 Samata E. Shetgaonkar
Samata E. Shetgaonkar Aleena Raju
Aleena Raju Hideyasu China2
Hideyasu China2 Naoko Takenaga
Naoko Takenaga Toshifumi Dohi
Toshifumi Dohi Fateh V. Singh
Fateh V. Singh- 1Chemistry Division, School of Advanced Science, VIT University, Chennai, India
- 2Department of Medical Bioscience, Nagahama Institute of Bio-Science and Technology, Nagahama, Japan
- 3Faculty of Pharmacy, Meijo University, Nagoya, Japan
- 4College of Pharmaceutical Sciences, Ritsumeikan University, Kusatsu, Japan
Transition metal-catalyzed direct oxidative coupling reactions via C–H bond activation have emerged as a straightforward strategy for the construction of complex molecules in organic synthesis. The direct transformation of C–H bonds into carbon–carbon and carbon–heteroatom bonds renders the requirement of prefunctionalization of starting materials and, therefore, represents a more efficient alternative to the traditional cross-coupling reactions. The key to the unprecedented progress made in this area has been the identification of an appropriate oxidant that facilitates oxidation and provides heteroatom ligands at the metal center. In this context, hypervalent iodine compounds have evolved as mainstream reagents particularly because of their excellent oxidizing nature, high electrophilicity, and versatile reactivity. They are environmentally benign reagents, stable, non-toxic, and relatively cheaper than inorganic oxidants. For many years, palladium catalysis has dominated these oxidative coupling reactions, but eventually, other transition metal catalysts such as gold, copper, platinum, iron, etc. were found to be promising alternate catalysts for facilitating such reactions. This review article critically summarizes the recent developments in non-palladium-catalyzed oxidative coupling reactions mediated by hypervalent iodine (III) reagents with significant emphasis on understanding the mechanistic aspects in detail.
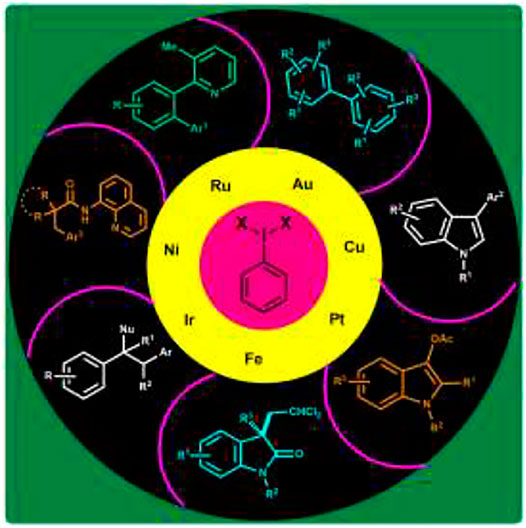
GRAPHICAL ABSTRACT
Introduction
Transition metal-catalyzed cross-coupling reactions have emerged as a powerful synthetic method for the construction of valuable organic molecules (Jana et al., 2011; Campeau, and Hazari, 2019). These represent a versatile strategy to connect nucleophilic organometallic compounds with electrophilic organic halides in the presence of palladium or other transition metal catalysts. A wide range of nucleophiles such as organo-boron, organo-zinc, and Grignard reagents have been used as coupling partners in these reactions (Jana et al., 2011; Campeau, and Hazari, 2019). However, a few drawbacks associated with traditional cross-coupling reactions include pre-activation of substrates and stoichiometric requirement of organometallic reagents that generate metallic salts as by-products leading to severe environmental concerns. From a green chemistry point of view, continuous efforts were directed toward the development of direct oxidative coupling reactions that could eliminate the requirement of prefunctionalization of substrates and also minimize waste material generation. In this aspect, new oxidative coupling reactions via transition metal-catalyzed direct C–H bond functionalization have evolved as a more efficient alternative to traditional cross-coupling reactions (Kuhl et al., 2012; Qiu and Wu, 2015; Henry et al., 2017). This method has enabled the effective formation of new carbon–carbon and carbon–heteroatom bonds by coupling non-prefunctionalized substrates via the C–H bond activation method. The key to the unprecedented progress made in this area has been the identification of appropriate catalysts, oxidants, additives, and ligands. Hypervalent iodine reagents have gained significant attention in this field owing to their excellent oxidizing and electrophilic properties.
Hypervalent iodine reagents are of utmost importance in modern organic synthesis as they are environmentally benign alternatives for toxic heavy metal oxidants (Dohi and Kita, 2009; Silva and Olofsson, 2011; Yusubov and Zhdankin, 2012; Kita and Dohi, 2015; Yoshimura and Zhdankin, 2016). They are highly stable, less toxic, readily available, easy to handle, and possess the ease of recovery from the reaction system. Various iodine(III) and iodine(V) reagents are existing in the literature and the structures of few representative examples are given in Figure 1. The first hypervalent iodine reagent, namely, (dichloroiodo)benzene 1 was prepared by Willgerodt (1886). After this, several iodine (III) compounds such as (difluoroiodo)benzene 2, phenyliodine (III) diacetate 3 (PIDA), phenyliodine (III) bis(trifluoroacetate) 4 (PIFA), phenyliodine (III) dipivaloate 5 (PIDP), [hydroxy (tosyloxy)iodo]-benzene HTIB 6 (Koser′s reagent), iodosylbenzene 7 (PhIO), 2-iodosobenzoic acid 8 (IBA), benziodoxol (on)e reagents 9–14, and diaryliodonium salts 15–17 were identified as powerful and effective oxidants and atom transfer reagents in different organic transformations (Tohma and Kita, 2004; Joshi and De, 2021). In addition to this, cyclic iodine(V) reagents, namely, Dess–Martin periodinane (DMP) 18 and 2-iodoxybenzoic acid (IBX) 19 are strong oxidizing agents (Uyanik and Ishihara, 2009). Several studies (Singh and Wirth, 2014a; Elsherbini and Wirth, 2018; Grelier et al., 2018; Singh et al., 2018; Hyatt et al., 2019; Shetgaonkar and Singh 2020; Shetgaonkar et al., 2021; Shetgaonkar et al., 2021; Singh and Wirth, 2021; Kumar et al., 2022) and books chapters (Singh and Wirth, 2014b; Singh and Wirth, 2017; Singh and Wirth, 2018) have been published covering the preparations and synthetic applications of these reagents.
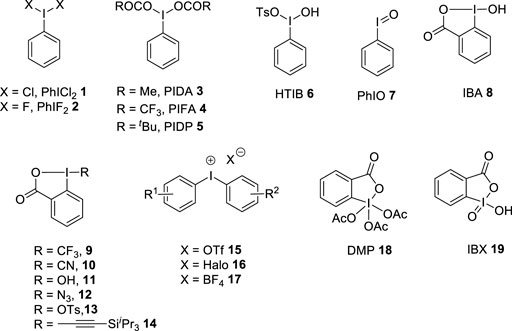
FIGURE 1. Hypervalent iodine (III)/(V) reagents 1–19.
Undoubtedly, hypervalent iodine reagents have led to the rapid growth in transition metal-catalyzed oxidative coupling reactions. In particular, they are used as selective oxidants and as ligand transfer reagents in these reactions. Hypervalent iodine reagents as oxidants facilitate the desired oxidation at the metal center and also provide heteroatom ligands such as acetate or chloride which are subsequently transferred to the substrates via reductive elimination. To date, palladium has been exclusively used as a catalyst in various oxidative coupling reactions involving the direct functionalization of C (sp2)–H and C (sp3)–H bonds (Deprez and Sanford, 2007; Silva et al., 2017; Shetgaonkar and Singh, 2020). On the other hand, other transition metals do catalyze such oxidative coupling reactions in the presence of hypervalent iodine reagents. To the best of our knowledge, studies on hypervalent iodine-mediated non-palladium-catalyzed oxidative coupling reactions remain rare (Silva et al., 2017). In the present review article, we will cover the recent advancements made in non-palladium-catalyzed oxidative coupling reactions using hypervalent iodine reagents. The article is classified on the basis of metal catalysts used such as gold, copper, platinum, iron, iridium, nickel, and ruthenium.
Gold
Gold has contributed a significant role in the development of oxidative coupling reactions. This is primarily because of its excellent ability as a carbophilic activator over other transition metals. Gold catalysis follows redox neutral pathways involving Au(I)/Au(III) catalytic cycles. Typically, reactions involving Au(I)/Au(III) catalytic cycles are rare because of the high barrier for the oxidation of Au(I) species to the Au(III) species (redox potential +1.41 V). Thus, the identification of powerful oxidants which can facilitate the oxidation of Au(I) to Au(III) would be crucial. In this context, hypervalent iodine reagents have emerged as the choice of oxidants because of their excellent oxidizing nature and high electrophilicity. This has led to the development of gold-catalyzed homo/cross-coupling, alkynylation, and alkene functionalization reactions using hypervalent iodine reagents as excellent oxidants and electrophilic functional group transfer reagents. In this section, we will highlight some of the significant studies accomplished in this area in great detail.
Gold-catalyzed cross-coupling reaction of arenes is an interesting method for the construction of biaryls. Although this area is less developed, a few examples have been reported using a hypervalent iodine reagent as an oxidant. In 2008, Tse et al. first reported a gold-catalyzed oxidative coupling of arenes 20 to obtain biaryls 21 via an electrophilic aromatic substitution pattern (Kar et al., 2008) (Figure 2). The reaction yielded homo-coupling products 21 in mild conditions using PhI(OAc)2 3 as an oxidant. The process used a variety of gold catalysts, including HAuCl4, Au(OAc)3, AuCl(PPh3), and generated products 21 with 74, 39, and 76% yields respectively. When acetic acid was used as a solvent, the process proceeded smoothly. Non-coordinative solvents, such as 1,2-dichloroethane, can also be used as a reaction medium. Notably, the present reaction does not require an inert environment, a pre-treated substrate, or silver salts to boost reactivity, and it can run under normal conditions. Furthermore, even if the starting material is used at a higher concentration, it can be reused. Arenes 20 containing diverse functional groups, such as esters and methoxides, were also tolerated. For halogenated arenes 20, homo-coupled products 21 are formed that are not fulfilled by Lewis acid or Pd-catalyzed reactions. It also produced considerable yields of electron-rich heterocycles such as thiophenes. The gold-mediated homo- and hetero-coupling of non-activated arenes was explored later by the same group (Kar et al., 2009).
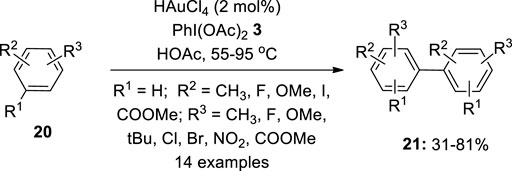
FIGURE 2. Gold-catalyzed oxidative coupling of arenes 20 for the synthesis of biaryls 21 using PhI(OAc)2 3 as an oxidant.
Another gold-based cross-coupling reaction of arenes was reported by Hofer and Nevado (2013). In this approach, they performed oxidative coupling of pentafluorophenyl gold(I) complexes 22 with electron-rich arenes 23 such as 1,3,5-trimethoxybenzene, 1,2,4-trimethoxybenzene, or 1-methylindole in the presence of hypervalent iodide oxidants such as PhI(OAc)2 3 and PhICl2 1 (Figure 3). Surprisingly, the complex generated a hetero-coupling product 24 with a 67% yield when treated with 1,3,5-trimethoxybenzene in the presence of PhI(OAc)2 3. Similarly, the reaction of 22 with 1,2,4-trimethoxybenzene gave 76% of hetero-coupling product 24 whereas with 1-methylindole, it yielded 80% of the product. In the absence of an oxidant, the reaction produced no product. Further replacing the oxidant with PhICl2 1 resulted in albeit lower yields of the hetero-coupled products 24. The stronger basic character of AcO- was posited as the reason for this alteration in the reaction pattern. Later, Cambeiro et al. (2013)designed a similar oxidative coupling method involving double C–H activation for the synthesis of biaryls by using PhI(OH)OTs 6 as an oxidant.
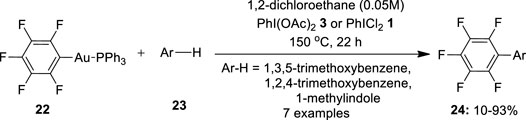
FIGURE 3. Gold-catalyzed synthesis of biaryls 24 via oxidative coupling of pentafluorophenyl gold(I) complexes 22 with electron-rich arenes 23 using PhI(OAc)2 3 as an oxidant.
Further hypervalent ethynyl benziodoxolone (EBX) reagents have evolved as a mainstream reagent in various gold-catalyzed electrophilic functional group transfer reactions (especially alkynylations, acetoxylations, and arylations) (Banerjee et al., 2020). The unique electrophilicity and oxidizing nature of these reagents have led to the rapid development in the direct C–H alkynylation reactions of (hetero)aromatic compounds using gold catalysis. In 2009, Waser et al. reported the first gold-catalyzed C–H alkynylation of indoles and pyrroles 25 with 1-[(triisopropylsilyl)ethynyl]-1,2-benziodoxol-3(1H)-one (TIPS-EBX) 14 for acetylene transfer (Figure 4) (Brand et al., 2009). The reaction proceeded in high yields using 5 mol% AuCl in Et2O at room temperature in air. Several indole derivatives were alkylated to deliver corresponding 3-alkynylation products 26 in good yields. Both electron-donating and electron-withdrawing groups were tolerated in the reaction. Interestingly, higher yields were obtained for substrates with Br and I substituents. Also, 2-substituted indoles 25 gave good yields of 3-alkynylation products 26. Moreover, the present protocol was successfully used for the alkynylation of 3-methylindole 25 to yield 76% of the 2-alkynylation product 27. Further alkynylation of pyrroles 25 was achieved for the first time under the same reaction conditions. Mono-, di-, and tri-substituted pyrroles 25 yielded 2-alkynylation products 27 in good yields. The two working hypotheses for the mechanism of the alkynylation reaction are depicted in Figure 4. In one hypothesis, the oxidation of Au(I) with 14 to give a Au(III)–acetylene complex 28, followed by indole metalation and reductive elimination yields a 3-alkynylation product 26. The other hypothesis involves Au-mediated addition of indole 25 to the triple bond of 14 to provide a vinyl-gold complex 31 or 32 followed by either β-elimination or α-elimination/1,2-shift sequence to give the desired alkynylation product 26.
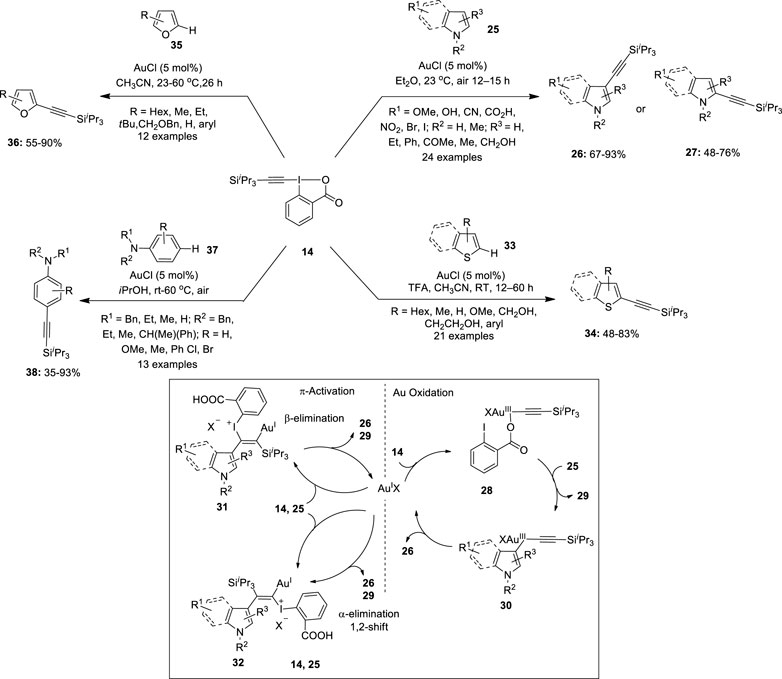
FIGURE 4. Gold-catalyzed C–H alkynylation of arenes/hetero-arenes using a benziodoxolone-derived hypervalent iodine reagent 14 as an acetylene transfer reagent.
Later, Brand and Waser (2010)demonstrated the alkynylation of thiophenes 33 by using benziodoxolone-derived hypervalent iodine reagent 14 as an acetylene transfer reagent (Figure 4). Notably, the activation of benziodoxole reagent 14 using a gold catalyst (AuCl) and Brønsted acids (TFA) was required for the success of this reaction. 2-Alkyl or 2-aryl-substituted thiophenes 33 were alkynylated in good yields. Moreover, mono-alkynylation of 2,2′-bithiophenes was achieved to yield important building blocks for oligothiophenes. In addition, alkynylation of less reactive benzothiophenes 33 was also investigated; however, no regioselectivity was observed and mixtures of 2- and 3-alkynylated thiophenes 34 were obtained in 65% yield. In continuation, Waser et al. reported the first example highlighting the direct alkynylation of furans 35 by using a Au(I) catalyst and TIPS-EBX 14 (Li, et al., 2013) (Figure 4). Initially, the standard conditions for thiophene alkynylation (TFA and acetonitrile) were used; however, it led to the decomposition of the starting material possibly due to the acid sensitivity of furan moiety. Thus, C2-alkynylation of furans 35 was performed in the absence of TFA and the desired alkynyl furans 36 were isolated in useful yields. Notably, this reaction was found to be highly selective toward the electron-rich C2 position of the furan ring. The alkynylation of 2-alkyl-substituted furans 35 proceeded well at room temperature to give 2-alkynylated products 36 in decent yields. Conversely, the reaction of 2-aryl furans 35 was slower and the corresponding 2-alkynylated furans 36 were isolated under heating conditions. Moreover, 2,5-disubstituted furan 35 yielded a 3-alkynylation product 36 in 45% yield.
Furthermore, Brand and Waser (2012)used TIPS-EBX 14 as an alkynylation reagent for the para-selective alkynylation of anilines 37 using a gold catalyst (Brand and Waser, 2012) (Figure 4). iPrOH as a solvent system provided the best result for the alkynylation reaction. A variety of anilines 37 were alkynylated regioselectively to the corresponding para-alkynyl anilines 38 in moderate to excellent yields. Interestingly, anilines 37 protected with benzyl, methyl, ethyl, and butyl groups as nitrogen substituents were tolerated. Moreover, the alkynylation of mono-protected anilines 37 was successful, indicating a tolerance toward free NH bonds on the aniline. Importantly, dibenzylated anilines 37 with meta-substitutions yielded 1,3,4-substituted anilines 38 in useful yields. In addition, the reaction attempted with a para-substituted aniline yielded an ortho-alkynylated product in 18%.
A gold-hypervalent iodine combination was further explored for the hetero-arylation of olefins 39 by Ball et al. (2012). They designed a three-component oxyarylation reaction by using a gold catalyst and 1-hydroxy-1,2-benziodoxol-3(1H)-one (IBA) 8 as an efficient oxidant (Figure 5). The reactions required the presence of p-TSA as an additive. Notably, benzotrifluoride (PhCF3) as solvent media substantially reduced the formation of homo-coupling products. A range of mono-substituted olefins 39 smoothly reacted with diverse arylsilanes 40 in the presence of alcohols 41 as O-nucleophiles to deliver corresponding products 42 in variable yields. O-nucleophiles such as methanol, ethanol, isopropanol, and water could be used, giving desired products in moderate yields. Arylsilanes 40 with electron-rich and electron-deficient substituents were well tolerated. However, electron-rich arylsilanes, carboxylic acids, and tertiary alcohols were unsuited to this reaction. Further oxyarylation of styrenes and gem-di-substituted olefins proceeded smoothly under similar conditions, although the reaction scope was limited to MeOH as an O-nucleophile.
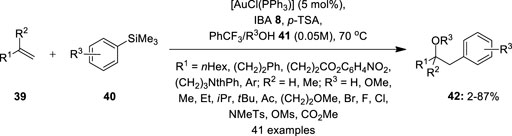
FIGURE 5. Gold-catalyzed oxyarylation of mono- and gem-di-substituted olefins 39 using IBA 8 as an oxidant.
Further significant work on the oxidative acyloxylation of an unactivated C (sp3)–H bond was achieved by Yang et al by gold catalysis (Guo et al., 2016). They reported the direct α-C (sp3)–H acyloxylation of methyl sulfides 43 in the presence of PPh3AuCl (0.5 mol%) as a catalyst, bis(acyloxy)iodobenzene 44 as an oxidant, and an acyloxy group source (Figure 6). A variety of thioanisoles 43 functionalized with both electron-rich and -deficient groups reacted with PhI(OAc)2 3 to yield α-thioaryl ester derivatives 45 in moderate to excellent yields. Notably, no product formation was observed in the case of –OH, –NH2, and –COOH-substituted thioanisole benzene rings. In the proposed reaction mechanism (Figure 6), the hypervalent iodine reagent 44 acts as a Lewis acid and activates the sulfur atom of the thioether 43 to form sulfonium salt 46 or 47. Subsequently, sulfonium salt 46 or 47 undergoes elimination in the presence of a gold catalyst to generate intermediate 48 and liberates R1COOH. Finally, the nucleophilic attack on the sulfanyl acetate intermediate 48 by–OCOR1 gives the desired product 45.
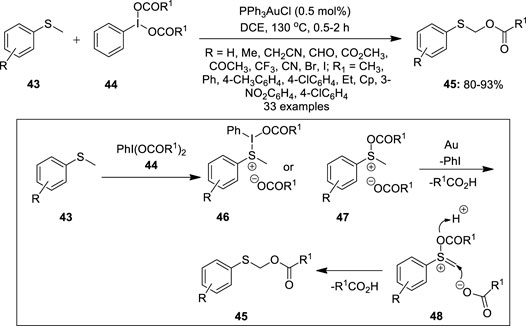
FIGURE 6. Au(II)-catalyzed α-C (sp3)–H acyloxylation of methyl sulfides 43 using bis(acyloxy)iodobenzene 44 as an oxidant.
Copper
In recent years, copper has evolved as the most promising alternative to costly metals such as Pd or Rh in metal-catalyzed oxidative coupling reactions. Significant progress has been accomplished in Cu(I)/Cu(III) catalysis and critical experimental studies revealed the clear existence of Cu(III) intermediates in a variety of catalytic transformations. The use of hypervalent iodine reagents as oxidants in copper-catalyzed reactions is a growing area of research. The present section of the review highlights recent examples of copper catalysis mediated by hypervalent iodine reagents. Gaunt et al. demonstrated a site-selective Cu(II)-catalyzed C–H bond arylation of indoles 49 using diaryliodonium salts 15 as a coupling partner (Figure 7) (Phipps et al., 2008). Treatment of (NH)-indoles or N-methylated indole 49 with [Ar-I-Ar]OTf salts 15 in the presence of Cu(OTf)2 (10 mol%) as a catalyst in dichloroethane yielded 3-arylindoles 50 in 38–86% yields with excellent selectivity. The addition of 2,6-di-tert-butylpyridine (dtbpy) as a base was necessary to avoid indole dimerization. Notably, electron-rich indoles smoothly underwent arylation at room temperature and the corresponding electron-deficient ones required a higher reaction temperature. The reaction scope was also administered with unsymmetrical diaryliodonium salts 15 enabling the efficient transfer of a variety of arenes and heteroarenes aryl motifs at the C3-position of indoles 49.
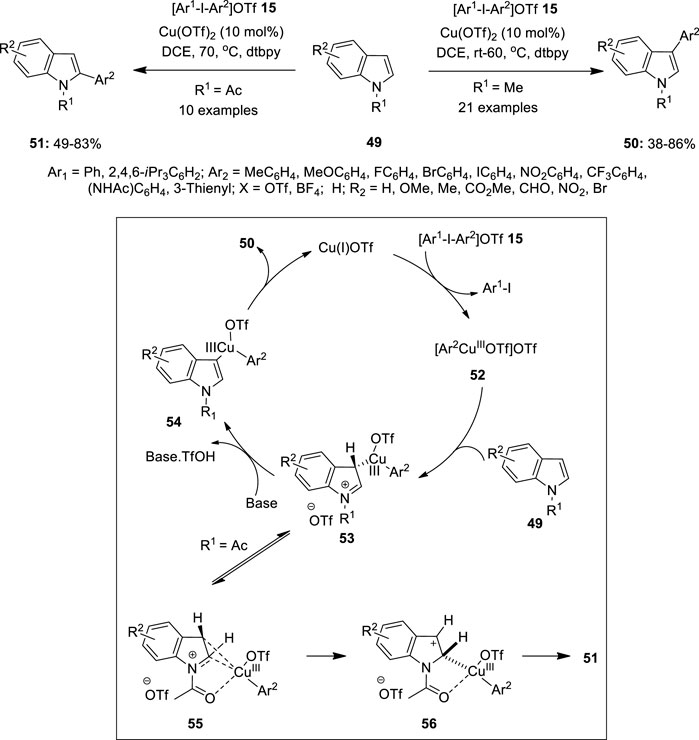
FIGURE 7. Cu(II)-catalyzed C–H bond arylation of indoles 49 using diaryliodonium salts 15 as a coupling partner.
The detailed mechanism of this arylation reaction is depicted in Figure 7. The reaction was proposed to initiate with the reduction of the Cu(II) catalyst to Cu(I) by indole 49, followed by the oxidative addition of diaryliodonium salts 15 to yield an electrophilic Cu(III)-aryl intermediate 52. The subsequent attack at the C3 position of indole 49 generates intermediate 53, which aromatizes via C–H bond cleavage forming intermediate 54. Finally, the reductive elimination of 54 liberates product 50 and re-forms the Cu(I) catalyst. Further reaction of N-acetylindole with 49 gave 2-arylindoles 51 selectively in excellent yields. It was speculated that the Cu(III)-aryl intermediate 53 would undergo migration of the Cu(III)-aryl group from the C3 to C2 position of the indole motif and subsequent re-aromatization to afford the C2-arylated indole 51. Based on the conclusions of Gaunt et al., Wu et al. investigated the site-selective nature of indoles with various directing groups (Wang, et al., 2018). The computational and mass spectrometric studies reveal that a neutral Heck-like mechanism is followed for a weak directing group such as N-acetyl indole leading to C2 site selection. This is because the electrophilic center tends to coordinate with the most electron-rich C3 carbon. However, in the case of indole with a N-P(O)tBu2 group and N-benzyl-3-pivaloyl indole, a cationic Heck-like reaction is preferred resulting in C6 and C5 selectivity, respectively (Wang, et al., 2018).
In continuation, Phipps and Gaunt (2009)designed a simple method to achieve selective meta C–H bond arylation by copper catalysis. They treated acetanilide 57 (R1 = Me and R2 = H) with an arylating agent, Ph2IOTf 15 in 1,2-dichloroethane in the presence of 10 mol% Cu(OTf)2 as the catalyst and isolated a meta arylated product 58 in low yield (Figure 8). With carbamate 57 (R1 = OMe and R2 = Me) and urea 57 (R1 = NEt2 and R2 = Me), the yield was modest, but with benzamides 57 (R1 = Ph and R2 = Me) and piv-anilides 57 (R1 = CMe3 and R2 = Me), the yield was excellent. It was observed that no arylation occurred in the absence of an amide group or a copper catalyst. Although the precise mechanism is not given, a possible reaction pathway could involve the activation of an aromatic ring by the highly electrophilic Cu(III)-aryl species, permitting the anti-oxy-cupration of the acetamide carbonyl group across the 2,3 position of the arene ring. This de-aromatization would give a Cu(III)-aryl species at the meta position and subsequent re-aromatizing deprotonation, followed by the reductive elimination which would deliver the anticipated meta product 58. While investigating the substrate scope, it was discovered that the electron-rich substrates provided excellent yields for meta arylation whereas the electron-deficient ones resulted in albeit lower yields. Remarkably, the reaction also produced good yields from unsymmetrical iodonium salts 15.
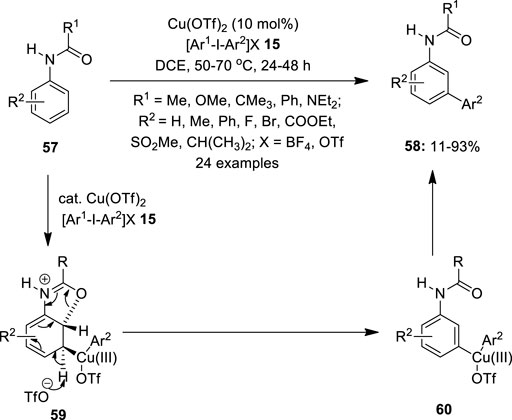
FIGURE 8. Cu(II)-catalyzed selective meta arylation of amides 57 using iodonium salts 15 as an arylating agent.
Recently, C–H arylation of fused pyrimidinone derivatives 61 was developed by Fruit et al using a CuI catalyst loaded with diaryliodonium triflates 15 as an aryl source under microwave irradiation (Pacheco-Benichou et al., 2021) (Figure 9). It was observed that the reactions perform well in CuI as the catalyst in 30 mol% and no product was formed in the absence of a base or catalyst. The addition of 3.5 equivalent of tBuOLi as the base and dioxane as the solvent media gave the best C2 arylation. The substrate scope showed that aromatic groups with para or meta CF3 groups, and halogens, all offered good yields of arylated compounds 62. In the case of unsymmetrical diaryliodonium salts 15, more sterically demanding groupings were shown to migrate preferentially over the others. However, in the absence of the steric effect, the most electron-donating aryl group transfer was favored. Finally, a mechanism was proposed for this reaction, in which a highly electrophilic CuIII intermediate 63 was formed by the oxidative addition of a diaryliodonium salt 15 to CuI species. Subsequent C–H activation leads to CuIII intermediate 64, which undergoes reductive elimination to give an arylated product 62 and releases CuI species for the next catalytic cycle.
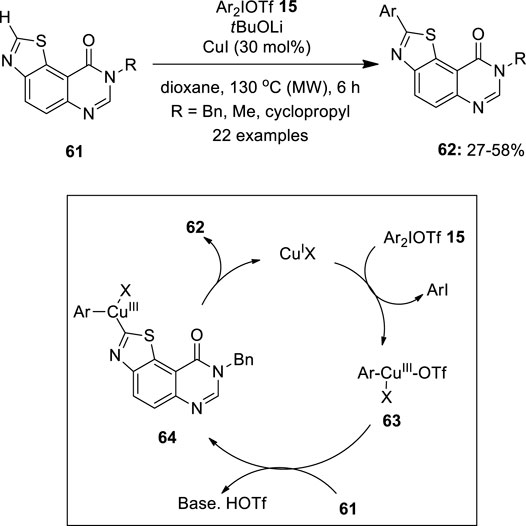
FIGURE 9. Copper-catalyzed C–H arylation of fused-pyrimidinone derivatives 61 using diaryliodonium triflates 15 as an aryl source.
Although diaryliodonium salts are gaining much attention as versatile arylating reagents, they always generate 1 equivalent of iodoarene as waste which needs to be separated from the desired product. Within this context, Modha and Greaney (2015) came up with an excellent atom economical protocol for harnessing discrete aryl groups in diaryliodonium salts 15 for double arylation of indoles 49 by copper catalysis
(Figure 10). They performed one-pot copper-catalyzed C−H arylation of indoles 49 followed by N-arylation using the in situ-generated aryl iodide using the same copper catalyst. The present tandem C−H/N−H process was found to be productive for a range of electron-rich/-poor symmetrical diaryliodonium salts 15. Further diarylation with unsymmetrical aryl-uracil iodonium triflate 15 was established successfully to furnish a novel indoyl uracil 65 in useful yields.
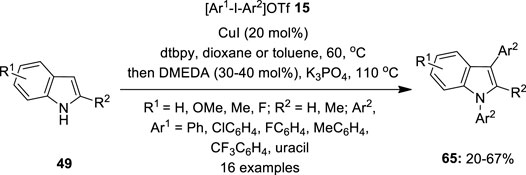
FIGURE 10. Copper-catalyzed C–H/N−H arylation of indoles 49 with diaryliodonium salts 65 as an aryl source.
In 2011, Chang et al. developed a copper-catalyzed synthesis of carbazoles 67 from N-substituted amidobiphenyls 66 using hypervalent iodine (III) reagent as an oxidant (Cho et al., 2011). The combination of copper (II) triflate (5 mol%) and PhI(OAc)2 3 significantly promotes this intramolecular oxidative C–N bond formation reaction to produce carbazoles 67 in good to excellent yields (Figure 11). Notably, substrates 66 with electron-rich substituents (R2 = OMe, tBu) at the “right” side of the nucleophilic phenyl part provided higher yields whereas those with electron-deficient ones (R2 = Cl) resulted in satisfactory product yields. Moreover, a reverse reactivity pattern was observed for the “left” amido-containing aryl part, wherein electron-withdrawing groups facilitate the cyclization. The reaction tolerated both N-sulfonyl and N-acetylamino substrates; however, lower product 67 yields were obtained in the latter case. The detailed mechanistic investigation revealed that the copper species catalytically activates the iodine (III) oxidant 3 used in the reaction. A plausible reaction pathway is depicted in Figure 11. Initially, 2 equiv. of substrate 66 binds reversibly to the copper to form a tetradentate copper species 68, followed by the formation of N-iodoamido species 69 through the release of acetic acid and Cu(OTf)2, which later participates in the next cycles. Furthermore, the ortho-phenyl group electrophilically attacks the amido moiety of 69 via a radical path to form a radical species 70. Finally, the in situ-generated acetoxy radical abstracts hydrogen from 70 and delivers carbazoles 67.
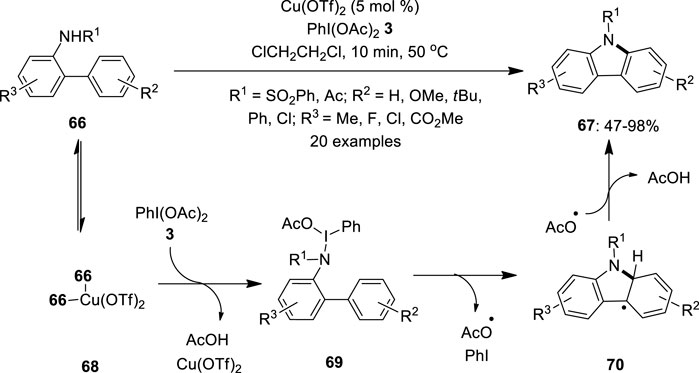
FIGURE 11. Copper-catalyzed intramolecular cyclization of N-substituted amidobiphenyls 66 using PhI(OAc)2 3 as an oxidant.
In 2012, Fossey et al. reported the copper-catalyzed C–H activation/intramolecular amination of 6-anilinopurine nucleosides 71 by using PhI(OAc)2 3 as an oxidant (Qu et al., 2012). Among the different catalysts screened, Cu(OTf)2 provided the best catalytic activity. This method enabled the facile synthesis of multi-heterocyclic compounds 72 from a variety of purines and its derivatives 71 (Figure 12). Substrates with N9-substituents such as sugar, alkyl, allyl, benzyl, etc. were evaluated successfully for reaction scope. In addition, substrates with electron-withdrawing groups on the aniline ring gave higher yields than those with electron-donating ones. Moreover, steric effect studies showed that ortho-substituted substrates 71 were found to be less reactive than those with para substituents. A plausible CuII/Cu0 catalytic cycle for this intramolecular cyclization is depicted in Figure 12. Initially, Cu(OTf)2 coordinates with 73, followed by an electrophilic substitution process to yield Cu(II) intermediate 75. Finally, reductive elimination releases the cyclized product 72 and delivers Cu0, which can be reoxidized to generate Cu(OTf)2.
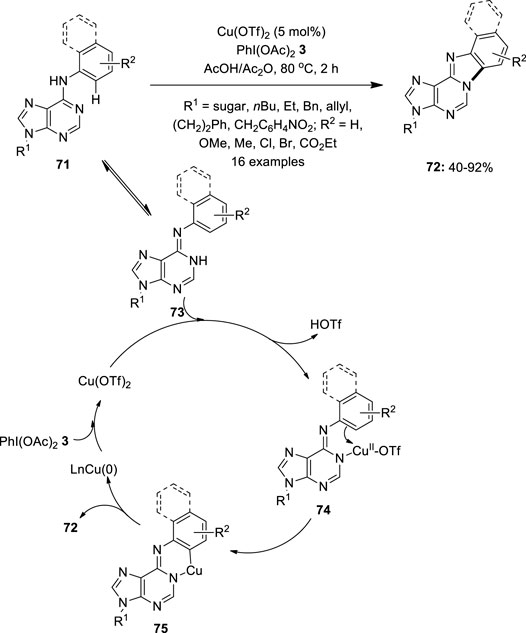
FIGURE 12. Copper-catalyzed synthesis of multi-heterocyclic compounds 72 from 6-anilinopurine nucleosides 71 using PIDA 3 as an oxidant.
Waser et al. used azidobenziodazolone 77 as an azide precursor for the ring-expansion of alkene-substituted cyclobutanol derivatives 76 (Alazet et al., 2018). This reaction was performed under photoredox conditions in acetonitrile in the presence of Cu(dap)2Cl as a catalyst (Figure 13). The reaction scope was significantly larger, as para-substituted styrenes 76 with alkyl-, methoxy-, chloro-, and phenyl groups provided excellent yields. Similarly, ortho or meta-substituted styrenes 76 produced cyclopentanones 78 in good yields. Boc-protected azetidine and styrene with 1,2 di-substituted olefins, on the other hand, were unresponsive. Unexpectedly, when para-CF3-substituted styrene 76 was reacted, the only result was epoxide.
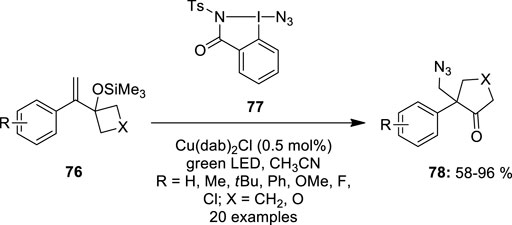
FIGURE 13. Copper-catalyzed ring expansion of alkene-substituted cyclobutanol derivatives 76 using azidobenziodazolone 77 as an azide precursor.
Parsons and Buchwald (2011) reported Cu(I)-catalyzed allylic trifluoromethylation of terminal alkenes 79 using Togni’s electrophilic trifluoromethylating reagent 9. This approach has notable advantages such as no harsh reaction conditions, free from toxic or expensive chemicals, and the reagent can be easily recycled. The promising result of these transformations was obtained with 15 mol% of [(MeCN)4Cu]PF6 as a copper source and methanol as a solvent (Figure 14). Trifluoromethylation of a series of alkenes with variable functional groups such as amides, protected amines, unprotected alcohols, esters, and alkyl bromides was achieved successfully to furnish allyl–CF3 products 80 with excellent E/Z selectivity having an average ratio of 94:6 under these conditions. However, 1,2-disubstituted olefins and branched terminal olefins were found unsuitable substrates because of the formation of undesired regioisomeric product mixtures.
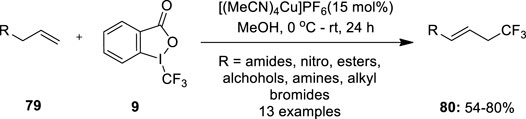
FIGURE 14. Cu(I)-catalyzed allylic trifluoromethylation of inactivated terminal olefins 79 using Togni’s electrophilic trifluoromethylating reagent 9.
Platinum
Platinum has great potential to offer in oxidative coupling reactions owing to its powerful reactivity. Recent developments in this area showed that platinum has remarkable reactivity and excellent selectivity than those with palladium-catalyzed reactions. In this section, we will discuss catalytic reports involving Pt (II)/Pt (IV) intermediates and hypervalent iodine reagents as oxidants. In 2009, Mutule et al. (2009)used a platinum species as an alternative catalyst for the regioselective C–H bond acetoxylation of indoles 49 to prepare biologically important 3-acetoxyindoles 81 via a C–H activation/oxidation strategy. In this reaction, C3-acetoxylation of indoles 49 was performed using 5 mol% PtCl2 as the catalyst in the presence of PhI(OAc)2 3 as the terminal oxidant. Optimization studies revealed that other oxidants such as K2S2O8, Cu(OAc)2, t-BuOOH, and m-CPBA were completely inefficient in promoting this acetoxylation reaction. Although the reaction also proceeded well with Pd(OAc)2, PtCl2 was found more efficient and high yielding. Straightforward access to a variety of 3-acetoxyindole-2-carboxylates 81 was achieved in moderate to good yields. Interestingly, C–H activation/oxidation occurred exclusively at the C-3 position, even in the case of 2,3-unsubstituted indole. The reaction tolerated bromo-, iodo-, and cyano-substituents on the indole moiety under these conditions. Notably, acetoxylation was sensitive to the nature of substituents on the pyrrole ring, particularly, substrates with the N-methyl group reacted faster than those with N-aryl substituents. In addition, C3-acetoxylation of N-unsubstituted indole 49 (R2 = H) was also achieved and the anticipated product 81 was isolated in 67% yield (Figure 15).
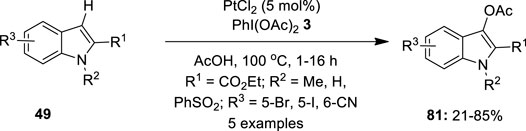
FIGURE 15. Pt(II)-catalyzed C3-acetoxylation of indoles 49 to afford C3-acetoxylated indoles 81 using PhI(OAc)2 3.
Furthermore, Sanford et al. used a Na2PtCl4 catalyst during the C−H arylation of arenes 82 with diaryliodonium salts 15 (Wagner et al., 2013). A variety of aryliodonium salts 15 with ortho-, meta-, and para-substituents were coupled successfully with naphthalene 82 to give biaryl products 84 with high selectivity for isomer 84 over 83 (Figure 16). The reaction produced the best yields in neat naphthalene as revealed by various optimization studies. Notably, C−H arylation of naphthalene 82 showed completely opposite site selectivity on changing the catalyst from Na2PdCl4 (Hickman and Sanford, 2011) to platinum salt under identical conditions. The site selectivity of this C−H cleavage reaction is predominantly controlled by steric factors, suggesting the feasibility of achieving high selectivity for isomer 84 in Pt-catalyzed naphthalene arylation. Further scope of the arylation reaction was evaluated with electronically diverse arene substrates 82. Most importantly, the reaction proceeded faster with electron-rich arenes 82 than with electron-deficient ones. A proposed Pt (II)/Pt (IV) catalytic cycle for this reaction is depicted in Figure 16. Initially, [Ar2I]TFA 15 oxidizes the Pt (II) catalyst to a Pt (IV)−aryl intermediate 85 followed by subsequent C−H activation of arene 82 to form diaryl Pt (IV) species 86. Finally, reductive elimination from the Pt (IV) species 86 releases a coupled product 84. Preliminary mechanistic studies revealed that reductive elimination is the rate-determining step in this Pt-catalyzed reaction.
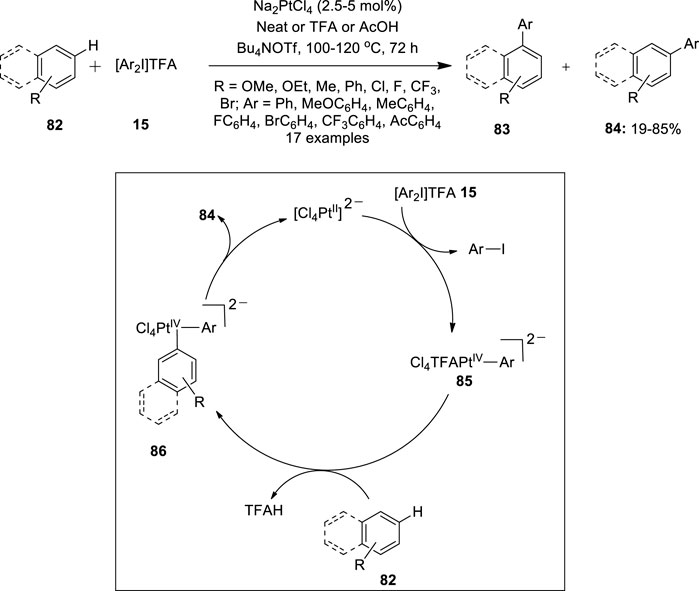
FIGURE 16. Pt(II)-catalyzed C−H arylation of arenes 82 with diaryliodonium salts 15.
Iron
Catalytic cycles involving iron as the catalyst in oxidative coupling reactions are rare. In 2014, Lu and Lu (2014)developed a novel protocol for the iron-catalyzed carbodi- and trichloromethylation/cyclization of N-arylacrylamides 87 with dichloro- and tetrachloromethane. This transformation used diaryliodonium salt 15 and triethylamine as an efficient oxidant and a base, respectively. Notably, the reaction of a wide range of N-arylacrylamides 87 bearing electron-donating and -withdrawing substituents proceeded smoothly to give the anticipated dichloromethylated oxindoles 88 and trichloromethylated oxindoles 89 in good-to-excellent yields (Figure 17). Substrates 87 with N-protection groups such as methyl, ethyl, benzyl, and isopropyl were tolerated; however, no product formation was observed with free N-H acrylamide. A plausible reaction mechanism for this reaction is depicted in Figure 17. Initially, diaryliodonium salt 15 generates an aryl radical 90 in the presence of an iron catalyst. Subsequently, aryl radical 90 abstracts hydrogen atoms from CH2Cl2 or chlorine atoms from CCl4 to generate the corresponding dichloromethyl radical (·CHCl2) 91 or trichloromethyl radical (·CCl3) 91, followed by the addition to the the carbon–carbon double bond of 87 to form a radical intermediate 92. Furthermore, the intramolecular cyclization of the radical intermediate 92 gives the intermediate 93, followed by the single-electron transfer (SET) to the Fe(III) intermediate, and the subsequent base-assisted proton abstraction provides the desired cyclized product 88 or 89.
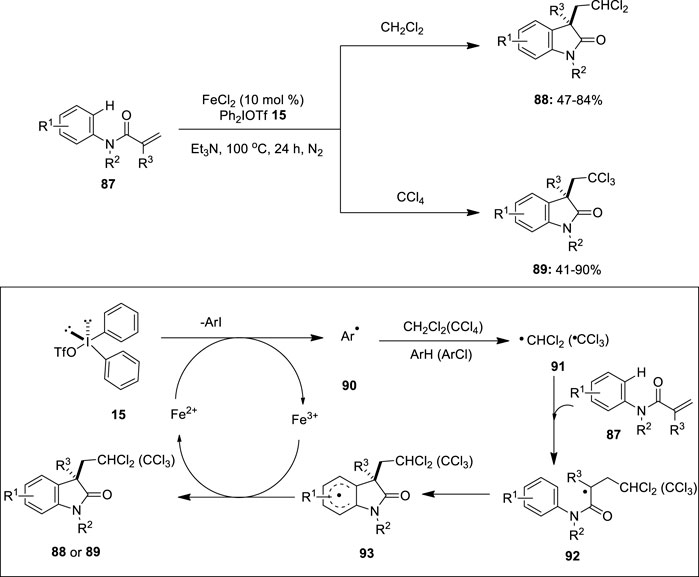
FIGURE 17. Iron(II)-catalyzed carbodi- and trichloromethylation/cyclization of N-arylacrylamides 87 using diaryliodonium salt 15 as an oxidant.
Iridium
Iridium has received great interest in oxidative cross-coupling reactions involving C–H activation as indicated by various literature reports. In 2013, Greaney et al. described a three-component coupling of styrenes 94 with diaryliodonium salts 15 and heteroatom nucleophiles 95 by photoredox catalysis (Fumagalli et al., 2013) (Figure 18). This method enabled the construction of doubly functionalized phenylethyl scaffolds 96 via simultaneous arylation and C–O or C–N bond formation. Reactions were performed in the presence of Ir(ppy)3 (1–5 mol%) as the photoredox catalyst and Zn(OAc)2 (20 mol%) as an effective additive. A range of styrene substrates 94 underwent smooth methoxyarylation in degassed methanol affording products 96 in moderate to good yields. The reaction scope was further examined with various heteroatom nucleophiles such as alcohols, water, or nitriles to afford corresponding coupled products 96 in variable yields. The proposed mechanistic pathway is shown in Figure 18. The reaction initiates with the reduction of the diaryliodonium salt 15 with photoexcited Ir(III)* and the resulting aryl radical 90 is trapped by styrene 94 to form a benzylic radical 97. Subsequently, the oxidation of radical 97 by Ir(IV) generates the cation 98 which can be trapped by an appropriate nucleophile 95 to afford the coupled product 96.
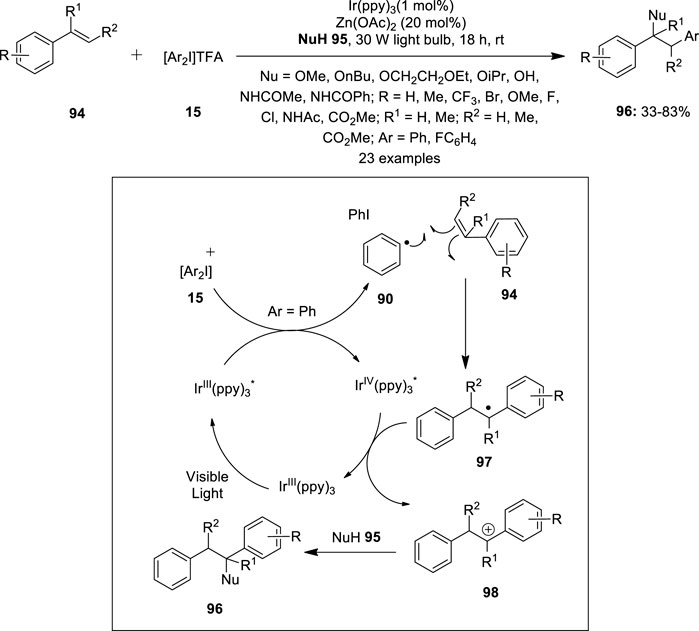
FIGURE 18. Iridium-catalyzed three-component coupling of styrenes 94 with diaryliodonium salts 15 and heteroatom nucleophiles 95.
Later, Shi et al. demonstrated a direct arylation of the sp3 C–H bond in ketoximes 99 and nitrogen-containing heterocycles 100 with appropriate diaryliodonium salts 15 using [(Cp*IrCl2)2] as the pre-catalyst (Gao et al., 2015) (Figure 19). The reaction was performed in the presence of AgNTf2 as the halide abstractor and PivOH as the additive. The ketoximes 99 containing α-hydrogens were found to be compatible with this reaction. Furthermore, ketoximes with a six- or seven-membered ring produce the desired product 101 with a high yield. Also, heterocycle-directed sp3 C–H bond arylation using pyridine, pyrazine, quinoline, pyrazole, and isoxazole as the directing groups was tested and the respective products 102 were obtained in variable yields. In addition, the reaction scope was successfully extended to the sp2 C–H bond arylation of a variety of substituted arenes and olefins under the same optimized conditions.
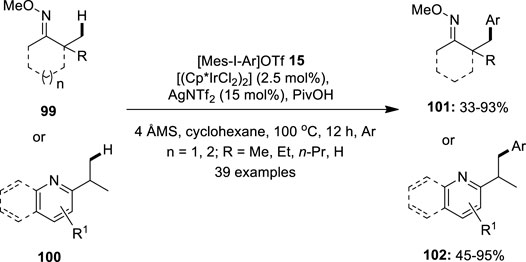
FIGURE 19. Iridium-catalyzed sp3 C–H bond arylation of ketoximes 99 and nitrogen-containing heterocycles 100 with diaryliodonium salts 15.
Moreover, the same catalytic condition was used to arylate the C–H bond of N-arylpyrrolidinones 103 and their analogs at the ortho position (Gao et al., 2016) (Figure 20). A wide spectrum of N-arylpyrrolidinones 103 and diaryliodonium salts 15 having electron-neutral, electron-donating, and halogen-containing motifs were effectively cross-coupled. Substrates containing electron-deficient trifluoromethyl and nitro substituents, on the other hand, produce lesser yields. Interestingly, moderate to excellent yields were obtained when polycyclic and heterocyclic aromatic motifs were coupled. In addition, phenylpyridines, indoles, arenes such as N-phenyl amide, benzamides, and sulphonamides, and enamide with both vinylic and allylic C–H linkages and vinyl carboxylic acids were also compatible with the reaction.
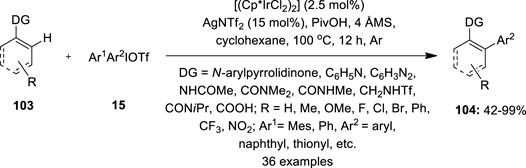
FIGURE 20. Iridium-catalyzed C–H arylation of arenes and olefins 103 using diaryliodonium salts 15 as an arylating agent.
In the next year, NH2-directed C–H alkenylation of 2-vinylanilines 105 with alkenyl-3-iodanes 106 as an electrophilic alkene-transfer reagent was developed using the same iridium catalyst (Boelke et al., 2017) (Figure 21). This method enabled the selective synthesis of 1,3-dienes 107 in excellent yields with high Z stereoselectivity. Optimization studies revealed that the electron-rich and electron-poor substrates all showed high reactivities similar to that of the un-substituted substrate. Notably, even after altering the different alkyl side chains (R2 = Et, iPr, Cy), the corresponding products with high selectivity were retained, whereas the reaction did not occur for highly substituted alkenylanilines 105 (R3 = Me, Ph). Further reaction failed with meta- and para-substituted anilines 105, thus showing the relevance of the free amino group at the ortho position to the exocyclic double-bond as the guiding group. The reaction scope with different vinyl benziodoxolones 106 was also investigated, wherein it was discovered that p-toloylvinyl benziodoxolone showed similar reactivity to that of phenyl vinyl benziodoxolone while p-methoxyphenylvinyl benziodoxolone displayed lower reactivity.
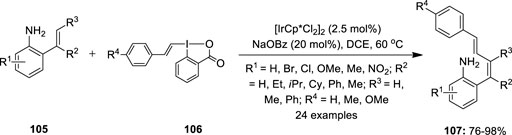
FIGURE 21. Iridium-catalyzed C–H alkenylation of 2-vinylanilines 105 using alkenyl-3-iodanes 106 as an electrophilic alkene-transfer reagent.
Recently, iridium-catalyzed C–H alkynylation of 2-(hetero)arylquinazolin-4-ones 108 has been investigated using ethynylbenziodoxolone 14 (TIPS-EBX) as an alkynylating reagent (Rohokale et al., 2019) (Figure 22). The reaction was found to be solvent-dependent and an exclusive formation of the monoalkynylated products 109 was observed when a polar protic solvent such as methanol was used as the solvent for this reaction. To form a di-alkynylated product 110 exclusively, dichloroethane at 70°C was used. Notably, selective mono-/dialkynylation of a variety of substrates 108 with different functional groups such as -F, -Cl, -Br, -NO2, -OMe, -CF3, alkyl, napthyl, etc. proceeded smoothly under these conditions to give mono-/dialkynylated products in good to excellent yields. The tentative mechanism for this reaction is shown in Figure 22. Initially, the dimeric iridium complex reacts with AgSbF6 to form a monomeric IrCp*(SbF6)2 complex, which undergoes a coordinative C−H insertion with 2-arylquinazolin-4-one 108 to give a cyclometalated Ir(III) complex 111. Authors proposed two pathways for the transfer of the alkyne group from the TIPS-EBX 14 to the aryl ring. In one approach, the oxidative addition of TIPS-EBX 14 results in the formation of an alkynyl-Ir(V) species 112, which undergoes reductive elimination to yield the crucial Ir(III)-alkyne intermediate 113 (path a). In another path, the complexation of the intermediate 111 with TIPS-EBX 14 followed by the subsequent regioselective migratory insertion of alkyne, resulting in the intermediate 114, which yields iridium vinylidene species 115 upon the α-elimination of 2-iodobenzoic acid. The intermediate 115 further undergoes concerted 1,2-R-group migration followed by elimination, resulting in the intermediate 113, a species seen in both pathways a and b. Finally, the dissociation of alkyne from 113 yields the alkynylated product 109 and the active Ir(III) species, which continue the catalytic cycle.
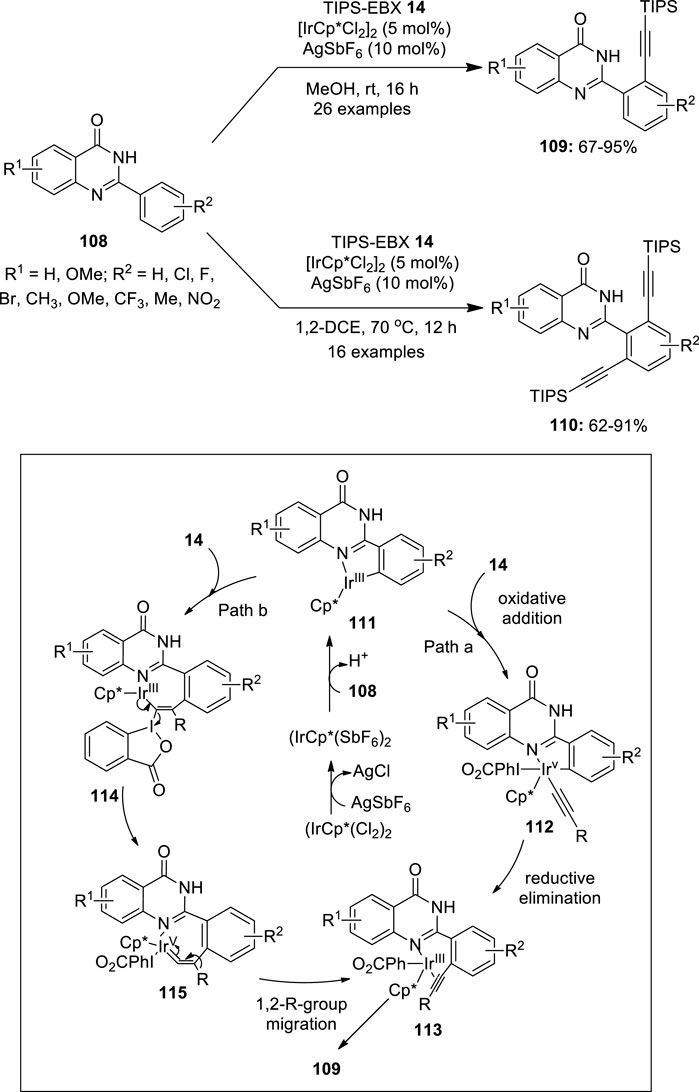
FIGURE 22. Iridium-catalyzed C–H alkynylation of 2-(hetero)arylquinazolin-4-ones 108 using TIPS-EBX 14 as an alkynylating reagent.
Nickel
Iyanaga et al. described the first example of Ni(II)-catalyzed 8-aminoquinoline directed C (sp3) –H arylation of aliphatic amides 116 using diaryliodonium salts 15 as an arylating agent (Iyanaga, et al., 2014) (Figure 23). The reaction used Ni(OTf)2 (10 mol%) as the catalyst and 4-methyltetrahydro-2H-pyran (MTHP) as the solvent system. Moreover, Na2CO3 as the base resulted in high product yields. Under this condition, β-arylation products 117 were obtained in good to excellent yields with broad functional group compatibility. Diaryliodonium triflates 15 with electron-donating aryl groups produced higher product yields than those with electron-withdrawing groups. Amides with no α-hydrogen reacted exclusively at the methyl group whereas those with methylene or benzene C–H bonds were not arylated. The proposed mechanistic pathway for the reaction is shown in Figure 23. Initially, amide 116 co-ordinates with the Ni center and followed by ligand exchange forms the Ni complex 118, which further undergoes reversible cyclometalation to give the intermediate 119 via a CMD mechanism. The oxidative addition of the diaryliodonium salt 15 to the intermediate 119 generates a high-valent Ni(IV) complex 120 and aryl iodide. Finally, reductive elimination of the complex 120 and subsequent protonation affords the desired arylation product 117 along with the regeneration of Ni(II) for the next catalytic cycle.
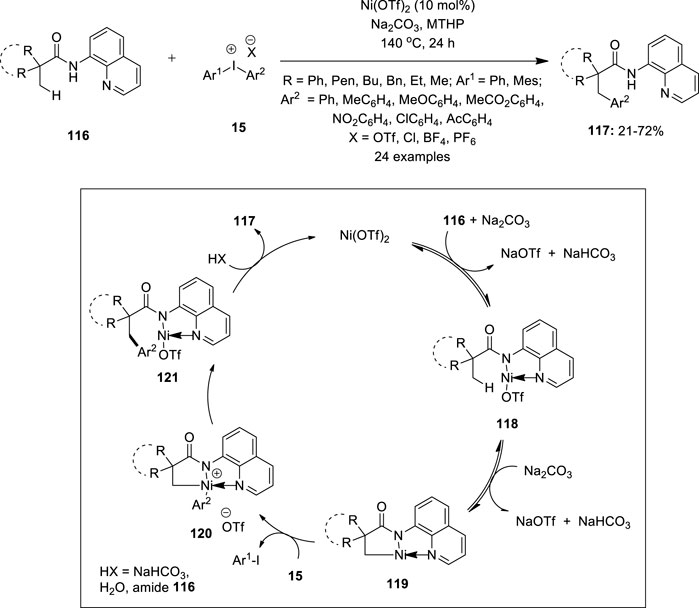
FIGURE 23. Ni(II)-catalyzed C (sp3) –H arylation of aliphatic amides 116 using diaryliodonium salts 15 as an arylating agent.
Ruthenium
In 2014, Chatani et al. reported the Ru(II)-catalyzed C–H arylation of 2-arylpyridine derivatives 122 using diaryliodonium salts 15 as an arylating reagent (Ho et al., 2014). The reaction used [Ru (OAc)2 (p-cymene)] as an active catalyst, K2CO3 as a base, and AcOH as a solvent (Figure 24). The substrates’ scope revealed that electron-rich 2-arylpyridines 122 gave the corresponding product 123 in lower yields than those with electron-deficient ones. A variety of mixed diaryliodonium salts 122 were used, particularly electron-rich and sterically less demanding aryl groups are selectively transferred to give 123 in useful yields. Notably, the reaction yielded single arylated products and tolerated a broad range of functional groups. The proposed reaction mechanism is discussed in Figure 24. Initially, 2-phenylpyridine 122 reacts with a Ru(II) catalyst to form a cyclometalated ruthenium complex 124, which upon oxidative addition with Ar1Ar2IOTf 15 gives the Ru(IV) complex 125. Finally, the complex 125 undergoes reductive elimination to give an arylated product 123 along with the regeneration of the Ru(II) catalyst. Another example of Ru-catalyzed C–H arylation of arenes and hetero-arenes using diaryliodonium salts 15 was reported previously by Xiao et al. (Liu et al., 2013).
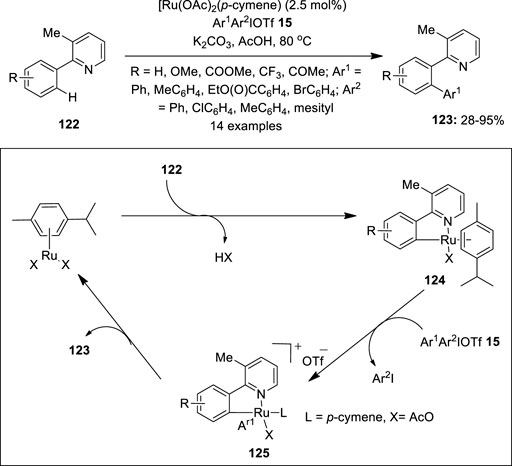
FIGURE 24. Ru2+-catalyzed arylation of 2-arylpyridines 122 with Ar1Ar2IOTf 15 as an arylation reagent.
Conclusion
A transition metal-catalyzed direct oxidative coupling reaction has been identified as a straightforward route for C–C and C–heteroatom bond formation. This strategy is most elegant, economic, and environmentally friendly than traditional cross-coupling reactions. For many years, most of these coupling reactions used palladium as a catalyst, but recent studies showed other transition metal catalysts such as gold, copper, platinum, iron, nickel, and ruthenium as potential alternative candidates for these reactions. Hypervalent iodine reagents have evolved as versatile oxidants and functional group transfer reagents in this area particularly because of their excellent oxidizing and electrophilic properties. This has led to the rapid development in transition metal-catalyzed homo/cross-coupling, arylation, alkenylation, alkynylation, acyloxylation, and alkene functionalization reactions. PhI(OAc)2 is the most commonly used oxidant whereas diaryliodonium salts and TIPS-EBX are widely used as arylating and alkynylating reagents, respectively, in these reactions. Despite the great success of hypervalent iodine reagents, the main drawback associated with them is the stoichiometric generation of byproducts, which needs to be addressed in future. In addition, there exists limited scope of hypervalent iodine (III) reagents for functional group transfer reactions. Moreover, the development of newer hypervalent iodine compounds that can be readily recycled or regenerated would be an active area of research to expand the versatility of this reaction manifold.
Author Contributions
SS has covered and compiled the literature regarding the gold and copper-catalyzed coupling reactions. AR has compiled the platinum-catalyzed oxidative coupling reactions. Iron and iridium-catalyzed coupling reactions were covered by HC while the remaining reactions (nickel and ruthenium-catalyzed) were compiled by NT. The idea of the manuscript was given by FS and the editing and final revision of the manuscript was given by FS and TD.
Funding
NT and TD acknowledge support from JSPS KAKENHI Grant Number 20K06980 (NT) and 19K05466 (TD), and the Ritsumeikan Global Innovation Research Organization (R-GIRO) project. HC also thanks support from JSPS via a Grant-in-Aid for Early-Career Scientists, KAKENHI (Grant No 20K15103).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Authors are thankful to VIT Chennai for providing infrastructure for preparing the review article.
References
Alazet, S., Preindl, J., Simonet-Davin, R., Nicolai, S., Nanchen, A., Meyer, T., et al. (2018). Cyclic Hypervalent Iodine Reagents for Azidation: Safer Reagents and Photoredox-Catalyzed Ring Expansion. J. Org. Chem. 83, 12334–12356. doi:10.1021/acs.joc.8b02068
Ball, L. T., Lloyd-Jones, G. C., and Russell, C. A. (2012). Gold-Catalysed Oxyarylation of Styrenes and Mono- Andgem-Disubstituted Olefins Facilitated by an Iodine(III) Oxidant. Chem. Eur. J. 18, 2931–2937. doi:10.1002/chem.201103061
Banerjee, S., Bhoyare, V. W., and Patil, N. T. (2020). Gold and Hypervalent Iodine(III): Liaisons over a Decade for Electrophilic Functional Group Transfer Reactions. Chem. Commun. 56, 2677–2690. doi:10.1039/D0CC00106F
Boelke, A., Caspers, L. D., and Nachtsheim, B. J. (2017). NH2-Directed C-H Alkenylation of 2-Vinylanilines with Vinylbenziodoxolones. Org. Lett. 19, 5344–5347. doi:10.1021/acs.orglett.7b02630
Brand, J. P., Charpentier, J., and Waser, J. r. m. (2009). Direct Alkynylation of Indole and Pyrrole Heterocycles. Angew. Chem. Int. Ed. 48, 9346–9349. doi:10.1002/anie.200905419
Brand, J. P., and Waser, J. (2010). Direct Alkynylation of Thiophenes: Cooperative Activation of TIPS-EBX with Gold and Brønsted Acids. Angew. Chem. 122, 7462–7465. doi:10.1002/ange.201003179
Brand, J. P., and Waser, J. (2012). Para-Selective Gold-Catalyzed Direct Alkynylation of Anilines. Org. Lett. 14 (3), 744–747. doi:10.1021/ol203289v
Cambeiro, X. C., Boorman, T. C., Lu, P., and Larrosa, I. (2013). Redox-Controlled Selectivity of CH Activation in the Oxidative Cross-Coupling of Arenes. Angew. Chem. 125, 1825–1828. doi:10.1002/ange.201209007
Campeau, L.-C., and Hazari, N. (2019). Cross-Coupling and Related Reactions: Connecting Past Success to the Development of New Reactions for the Future. Organometallics 38, 3–35. doi:10.1021/acs.organomet.8b00720
Cho, S. H., Yoon, J., and Chang, S. (2011). Intramolecular Oxidative C−N Bond Formation for the Synthesis of Carbazoles: Comparison of Reactivity between the Copper-Catalyzed and Metal-free Conditions. J. Am. Chem. Soc. 133, 5996–6005. doi:10.1021/ja111652v
Deprez, N. R., and Sanford, M. S. (2007). Reactions of Hypervalent Iodine Reagents with Palladium: Mechanisms and Applications in Organic Synthesis. Inorg. Chem. 46, 1924–1935. doi:10.1021/ic0620337
Dohi, T., and Kita, Y. (20092073–2085). Hypervalent Iodine Reagents as a New Entrance to Organocatalysts. Chem. Commun., 2073. doi:10.1039/B821747E
Elsherbini, M., and Wirth, T. (2018). Hypervalent Iodine Reagents by Anodic Oxidation: A Powerful Green Synthesis. Chem. Eur. J. 24, 13399–13407. doi:10.1002/chem.201801232
Fumagalli, G., Boyd, S., and Greaney, M. F. (2013). Oxyarylation and Aminoarylation of Styrenes Using Photoredox Catalysis. Org. Lett. 15 (17), 4398–4401. doi:10.1021/ol401940c
Gao, P., Guo, W., Xue, J., Zhao, Y., Yuan, Y., Xia, Y., et al. (2015). Iridium(III)-Catalyzed Direct Arylation of C-H Bonds with Diaryliodonium Salts. J. Am. Chem. Soc. 137, 12231–12240. doi:10.1021/jacs.5b06758
Gao, P., Liu, L., Shi, Z., and Yuan, Y. (2016). Iridium(iii)-catalyzed Regioselective Direct Arylation of Sp2 C-H Bonds with Diaryliodonium Salts. Org. Biomol. Chem. 14, 7109–7113. doi:10.1039/c6ob01145d
Grelier, G., Darses, B., and Dauban, P. (2018). Hypervalent Organoiodine Compounds: from Reagents to Valuable Building Blocks in Synthesis. Beilstein J. Org. Chem. 14, 1508–1528. doi:10.3762/bjoc.14.128
Guo, S.-r., Santhosh Kumar, P., Yuan, Y.-q., and Yang, M.-h. (2016). Gold-Catalyzed, Iodine(III)-Mediated Direct Acyloxylation of the Unactivated C(sp3)-H Bonds of Methyl Sulfides. Eur. J. Org. Chem. 2016, 4260–4264. doi:10.1002/ejoc.201600632
Henry, M., Mostafa, M., and Sutherland, A. (2017). Recent Advances in Transition-Metal-Catalyzed, Directed Aryl C-H/N-H Cross-Coupling Reactions. Synthesis 49, 4586–4598. doi:10.1055/s-0036-1588536
Hickman, A. J., and Sanford, M. S. (2011). Catalyst Control of Site Selectivity in the PdII/IV-Catalyzed Direct Arylation of Naphthalene. ACS Catal. 1, 170–174. doi:10.1021/cs1001543
Ho, J. S., Misal Castro, L. C., Aihara, Y., Tobisu, M., and Chatani, N. (2014). Ruthenium(II)-Catalyzed Chelation-Assisted Arylation of CH Bonds with Diaryliodonium Salts. Asian J. Org. Chem. 3, 48–51. doi:10.1002/ajoc.201300199
Hofer, M., and Nevado, C. (2013). Cross-coupling of arene-Gold(III) Complexes. Tetrahedron 69, 5751–5757. doi:10.1016/j.tet.2013.04.029
Hyatt, I. F. D., Dave, L., David, N., Kaur, K., Medard, M., and Mowdawalla, C. (2019). Hypervalent Iodine Reactions Utilized in Carbon-Carbon Bond Formations. Org. Biomol. Chem. 17, 7822–7848. doi:10.1039/C9OB01267B
Iyanaga, M., Aihara, Y., and Chatani, N. (2014). Direct Arylation of C(sp3)-H Bonds in Aliphatic Amides with Diaryliodonium Salts in the Presence of a Nickel Catalyst. J. Org. Chem. 79 (24), 11933–11939. doi:10.1021/jo501691f
Jana, R., Pathak, T. P., and Sigman, M. S. (2011). Advances in Transition Metal (Pd,Ni,Fe)-Catalyzed Cross-Coupling Reactions Using Alkyl-Organometallics as Reaction Partners. Chem. Rev. 111, 1417–1492. doi:10.1021/cr100327p
Joshi, A., and De, S. R. (2021). Diaryliodonium Salts in Transition‐Metal‐Catalyzed Chelation‐Induced C(sp 2/sp 3 )−H Arylations. Eur. J. Org. Chem. 2021, 1837–1858. doi:10.1002/ejoc.202100066
Kar, A., Mangu, N., Kaiser, H. M., Beller, M., and Tse, M. K. (2008). A General Gold-Catalyzed Direct Oxidative Coupling of Non-activated Arenes. Chem. Commun., 386–388. doi:10.1039/b714928j
Kar, A., Mangu, N., Kaiser, H. M., and Tse, M. K. (2009). Gold-catalyzed Direct Oxidative Coupling Reactions of Non-activated Arenes. J. Organomet. Chem. 694, 524–537. doi:10.1016/j.jorganchem.2008.11.016
Kita, Y., and Dohi, T. (2015). Pioneering Metal-free Oxidative Coupling Strategy of Aromatic Compounds Using Hypervalent Iodine Reagents. Chem. Rec. 15, 886–906. doi:10.1002/tcr.201500020
Kuhl, N., Hopkinson, M. N., Wencel-Delord, J., and Glorius, F. (2012). Beyond Directing Groups: Transition-Metal-Catalyzed CH Activation of Simple Arenes. Angew. Chem. Int. Ed. 51, 10236–10254. doi:10.1002/anie.201203269
Kumar, R., Singh, F. V., Takenaga, N., and Dohi, T. (2022). Asymmetric Direct/Stepwise Dearomatization Reactions Involving Hypervalent Iodine Reagents. Chem. Asian J. 17. article in press. doi:10.1002/asia.202101334
Li, Y., Brand, J. P., and Waser, J. (2013). Gold-Catalyzed Regioselective Synthesis of 2- and 3-Alkynyl Furans. Angew. Chem. Int. Ed. 52, 6743–6747. doi:10.1002/anie.201302210
Liu, Y.-X., Xue, D., Wang, J.-D., Zhao, C.-J., Zou, Q.-Z., Wang, C., et al. (2013). Room-Temperature Arylation of Arenes and Heteroarenes with Diaryl-Iodonium Salts by Photoredox Catalysis. Synlett. 24 (4), 507–513. doi:10.1055/s-0032-1318155
Lu, M.-Z., and Lu, T. P. (2014). Iron-Catalyzed Cascade Carbochloromethylation of Activated Alkenes: Highly Efficient Access to Chloro-Containing Oxindoles. Org. Lett. 16, 4698–4701. doi:10.1021/ol502411c
Mekhman S. Yusubov, M. S., and Viktor V. Zhdankin, V. V. (2012). Hypervalent Iodine Reagents and Green Chemistry. Cos 9, 247–272. doi:10.2174/157017912799829021
Modha, S. G., and Greaney, M. F. (2015). Atom-Economical Transformation of Diaryliodonium Salts: Tandem C-H and N-H Arylation of Indoles. J. Am. Chem. Soc. 137 (4), 1416–1419. doi:10.1021/ja5124754
Mutule, I., Suna, E., Olofsson, K., and Pelcman, B. (2009). Catalytic Direct Acetoxylation of Indoles. J. Org. Chem. 74, 7195–7198. doi:10.1021/jo901321b
Pacheco-Benichou, A., Ivendengani, E., Kostakis, I. K., Besson, T., and Fruit, C. (2021). Copper-Catalyzed C-H Arylation of Fused-Pyrimidinone Derivatives Using Diaryliodonium Salts. Catalysts 11, 28. doi:10.3390/catal11010028
Parsons, A. T., and Buchwald, S. L. (2011). Copper-Catalyzed Trifluoromethylation of Unactivated Olefins. Angew. Chem. 123, 9286–9289. doi:10.1002/ange.201104053
Phipps, R. J., and Gaunt, M. J. (2009). A Meta-Selective Copper-Catalyzed C-H Bond Arylation. Science 323, 1593–1597. doi:10.1126/science.1169975
Phipps, R. J., Grimster, N. P., and Gaunt, M. J. (2008). Cu(II)-Catalyzed Direct and Site-Selective Arylation of Indoles under Mild Conditions. J. Am. Chem. Soc. 130, 8172–8174. doi:10.1021/ja801767s
Qiu, G., and Wu, J. (2015). Transition Metal-Catalyzed Direct Remote C-H Functionalization of Alkyl Groups via C(sp3)-H Bond Activation. Org. Chem. Front. 2, 169–178. doi:10.1002/chin.201517312
Qu, G.-R., Liang, L., Niu, H.-Y., Rao, W.-H., Guo, H.-M., and Fossey, J. S. (2012). Copper-Catalyzed Synthesis of Purine-Fused Polycyclics. Org. Lett. 14, 4494–4497. doi:10.1021/ol301848v
Rohokale, R. S., Kalshetti, R. G., and Ramana, C. V. (2019). Iridium(III)-Catalyzed Alkynylation of 2-(Hetero)arylquinazolin-4-One Scaffolds via C-H Bond Activation. J. Org. Chem. 84, 2951–2961. doi:10.1021/acs.joc.8b02738
Shetgaonkar, S. E., Krishnan, M., and Singh, F. V. (2021). Hypervalent Iodine Reagents for Oxidative Rearrangements. Mroc 18, 138–158. doi:10.2174/1570193X17999200727204349
Shetgaonkar, S. E., and Singh, F. V. (2020). Hypervalent Iodine Reagents in Palladium-Catalyzed Oxidative Cross-Coupling Reactions. Front. Chem. 8, 705. doi:10.3389/fchem.2020.00705
Shetgaonkar, S. E., and Singh, F. V. (2000). Hypervalent Iodine-Mediated Synthesis and Late-Stage Functionalization of Heterocycles. Arkivoc, 86–161. doi:10.1016/B978-0-08-097742-3.00735-7
Silva, F. C. S., Tierno, A., and Wengryniuk, S. (2017). Hypervalent Iodine Reagents in High Valent Transition Metal Chemistry. Molecules 22, 780. doi:10.3390/molecules22050780
Silva, L. F., and Olofsson, B. (2011). Hypervalent Iodine Reagents in the Total Synthesis of Natural Products. Nat. Prod. Rep. 28, 1722–1754. doi:10.1039/C1NP00028D
Singh, F. V., Kole, P. B., Mangaonkar, S. R., and Shetgaonkar, S. E. (2018). Synthesis of Spirocyclic Scaffolds Using Hypervalent Iodine Reagents. Beilstein J. Org. Chem. 14, 1778–1805. doi:10.3762/bjoc.14.152
Singh, F. V., and Wirth, T. (2014b). “7.29 Oxidative Functionalization with Hypervalent Halides,” in Oxidative Functionalization with Hypervalent Iodine Reagents in Comprehensive Organic Synthesis II. Editors P Knochel, and G Molander (Elsevier), 880–933. doi:10.1016/b978-0-08-097742-3.00735-7
Singh, F. V., and Wirth, T. (2017). Catalytic Oxidation with Hypervalent Iodine in: Catalytic Oxidation In Organic Synthesis. Thieme 1, 29. doi:10.1055/sos-SD-225-23
Singh, F. V., and Wirth, T. (2021). Hypervalent Iodine Chemistry and Light: Photochemical Reactions Involving Hypervalent Iodine Chemistry. Arkivoc 2021, 12–47. doi:10.24820/ark.5550190.p011.483
Singh, F. V., and Wirth, T. (2014a). Hypervalent Iodine-Catalyzed Oxidative Functionalizations Including Stereoselective Reactions. Chem. Asian J. 9, 950–971. doi:10.1002/asia.201301582
Singh, F. V., and Wirth, T. (2018). “Stereoselective Reactions,” in Patai’s Chemistry Of Functional Groups. Editor I. John (Chichester: Wiley & Sons, Ltd).
Tohma, H., and Kita, Y. (2004). Hypervalent Iodine Reagents for the Oxidation of Alcohols and Their Application to Complex Molecule Synthesis. Adv. Synthesis Catal. 346, 111–124. doi:10.1002/adsc.200303203
Uyanik, M., and Ishihara, K. (2009). Hypervalent Iodine-Mediated Oxidation of Alcohols. Chem. Commun., 2086–2099. doi:10.1039/b823399c
Wagner, A. M., Hickman, A. J., and Sanford, M. S. (2013). Platinum-Catalyzed C-H Arylation of Simple Arenes. J. Am. Chem. Soc. 135, 15710–15713. doi:10.1021/ja408112j
Wang, T., Zhou, L., Yang, Y., Zhang, X., Shi, Z., and Wu, Y.-D. (2018). Directing Effects on the Copper-Catalyzed Site-Selective Arylation of Indoles. Org. Lett. 20, 6502–6505. doi:10.1021/acs.orglett.8b02825
Willgerodt, C. (1886). Ueber Einige Aromatische Jodidchloride. J. Prakt. Chem. 33, 154–160. doi:10.1002/prac.18860330117
Keywords: hypervalent iodine reagents, oxidative coupling, gold, copper, oxidant, catalyst
Citation: Shetgaonkar SE, Raju A, China H, Takenaga N, Dohi T and Singh FV (2022) Non-Palladium-Catalyzed Oxidative Coupling Reactions Using Hypervalent Iodine Reagents. Front. Chem. 10:909250. doi: 10.3389/fchem.2022.909250
Received: 31 March 2022; Accepted: 20 May 2022;
Published: 01 July 2022.
Edited by:
Santosh Gaonkar, Manipal Institute of Technology, IndiaReviewed by:
Rajasekar Reddy Annapureddy, Ludwig Maximilian University of Munich, GermanyZhenhua Ding, China Pharmaceutical University, China
Copyright © 2022 Shetgaonkar, Raju, China, Takenaga, Dohi and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Toshifumi Dohi, dGQxMjAzQHBoLnJpdHN1bWVpLmFjLmpw; Fateh V. Singh, ZmF0ZWh2ZWVyLnNpbmdoQHZpdC5hYy5pbg==