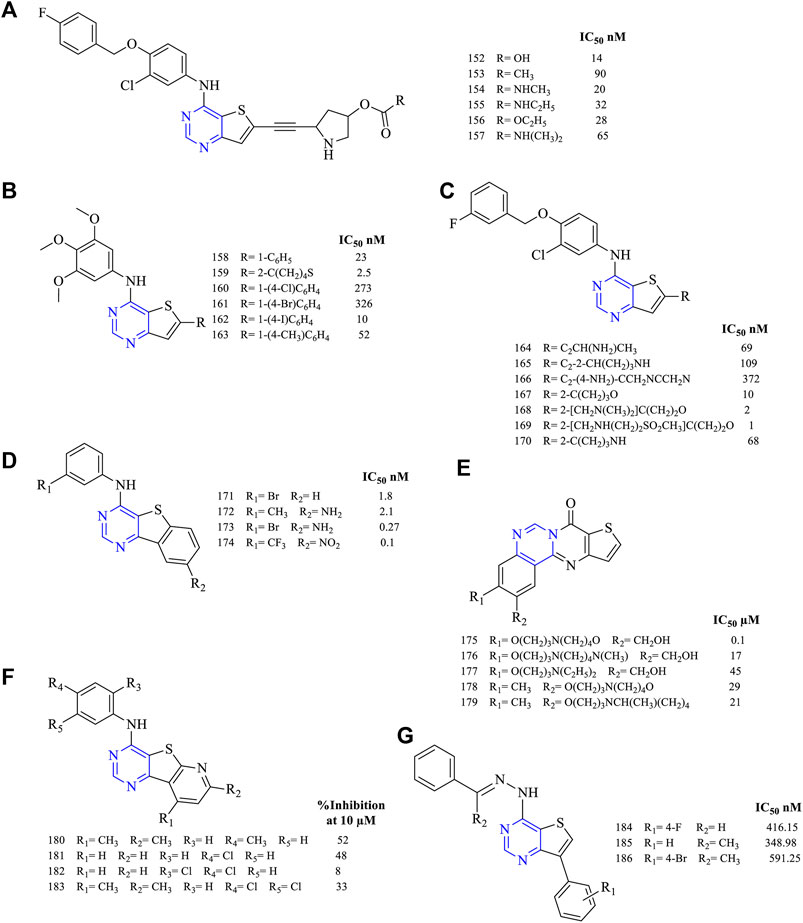
FIGURE 13. Chemical structure of (A–C) 4,6-disubstituted thieno[3,2-d]pyrimidines, (D) phenyl fused thieno[2,3-d]pyrimidines, (E) quinazoline fused thieno[2,3-d]pyrimidines, and (F) pyrido fused thieno[2,3-d]pyrimidines. (G): 4,7-disubstitued thieno [3,2-d]pyrimidine.
The presence of substituted aniline (158–163, Figure 13B) (EGFR IC50 23 nM for 158, 2.5 nM for 159, 273 nM for 160, 326 nM for 161, 10 nM for 162, and 52 nM for 163) was reported to show good inhibitory activity against EGFR but lacked activity toward VEGFR. Moreover, 159 has shown promising results in inhibiting EGFR with slight activity toward tubulin inhibition. Western blot has proved that 163 was a selective inhibitor of VEGFR (Romagnoli et al., 2019).
The alkynyl group helps in improving the inhibitory activity (164–170, Figure 13C) (EGFR IC50 69 nM for 164, 109 nM for 165, 372 nM for 166, 10 nM for 167, 2 nM for 168, 1 nM for 169, and 68 nM for 170). Depending on the type of group attached, the activity varies. The presence of a linker between the aryl and the parent rings helps in bringing selectivity to the compounds. Compound 165 exhibited only 24% alkylation to EGFR, whereas the mesylate group was found to be a potent EGFR inhibitor (Wood et al., 2008; Rheault et al., 2009).
From the literature, it was found that the fused thieno [2,3-d]pyrimidines were investigated for their inhibitory potential. The fusion of phenyl rings (171–174, Figure 13D) (EGFR IC50 1.8 nM for 171, 2.1 nM for 172, 0.27 nM for 173, and 0.1 nM for 174) resulted in compounds with an IC50 range of 0.27–2.1 nM, suggesting that the compounds were highly effective in inhibiting EGFR. Further, 173 was taken in preclinical studies, which was found to be disappointing due to the ineffective dose (50 mg/kg) and less solubility (less than 30 μg/ml) (Showalter et al., 1999).
Quinazoline-fused thieno [3,2-d]pyrimidine (175–179, Figure 13E) (DU-145 IC50 0.1 µM for 175, 17 µM for 176, 45 µM for 177, 29 µM for 178, and 21 µM for 179) compounds reported EGFR inhibitory activity. Their inhibitory potential was determined using DU-145 and MiaPaCa-2 cell lines but was found to be less effective as they showed the inhibition of a maximum of 10% (Zheng et al., 2010).
Studies on pyrido-fused thieno [3,2-d]pyrimidine (180–183, Figure 13F) (percent EGFR inhibition at 10 µM; 52% for 180, 48% for 181, 8% for 182, and 33% for 183) reported showing EGFR inhibitory activity. Compound 181 was very potent as it exhibited an 81% inhibitory potential when compared to doxorubicin. It was further taken up for preclinical studies but failed due to a lack of specificity in the study model (Aziz et al., 2015).
It was found that thieno [3,2-d]pyrimidine (184–186, Figure 13G) (EGFR IC50 416.15 nM for 184, 348.98 nM for 185, and 591.25 nM for 186) are EGFR inhibitors. These compounds were found to be less potent in inhibiting EGFR when compared to the reference erlotinib (EGFR IC50 166.92 nM) (Traxler et al., 1997).
3.3.3 SAR of Thienopyrimidine
Depending on the type of substituents, thienopyrimidine exhibits EGFR inhibitory activity. It was also reported that thieno [2,3-d]pyrimidine showed inhibitory activity against EGFR, whereas thieno [3,2-d]pyrimidine shows a dual inhibitory activity toward EGFR and ErbB-2. The following is the compilation of the SAR:
1) At R1: the presence of a nitrogen atom is essential for inhibitory activity. The presence of substituted phenyl improves the activity. The substitution of F and Cl showed optimal activity (Ji et al., 2014). Increasing the distance between the N and phenyl groups helps in improving the selectivity toward EGFR (Bugge et al., 2015, 2016). The substitution with the phenoxy phenyl group shows very potent inhibitory activity (Hubbard et al., 2008; Wood et al., 2008; Rheault et al., 2009; Stevens et al., 2009; Pédeboscq et al., 2010; Wu et al., 2010; Milik et al., 2018; Mghwary et al., 2019). The presence of CH2OH increased its selectivity and activity (Wu et al., 2010). Substitutions with CH3 and OCH3 showed a strong blocking of EGFR (Pédeboscq et al., 2010; Romagnoli et al., 2019), whereas substitution by the NH2 group showed very potent activity and strong inhibition (Mghwary et al., 2019). The presence of the CH3 group helps in EGFR inhibitory activity. The presence of Br on the phenyl ring has shown enhanced potency in inhibiting EGFR as compared to that of F (Traxler et al., 1997).
2) At R2: the presence of an indole ring makes the compound show a dual inhibitory activity (Beckers et al., 2012). The presence of an amide linkage makes the molecule more active (Ji et al., 2014). The addition of acrylamide in thieno [2,3-d]pyrimidine makes the compound very potent. The substituted aryl showed increased activity and selectivity to EGFR (Bugge et al., 2015, 2016; Milik et al., 2018), whereas in the case of thieno [3,2-d]pyrimidine, it showed the loss of activity (Romagnoli et al., 2019). The methoxy group exhibits inhibitory potential (Bugge et al., 2016), whereas the nitro group decreases the activity due to its strong electron-withdrawing abilities (Milik et al., 2018). The thiophene-substituted derivatives were also effective in inhibiting EGFR and VEGFR (Romagnoli et al., 2019). The alkynyl group of thieno [2,3-d]pyrimidine was dually effective in inhibiting EGFR and ErbB2 (Wood et al., 2008; Rheault et al., 2009), but in the case of thieno [3,2-d]pyrimidine, the selectivity for ErbB2 decreased significantly (Wood et al., 2008; Rheault et al., 2009). The smaller heteroaryl-like pyrrole showed good dual inhibitory potential (Hubbard et al., 2008), but replacing it with a morpholine group reduced its activity (Stevens et al., 2009).
3) The fusion of thienopyrimidine with other rings helps in improving its activity. The fusion with piperidine (Wu et al., 2010), phenyl (Showalter et al., 1999; Pédeboscq et al., 2010), and cycloheptane (Mghwary et al., 2019) was found to exhibit a dual inhibitory activity. The presence of quinazoline fusion also improved the activity of the compound (Zheng et al., 2010). The pyrido fused molecules were found to be weakly active (Aziz et al., 2015).
The summary of the SAR is depicted in Figure 14.
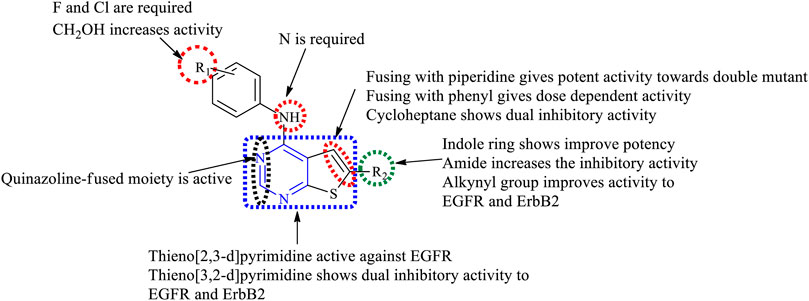
FIGURE 14. Summary SAR of thienopyrimidine.
3.4 Pyrazolopyrimidine
Pyrazole is a five-membered heterocycle that consists of two nitrogen atoms in the system. The literature reports that pyrazolo [3,4-d]pyrimidines have been synthesized and their EGFR inhibition studies were carried out.
Based on the pharmacophoric studies, 4-(phenylamino) pyrazolo [3,4-d]pyrimidine (187–190, Figure 15A) (EGFR IC50 0.033 µM for 187, 0.008 µM for 188, 0.13 µM for 189, and 0.005 µM for 190) was designed and synthesized. These compounds were subjected to cellular study using MK cell lines and A-431 cell lines and found to be highly active. It was further reported that the compounds have a pronounced effect on the blockage of the EGFR signaling pathway. Furthermore, the in vivo studies revealed that the compounds were effective in the mice model when the animal was intraperitoneally administered with 12.5 mg/kg and 50 mg/kg (Traxler et al., 1997).
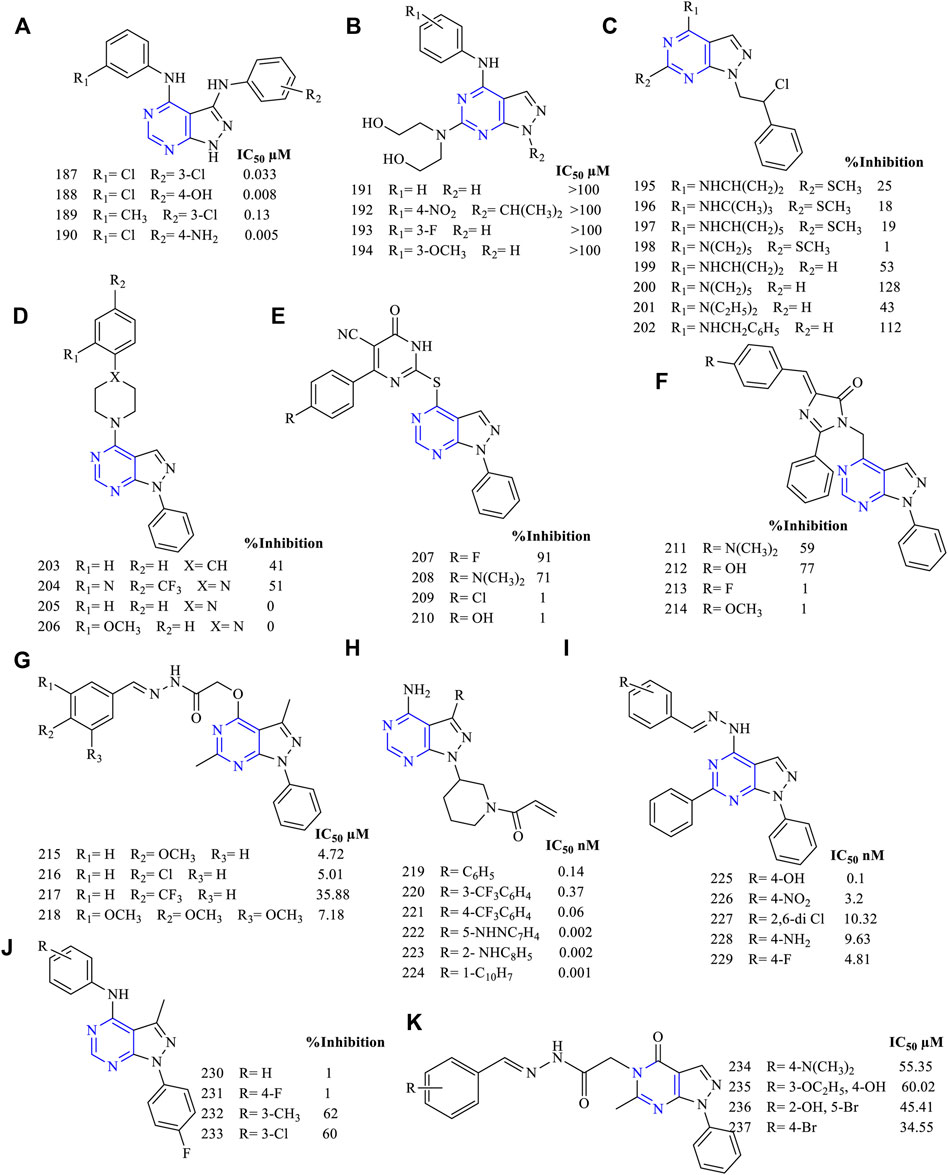
FIGURE 15. Chemical structure of (A) 4-(phenylamino) pyrazolo[3,4-d]pyrimidine pyrazolopyrimidine, (B) 4-substituted anilino-6-hydroxylaminopyrazolo[3,4-d]pyrimidine, (C) 1-(phenyl ethyl)-4-substituted amino-6-thiopyrazolo[3,4-d]pyrimidine, (D) piperidine-substituted pyrazolo[3,4-d]pyrimidine, (E) pyrimidinone-substituted pyrazolo[3,4-d]pyrimidine, (F) imidazole-substituted pyrazolo[3,4-d]pyrimidine, (G) acetohydride derivatives of pyrazolo[3,4-d]pyrimidine, (H) propanone derivatives of pyrazolo[3,4-d]pyrimidine, (I) benzylidene derivatives of pyrazolo[3,4-d]pyrimidine (J) 1-phenyl-3-methylpyrazolo[3,4-d]pyrimidin-4-amine, and (K) N′-substituted benzylidene-2-(6-methyl-4-oxo-1-phenyl-1H-pyrazolo[3,4-d]pyrimidin-5(4H)-yl)acetohydrazide.
It was reported that 4-substituted anilino-6-hydroxylaminopyrazolo [3,4-d]pyrimidine derivatives (191–194, Figure 15B) were synthesized as anti-EGFR inhibitors. However, they lacked specific anti-EGFR properties (IC50 of synthesized compounds was more than 100 µM, whereas for the standard compound, it was found to be 0.0008 µM). Instead, they were found to inhibit CDK2, which plays an important role in the cell cycle (Kim et al., 2003).
Schenone et al. were the first in designing 6-substituted pyrazolo [3,4-d]pyrimidine as an antiproliferative agent. They designed and synthesized various 1-(phenyl ethyl)-4-substituted amino-6-thiopyrazolo [3,4-d]pyrimidine derivatives (195–198, Figure 15C) (percentage EGFR inhibition: 25% for 195, 18% for 196, 19% for 197, and 0% for 198). These compounds were less cytotoxic and ineffective compared to the reference molecule AG1478 (Schenone et al., 2004b). Further, they removed the substitution on the sixth position (199–202, Figure 15C) (53% for 199, 128% for 198, 43% for 199, and 112% for 200), and it was found that the compounds were effective in inhibiting EGFR. Compound 200 was the most potent and showed its activity against EGFR by blocking ERK and SRC that was required for its downstream signaling pathway (Schenone et al., 2004a).
The activity of these compounds also varied depending on the type of moiety that is substituted on the pyrazolo [3,4-d]pyrimidine. Abbas et al. synthesized pyrazolo [3,4-d]pyrimidine with three different attachments on the piperidine (203–206, Figure 15D) (percentage EGFR inhibition: 41% for 203, 51% for 204, 0% for 205–206), pyrimidinone (207–210, Figure 15E) (percentage EGFR inhibition: 91% for 207, 71% for 208), and imidazole (211–214, Figure 15F) (percentage EGFR inhibition: 59% for 211, 77% for 212) moieties. On comparing their inhibitory potential, it was found that pyrimidinone was the most active as they gave a maximum inhibition of 91% when compared to gefitinib. The docking was carried out using protein PDB ID: 3W2O. The co-crystallized ligand was able to form a hydrogen bond with the MET793 residue, whereas the pyrimidinone 203–206 was binding not only to MET793 but also to the LYS745 residue. These resulted in stronger binding between the molecules and the protein, exhibiting higher EGFR inhibitory potential (Abbas et al., 2015).
The acetohydride derivatives of pyrazolo [3,4-d]pyrimidine were designed and synthesized (215–218, Figure 15G) (EGFR IC50 4.72 µM for 215, 5.01 µM for 216, 35.88 µM for 217, and 7.18 µM for 218). These compounds were highly active against the MCF-7 cell lines with IC50 values in the range of 6.14–15.21 µM. These compounds were effective against breast cancer cell lines. Furthermore, the docking analysis predicted stronger binding interactions of compound 218 with the protein as compared to the reference erlotinib, suggesting that it can be further modified to improve its activity and enter into preclinical studies (Abdelgawad et al., 2016).
A structure-guided synthesis of propanone derivatives (219–224, Figure 15H) (EGFR IC50 0.14 nM for 219, 0.37 nM for 220, 0.06 nM for 221, 0.002 nM for 222, 0.002 nM for 223, and 0.001 nM for 224) was carried out. The compounds have shown greater inhibition potential to EGFRL858R and EGFRL858R/T790M. These compounds were further screened against the third-generation EGFR inhibitor gefitinib and were found to be superior, concluding that the compounds synthesized were selective third-generation EGFR inhibitors. Further, 223 was taken up for preclinical study using the mice model and results showed good activity by the intraperitoneal route compared to that by intravenous (Engel et al., 2017).
Making use of rational drug design tools and methods, benzylidene derivatives of pyrazolo [3,4-d]pyrimidine (225–229, Figure 15I) (EGFR IC50 0.1 nM for 225, 3.2 nM for 226, 10.32 nM for 227, 9.63 nM for 228, and 4.81 nM for 229) were designed, synthesized, and evaluated. The compounds were tested against the MCF-7, A-549, and mutant T790M. All the synthesized molecules were effective against most of the cell lines, whereas 229 was the most potent. Compound 229 showed good binding interaction in the docking studies but failed to exhibit selectivity. Moreover, the cell cycle analysis has shown that these compounds were capable of inhibiting the G0/G1 phase of the cell division (Gaber et al., 2018).
The design and synthesis of 1-phenyl-3-methylpyrazolo [3,4-d] pyrimidin-4-amine (230–233, Figure 15J) (A549 inhibition: 1% for 230–231, 62% for 232, and 60% for 233) derivatives were carried out, and their inhibitory studies were revealed. The compounds showed a dual inhibitory action against EGFR as well as to ErbB2, which was further confirmed by performing caspase-3 analysis. Also, cell cycle analysis revealed that compound 233 showed the cell division arrest by the G0/G1 phase (Maher et al., 2019).
The design of carbohydrazide-based N′-substituted benzylidene-2-(6-methyl-4-oxo-1-phenyl-1H-pyrazolo [3,4-d]pyrimidin-5(4H)-yl)acetohydrazide (234–237, Figure 15K) (MCF IC50 55.35 ± 7.711 µM for 234, 60.02 ± 2.716 µM for 235, 45.41 ± 5.376 µM for 236, and 34.55 ± 2.381 µM for 237; EGFR IC50 0.186 µM for 237 and 0.03 µM for erlotinib) has been reported. The compounds have displayed EGFR inhibition using cell lines. These compounds were also subjected to docking analysis using PDB ID: 1M17. It was found that these compounds could inhibit the ATP binding site of the protein when compared to the binding interaction of the co-crystallized ligand. Flow-cytometry studies have found that 235 exhibited the highest apoptosis activity followed by 234, 236, and 237 when compared to the control 0.5% DMSO vehicle. This represents the apoptotic activity of these compounds (Horchani et al., 2021).
3.4.1 SAR of Pyrazolopyrimidine
Depending on the types of substitutions, pyrazolo [3,4-d]pyrimidine, the following is the compiled data of SAR:
1) Pyrazolo [3,4-d]pyrimidine showed good inhibitory activity against EGFR.
2) At N3: the presence of the carbohydrazide group results in decreased EGFR inhibitory activity but improves selectivity in binding to EGFR (Horchani et al., 2021).
3) At R1: the presence of a phenyl group showed good inhibitory properties (Abbas et al., 2015; Gaber et al., 2018). Replacing with the dialkylamino group exhibited reduced activity (Kim et al., 2003), whereas replacing with the thio group resulted in a significant loss of activity (Schenone et al., 2004b).
4) At R2: the linker can be either S or N; this resulted in compounds with good inhibitory activity (Abbas et al., 2015). The presence of the NH2 group makes the compound active toward the double mutant EGFR (Engel et al., 2017). The N-phenyl compounds were also active (Traxler et al., 1997; Kim et al., 2003; Maher et al., 2019). Replacing phenyl with piperidine or imidazole increased the potency (Abbas et al., 2015). The presence of acetohydride linkage results in the enhanced binding of the compounds to EGFR (Abdelgawad et al., 2016), whereas the hydrazine reduces the selectivity of the compound to EGFR (Gaber et al., 2018).
5) At R3: the presence of an aniline group is responsible for the potent activity toward EGFR (Engel et al., 2017). Replacing the aniline with methyl exhibited a dual inhibitory activity to EGFR and ErbB2 (Maher et al., 2019).
6) At R4: the presence of a piperidine ring gives potent activity (Engel et al., 2017). Substituting with phenyl decreases the activity of the compound (Abbas et al., 2015; Abdelgawad et al., 2016; Engel et al., 2017; Gaber et al., 2018; Maher et al., 2019), whereas the phenyl ethyl group improves the inhibitory activity (Schenone et al., 2004b; 2004a).
The summary of the SAR is depicted in Figure 16.
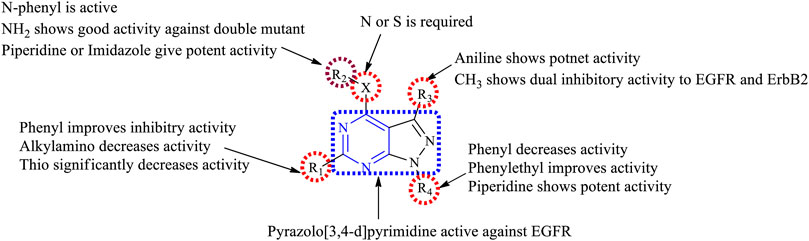
FIGURE 16. Summary SAR of pyrazolopyrimidine.
3.5 Furopyrimidines
Furan is a five-membered heterocycle that consists of one oxygen atom in the system. Substitution can occur in a total of four positions, depending on the fusion of furan and pyrimidine.
It was reported that 4-amino-5-methylfuro [2,3-d]pyrimidine (238–241, Figure 17A) (EGFR IC50 15.5 nM for 238, 283 nM for 239, 7.1 nM for 240, and 3.1 nM for 241) has been designed and studied for anti-EGFR activity. The compounds exhibited good kinase activity. The compounds were further tested for tubulin inhibitory activity, and 241 was found to be the most potent (Devambatla et al., 2018).
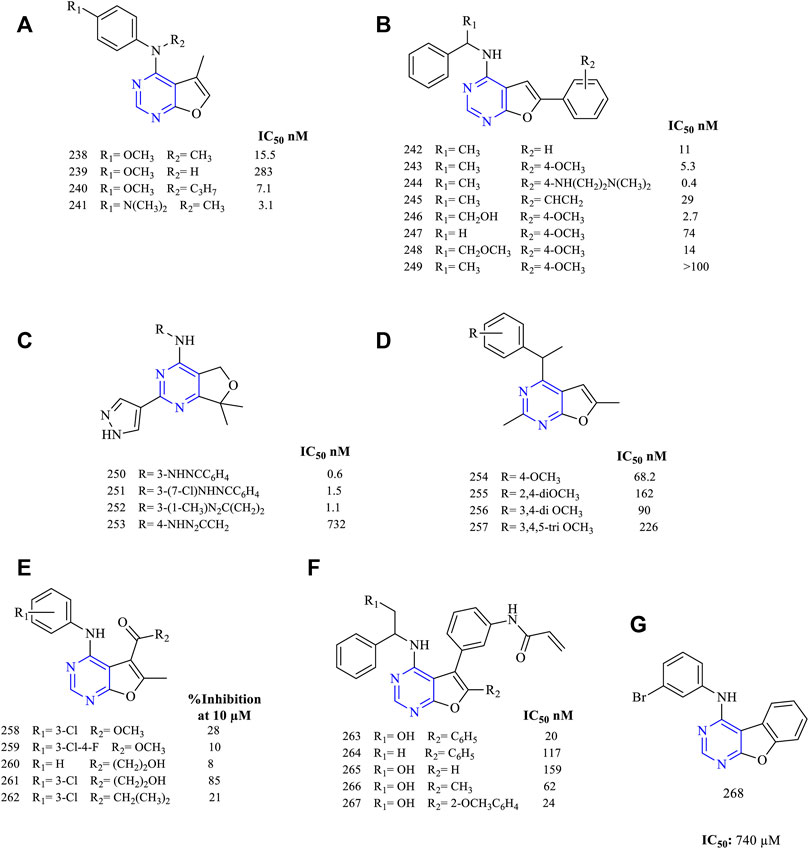
FIGURE 17. Chemical structure of (A) 4-amino-5-methylfuro[2,3-d]pyrimidine, (B) 6-arylsubstituted furo[2,3-d] pyrimidin-4-amine, (C) 2-indazolesubstituted furo[2,3-d] pyrimidin-4-amine, (D) 4-substituted-2,6-dimethylfuro[2,3-d]pyrimidine, (E) 4-anilino-6-methylfuro[2,3-d] pyrimidin-5-carboxylate, (F) propenamide derivatives of furo[2,3-d]pyrimidine, and (G) phenyl fused furo[3,2-d]pyrimidine.
The 6-arylsubstituted furo [2,3-d] pyrimidin-4-amine (242–249, Figure 17B) (EGFR IC50 11 nM for 242, 5.3 nM for 243, 0.4 nM for 244, 29 nM for 245, 2.7 nM for 246, 74 nM for 247, 14 nM for 248)-based derivatives were synthesized with varied substitutions on the sixth position. The kinase inhibition studies found that the presence of ethyl amino linker in 243 at the sixth position is very effective and potent (IC50 0.4 nM), whereas other substitutions decreased the activity. Compound 243 was subjected to further studies to identify its cellular potency but showed a significant loss in cellular activity (IC50 217 nM) when compared to erlotinib (IC50 87 nM) (Han et al., 2016b).
Based on the design and pharmacophoric groups, the indazole group has been tried (250–253, Figure 17C) (EGFR IC50 0.6 nM for 250, 1.5 nM for 251, 1.1 nM for 252, and 732 nM for 253). These compounds with the triazole moiety exhibited great inhibitory potential toward EGFR with an IC50 in the range of 0.6–1.5 nM. Also, these compounds were found to be potent against the mutant cell line EGFRL858R/T790M (Hanan et al., 2016).
Depending on the conformational studies, 4-substituted-2,6-dimethylfuro [2,3-d]pyrimidine (254–257 Figure 17D) (EGFR IC50 68.2 nM for 254, 162 nM for 255, 90 nM for 256, and 226 nM for 257) were synthesized and studied. These compounds were less effective against EGFR and VEGFR but were capable of inhibiting the tubulin network (Zhang X. et al., 2015).
A series of 4-anilino-6-methylfuro [2,3-d] pyrimidin-5-carboxylate (258–262, Figure 17E) (percent EGFR inhibition at 10 µM: 28% for 258, 10% for 259, 8% for 260, 85% for 261, and 21% for 262)-based derivatives were synthesized. These compounds were capable of showing a weak dual inhibitory activity toward EGFR and ErbB2. Also, the antiproliferative studies proved that the compounds exhibited a significant loss of activity (Hossam et al., 2018).
The literature review suggests that the propenamide derivatives (263–267, Figure 17F) (EGFR IC50 20 nM for 263, 117 nM for 264, 159 nM for 265, 62 nM for 266, and 24 nM for 267) were designed and synthesized and these were highly effective against the EGFRWT and the double mutant EFGRL828R/T790M. Moreover, these compounds have been tested with poziotinib (a potent inhibitor of HER2 exon 20 insertions under the clinical trial phase II (NCT03066206). It was found that the propenamide derivatives exhibited equal potency as compared to poziotinib in inhibiting EGFR and HER2 (Lin et al., 2019).
From the literature, it was found that the furo [3,2-d]pyrimidine can be fused with a phenyl ring (268, Figure 17G). This fusion led to a significant loss in the inhibitory activity toward EGFR with an IC50 of 740 µM (Showalter et al., 1999). These types of compounds were not showing much biological activity.
3.5.1 SAR of Furopyrimidine
Various furopyrimidines have been studied. Based on the results reported in the literature, most of these compounds showed the presence of [2,3-d] fusion between furan and pyrimidine. This section provides an overview of the SAR:
1) The furo [2,3-d]pyrimidine derivatives were active against EGFR but modifying to furo [3,4-d]pyrimidine produced potent and selective EGFR inhibition (Hanan et al., 2016).
2) At R1: the presence of the CH3 group resulted in a decreased EGFR inhibitory activity (Zhang X. et al., 2015), whereas substituting R1 with the imidazolyl ring resulted in potent EGFR inhibitors that were active against the double mutant cell lines (Hanan et al., 2016).
3) At R2: the presence of a secondary N is required. Converting it into tertiary N, the activity was found to decrease (Zhang X. et al., 2015). Amino phenyl and methoxy-substituted phenyl derivatives were found to be active (Devambatla et al., 2018), but di- and tri-substituted methoxy phenyl compounds showed significant loss of activity (Zhang X. et al., 2015). A carbon linker present between the phenyl and N helps in improving its activity (Han et al., 2016b; Lin et al., 2019). Substituting with triazole shows a significant loss in anticancer activity, whereas indazole is a potent inhibitor (Hanan et al., 2016).
4) At R3: methyl substituents were found to be active (Devambatla et al., 2018). The carboxylate derivatives showed a significant loss of activity (Hossam et al., 2018). The presence of a phenyl ring decreases anticancer activity (Lin et al., 2019).
5) At R4: substitution with a phenyl ring showed good inhibitor activity (Han et al., 2016b). Fusing with benzene slightly reduces its activity (Showalter et al., 1999). Replacing with the methyl group significantly decreases the inhibitory activity (Hossam et al., 2018; Lin et al., 2019).
The summary of the SAR is depicted in Figure 18.
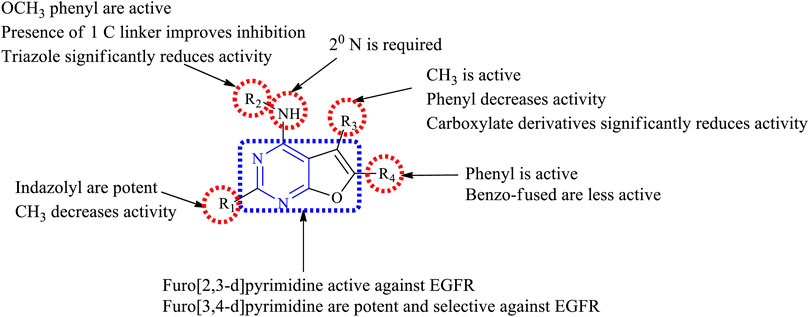
FIGURE 18. Summary SAR of furopyrimidine.
3.6 Pyrimidopyrimidines
Pyrimidine is a six-membered heterocycle that is fused with its counterpart pyrimidine with [4,5-d] fusion and [5,4-d] fusion. This fusion greatly influences the EGFR inhibitory activity of the compounds.
The 4-amino-6-substituted pyrimido [5,4-d]pyrimidine 269–276, Figure 19A) (EGFR IC50 2,550 nM for 269, 0.78 nM for 270, 0.81 nM for 271, 380 nM for 272, 3 nM for 273, 3 nM for 274–275)-based derivatives were synthesized and reported to have an anti-EGFR activity. These compounds were tested against the A431 cancer cell line for their antiproliferative activity, and the results indicated that they were potent with IC50 values in the range of 0.78–3 nM (Rewcastle et al., 1997; Solca et al., 2004). The nitrogen at position 5 resulted in enhancing its activity by increasing the interaction with the target (Rewcastle et al., 1997). Compounds 274 and 275 were studied further for their antitumor activity using the mice xenograft model. Compound 274 was found to be more effective in reducing the tumor size, and Western blot analysis revealed that it was selective in inhibiting EGFR phosphorylation (Solca et al., 2004).
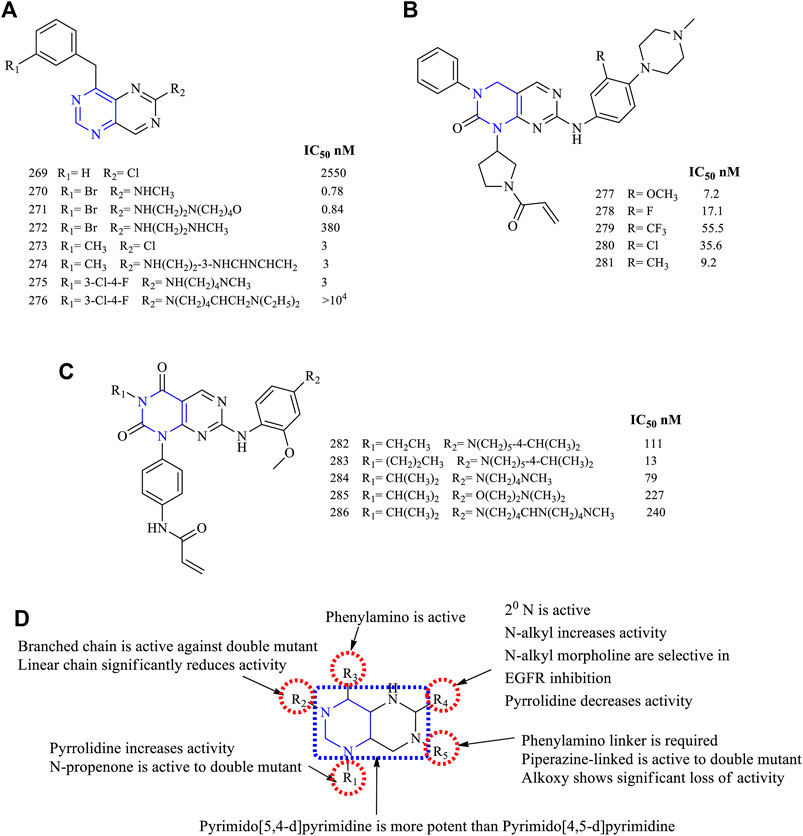
FIGURE 19. Chemical structure of (A) 4-amino-6-substituted pyrimido[5,4-d]pyrimidine, (B) 2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidine, (C) pyrimido[4,5-d] pyrimidin-2,4-(1H,3H)-dione, and (D) summary SAR of pyrimido[5,4-d]pyrimidine.
A series of 2-oxo-3,4-dihydropyrimido [4,5-d]pyrimidine (277–281, Figure 19B) (EGFR IC50 7.2 nM for 277, 17.1 nM for 278, 55.5 nM for 279, 35.6 nM for 280, and 9.2 nM for 281)-based derivatives were synthesized and its inhibitory properties were investigated. These compounds were found to inhibit EGFR in a range of IC50 7.2–35.6 nM. Also, it was found that the compound with a bulkier substituent CF3 was less potent than the others. Further, compound 280 was found to be better in terms of the pharmacokinetic properties and antitumor activities in the Sprague-Dawley rat model, whereas the Western blot analysis proved that 280 was an irreversible inhibitor of EGFR and resulted in the complete blockage of the EGFR pathway (Xu et al., 2013).
Based on the molecular hopping, mutant selective pyrimido [4,5-d] pyrimidin-2,4-(1H, 3H)-dione was synthesized (282–286, Figure 19C) (EGFR IC50 111 nM for 282, 13 nM for 283, 79 nM for 284, 227 nM for 285, and 240 nM for 286). The compounds were investigated against EGFRWT and EGFRL858R/T790M, and the results indicated that they were more selective to the mutant form. When compared using the reference AZD9291, the time of onset and duration of action were similar to 284, but in the A431 xenograft model, 286 showed less inhibition in tumor growth (Hao et al., 2018).
3.6.1 SAR of Pyrimidopyrimidines
Based on the available literature, pyrimidopyrimidines can undergo two types of fusion, mainly pyrimido [4,5-d] pyrimidine and pyrimido [5,4-d]pyrimidine. The following section gives the compilation of the SAR:
1) The fused pyrimido [5,4-d]pyrimidine was found to be more potent than the pyrimido [4,5-d]pyrimidine fused.
2) At R1: the presence of pyrrolidine helps in improving the solubility and pharmacokinetics of the compounds (Xu et al., 2013). Substitution with N-propenone makes the compound selective against double mutant EGFR (Hao et al., 2018).
3) At R2: the short alkyl chain of two to three carbons was investigated. The linear chains showed a significant loss in inhibitory activity, whereas the branched-chain (Hao et al., 2018) and phenyl substituents (Xu et al., 2013) were found to be potent against the double mutant.
4) At R3: the phenylamino is required for its anti-EGFR activity (Rewcastle et al., 1997; Solca et al., 2004).
5) At R4: chlorine decreases the activity (Rewcastle et al., 1997). The secondary N is required for the activity (Rewcastle et al., 1997; Solca et al., 2004). When N is a part of the heterocycle system, the activity decreases (Solca et al., 2004). The N-alkyl substituents were active. Also, N-alkylmorpholine substituents were selective in inhibiting EGFR, whereas alkyl diamino was less active (Rewcastle et al., 1997).
6) At R5: the phenylamino was linked with heterocycle piperidine, and the inhibitory activity decreased (Hao et al., 2018). Replacing piperidine with piperazine, the activity of compounds increased and they are more selective against the double mutant EGFR (Xu et al., 2013; Hao et al., 2018), whereas replacing with alkoxy resulted in significant loss of activity (Hao et al., 2018).
The summary of SAR is depicted in Figure 19D.
3.7 Pyrimidoindole
Indole is a heterocycle that consists of benzene fused with pyrrole. Based on the literature, pyrimidoindole was synthesized; also, the available literature suggests that pyrimidoindole is less active against EGFR.
A series of pyrimido [5,4-b] indol-4-amine (287–291, Figure 20A) (EGFR IC50 2,110 nM for 287, 72 nM for 288, 460 nM for 289, 419 nM for 290, and >105 nM for 291) derivatives were synthesized. These compounds were tested against the A431 cell lines, and studies showed that these molecules had inhibitory potential. These compounds further exhibited a significant loss of activity toward EGFR because of the lack of interaction with the target cell (Showalter et al., 1999). Similarly, N-phenylpyrimido [5,4-b] indol-4-amine (292–297, Figure 20B) (EGFR IC50 31 nM for 292, 742 nM for 293, 4,100 nM for 294, >104 nM for 295, 147 nM for 296, and 1.2 nM for 297)-based derivatives were also synthesized. Among them, compound 297 was the only potent molecule found (IC50 1.2 nM), whereas others lacked activity toward EGFR. This loss of activity was due to the presence of an electron-withdrawing group present in the ring (Showalter et al., 1999).
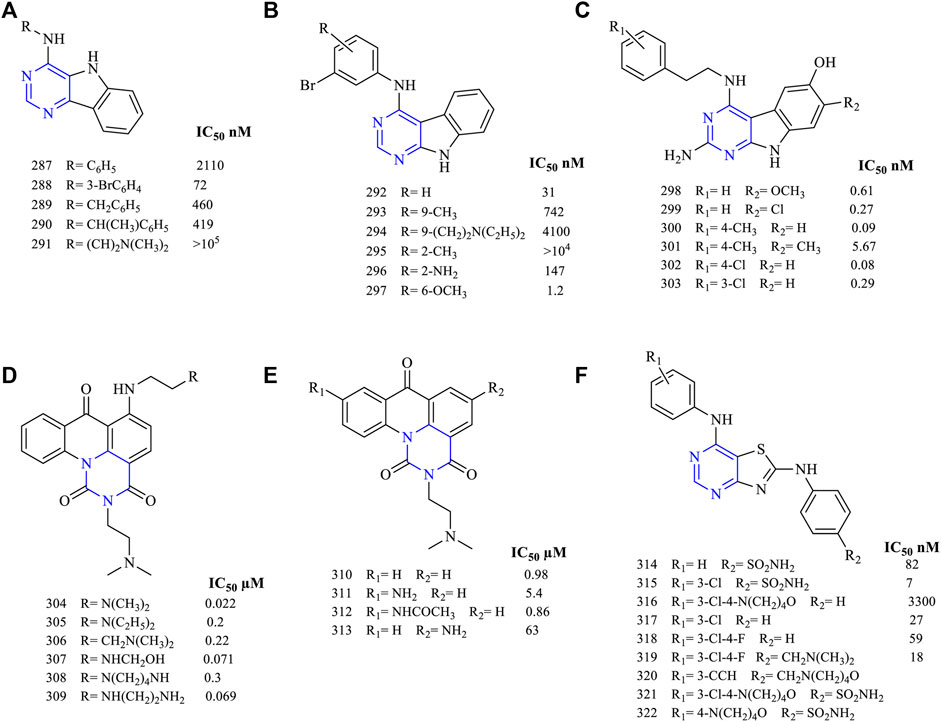
FIGURE 20. Chemical structures of (A) pyrimido [5,4-b]indol-4-amine, (B) N-phenylpyrimido [5,4-b]indol-4-amine, (C) 2-amino-4-(phenylethylamino) pyrimido [4,5-b]indole, (D) 6-substituted pyrimido [5,6,1-de]acridin-1,3,7-trione, (E) 9-substituted pyrimido [5,6,1-de]acridin-1,3,7-trione), and (F) 4,6-diaminothiazolo [4,5-d]pyrimidine.
The compound 2-amino-4-(phenylethylamino) pyrimido [4,5-b] indole (298–303, Figure 20C) (EGFR IC50 0.61 nM for 298, 0.27 nM for 299, 0.09 nM for 300, 5.67 nM for 301, 0.08 nM for 302, and 0.29 nM for 303) has been designed and studied. These compounds were found to inhibit EGFR in the nanomolar range, making them very potent and selective in inhibition. Further, compounds 300 and 302 were found to be active against VEGFR2. The growth inhibition of 298 against MDA-MB-231 cell lines is 34%, whereas that for 300 is 65%. These compounds were highly active due to the presence of the phenylamino substituent in the fourth position (Fischer et al., 2017).
3.8 Pyrimido Acridine
Acridine was fused with pyrimidines, and the inhibitory potential was determined against the EGFR receptor. In order to inhibit DNA replication, 6-substituted pyrimido [5,6,1-de] acridin-1,3,7-trione (304–309, Figure 20D) (HT29 IC50,0.022 µM for 304, 0.2 µM for 305, 0.22 µM for 306, 0.071 µM for 307, 0.3 µM for 308, and 0.069 µM for 309) has been designed. They have been tested for their antitumor activity against HT29 and LoVo/DX resistant cell lines. These compounds were found to be effective in inhibiting both the cell lines, whereas 302 was the most potent against HT29 (IC50 0.022 µM) and LoVo/DX (IC50 0.029 µM). Further, the cytotoxicity studies revealed that the basic side chain was essential for its antitumor activity. Also, the presence of hydrogen bonding between the 4-N and 5-O improved the cytotoxicity profile and DNA binding (Antonini, 2002).
Further, it was reported that the 9-substituted pyrimido [5,6,1-de] acridin-1,3,7-trione (310–313 Figure 20E) (P388 IC50 0.98 µM for 310, 5.4 µM for 311, 0.86 µM for 312, and 63 µM for 313) derivatives were tested for their cytotoxicity against various cancer cell lines. Based on the results, the substitution at the ninth position exhibited potent to weak activity. Further in vivo studies were carried out using P388 leukemia in mice model, and 310 was found to be capable of increasing the life expectancy at a dose of 25 mg/kg/day but the animals died due to toxicity at such a high dose (Kamata et al., 2004). But the acridine-based derivatives were not recommended anymore to their high toxic nature.
3.9 Thiazolopyrimidines
Thiazole is a five-membered heterocycle consisting of heteroatoms sulfur and nitrogen. Based on the literature, thiazole is fused with pyrimidine by [4,5-d] fusion. Using molecular dynamics and molecular modeling studies, 4,6-diaminothiazolo [4,5-d]pyrimidine (314–322, Figure 20F) (IC50 82 nM for 314, 7 nM for 315, 3,300 nM for 316, 27 nM for 317, 59 nM for 318, 15 nM for 319, 21 nM for 320) (Dong et al., 2011; Mitrasinovic, 2014) were designed and studied for their anti-EGFR inhibition. Molecules 314–318 have been studied using docking analysis and were found to inhibit the ATP-binding site of EGFR. The SAR study further revealed that the 3′ and 4’ positions on R2 were beneficial in binding and the presence of chlorine enhanced its selectivity (Dong et al., 2011). Molecules 321 and 322 were structurally similar except with the chlorine substitution, but results indicated a difference in the activity of both the compounds. Also, it was found that 321 was a non-specific intercalator, whereas 322 was an intercalator and grove inhibitor as compared to lapatinib (Mitrasinovic, 2014).
3.10 Miscellaneous Pyrimidine-Based EGFR Inhibitor
This section provides details of the pyrimidine-based EGFR inhibitors that are not fused with a heterocyclic system. The literature suggests that the NH group on the pyrimidine ring is an important substituent for exhibiting EGFR inhibitory activity.
A series of 4-amino-5-((2-methoxyphenyl) amino)-6-(4-substituted) phenylpyrimidin-2(1H)-one (323–326, Figure 21A) have been synthesized and studied for the EGFR inhibitory activity (EGFRWT IC50: 0.087 ± 0.013 µM for 323, 0.11 ± 0.014 µM for 324). It was reported that these molecules were cytotoxic when analyzed using the MCF-7 cell line (0.01 ± 0.003 µM for 323, 0.02 ± 0.001 µM for 324, 0.04 ± 0.04 µM for 325, 0.08 ± 0.02 µM for 326, and 0.42 ± 0.2 µM for erlotinib, which was used as standard). The EGFR kinase inhibitory activity revealed that the molecules have displayed very little activity in inhibiting the EGFRWT (Othman et al., 2021).
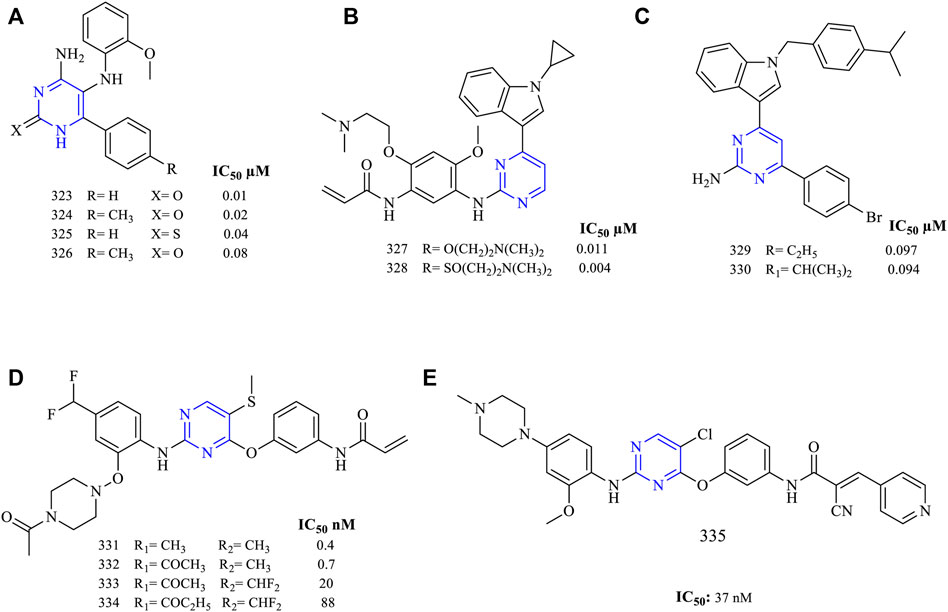
FIGURE 21. Chemical structures of (A) 4-amino-5-((2-methoxyphenyl)amino)-6-(4-substituted) phenylpyrimidin-2(1H)-one, (B) N-(5-((4-(1-cyclopropyl-1H-indol-3-yl)pyrimidin-2-yl)amino)-2-(substituted)-4-methoxyphenyl)acrylamide, (C) 4-(4-bromophenyl)-6-(1-(substituted)-1H-indol-3-yl)pyrimidin-2-amine, (D) N-(3-((2-((4-substituted methyl-2-((4-substituted piperazin-1-yl)oxy)phenyl)amino)-5-(methylthio)pyrimidin-4-yl)oxy)phenyl)acrylamide, and (E) N-(3-((5-chloro-2-((2-methoxy-4-(4-methylpiperazin-1-yl)phenyl)amino)pyrimidin-4-yl)oxy)phenyl)-2-cyano-3-(pyridin-4-yl)acrylamide.
Another series of N-(5-((4-(1-cyclopropyl-1H-indol-3-yl)pyrimidin-2-yl)amino)-2-(substituted)-4-methoxyphenyl)acrylamide (327–328, Figure 21B) have been synthesized and their EGFR inhibitory potential was determined (H1975 IC50: 0.011 ± 0.001 µM for 327, 0.004 ± 0.002 µM for 328, and 0.01 ± 0.003 µM for erlotinib). The presence of the S atom in 328 has resulted in displaying a selectivity of 15.8 times toward EGFRWT as compared to the erlotinib with a selectivity of 6.5. This inhibitory activity was a result of the presence of an indole ring in the molecule (Li et al., 2021).
A novel series of substituted indole were designed, as shown in Figure 21C (329–330). Their IC50 values were determined by using EGFRWT (0.097 µM for 329, 0.094 µM for 330, and 0.9 µM for erlotinib). It was found that the molecules with two to three carbons in the substituent side chain of indole have exhibited higher EGFR inhibitory activity. Moreover, their EGFR kinase activity was found to be similar to that of erlotinib (Singh and Silakari, 2018) and hence such molecules are better antiproliferative agents rather than potent EGFR inhibitors.
A series of trisubstituted pyrimidine have been designed (Figures 21D,E). Both series differ in the position of the substituent, whereas the parent structure remains constant (EGFRL858R/T790M IC50 values: 0.4 nM for 331, 0.7 nM for 332, 20 nM for 333, 88 nM for 334, and 37 nM for 335). The higher EGFR inhibitory activity found for compounds 331 and 332 was due to the presence of “S” in the structure, whereas replacing “S” to “Cl” results in a drastic decrease in the EGFR inhibitory activity. This is due to the formation of stronger hydrophobic interactions with the MET790 residue present at the binding site of the protein. Moreover, the presence of the Michael acceptor helps improve the activity of the pyrimidine derivatives containing a heterocycle. These attributes have resulted in molecules that are selective in the inhibition of EGFR, as determined by using the Western blot analysis (Basu et al., 2015; Xiao et al., 2016).
3.11 Computational Aspects of Fused Pyrimidine as EGFR Inhibitor
Pyrimidine is essential for binding interaction with the ligand-binding site, as the ring fits into the ATP binding site forming H-bond with the MET793 residue (Aboulwafa et al., 2020). The fused-pyrimidine system consisting of an aromatic substitution results in the π–π interaction between the phenyl ring and the aromatic or hetero-aromatic amino acid residue. The presence of an electron-donating group on the pyrimidine ring is an important pharmacophore feature of the fused-pyrimidine to exhibit EGFR inhibitory activity. Moreover, the presence of the Michael acceptor results in the formation of a covalent bond to CYS773 present in the ATP binding site (Li and Li, 2014). See Figure 22.
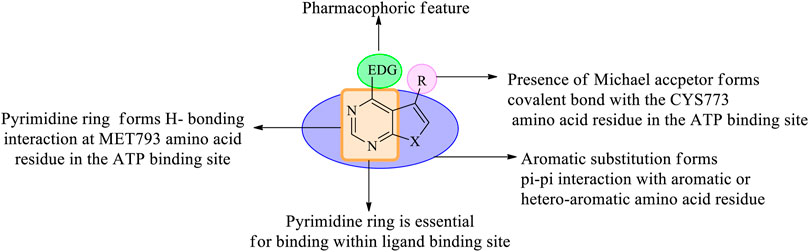
FIGURE 22. Computational aspects of fused pyrimidine as EGFR inhibitor.
3.12 Mechanism of Pyrimidine-Based Hybrids in Inhibiting EGFR
From the in vitro studies, it is obvious that the pyrimidine derivatives display good inhibitory potential in inhibiting EGFR’s overexpression. The pyrimidine-fused hybrids mainly act on the mutation that takes place at L858R and T790M as mentioned above in this review, but their activities vary greatly with the structure and various changes in the heterocyclic ring and their respective substitution (see Figure 23). EGFR’s overexpression is a result of a mutation in the exon at the 20th position (T790M) and 21st position (L858R). The pyrimidine hybrids act on these two positions with different potencies. Furan shows inhibitory activity not only to single mutant L858R but also to the double mutant T790M and L858R. Pyridine, thiophene, pyrrole, pyrimidine, acridone, pyrazole, indole, and thiazole are active against the double mutant T790M and L858R. Pyridine is the irreversible inhibitor, whereas pyrimidine is a selective inhibitor.
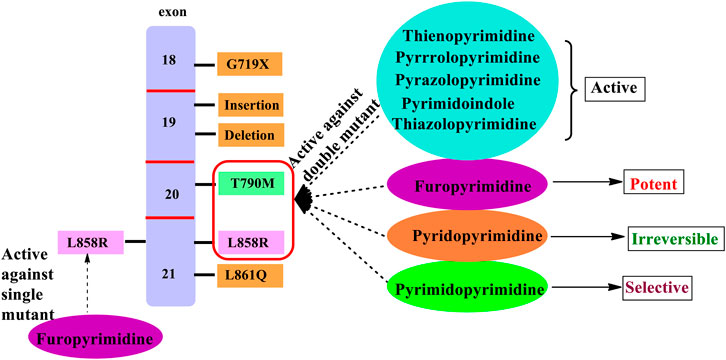
FIGURE 23. Mechanism of pyrimidine-based hybrids in inhibiting EGFR. The mutation mainly occurs at the T790M and L858R exons. From the known chemistry, various hybrids act differently on this mutated exon. Furopyrimidine shows inhibitory activity not only to L858R but also to the double mutant T790M and L858R. Pyridopyrimidine, thienopyrimidine, pyrrolopyrimidine, pyrimidopyrimidine, pyrimidoacridone, pyrazolopyrimidine, pyrimidoindole, and thiazolopyrimidine are active against the double mutant T790M and L858R. Pyridopyrimidine is the irreversible inhibitor, whereas pyrimidopyrimidine is a selective inhibitor.
4 Conclusion
EGFR is an important receptor that controls the cell growth and its proliferation. The uncontrolled downstream signaling of EGFR is the main cause underlying for uncontrolled cell growth leading to NSCLC. Various EGFR inhibitors like lazertinib, avitinib, nazartinib, osimertinib, erlotinib, gefitinib, afatinib, and lapatinib have been approved. They reduce the downstream signaling by inhibiting the PTK, and hence, there’s reduction in the cell proliferation. Based on the pharmacophoric features and SAR analysis, various researchers have tried exploring various chemical moieties for inhibiting EGFR. The most widely studied chemical structure is a pyrimidine. This review mainly focuses on the pyrimidine-fused heterocycle system. Pyridine, pyrrole, thiophene, pyrazole, furan, indole, acridone, thiazole, and pyrimidine are among these heterocycles. Certain heterocycles (pyridine, pyrrole, furan, and pyrazole) show stronger effects toward EGFR inhibition, while pyrrole and pyrazole have good potency against ErbB2, making them dual inhibitors, based on their inhibitory values and kinase studies. However, it is clear that thiophene, indole, and thiazole have a lower inhibitory potential for EGFR. In comparison to the merely substituted pyrimidine, the EGFR inhibitory efficacy of pyrimidine-based heterocyclic hybrids differs dramatically. It is also reported that pyrimidine hybrids shows inhibition toward EGFR and CDK. Therefore, the selectivity aspects come into picture. Selectivity in inhibiting EGFR is due to the presence of O in furan, S in thieophene, N in thiazole and indole, acridone (tricyclic ring system). Pyrimidine fused with such monocyclic heterocycle system and containing N, namely, pyrazole, pyrrole, pyrimidine, and pyridine, shows a dual inhibitory activity toward EGFR and CDK. Furthermore, because pyrimidine-based heterocyclic hybrid inhibitors have been proven to be very hazardous in in vivo investigations, it is critical to understand their toxicity profiles. SAR is an important tool in designing the molecules. Based on the review, it is quite evident that the studies should aim at bringing newer and better molecules using the available SAR of such pyrimidine analogs. Moreover, they should be thoroughly investigated for acute and chronic toxicity before being considered as potential EGFR inhibitors in the future. Using SAR of compounds will aid the researchers in the better designing of the medicinal molecules and greater insights into the development of powerful molecules with higher activity and fewer side effects, hence increasing the quality of life of patients with cancer.
Author Contributions
TTY and GMS: They have contributed equally to writing the manuscript, MSK, MC and MYC: They have reviewed the manuscript and suggested necessary changes in the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors are grateful to SVKM’s NMIMS University, Mumbai, India, for providing the necessary facilities for writing this review article.
Reference
Abbas, S. E.-S., Aly, E. I., Awadallah, F. M., and Mahmoud, W. R. (2015). 4-Substituted-1-phenyl-1H-pyrazolo[3,4-d]pyrimidine Derivatives: Design, Synthesis, Antitumor and EGFR Tyrosine Kinase Inhibitory Activity. Chem. Biol. Drug Des. 85, 608–622. doi:10.1111/cbdd.12451
Abbas, S. E. S., George, R. F., Samir, E. M., Aref, M. M. A., and Abdel-Aziz, H. A. (2019). Synthesis and Anticancer Activity of Some Pyrido[2,3-d]Pyrimidine Derivatives as Apoptosis Inducers and Cyclin-Dependent Kinase Inhibitors. Future. Med. Chem. 11, 2395–2414. doi:10.4155/fmc-2019-0050
Abdelgalil, A. A., Al-Kahtani, H. M., and Al-Jenoobi, F. I. (2020). Erlotinib. Profiles Drug Subst. Excipients Relat. Methodol. 45, 93–117. doi:10.1016/bs.podrm.2019.10.004
Abdelgawad, M. A., Bakr, R. B., Alkhoja, O. A., and Mohamed, W. R. (2016). Design, Synthesis and Antitumor Activity of Novel Pyrazolo[3,4-D]pyrimidine Derivatives as EGFR-TK Inhibitors. Bioorg. Chem. 66, 88–96. doi:10.1016/j.bioorg.2016.03.011
Abdelgawad, M. A., Bakr, R. B., and Azouz, A. A. (2018). Novel Pyrimidine-Pyridine Hybrids: Synthesis, Cyclooxygenase Inhibition, Anti-inflammatory Activity and Ulcerogenic Liability. Bioorg. Chem. 77, 339–348. doi:10.1016/j.bioorg.2018.01.028
Abdellatif, K. R. A., and Bakr, R. B. (2021). Pyrimidine and Fused Pyrimidine Derivatives as Promising Protein Kinase Inhibitors for Cancer Treatment. Med. Chem. Res. 30, 31–49. doi:10.1007/s00044-020-02656-8
Aboulwafa, O. M., Daabees, H. M. G., and Badawi, W. A. (2020). 2-Anilinopyrimidine Derivatives: Design, Synthesis, In Vitro Anti-proliferative Activity, EGFR and ARO Inhibitory Activity, Cell Cycle Analysis and Molecular Docking Study. Bioorg. Chem. 99, 103798. doi:10.1016/j.bioorg.2020.103798
Antonini, I. (2002). DNA-binding Antitumor Agents: from Pyrimido[5,6,1-De]acridines to Other Intriguing Classes of Acridine Derivatives. cmc 9, 1701–1716. doi:10.2174/0929867023369268
Aziz, Y. M. A., Said, M. M., El Shihawy, H. A., Tolba, M. F., and Abouzid, K. A. M. (2015). Discovery of Potent Antiproliferative Agents Targeting EGFR Tyrosine Kinase Based on the Pyrido[3′,2′:4,5]thieno[3,2-D]pyrimidin-4-Amine Scaffold. Chem. Pharm. Bull. 63, 1015–1028. doi:10.1248/cpb.c15-00592
Barvian, M., Boschelli, D. H., Cossrow, J., Dobrusin, E., Fattaey, A., Fritsch, A., et al. (2000). Pyrido[2,3-d]Pyrimidin-7-One Inhibitors of Cyclin-Dependent Kinases. J. Med. Chem. 43, 4606–4616. doi:10.1021/jm000271k
Basu, D., Richters, A., and Rauh, D. (2015). Structure-based Design and Synthesis of Covalent-Reversible Inhibitors to Overcome Drug Resistance in EGFR. Bioorg. Med. Chem. 23, 2767–2780. doi:10.1016/j.bmc.2015.04.038
Beckers, T., Sellmer, A., Eichhorn, E., Pongratz, H., Schächtele, C., Totzke, F., et al. (2012). Novel Inhibitors of Epidermal Growth Factor Receptor: (4-(Arylamino)-7h-Pyrrolo[2,3-D]pyrimidin-6-yl)(1H-Indol-2-Yl)methanones and (1H-Indol-2-Yl)(4-(phenylamino)thieno[2,3-D]pyrimidin-6-Yl)methanones. Bioorg. Med. Chem. 20, 125–136. doi:10.1016/j.bmc.2011.11.023
Beyzaei, H., Aryan, R., Moghaddam-Manesh, M., Ghasemi, B., Karimi, P., Samareh Delarami, H., et al. (2017). Evaluation and Structure-Activity Relationship Analysis of a New Series of 4-Imino-5h-Pyrazolo[3,4-D]pyrimidin-5-Amines as Potential Antibacterial Agents. J. Mol. Struct. 1144, 273–279. doi:10.1016/j.molstruc.2017.05.050
Bugge, S., Buene, A. F., Jurisch-Yaksi, N., Moen, I. U., Skjønsfjell, E. M., Sundby, E., et al. (2016). Extended Structure-Activity Study of Thienopyrimidine-Based EGFR Inhibitors with Evaluation of Drug-like Properties. Eur. J. Med. Chem. 107, 255–274. doi:10.1016/j.ejmech.2015.11.012
Bugge, S., Moen, I. U., Kragseth Sylte, K.-O., Sundby, E., and Hoff, B. H. (2015). Truncated Structures Used in Search for New Lead Compounds and in a Retrospective Analysis of Thienopyrimidine-Based EGFR Inhibitors. Eur. J. Med. Chem. 94, 175–194. doi:10.1016/j.ejmech.2015.03.004
Caballero, J., Fernández, M., and González-Nilo, F. D. (2008). Structural Requirements of Pyrido[2,3-d]Pyrimidin-7-One as CDK4/D Inhibitors: 2D Autocorrelation, CoMFA and CoMSIA Analyses. Bioorg. Med. Chem 16, 6103–6115. doi:10.1016/j.bmc.2008.04.048
Chen, Z., Fillmore, C. M., Hammerman, P. S., Kim, C. F., and Wong, K.-K. (2014). Non-small-cell Lung Cancers: a Heterogeneous Set of Diseases. Nat. Rev. Cancer 14, 535–546. doi:10.1038/nrc3775
Cheng, H., Nair, S. K., Murray, B. W., Almaden, C., Bailey, S., Baxi, S., et al. (2016). Discovery of 1-{(3r,4r)-3-[({5-Chloro-2-[(1-Methyl-1h-Pyrazol-4-Yl)amino]-7h-Pyrrolo[2,3-D]pyrimidin-4-Yl}oxy)methyl]-4-Methoxypyrrolidin-1-Yl}prop-2-En-1-One (PF-06459988), a Potent, WT Sparing, Irreversible Inhibitor of T790M-Containing EGFR Mutants. J. Med. Chem. 59, 2005–2024. doi:10.1021/acs.jmedchem.5b01633
Cockerill, S., Stubberfield, C., Stables, J., Carter, M., Guntrip, S., Smith, K., et al. (2001). Indazolylamino Quinazolines and Pyridopyrimidines as Inhibitors of the EGFr and C-erbB-2. Bioorg. Med. Chem. Lett. 11, 1401–1405. doi:10.1016/S0960-894X(01)00219-0
Collins, D. M., Conlon, N. T., Kannan, S., Verma, C. S., Eli, L. D., Lalani, A. S., et al. (2019). Preclinical Characteristics of the Irreversible Pan-HER Kinase Inhibitor Neratinib Compared with Lapatinib: Implications for the Treatment of HER2-Positive and HER2-Mutated Breast Cancer. Cancers 11, 737–764. doi:10.3390/cancers11060737
Devambatla, R. K. V., Choudhary, S., Ihnat, M., Hamel, E., Mooberry, S. L., and Gangjee, A. (2018). Design, Synthesis and Preclinical Evaluation of 5-Methyl-N4-Aryl-Furo[2,3-D]pyrimidines as Single Agents with Combination Chemotherapy Potential. Bioorg. Med. Chem. Lett. 28, 3085–3093. doi:10.1016/j.bmcl.2018.07.039
Dong, B.-L., Liao, Q.-H., and Wei, J. (2011). Docking and Molecular Dynamics Study on the Inhibitory Activity of N, N-Disubstituted-Trifluoro-3-Amino-2-Propanols-Based Inhibitors of Cholesteryl Ester Transfer Protein. J. Mol. Model 17, 1727–1734. doi:10.1007/s00894-010-0881-7
Downward, J., Yarden, Y., Mayes, E., Scrace, G., Totty, N., Stockwell, P., et al. (1984). Close Similarity of Epidermal Growth Factor Receptor and V-Erb-B Oncogene Protein Sequences. Nature 307, 521–527. doi:10.1038/307521a0
El-Hamouly, W. S., El-Khamry, A.-M. A., and Abbas, E. M. H. (2007). Synthesis of New 4-Aryl-Isoxazolo[5,4-D]pyrimidin-6-One(thione) and 4-Aryl-Pyrazolo[3,4-D]pyrimidin-6-One Derivatives of Potential Antihypertensive Activity. ChemInform 38, 2091–2098. doi:10.1002/chin.200703140
El-Moghazy, S. M., Ibrahim, D. A., Abdelgawad, N. M., Farag, N. A. H., and El-Khouly, A. S. (2011). Design, Synthesis and Biological Evaluation of Novel Pyrimido[4,5-D]pyrimidine CDK2 Inhibitors as Anti-tumor Agents. Sci. Pharm. 79, 429–447. doi:10.3797/scipharm.1103-16
Elzahabi, H. S. A., Nossier, E. S., Khalifa, N. M., Alasfoury, R. A., and El-Manawaty, M. A. (2018). Anticancer Evaluation and Molecular Modeling of Multi-Targeted Kinase Inhibitors Based Pyrido[2,3-D]pyrimidine Scaffold. J. Enzyme Inhibition Med. Chem. 33, 546–557. doi:10.1080/14756366.2018.1437729
Engel, J., Smith, S., Lategahn, J., Tumbrink, H. L., Goebel, L., Becker, C., et al. (2017). Structure-Guided Development of Covalent and Mutant-Selective Pyrazolopyrimidines to Target T790M Drug Resistance in Epidermal Growth Factor Receptor. J. Med. Chem. 60, 7725–7744. doi:10.1021/acs.jmedchem.7b00515
Fang, Y., Xu, J., Li, Z., Yang, Z., Xiong, L., Jin, Y., et al. (2018). Design and Synthesis of Novel Pyrimido[5,4-D]pyrimidine Derivatives as GPR119 Agonist for Treatment of Type 2 Diabetes. Bioorg. Med. Chem. 26, 4080–4087. doi:10.1016/j.bmc.2018.06.035
Fischer, T., Krüger, T., Najjar, A., Totzke, F., Schächtele, C., Sippl, W., et al. (2017). Discovery of Novel Substituted Benzo-Anellated 4-benzylamino Pyrrolopyrimidines as Dual EGFR and VEGFR2 Inhibitors. Bioorg. Med. Chem. Lett. 27, 2708–2712. doi:10.1016/j.bmcl.2017.04.053
Gaber, A. A., Bayoumi, A. H., El-morsy, A. M., Sherbiny, F. F., Mehany, A. B. M., and Eissa, I. H. (2018). Design, Synthesis and Anticancer Evaluation of 1H-Pyrazolo[3,4-D]pyrimidine Derivatives as Potent EGFRWT and EGFRT790M Inhibitors and Apoptosis Inducers. Bioorg. Chem. 80, 375–395. doi:10.1016/j.bioorg.2018.06.017
Galmarini, C. M., Jordheim, L., and Dumontet, C. (2003). Pyrimidine Nucleoside Analogs in Cancer Treatment. Expert Rev. Anticancer Ther. 3, 717–728. doi:10.1586/14737140.3.5.717
Gan, H. K., Cvrljevic, A. N., and Johns, T. G. (2013). The Epidermal Growth Factor Receptor Variant III (EGFRvIII): where Wild Things Are Altered. Febs J. 280, 5350–5370. doi:10.1111/febs.12393
Gangjee, A., Kurup, S., Ihnat, M. A., Thorpe, J. E., and Disch, B. (2012). N4-Aryl-6-substitutedphenylmethyl-7H-pyrrolo[2,3-d]pyrimidine-2,4-diamines as Receptor Tyrosine Kinase Inhibitors. Bioorg. Med. Chem. 20, 910–914. doi:10.1016/j.bmc.2011.11.058
Gangjee, A., Namjoshi, O. A., Ihnat, M. A., and Buchanan, A. (2010a). The Contribution of a 2-amino Group on Receptor Tyrosine Kinase Inhibition and Antiangiogenic Activity in 4-anilinosubstituted Pyrrolo[2,3-D]pyrimidines. Bioorg. Med. Chem. Lett. 20, 3177–3181. doi:10.1016/j.bmcl.2010.03.064
Gangjee, A., Namjoshi, O. A., Yu, J., Ihnat, M. A., Thorpe, J. E., and Warnke, L. A. (2008). Design, Synthesis and Biological Evaluation of Substituted Pyrrolo[2,3-D]pyrimidines as Multiple Receptor Tyrosine Kinase Inhibitors and Antiangiogenic Agents. Bioorg. Med. Chem. 16, 5514–5528. doi:10.1016/j.bmc.2008.04.019
Gangjee, A., Zhao, Y., Raghavan, S., Ihnat, M. A., and Disch, B. C. (2010b). Design, Synthesis and Evaluation of 2-Amino-4-M-Bromoanilino-6-Arylmethyl-7h-Pyrrolo[2,3-D]pyrimidines as Tyrosine Kinase Inhibitors and Antiangiogenic Agents1. Bioorg. Med. Chem. 18, 5261–5273. doi:10.1016/j.bmc.2010.05.049
Garg, G., Pande, M., Agrawal, A., Li, J., and Kumar, R. (2016). Investigation of 4-Amino-5-Alkynylpyrimidine-2(1h)-Ones as Anti-mycobacterial Agents. Bioorg. Med. Chem. 24, 1771–1777. doi:10.1016/j.bmc.2016.03.003
Giaccone, G. (2004). The Role of Gefitinib in Lung Cancer Treatment. Clin. Cancer Res. 10, 4233s–4237s. doi:10.1158/1078-0432.CCR-040005
Han, J., Henriksen, S., Nørsett, K. G., Sundby, E., and Hoff, B. H. (2016a). Balancing Potency, Metabolic Stability and Permeability in Pyrrolopyrimidine-Based EGFR Inhibitors. Eur. J. Med. Chem. 124, 583–607. doi:10.1016/j.ejmech.2016.08.068
Han, J., Kaspersen, S. J., Nervik, S., Nørsett, K. G., Sundby, E., and Hoff, B. H. (2016b). Chiral 6-Aryl-Furo[2,3-D]pyrimidin-4-Amines as EGFR Inhibitors. Eur. J. Med. Chem. 119, 278–299. doi:10.1016/j.ejmech.2016.04.054
Hanan, E. J., Baumgardner, M., Bryan, M. C., Chen, Y., Eigenbrot, C., Fan, P., et al. (2016). 4-Aminoindazolyl-dihydrofuro[3,4- D ]pyrimidines as Non-covalent Inhibitors of Mutant Epidermal Growth Factor Receptor Tyrosine Kinase. Bioorg. Med. Chem. Lett. 26, 534–539. doi:10.1016/j.bmcl.2015.11.078
Hao, Y., Lyu, J., Qu, R., Tong, Y., Sun, D., Feng, F., et al. (2018). Design, Synthesis, and Biological Evaluation of Pyrimido[4,5- D]pyrimidine-2,4(1 H,3 H)-diones as Potent and Selective Epidermal Growth Factor Receptor (EGFR) Inhibitors against L858R/T790M Resistance Mutation. J. Med. Chem. 61, 5609–5622. doi:10.1021/acs.jmedchem.8b00346
Harvey, R. D., Adams, V. R., Beardslee, T., and Medina, P. (2020). Afatinib for the Treatment of EGFR Mutation-Positive NSCLC: A Review of Clinical Findings. J. Oncol. Pharm. Pract. 26, 1461–1474. doi:10.1177/1078155220931926
Horchani, M., Della Sala, G., Caso, A., D’Aria, F., Esposito, G., Laurenzana, I., et al. (2021). Molecular Docking and Biophysical Studies for Antiproliferative Assessment of Synthetic Pyrazolo-Pyrimidinones Tethered with Hydrazide-Hydrazones. Ijms 22, 2742–2762. doi:10.3390/ijms22052742
Hossam, M., Lasheen, D. S., Ismail, N. S. M., Esmat, A., Mansour, A. M., Singab, A. N. B., et al. (2018). Discovery of Anilino-Furo[2,3- D ]pyrimidine Derivatives as Dual Inhibitors of EGFR/HER2 Tyrosine Kinase and Their Anticancer Activity. Eur. J. Med. Chem. 144, 330–348. doi:10.1016/j.ejmech.2017.12.022
Hou, J., Wan, S., Wang, G., Zhang, T., Li, Z., Tian, Y., et al. (2016). Design, Synthesis, Anti-tumor Activity, and Molecular Modeling of Quinazoline and Pyrido[2,3-D]pyrimidine Derivatives Targeting Epidermal Growth Factor Receptor. Eur. J. Med. Chem. 118, 276–289. doi:10.1016/j.ejmech.2016.04.026
Hubbard, R. D., Dickerson, S. H., Emerson, H. K., Griffin, R. J., Reno, M. J., Hornberger, K. R., et al. (2008). Dual EGFR/ErbB-2 Inhibitors from Novel Pyrrolidinyl-Acetylenic Thieno[3,2-D]pyrimidines. Bioorg. Med. Chem. Lett. 18, 5738–5740. doi:10.1016/j.bmcl.2008.09.090
Ishikawa, T., Seto, M., Banno, H., Kawakita, Y., Oorui, M., Taniguchi, T., et al. (2011). Design and Synthesis of Novel Human Epidermal Growth Factor Receptor 2 (HER2)/epidermal Growth Factor Receptor (EGFR) Dual Inhibitors Bearing a Pyrrolo[3,2-D]pyrimidine Scaffold. J. Med. Chem. 54, 8030–8050. doi:10.1021/jm2008634
Ismail, M., El-Sayed, N., Rateb, H., Ellithey, M., and Ammar, Y. (2006). Synthesis and Evaluation of Some 1,2,3,4-Tetrahydropyrimidine-2-Thione and Condensed Pyrimidine Derivatives as Potential Antihypertensive Agents. Arzneimittelforschung 56, 322–327. doi:10.1055/s-0031-1296729
Ji, X., Peng, T., Zhang, X., Li, J., Yang, W., Tong, L., et al. (2014). Design, Synthesis and Biological Evaluation of Novel 6-alkenylamides Substituted of 4-Anilinothieno[2,3-D]pyrimidines as Irreversible Epidermal Growth Factor Receptor Inhibitors. Bioorg. Med. Chem. 22, 2366–2378. doi:10.1016/j.bmc.2014.01.035
Jing, T., Miao, X., Jiang, F., Guo, M., Xing, L., Zhang, J., et al. (2018). Discovery and Optimization of Tetrahydropyrido[4,3-D]pyrimidine Derivatives as Novel ATX and EGFR Dual Inhibitors. Bioorg. Med. Chem. 26, 1784–1796. doi:10.1016/j.bmc.2018.02.023
Jorda, R., Havlíček, L., Šturc, A., Tušková, D., Daumová, L., Alam, M., et al. (2019). 3,5,7-Substituted Pyrazolo[4,3-D]pyrimidine Inhibitors of Cyclin-dependent Kinases and Their Evaluation in Lymphoma Models. J. Med. Chem. 62, 4606–4623. doi:10.1021/acs.jmedchem.9b00189
Kamata, J., Okada, T., Kotake, Y., Niijima, J., Nakamura, K., Uenaka, T., et al. (2004). Synthesis and Evaluation of Novel Pyrimido-Acridone, -phenoxadine, and -carbazole as Topoisomerase II Inhibitors. Chem. Pharm. Bull. 52, 1071–1081. doi:10.1248/cpb.52.1071
Kaspersen, S. J., Han, J., Nørsett, K. G., Rydså, L., Kjøbli, E., Bugge, S., et al. (2014). Identification of New 4-N-Substituted 6-Aryl-7h-Pyrrolo[2,3-D]pyrimidine-4-Amines as Highly Potent EGFR-TK Inhibitors with Src-Family Activity. Eur. J. Pharm. Sci. 59, 69–82. doi:10.1016/j.ejps.2014.04.011
Kaspersen, S. J., Sørum, C., Willassen, V., Fuglseth, E., Kjøbli, E., Bjørkøy, G., et al. (2011). Synthesis and In Vitro EGFR (ErbB1) Tyrosine Kinase Inhibitory Activity of 4-N-Substituted 6-Aryl-7h-Pyrrolo[2,3-D]pyrimidine-4-Amines. Eur. J. Med. Chem. 46, 6002–6014. doi:10.1016/j.ejmech.2011.10.012
Kawakita, Y., Banno, H., Ohashi, T., Tamura, T., Yusa, T., Nakayama, A., et al. (2012a). Design and Synthesis of Pyrrolo[3,2-D]pyrimidine Human Epidermal Growth Factor Receptor 2 (HER2)/Epidermal Growth Factor Receptor (EGFR) Dual Inhibitors: Exploration of Novel Back-Pocket Binders. J. Med. Chem. 55, 3975–3991. doi:10.1021/jm300185p
Kawakita, Y., Miwa, K., Seto, M., Banno, H., Ohta, Y., Tamura, T., et al. (2012b). Design and Synthesis of Pyrrolo[3,2-D]pyrimidine HER2/EGFR Dual Inhibitors: Improvement of the Physicochemical and Pharmacokinetic Profiles for Potent In Vivo Anti-tumor Efficacy. Bioorg. Med. Chem. 20, 6171–6180. doi:10.1016/j.bmc.2012.08.002
Khandazhinskaya, A., Alexandrova, L., Matyugina, E., Solyev, P., Efremenkova, O., Buckheit, K., et al. (2018). Novel 5′-Norcarbocyclic Pyrimidine Derivatives as Antibacterial Agents. Molecules 23, 3069–3087. doi:10.3390/molecules23123069
Kim, D. C., Lee, Y. R., Yang, B.-S., Shin, K. J., Kim, D. J., Chung, B. Y., et al. (2003). Synthesis and Biological Evaluations of Pyrazolo[3,4-D]pyrimidines as Cyclin-dependent Kinase 2 Inhibitors. Eur. J. Med. Chem. 38, 525–532. doi:10.1016/S0223-5234(03)00065-5
Klutchko, S. R., Zhou, H., Winters, R. T., Tran, T. P., Bridges, A. J., Althaus, I. W., et al. (2006). Tyrosine Kinase Inhibitors. 19. 6-alkynamides of 4-anilinoquinazolines and 4-Anilinopyrido[3,4-D]pyrimidines as Irreversible Inhibitors of the erbB Family of Tyrosine Kinase Receptors. J. Med. Chem. 49, 1475–1485. doi:10.1021/jm050936o
Kurup, S., McAllister, B., Liskova, P., Mistry, T., Fanizza, A., Stanford, D., et al. (2018). Design, Synthesis and Biological Activity of N4-Phenylsubstituted-7h-Pyrrolo[2,3-D]pyrimidin-4-Amines as Dual Inhibitors of Aurora Kinase A and Epidermal Growth Factor Receptor Kinase. J. Enzyme Inhibition Med. Chem. 33, 74–84. doi:10.1080/14756366.2017.1376666
Lazzari, C., Gregorc, V., Karachaliou, N., Rosell, R., and Santarpia, M. (2020). Mechanisms of Resistance to Osimertinib. J. Thorac. Dis. 12, 2851–2858. doi:10.21037/jtd.2019.08.30
Lee, Y. J., and Han, H. J. (2008). Albumin-stimulated DNA Synthesis Is Mediated by Ca2+/PKC as Well as EGF Receptor-dependent P44/42 MAPK and NF-kappaB Signal Pathways in Renal Proximal Tubule Cells. Am. J. Physiol. Ren. Physiol. 294, F534–F541. doi:10.1152/ajprenal.00408.2007.-It
Lemmon, M. A., and Schlessinger, J. (2010). Cell Signaling by Receptor Tyrosine Kinases. Cell 141, 1117–1134. doi:10.1016/j.cell.2010.06.011
Levitzki, A., and Mishani, E. (2006). Tyrphostins and Other Tyrosine Kinase Inhibitors. Annu. Rev. Biochem. 75, 93–109. doi:10.1146/annurev.biochem.75.103004.142657
Li, J., An, B., Song, X., Zhang, Q., Chen, C., Wei, S., et al. (2021). Design, Synthesis and Biological Evaluation of Novel 2,4-diaryl Pyrimidine Derivatives as Selective EGFRL858R/T790M Inhibitors. Eur. J. Med. Chem. 212, 113019. doi:10.1016/j.ejmech.2020.113019
Li, S.-N., and Li, H.-Q. (2014). Epidermal Growth Factor Receptor Inhibitors: a Patent Review (2010 - Present). Expert Opin. Ther. Pat. 24, 309–321. doi:10.1517/13543776.2014.871527
Lin, S.-Y., Chang Hsu, Y., Peng, Y.-H., Ke, Y.-Y., Lin, W.-H., Sun, H.-Y., et al. (2019). Discovery of a Furanopyrimidine-Based Epidermal Growth Factor Receptor Inhibitor (DBPR112) as a Clinical Candidate for the Treatment of Non-small Cell Lung Cancer. J. Med. Chem. 62, 10108–10123. doi:10.1021/acs.jmedchem.9b00722
Liu, Q., Yu, S., Zhao, W., Qin, S., Chu, Q., and Wu, K. (2018). EGFR-TKIs Resistance via EGFR-independent Signaling Pathways. Mol. Cancer 17, 1–9. doi:10.1186/s12943-018-0793-1
Maher, M., Kassab, A. E., Zaher, A. F., and Mahmoud, Z. (2019). Novel Pyrazolo[3,4-D]pyrimidines: Design, Synthesis, Anticancer Activity, Dual EGFR/ErbB2 Receptor Tyrosine Kinases Inhibitory Activity, Effects on Cell Cycle Profile and Caspase-3-Mediated Apoptosis. J. Enzyme Inhibition Med. Chem. 34, 532–546. doi:10.1080/14756366.2018.1564046
Marepu, N., Yeturu, S., and Pal, M. (2018). 1,2,3-Triazole Fused with Pyridine/pyrimidine as New Template for Antimicrobial Agents: Regioselective Synthesis and Identification of Potent N-Heteroarenes. Bioorg. Med. Chem. Lett. 28, 3302–3306. doi:10.1016/j.bmcl.2018.09.021
Massaro, M., Barone, G., Barra, V., Cancemi, P., di Leonardo, A., Grossi, G., et al. (2021). Pyrazole[3,4-d]pyrimidine Derivatives Loaded into Halloysite as Potential CDK Inhibitors. Int. J. Pharm. 599, 120281. doi:10.1016/j.ijpharm.2021.120281
Merlino, G. T., Xu, Y. H., Richert, N., Clark, A. J., Ishii, S., Banks-Schlegel, S., et al. (1985). Elevated Epidermal Growth Factor Receptor Gene Copy Number and Expression in a Squamous Carcinoma Cell Line. J. Clin. Invest. 75, 1077–1079. doi:10.1172/JCI111770
Mghwary, A. E.-S., Gedawy, E. M., Kamal, A. M., and Abuel-Maaty, S. M. (2019). Novel Thienopyrimidine Derivatives as Dual EGFR and VEGFR-2 Inhibitors: Design, Synthesis, Anticancer Activity and Effect on Cell Cycle Profile. J. Enzyme Inhibition Med. Chem. 34, 838–852. doi:10.1080/14756366.2019.1593160
Milik, S. N., Abdel-Aziz, A. K., Lasheen, D. S., Serya, R. A. T., Minucci, S., and Abouzid, K. A. M. (2018). Surmounting the Resistance against EGFR Inhibitors through the Development of Thieno[2,3-D]pyrimidine-Based Dual EGFR/HER2 Inhibitors. Eur. J. Med. Chem. 155, 316–336. doi:10.1016/j.ejmech.2018.06.011
Mitrasinovic, P. (2013). Structural Elucidation of Unique Inhibitory Activities of Two Thiazolo[ 4,5-d]pyrimidines against Epidermal Growth Factor Receptor (EGFR): Implications for Successful Drug Design. Mc 10, 46–58. doi:10.2174/157340641001131226122124
Murtuza, A., Bulbul, A., Shen, J. P., Keshavarzian, P., Woodward, B. D., Lopez-Diaz, F. J., et al. (2019). Novel Third-Generation EGFR Tyrosine Kinase Inhibitors and Strategies to Overcome Therapeutic Resistance in Lung Cancer. Cancer Res. 79, 689–698. doi:10.1158/0008-5472.CAN-18-1281
Nagasaka, M., Zhu, V. W., Lim, S. M., Greco, M., Wu, F., and Ou, S.-H. I. (2021). Beyond Osimertinib: The Development of Third-Generation EGFR Tyrosine Kinase Inhibitors for Advanced EGFR+ NSCLC. J. Thorac. Oncol. 16, 740–763. doi:10.1016/j.jtho.2020.11.028
Okasha, R., Albalawi, F., Afifi, T., Fouda, A., Al-Dies, A.-A., and El-Agrody, A. (2016). Structural Characterization and Antimicrobial Activities of 7H-Benzo[h]chromeno[2,3-D]pyrimidine and 14H-Benzo[h]chromeno[3,2-E][1,2,4]triazolo[1,5-C] Pyrimidine Derivatives. Molecules 21, 14501–14515. doi:10.3390/molecules21111450
Othman, I. M. M., Alamshany, Z. M., Tashkandi, N. Y., Gad-Elkareem, M. A. M., Anwar, M. M., and Nossier, E. S. (2021). New Pyrimidine and Pyrazole-Based Compounds as Potential EGFR Inhibitors: Synthesis, Anticancer, Antimicrobial Evaluation and Computational Studies. Bioorg. Chem. 114, 105078. doi:10.1016/j.bioorg.2021.105078
Paez, J. G., Jänne, P. A., Lee, J. C., Tracy, S., Greulich, H., Gabriel, S., et al. (2004). EGFR Mutations in Lung Cancer: Correlation with Clinical Response to Gefitinib Therapy. Science 304, 1497–1500. doi:10.1126/science.1099314
Park, S., Ku, B. M., Jung, H. A., Sun, J.-M., Ahn, J. S., Lee, S.-H., et al. (2020). EGFR C797S as a Resistance Mechanism of Lazertinib in Non-small Cell Lung Cancer with EGFR T790M Mutation. Cancer Res. Treat. 52, 1288–1290. doi:10.4143/crt.2020.278
Pédeboscq, S., Gravier, D., Casadebaig, F., Hou, G., Gissot, A., De Giorgi, F., et al. (2010). Synthesis and Study of Antiproliferative Activity of Novel Thienopyrimidines on Glioblastoma Cells. Eur. J. Med. Chem. 45, 2473–2479. doi:10.1016/j.ejmech.2010.02.032
Perlíková, P., and Hocek, M. (2017). Pyrrolo[2,3-d]pyrimidine (7-deazapurine) as a Privileged Scaffold in Design of Antitumor and Antiviral Nucleosides. Med. Res. Rev. 37, 1429–1460. doi:10.1002/med.21465
Rashad, A. E., and Ali, M. A. (2006). Synthesis and Antiviral Screening of Some Thieno[2,3-d]Pyrimidine Nucleosides. Nucleosides, Nucleotides Nucleic Acids 25, 17–28. doi:10.1080/15257770500377730
Rashad, A. E., Hegab, M. I., Abdel-Megeid, R. E., Fathalla, N., and Abdel-Megeid, F. M. E. (2009). Synthesis and Anti-HSV-1 Evaluation of Some Pyrazoles and Fused Pyrazolopyrimidines. Eur. J. Med. Chem. 44, 3285–3292. doi:10.1016/j.ejmech.2009.02.012
Raymond, E., Faivre, S., and Armand, J. P. (2000). Epidermal Growth Factor Receptor Tyrosine Kinase as a Target for Anticancer Therapy. Drugs 60, 15–23. doi:10.2165/00003495-200060001-00002
Rewcastle, G. W., Bridges, A. J., Fry, D. W., Rubin, J. R., and Denny, W. A. (1997). Tyrosine Kinase Inhibitors. 12. Synthesis and Structure−Activity Relationships for 6-Substituted 4-(Phenylamino)pyrimido[5,4-D]pyrimidines Designed as Inhibitors of the Epidermal Growth Factor Receptor. J. Med. Chem. 40, 1820–1826. doi:10.1021/jm960879m
Rheault, T. R., Caferro, T. R., Dickerson, S. H., Donaldson, K. H., Gaul, M. D., Goetz, A. S., et al. (2009). Thienopyrimidine-based Dual EGFR/ErbB-2 Inhibitors. Bioorg. Med. Chem. Lett. 19, 817–820. doi:10.1016/j.bmcl.2008.12.011
Romagnoli, R., Prencipe, F., Oliva, P., Baraldi, S., Baraldi, P. G., Schiaffino Ortega, S., et al. (2019). Design, Synthesis, and Biological Evaluation of 6-Substituted Thieno[3,2-D]pyrimidine Analogues as Dual Epidermal Growth Factor Receptor Kinase and Microtubule Inhibitors. J. Med. Chem. 62, 1274–1290. doi:10.1021/acs.jmedchem.8b01391
Roskoski, R. (2014). The ErbB/HER Family of Protein-Tyrosine Kinases and Cancer. Pharmacol. Res. 79, 34–74. doi:10.1016/j.phrs.2013.11.002
Schenone, S., Bruno, O., Bondavalli, F., Ranise, A., Mosti, L., Menozzi, G., et al. (2004a). Antiproliferative Activity of New 1-Aryl-4-Amino-1h-Pyrazolo[3,4-D]pyrimidine Derivatives toward the Human Epidermoid Carcinoma A431 Cell Line. Eur. J. Med. Chem. 39, 939–946. doi:10.1016/j.ejmech.2004.07.010
Schenone, S., Bruno, O., Bondavalli, F., Ranise, A., Mosti, L., Menozzi, G., et al. (2004b). Synthesis of 1-(2-Chloro-2-Phenylethyl)-6-Methylthio-1h-Pyrazolo[3,4-D]pyrimidines 4-amino Substituted and Their Biological Evaluation. Eur. J. Med. Chem. 39, 153–160. doi:10.1016/j.ejmech.2003.11.007
Schroeder, M. C., Hamby, J. M., Connolly, C. J. C., Grohar, P. J., Winters, R. T., Barvian, M. R., et al. (2001). Soluble 2-Substituted Aminopyrido[2,3-D]pyrimidin-7-Yl Ureas. Structure−Activity Relationships against Selected Tyrosine Kinases and Exploration of In Vitro and In Vivo Anticancer Activity. J. Med. Chem. 44, 1915–1926. doi:10.1021/jm0004291
Shi, X., Quan, Y., Wang, Y., Wang, Y., and Li, Y. (2021). Design, Synthesis, and Biological Evaluation of 2,6,7-substituted Pyrrolo[2,3-D]pyrimidines as Cyclin Dependent Kinase Inhibitor in Pancreatic Cancer Cells. Bioorg. Med. Chem. Lett. 33, 127725. doi:10.1016/j.bmcl.2020.127725
Showalter, H. D. H., Bridges, A. J., Zhou, H., Sercel, A. D., McMichael, A., and Fry, D. W. (1999). Tyrosine Kinase Inhibitors. 16. 6,5,6-Tricyclic Benzothieno[3,2-D]pyrimidines and Pyrimido[5,4-B]- and -[4,5-b]indoles as Potent Inhibitors of the Epidermal Growth Factor Receptor Tyrosine Kinase. J. Med. Chem. 42, 5464–5474. doi:10.1021/jm9903949
Singh, P. K., and Silakari, O. (2018). Molecular Dynamics Guided Development of Indole Based Dual Inhibitors of EGFR (T790M) and C-MET. Bioorg. Chem. 79, 163–170. doi:10.1016/j.bioorg.2018.04.001
Smaill, J. B., Gonzales, A. J., Spicer, J. A., Lee, H., Reed, J. E., Sexton, K., et al. (2016). Tyrosine Kinase Inhibitors. 20. Optimization of Substituted Quinazoline and Pyrido[3,4-D]pyrimidine Derivatives as Orally Active, Irreversible Inhibitors of the Epidermal Growth Factor Receptor Family. J. Med. Chem. 59, 8103–8124. doi:10.1021/acs.jmedchem.6b00883
Smaill, J. B., Rewcastle, G. W., Loo, J. A., Greis, K. D., Chan, O. H., Reyner, E. L., et al. (2000). Tyrosine Kinase Inhibitors. 17. Irreversible Inhibitors of the Epidermal Growth Factor Receptor: 4-(Phenylamino)quinazoline- and 4-(Phenylamino)pyrido[3,2-D]pyrimidine-6-Acrylamides Bearing Additional Solubilizing Functions. J. Med. Chem. 43, 1380–1397. doi:10.1021/jm990482t
Smaill, J. B., Showalter, H. D. H., Zhou, H., Bridges, A. J., McNamara, D. J., Fry, D. W., et al. (2001). Tyrosine Kinase Inhibitors. 18. 6-Substituted 4-anilinoquinazolines and 4-Anilinopyrido[3,4-D]pyrimidines as Soluble, Irreversible Inhibitors of the Epidermal Growth Factor Receptor. J. Med. Chem. 44, 429–440. doi:10.1021/jm000372i
Solca, F. F., Baum, A., Langkopf, E., Dahmann, G., Heider, K. H., Himmelsbach, F., et al. (2004). Inhibition of Epidermal Growth Factor Receptor Activity by Two Pyrimidopyrimidine Derivatives. J. Pharmacol. Exp. Ther. 311, 502–509. doi:10.1124/jpet.104.069138
Song, X. J., Shao, Y., and Dong, X. G. (2011). Microwave-assisted Synthesis of Some Novel Fluorinated Pyrazolo[3,4-D]pyrimidine Derivatives Containing 1,3,4-thiadiazole as Potential Antitumor Agents. Chin. Chem. Lett. 22, 1036–1038. doi:10.1016/j.cclet.2011.05.012
Soria, J.-C., Ohe, Y., Vansteenkiste, J., Reungwetwattana, T., Chewaskulyong, B., Lee, K. H., et al. (2018). Osimertinib in UntreatedEGFR-Mutated Advanced Non-small-cell Lung Cancer. N. Engl. J. Med. 378, 113–125. doi:10.1056/NEJMoa1713137
Spasov, A. A., Babkov, D. A., Sysoeva, V. A., Litvinov, R. A., Shamshina, D. D., Ulomsky, E. N., et al. (2017). 6-Nitroazolo[1,5-a ]pyrimidin-7(4H )-ones as Antidiabetic Agents. Arch. Pharm. Chem. Life Sci. 350, 1700226–1700238. doi:10.1002/ardp.201700226
Stevens, K. L., Alligood, K. J., Alberti, J. G. B., Caferro, T. R., Chamberlain, S. D., Dickerson, S. H., et al. (2009). Synthesis and Stereochemical Effects of Pyrrolidinyl-Acetylenic Thieno[3,2-D]pyrimidines as EGFR and ErbB-2 Inhibitors. Bioorg. Med. Chem. Lett. 19, 21–26. doi:10.1016/j.bmcl.2008.11.023
Sun, L., Cui, J., Liang, C., Zhou, Y., Nematalla, A., Wang, X., et al. (2002). Rational Design of 4,5-Disubstituted-5,7-Dihydro-Pyrrolo[2,3-D]pyrimidin-6-Ones as a Novel Class of Inhibitors of Epidermal Growth Factor Receptor (EGF-R) and Her2(p185erbB) Tyrosine Kinases. Bioorg. Med. Chem. Lett. 12, 2153–2157. doi:10.1016/S0960-894X(02)00364-5
Sundby, E., Han, J., Kaspersen, S. J., and Hoff, B. H. (2015). In Vitro baselining of New Pyrrolopyrimidine EGFR-TK Inhibitors with Erlotinib. Eur. J. Pharm. Sci. 80, 56–65. doi:10.1016/j.ejps.2015.08.003
Tan, D. S.-W., Leighl, N. B., Riely, G. J., Yang, J. C.-H., Sequist, L. V., Wolf, J., et al. (2020). Safety and Efficacy of Nazartinib (EGF816) in Adults with EGFR-Mutant Non-small-cell Lung Carcinoma: a Multicentre, Open-Label, Phase 1 Study. Lancet Respir. Med. 8, 561–572. doi:10.1016/S2213-2600(19)30267-X
Teramura, Y., Ichinose, J., Takagi, H., Nishida, K., Yanagida, T., and Sako, Y. (2006). Single-molecule Analysis of Epidermal Growth Factor Binding on the Surface of Living Cells. Embo J. 25, 4215–4222. doi:10.1038/sj.emboj.7601308
Thiriveedhi, A., Nadh, R. V., Srinivasu, N., Bobde, Y., Ghosh, B., and Sekhar, K. V. G. C. (2019). Design, Synthesis and Anti-tumour Activity of New Pyrimidine-Pyrrole Appended Triazoles. Toxicol. Vitro 60, 87–96. doi:10.1016/j.tiv.2019.05.009
Traxler, P., Bold, G., Frei, J., Lang, M., Lydon, N., Mett, H., et al. (1997). Use of a Pharmacophore Model for the Design of EGF-R Tyrosine Kinase Inhibitors: 4-(Phenylamino)pyrazolo[3,4-D]pyrimidines. J. Med. Chem. 40, 3601–3616. doi:10.1021/jm970124v
Ullrich, A., Coussens, L., Hayflick, J. S., Dull, T. J., Gray, A., Tam, A. W., et al. (1984). Human Epidermal Growth Factor Receptor cDNA Sequence and Aberrant Expression of the Amplified Gene in A431 Epidermoid Carcinoma Cells. Nature 309, 418–425. doi:10.1038/309418a0
Wang, H., Pan, R., Zhang, X., Si, X., Wang, M., and Zhang, L. (2020). Abivertinib in Patients with T790M‐positive Advanced NSCLC and its Subsequent Treatment with Osimertinib. Thorac. Cancer 11, 594–602. doi:10.1111/1759-7714.13302
Wang, J., Zhang, L., Zhao, J., Zhang, Y., Liu, Q., Tian, C., et al. (2018). 2-Arylthio-5-iodo Pyrimidine Derivatives as Non-nucleoside HBV Polymerase Inhibitors. Bioorg. Med. Chem. 26, 1573–1578. doi:10.1016/j.bmc.2018.02.003
Widler, L., Green, J., Missbach, M., Šuša, M., and Altmann, E. (2001). 7-Alkyl- and 7-Cycloalkyl-5-Aryl-Pyrrolo[2,3-D]pyrimidines-Potent Inhibitors of the Tyrosine Kinase C-Src. Bioorg. Med. Chem. Lett. 11, 849–852. doi:10.1016/S0960-894X(01)00079-8
Wind, S., Schnell, D., Ebner, T., Freiwald, M., and Stopfer, P. (2017a). Clinical Pharmacokinetics and Pharmacodynamics of Afatinib. Clin. Pharmacokinet. 56, 235–250. doi:10.1007/s40262-016-0440-1
Wind, S., Schnell, D., Ebner, T., Freiwald, M., and Stopfer, P. (2017b). Clinical Pharmacokinetics and Pharmacodynamics of Afatinib. Clin. Pharmacokinet. 56, 235–250. doi:10.1007/s40262-016-0440-1
Wood, E. R., Shewchuk, L. M., Ellis, B., Brignola, P., Brashear, R. L., Caferro, T. R., et al. (2008). 6-Ethynylthieno[3,2-d]- and 6-Ethynylthieno[2,3-D]pyrimidin-4-Anilines as Tunable Covalent Modifiers of ErbB Kinases. Proc. Natl. Acad. Sci. U.S.A. 105, 2773–2778. doi:10.1073/pnas.0708281105
World Health Organization (2020). WHO Report on Cancer: Setting Priorities Investing Wisely and Providing Care for All. Cambridge: WHO.
Wu, C.-H., Coumar, M. S., Chu, C.-Y., Lin, W.-H., Chen, Y.-R., Chen, C.-T., et al. (2010). Design and Synthesis of Tetrahydropyridothieno[2,3-D]pyrimidine Scaffold Based Epidermal Growth Factor Receptor (EGFR) Kinase Inhibitors: The Role of Side Chain Chirality and Michael Acceptor Group for Maximal Potency. J. Med. Chem. 53, 7316–7326. doi:10.1021/jm100607r
Wu, M., and Zhang, P. (2020). EGFR-mediated Autophagy in Tumourigenesis and Therapeutic Resistance. Cancer Lett. 469, 207–216. doi:10.1016/j.canlet.2019.10.030
Xiao, Q., Qu, R., Gao, D., Yan, Q., Tong, L., Zhang, W., et al. (2016). Discovery of 5-(methylthio)pyrimidine Derivatives as L858R/T790M Mutant Selective Epidermal Growth Factor Receptor (EGFR) Inhibitors. Bioorg. Med. Chem. 24, 2673–2680. doi:10.1016/j.bmc.2016.04.032
Xu, S., Xu, T., Zhang, L., Zhang, Z., Luo, J., Liu, Y., et al. (2013). Design, Synthesis, and Biological Evaluation of 2-Oxo-3,4-Dihydropyrimido[4,5-D]pyrimidinyl Derivatives as New Irreversible Epidermal Growth Factor Receptor Inhibitors with Improved Pharmacokinetic Properties. J. Med. Chem. 56, 8803–8813. doi:10.1021/jm4012388
Yasuda, H., Park, E., Yun, C.-H., Sng, N. J., Lucena-Araujo, A. R., Yeo, W.-L., et al. (2013). Structural, Biochemical, and Clinical Characterization of Epidermal Growth Factor Receptor (EGFR) Exon 20 Insertion Mutations in Lung Cancer. Sci. Transl. Med. 5, 177. doi:10.1126/scitranslmed.3007205
Yu, L., Huang, M., Xu, T., Tong, L., Yan, X.-e., Zhang, Z., et al. (2017). A Structure-Guided Optimization of Pyrido[2,3-D]pyrimidin-7-Ones as Selective Inhibitors of EGFRL858R/T790M Mutant with Improved Pharmacokinetic Properties. Eur. J. Med. Chem. 126, 1107–1117. doi:10.1016/j.ejmech.2016.12.006
Yun, C.-H., Mengwasser, K. E., Toms, A. V., Woo, M. S., Greulich, H., Wong, K.-K., et al. (2008). The T790M Mutation in EGFR Kinase Causes Drug Resistance by Increasing the Affinity for ATP. Proc. Natl. Acad. Sci. U.S.A. 105, 2070–2075. doi:10.1073/pnas.0709662105
Zhang, D., Yan, Y., Jin, G., Liu, B., Ma, X., Han, D., et al. (2018a). Synthesis and Antitumor Evaluation of Novel 4-Anilino-7,8-Dihydropyrido[4,3-D ]pyrimidine-6(5H )-carboxylate Derivatives as Potential EGFR Inhibitors. Arch. Pharm. Chem. Life Sci. 351, 1800110–1800111. doi:10.1002/ardp.201800110
Zhang, H., Wang, J., Shen, Y., Wang, H.-Y., Duan, W.-M., Zhao, H.-Y., et al. (2018b). Discovery of 2,4,6-trisubstitued Pyrido[3,4-D]pyrimidine Derivatives as New EGFR-TKIs. Eur. J. Med. Chem. 148, 221–237. doi:10.1016/j.ejmech.2018.02.051
Zhang, H., Wang, J., Zhao, H.-Y., Yang, X.-Y., Lei, H., Xin, M., et al. (2018c). Synthesis and Biological Evaluation of Irreversible EGFR Tyrosine Kinase Inhibitors Containing Pyrido[3,4-D]pyrimidine Scaffold. Bioorg. Med. Chem. 26, 3619–3633. doi:10.1016/j.bmc.2018.05.039
Zhang, S., Ulrich, M., Gromnicka, A., Havlíček, L., Kryštof, V., Jorda, R., et al. (2016). Anti-angiogenic Effects of Novel Cyclin-dependent Kinase Inhibitors with a Pyrazolo[4,3-D]pyrimidine Scaffold. Br. J. Pharmacol. 173, 2645–2656. doi:10.1111/bph.13546
Zhang, X., Raghavan, S., Ihnat, M., Hamel, E., Zammiello, C., Bastian, A., et al. (2015a). The Design, Synthesis and Biological Evaluation of Conformationally Restricted 4-Substituted-2,6-Dimethylfuro[2,3-D]pyrimidines as Multi-Targeted Receptor Tyrosine Kinase and Microtubule Inhibitors as Potential Antitumor Agents. Bioorg Med. Chem. 23, 2408–2423. doi:10.1016/j.bmc.2015.03.061
Zhang, Y., Luo, L., Han, C., Lv, H., Chen, D., Shen, G., et al. (2017). Design, Synthesis, and Biological Activity of Tetrahydrobenzo[4,5]thieno[2,3-D]pyrimidine Derivatives as Anti-inflammatory Agents. Molecules 22, 1960. doi:10.3390/molecules22111960
Zhang, Y., Zhang, K., Zhao, M., Zhang, L., Qin, M., Guo, S., et al. (2015b). Discovery of a Novel Class Anti-proliferative Agents and Potential Inhibitors of EGFR Tyrosine Kinases Based on 4-Anilinotetrahydropyrido[4,3-D]pyrimidine Scaffold: Design, Synthesis and Biological Evaluations. Bioorg. Med. Chem. 23, 4591–4607. doi:10.1016/j.bmc.2015.05.059
Keywords: EGFR, pyrimidine, antiproliferative, SAR, fused pyrimidine
Citation: Yadav TT, Moin Shaikh G, Kumar MS, Chintamaneni M and YC M (2022) A Review on Fused Pyrimidine Systems as EGFR Inhibitors and Their Structure–Activity Relationship. Front. Chem. 10:861288. doi: 10.3389/fchem.2022.861288
Received: 24 January 2022; Accepted: 28 April 2022;
Published: 13 June 2022.
Edited by:
Steven De Jonghe, KU Leuven, BelgiumReviewed by:
Suresh Narva, Baylor University, United StatesSandip Bharate, Indian Institute of Integrative Medicine (CSIR), India
Copyright © 2022 Yadav, Moin Shaikh, Kumar, Chintamaneni and YC. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mayur YC, bWF5dXI8YSBocmVmPSJtYWlsdG86eWNAcmVkaWZmbWFpbC5jb20iPnljQHJlZGlmZm1haWwuY29tPC9hPg==
†These authors have contributed equally to this work