 Kristina Spektor1,2*
Kristina Spektor1,2* Holger Kohlmann1
Holger Kohlmann1 Dmitrii Druzhbin3
Dmitrii Druzhbin3 Wilson A. Crichton3
Wilson A. Crichton3 Shrikant Bhat2
Shrikant Bhat2 Sergei I. Simak4,5Olga Yu Vekilova6
Sergei I. Simak4,5Olga Yu Vekilova6 Ulrich Häussermann6*
Ulrich Häussermann6*- 1Inorganic Chemistry, Faculty for Chemistry and Mineralogy, Leipzig University, Leipzig, Germany
- 2Deutsches Elektronen-Synchrotron DESY, Hamburg, Germany
- 3ESRF-The European Synchrotron Radiation Facility, Grenoble, France
- 4Theoretical Physics Division, Department of Physics, Chemistry and Biology (IFM), Linköping University, Linköping, Sweden
- 5Department of Physics and Astronomy, Uppsala University, Uppsala, Sweden
- 6Department of Materials and Environmental Chemistry, Stockholm University, Stockholm, Sweden
Hydrogenation reactions at gigapascal pressures can yield hydrogen-rich materials with properties relating to superconductivity, ion conductivity, and hydrogen storage. Here, we investigated the ternary Na–Si–H system by computational structure prediction and in situ synchrotron diffraction studies of reaction mixtures NaH–Si–H2 at 5–10 GPa. Structure prediction indicated the existence of various hypervalent hydridosilicate phases with compositions NamSiH(4+m) (m = 1–3) at comparatively low pressures, 0–20 GPa. These ternary Na–Si–H phases share, as a common structural feature, octahedral SiH62− complexes which are condensed into chains for m = 1 and occur as isolated species for m = 2, 3. In situ studies demonstrated the formation of the double salt Na3[SiH6]H (Na3SiH7, m = 3) containing both octahedral SiH62− moieties and hydridic H−. Upon formation at elevated temperatures (>500°C), Na3SiH7 attains a tetragonal structure (P4/mbm, Z = 2) which, during cooling, transforms to an orthorhombic polymorph (Pbam, Z = 4). Upon decompression, Pbam-Na3SiH7 was retained to approx. 4.5 GPa, below which a further transition into a yet unknown polymorph occurred. Na3SiH7 is a new representative of yet elusive hydridosilicate compounds. Its double salt nature and polymorphism are strongly reminiscent of fluorosilicates and germanates.
1 Introduction
The alkali metal A–Si–H systems (A = Li, Na, and K) have recently attracted attention because computational predictions suggested the existence of hydrogen-rich ternary phases at high pressures with potentially superconducting, superionic, and/or hydrogen storage properties (Liang et al., 2020; Zhang et al., 2020; Liang et al., 2021; Wu et al., 2022; Xie et al., 2022). According to these predictions, at lower pressures (up to 50 GPa), A–Si–H systems commonly possess a stable compound, A2SiH6, featuring octahedral SiH62− species in which Si attains a hypervalent bonding situation. Na2SiH6 was suggested being a H− superionic conductor in the pressure range 4–10 GPa (Liang et al., 2021) and at temperatures around 1,000°C, and K2SiH6 has been attributed favorable H-storage properties (Xie et al., 2022).
At higher pressure (50–200 GPa), more varied compositions become stable, with structures where octahedral units are connected and/or H2 molecules are additionally incorporated (i.e., KSiH7, KSiH8, K2SiH8, Na2SiH14, and Na3SiH10) (Liang et al., 2021; Xie et al., 2022). For A = Li, the structures of the predicted phases deviate frequently from the octahedral theme (Liang et al., 2020; Zhang et al., 2020). Also, for Li–Si–H, there seems to be a greater variety of stable hydrogen-rich structures and compositions at high pressures (e.g., LiSiH5, LiSiH6, Li3SiH10, Li2SiH10, and Li2SiH12). LiSi2H9 and LiSiH8 were predicted to become good phonon-mediated superconductors at pressures above 170 GPa (Liang et al., 2020).
Despite the interesting results from computational structure prediction, hitherto only polymorphic K2SiH6 has been reported from experimental high-pressure investigations (Puhakainen et al., 2012; Vekilova et al., 2023). This compound is stable even at ambient pressure where it adopts the cubic K2PtCl6 structure. At pressures above 8 GPa, K2SiH6 crystallizes in a trigonal structure (Vekilova et al., 2023).
Here, we report on the re-examination of the Na–Si–H system by crystal structure prediction and in situ studies of reactions m NaH + Si + 4 H2 (m = 1, 2) at pressures up to 10 GPa. For this, we employed a large-volume press (LVP) high pressure methodology which provides well-controlled p, T environments for high pressure hydrogenation reactions (Spektor et al., 2020a; Spektor et al., 2020b). Our initial intention was to obtain the predicted (superionic) compound Na2SiH6 with a simple P¯3m1 structure (Liang et al., 2021), which is also the structure of the high pressure polymorph of K2SiH6 (Vekilova et al., 2023). However, in contrast with earlier reports, we find evidence for the existence of multiple Na–Si–H phases along the composition line NamSiH(4+m) (m = 1, 2, 3) at comparatively low pressures up to 20 GPa, among which polymorphic Na3SiH7 was experimentally observed.
2 Materials and methods
2.1 High pressure experiments and data analysis
All steps of sample preparation and recovery were performed in a glove box under argon atmosphere. Powdered NaH (Sigma-Aldrich, 90%) and powdered Si [325 mesh, 99.999% (metals basis), Thermo Scientific] were mixed at a molar ratio of 1:1 and 2:1 (NaH:Si) and compressed into pellets with an outer diameter (OD) of 2 mm. Ammonia borane (BH3NH3, Sigma-Aldrich, 90%) served as a hydrogen source since it has a well-defined decomposition behavior at high pressures and produces chemically inert BN as a residue (Nylén et al., 2009). The amount of BH3NH3 used for each sample corresponded to an approx. 4× molar excess of H2 with respect to Si. NaH/Si sample pellets were sandwiched between pelletized BH3NH3 and sealed inside NaCl capsules with 3.0 mm OD.
In situ synchrotron diffraction high-pressure experiments were performed at beamline ID06-LVP at the ESRF and employed 14/7 multi-anvil assemblies, which are described in detail elsewhere (Vekilova et al., 2023). Amorphous SiBCN rods and either MgO or amorphous BCN epoxy were used as X-ray windows in the octahedra and gaskets, respectively, along the beam direction. Assemblies were compressed to target pressures ≈5 and ≈9 GPa and heated in a Voggenreiter-built modified-cubic press (Guignard and Crichton, 2015). Pressure was estimated in situ from PXRD diffraction patterns using the equation of state of NaCl by Matsui et al. (2012). The temperature was evaluated from power–T calibration curves. Angle-dispersive powder X-ray diffraction patterns were collected in the 1.27°–15.26° 2θ range at a constant wavelength (λ = 0.233933 Å). Data were acquired using the Pilatus3X-900 kW CdTe high-resolution 2D detector. The in situ data were integrated, visualized, and manipulated using Fit2D software (Hammersley, 2016). Indexing of the powder patterns was performed using DICVOL and TAUP algorithms within the CRYSFIRE package (Shirley, 2004). Le Bail fitting (Le Bail et al., 1988) and Rietveld refinement (Rietveld, 1969) against the in situ data were performed in Jana 2006 (Petříček et al., 2014). A detailed description of the high-pressure experiments and data analysis is given in Supplementary Material.
2.2 Theoretical calculations
The Na–Si–H system was studied with a crystal structure prediction methodology using Ab initio random structure searching implemented in the code AIRSS (Pickard and Needs, 2006; Pickard and Needs, 2011) and an evolutionary algorithm implemented in the Universal Structure Predictor: Evolutionary Xtallography (USPEX) code (Oganov and Glass, 2006; Oganov et al., 2011; Lyakhov et al., 2013), both coupled with the Vienna Ab Initio Simulation Package (VASP) (Kresse and Hafner, 1993; Kresse and Furthmüller, 1996). VASP calculations were based on a first-principles projector-augmented wave (PAW) method (Blöchl, 1994) within the density functional theory (DFT) (Hohenberg and Kohn, 1964; Kohn and Sham, 1965). The generalized gradient approximation for exchange and correlation potential and energy was used in its Perdew–Burke–Erzernhof (PBE) (Perdew et al., 1996; Perdew et al., 1997) flavor. H at zero pressure was calculated as a molecule (H2) and at higher pressures as a solid with an I4/mmm structure (according to the work of Pickard and Needs (2007)).
All NamSiH(4+m) (m = 1, 2, 3) model structures were fully relaxed; i.e., volume, shape, and internal atomic positions were adjusted to get nearly zero forces and stresses (stress components of order 0.1 GPa and forces of order 0.01 eV/Å maximum). The considered pressures were 0, 10, and 20 GPa. The Monkhorst–Pack (Monkhorst and Pack, 1976) k-point density for integrations over the Brillouin zone was set with 0.2. The energy cutoff for plane waves was set to 320 eV. All the static calculations were conducted at temperature T = 0 K.
Phonons were calculated from the force constants in real space obtained by VASP using density functional perturbation theory (DFPT) used as an input for the PHONOPY program (Togo and Tanaka, 2015). A plane-wave energy cutoff of 500 eV was used in all the corresponding calculations. A 2 × 2 × 2 supercell of each structure was used. To investigate the dynamical stability of P4/mbm Na3SiH7 at experimentally relevant temperature of 600 K and pressure 10 GPa, ab initio molecular dynamics (AIMD) calculations, as implemented in VASP, were conducted and then post-processed by the temperature-dependent effective potential method (TDEP) (Hellman et al., 2011; Hellman et al., 2013). TDEP maps the AIMD data onto a model Hamiltonian and provides the best harmonic fit to the system of anharmonic vibrating atoms at a particular temperature. Therefore, effective temperature-dependent phonon dispersions are obtained, and in particular, dynamical stability due to anharmonic vibrations can be studied. AIMD simulation were performed in 2 × 2 × 2 supercells (176 atoms) for P4/mbm Na3SiH7, with 2 × 2 × 2 k points, and 500 eV energy cutoff for the plane waves, with the same PAW potentials and exchange-correlation as in the static calculations. The canonical NVT ensemble using a Nosé–Hoover thermostat with the default Nosé mass as set by VASP and a 0.5 fs time step was applied. The data from 600 time steps after equilibration were used for the extraction of temperature-dependent force constants.
3 Results and discussion
3.1 Revisiting structure prediction for the Na–Si–H system
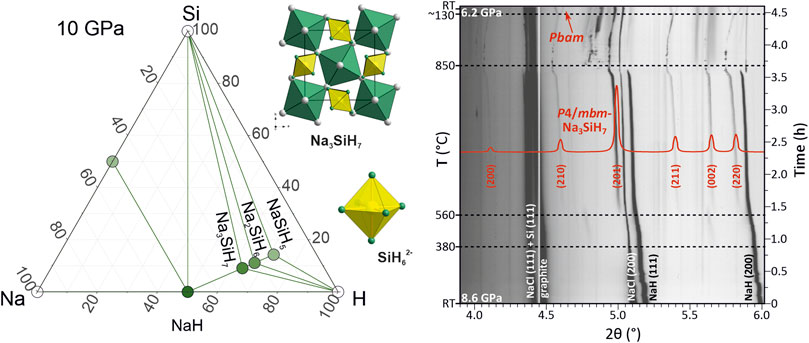
A recent exploration of the Na–Si–H system for stable ternary high-pressure phases yielded only one compound, trigonal P¯3m1 Na2SiH6, up to 50 GPa (Liang et al., 2021). At pressures below 3 GPa, the proposed Na2SiH6 is not dynamically stable, i.e., its phonon dispersion has branches with imaginary frequencies. This indicates lattice instability, and as a consequence, there should be a more stable structure with this composition, or decomposition into a different composition. We re-examined structure predictions (and also introduced a number of trial structures from chemical intuition) allowing for Z = 4 (Z = number of formula units) and focused on the low-pressure region up to 20 GPa which would be accessible in LVP hydrogenations. The dominant feature of SiH62− octahedral units in low-pressure structures, as established from previous A–Si–H structure prediction work (Liang et al., 2020; Liang et al., 2021; Zhang et al., 2020; Wu et al., 2022; Xie et al., 2022), together with chemical reasoning, suggested that compositions NamSiH(4+m) (m = 1, 2, 3) are most plausible, and we restricted our search to these compositions. Phase diagrams were established for 0, 10, and 20 GPa. Compared to earlier work, a radically different picture evolved.
Figure 1 shows the phase diagrams for ambient pressure and 10 GPa (the phase diagram for 20 GPa is virtually identical to that for 10 GPa). In Supplementary Figure S5, phonon dispersions of some of the predicted structures are depicted. At ambient pressure (Figure 1A), a compound Na3SiH7 with an orthorhombic Pbam structure appeared to be stable with respect to decomposition into NaH, NaSi, Si, and H2 but was not dynamically stable. This suggests that the composition Na3SiH7 is stable at ambient pressure but should occur in a different, most likely more complex, structure. As a matter of fact, identifying Na3SiH7 as a potential stable composition at ambient pressure was rather inferred from the experimental results than initially obtained from computational structure prediction. We will discuss the structures for Na3SiH7 and their stability in detail later. Furthermore, at ambient pressure, we found a hexagonal form of Na2SiH6 with P63mc symmetry slightly (by ≈0.05 eV/Z) more stable than P¯3m1-Na2SiH6. However, the enthalpy of Na2SiH6 is still above the convex hull, and both polymorphs are not dynamically stable at ambient pressure (cf. Supplementary Figures 5D, E). The Na2SiH6 structures are depicted in Figure 2.
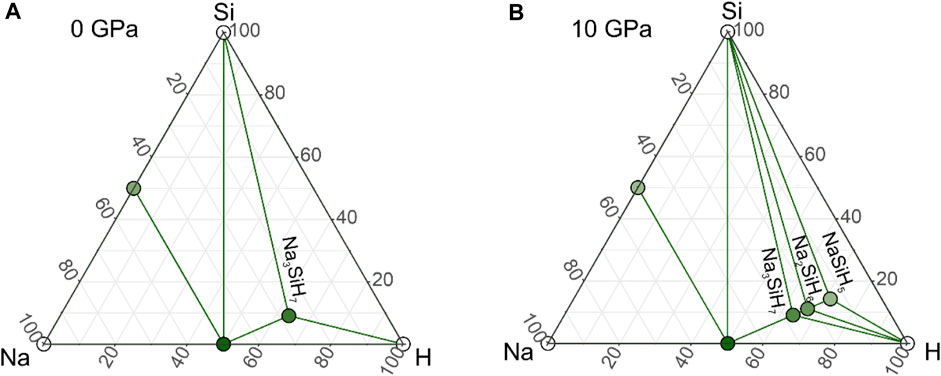
FIGURE 1. Na–Si–H phase diagram at (A) 1 atm and (B) 10 GPa. Large green circles represent compounds along the composition line mNaH + SiH4 (m = 1, 2, 3) which are located on the convex hull. Note that Na3SiH7 in the here-established Pbam structure is not dynamically stable at 0 GPa.

FIGURE 2. Compilation of predicted structures for NamSiH(4+m) (m = 1, 2) compositions. Na, Si, and H atoms are depicted as gray, yellow, and green circles, respectively. SiH62− octahedra are shown in yellow.
At 10 GPa, we find that the complete sequence of compositions NamSiH(4+m) (m = 1, 2, 3) is stable, both thermodynamically and dynamically. Pbam-Na3SiH7, which was found only enthalpically stable at ambient pressure, is now also dynamically stable. With pressure, the trigonal Na2SiH6 polymorph stabilizes over the hexagonal one and also becomes dynamically stable. NaSiH5 structures are not expected to realize separated SiH62− octahedral units but chains of corner-condensed or dimers of edge-condensed octahedra, [SiH4H2/2−]∞ and Si2H102−, respectively. Indeed, Pbcm-NaSiH5, which is isostructural to BaAlH5 (Figure 2) (Zhang et al., 2002), was found on the convex hull. At the same time, a different octahedral chain structure, Pna21-NaSiH5 [isostructural to SrAlH5 (Sato et al., 2018)], is only about 0.15 eV/Z less stable (Figure 2). It can be speculated that the composition NaSiH5 gives rise to a manifold of enthalpically close-lying polymorphic structures (Weidenthaler et al., 2006; Shlyapnikov et al., 2018) and that, most likely, the most stable one has yet to be identified. Supplementary Tables S3–S5 in Supplementary Material list the parameters of some NamSiH(4+m) structures. It is also easy to understand that computational structure prediction meets severe difficulties when tackling ternary compositions with complex structural features (Kvashnin et al., 2020).
3.2 In situ experiments and identification of Na3SiH7
Reactions m NaH + Si + 4 H2 (m = 1, 2), targeting originally proposed Na2SiH6, were performed at around 5 and 9 GPa. After compressing to target pressure, the reaction mixtures were initially heated to a temperature around 400°C at which the H-source BH3NH3 is expected to be completely decomposed into h-BN and hydrogen fluid (Nylén et al., 2009). The samples were then equilibrated for about 15–20 min. The reaction mixtures behaved very similar, independent of starting composition (m = 1 or 2) or pressure (5 or 9 GPa). In the following, reported results refer to m = 2 (2NaH:1Si) mixtures.
Figure 3 shows the evolution of diffraction patterns for the p ≈ 9 GPa run. In this experiment, one can recognize the growth of an intermediate phase above ≈380°C, which manifests as broad low-intensity diffraction peaks. The peaks could be indexed to a hexagonal unit cell (a ≈ 4.75 Å, c ≈ 7.65 Å), yet this phase remains uncharacterized due to the diffuse character and poor intensity of its reflections. Above ≈560°C, the peaks of the intermediate phase are superseded by another set of reflections, which were indexed to a primitive tetragonal unit cell with a ≈ 6.59 and c ≈ 4.78 Å (at ≈7.2 GPa and 770°C, as observed in the p ≈ 5 GPa run). P4/mbm was derived as the highest applicable space group from the extinction symbol P–b–. The symmetry and lattice parameter ratio (c/a ≈ 0.725) suggested an isostructural relation to K3SiF7 (c/a = 0.719 at ambient pressure) (Deadmore and Bradley, 1962; Hofmann and Hoppe, 1979) and, thus, a composition Na3SiH7. Calculated reflection intensities for K3SiF7-type Na3SiH7 matched closely with the experiment. Furthermore, with the knowledge of composition and formula units, P4/mbm-Na3SiH7 could be obtained as a low-enthalpy phase from the computational structure prediction methodology. The DFT optimized structure (Supplementary Tables S5D, E) was then used in Rietveld analysis of the in situ PXRD data (≈7.2 GPa, 770°C), yielding a reasonable fit (Robs≈5.5%) despite the low phase fraction of Na3SiH7 (Supplementary Figure S3). Details of the refinement process as well as the corresponding plot and extracted structural data are provided in Supplementary Material (Supplementary Tables S1, S2).
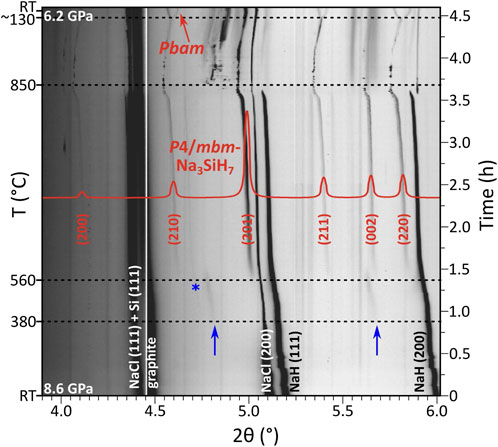
FIGURE 3. Compilation of PXRD patterns (λ = 0.233933 Å) acquired during the p ≈ 9 GPa experiment (2NaH:1Si starting mixture) (observed intensities are shown on the logarithmic scale). Overlain is the simulated pattern of P4/mbm-Na3SiH7 (shown in red, linear scale) based on the DFT optimized model structure (Supplementary Table S5D). Blue arrows mark the appearance of an intermediate hexagonal phase (a ≈ 4.75 Å, c ≈ 7.65 Å). The blue asterisk marks a peak belonging to an additional unidentified phase, growing in parallel with Na3SiH7 and disappearing upon heating above ≈830°C. Splitting of the tetragonal reflections upon transition to the Pbam phase is visible on cooling below 130°C (at the very top of the figure).
The formation of P4/mbm-Na3SiH7 during hydrogenations of NaH–Si mixtures requires pressures above 5 GPa (cf. Supplementary Figures S2, S4). In the p ≈ 5 GPa hydrogenation experiment, the intermediate hexagonal phase was not seen. Diffraction peaks from P4/mbm-Na3SiH7 become noticeable above ≈490°C. Irrespective of pressure, the P4/mbm-Na3SiH7 phase grows very sluggishly even when raising temperatures up to 850°C. This indicates a rather high thermal stability of P4/mbm-Na3SiH7 at high-pressure conditions and shows at the same time that elemental Si represents a rather unreactive precursor.
Upon cooling from (high) synthesis temperatures to below 120°C, the tetragonal diffraction pattern showed a clear splitting into an orthorhombic one, which appeared continuous and was accompanied with a doubling of unit cell (ao ≈ bo = √2at, co ≈ ct). For the p ≈ 9 GPa run, this feature is included in Figure 3. For the p ≈ 5 GPa experiment, this tetragonal-to-orthorhombic transition is shown in Figure 4. The reflections were indexed to a unit cell a ≈ 9.23 Å, b ≈ 9.36 Å, and c ≈ 4.76 Å (at p ≈ 5.2 GPa, ≈RT). Again, suspecting analogy with fluorides, the (NH4)3GeF7 structure (Pbam space group symmetry, Z = 4) (Mel’nikova et al., 2016; Mel’nikova et al., 2017; Bogdanov et al., 2019) was assigned to the orthorhombic phase.
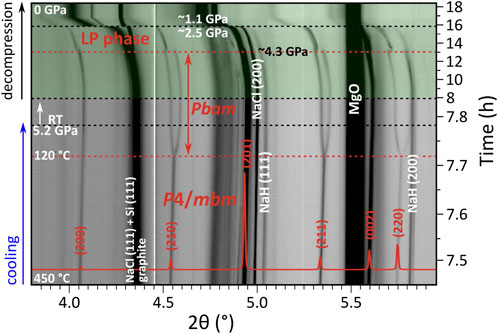
FIGURE 4. Compilation of PXRD patterns (λ = 0.233933 Å) acquired during the p ≈ 5 GPa experiment (2NaH:1Si starting mixture) during cooling and decompression. Observed intensities are shown on the logarithmic scale. Data collected upon decompression (1 pattern/min) are highlighted in green. The transition of P4/mbm-Na3SiH7 to a Pbam polymorph occurs below 120°C, and a further transition to an unidentified low-pressure polymorph is visible below ≈4.3 GPa (both events are marked with red dashed lines). Overlain is the simulated pattern of P4/mbm-Na3SiH7 (shown in red, linear scale) based on the refined structure model (Supplementary Tables S1, S2). Mismatch between observed and calculated intensities is due to texture which developed in Na3SiH7 above ≈770°C (cf. Supplementary Figure S4).
In both 5 and 9 GPa runs, a further transition was noticeable upon decompression below ≈4.5 GPa, see again Figure 4 (and Supplementary Material for details). Indexing of the diffraction peaks arising at ≈4 GPa suggested as best-fitting candidates two primitive tetragonal unit cells (parameters are given relative to the initial tetragonal phase): a) a ≈ √2at, c ≈ 2ct (a ≈ 9.42 Å, c ≈ 9.61 Å, Z = 8); b) a ≈ at, c ≈ 2ct (a ≈ 6.66 Å, c ≈ 9.61 Å, Z = 4). The structure was difficult to resolve due to peak overlap as well as the occurrence of sudden pressure drops during decompression which obscured analysis. Further structure prediction work would be required to establish the structure and corresponding space group. Also, it remains unclear whether Na3SiH7 is recoverable to ambient pressure. The ex situ analysis of the only partially reacted run products was inconclusive.
3.3 Phase relations in Na3SiH7
Figure 5 shows the structures of polymorphic Na3SiH7 along with the p, T conditions for the observed phases. As mentioned, Na3SiH7 represents a double salt Na3[SiH6]H containing both octahedral SiH62− moieties and hydridic H. Hydridic H is octahedrally coordinated by six Na atoms, and HNa6 octahedra build up a corner-connected framework similar to the BO3 framework in perovskites ABO3. SiH62− octahedra are located in the voids accommodating the (larger-sized) A constituent of perovskites. Thus, the Na3SiH7 structures may be considered as an anti-perovskite arrangement (SiH62−)[HNa3]2+, and as for perovskites, there is inherent structural flexibility from rotations and tilts of octahedra. Pbam is a subgroup of P4/mbm. The group-subgroup relationship is indicated in Figure 5. Analogous and also more extended sequences of phase transitions have been reported for fluorosilicate and fluorogermanate double salts [e.g., (NH4)3SiF7 and (NH4)3GeF7] as a function of temperature (Mel’nikova et al., 2014; Molokeev et al., 2014; Pogoreltsev et al., 2014; Mel’nikova et al., 2016; Mel’nikova et al., 2017; Bogdanov et al., 2019). These may potentially also include the yet unknown Na3SiH7 structure at room temperature and low pressures.
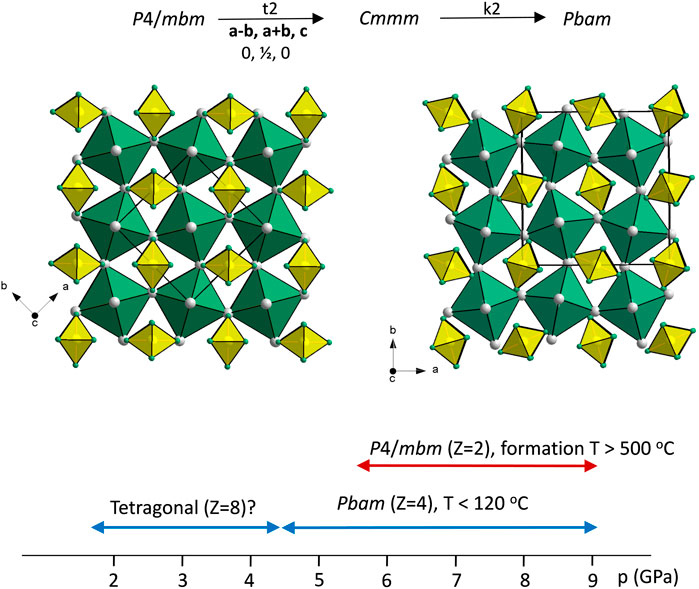
FIGURE 5. Structures of P4/mbm-Na3SiH7 (left) and Pbam-Na3SiH7 (right). Their group–subgroup relation is depicted at the top, and the experimentally observed pressure stability ranges are shown at the bottom. Na, Si, and H atoms are shown as gray, yellow, and green circles, respectively. SiH62− and HNa6 octahedra are shown in yellow and green, respectively.
As previously mentioned, Pbam-Na3SiH7 was found enthalpically stable but dynamically unstable at ambient pressure. According to total energy calculations, this polymorph is by 0.4 eV/Z more stable than the P4/mbm structure. At 10 GPa, Pbam-Na3SiH7 is by about 0.24 eV more stable than the tetragonal form and dynamical stable, whereas P4/mbm-Na3SiH7 appears to be dynamically unstable at all pressures (cf. Supplementary Figures S5G, H).
To investigate the dynamical stability of the P4/mbm-Na3SiH7 phase at experimentally relevant conditions, phonon dispersions were calculated in the framework of the TDEP method (Hellman et al., 2011; Hellman et al., 2013) at 10 GPa and 600 K. Figures 6A, B compare the phonon dispersions for 10 GPa at zero and 600 K. Si–H stretching and bending modes of the entities SiH62− (i.e., internal modes) and the (translational) modes of the hydridic H within Na6 octahedra are above around 500 cm−1, whereas SiH62− libration modes (i.e., rotation of octahedral units against each other) and optic translation modes of Na+ and SiH62− units (i.e., external modes) are below 500 cm−1.

FIGURE 6. Phonon dispersion relations for P4/mbm and Pbam Na3SiH7 at different p, T conditions. (A) P4/mbm-Na3SiH7 at 10 GPa and 0 K. (B) P4/mbm-Na3SiH7 at 10 GPa and 600 K. (C) Pbam-Na3SiH7 at 5 GPa and 0 K. (D) Pbam-Na3SiH7 at 10 GPa and 0 K.
At 0 K, P4/mbm-Na3SiH7 has two imaginary branches (Figure 6A). These originate from libration and acoustic modes which, according to the atom-decomposed phonon density of states (shown in Supplementary Figure S6), involve Na1 atoms (which are situated on the 4-fold axes 0,0,z and ½,½,z, cf. Figure 5) and the H atoms being part of SiH6 octahedra. The imaginary modes stabilize with temperature, which is attributed to the anharmonicity of atomic vibrations, causing renormalizing of the phonon modes due to phonon–phonon interaction (Figure 6B). Thus, P4/mbm-Na3SiH7 is stable at high-pressure and high-temperature conditions. Conversely, lowering temperature introduces dynamical instability, driving the distortion to the orthorhombic Pbam structure (ao ≈ √2at, bo ≈ √2at) which is dynamically stable at 10 GPa (Figure 6D). Reducing pressure to about 5 GPa introduces dynamical instability for the Pbam polymorph (Figure 6C), indicating a transition to yet another polymorph.
Thus, total energy and phonon calculations support the experimental findings described in the previous section. The structural chemistry and polymorphism of Na3SiH7 reminds of fluorosilicates and germanates A3Si/GeF7 (A = K, Rb, Cs, NH4). These materials have wide band gaps in the UV region (≈6 eV) in combination with interesting birefringent optical properties (Mel’nikova et al., 2016; Huang et al., 2020). In contrast, the calculated band structure of Na3SiH7 (Supplementary Figure S7) suggests a semiconductor with a direct band gap of around 2 eV. An interesting aspect, which has not been investigated in the course of this work, is a potentially superionic behavior of Na3SiH7. Superionicity of H− was proposed in (yet) hypothetical Na2SiH6 where extraordinary diffusive H seems to be promoted by the hypervalent nature of SiH62− units (Liang et al., 2021). Against this background, it appears worthwhile to study the dynamical properties of Na3SiH7 over a wider temperature range by dedicated AIMD simulations.
4 Conclusion
In situ studies of reactions m NaH + Si + 2.5 H2 (m = 1, 2) at pressures up to 10 GPa revealed a new hypervalent hydridosilicate Na3SiH7 which corresponds to a double salt Na3[SiH6]H, featuring isolated octahedral SiH62− complexes and H− anions. In the pressure range 5–10 GPa, Na3SiH7 occurs in a tetragonal high-temperature and orthorhombic low-temperature form. It is not yet clear whether and in which polymorphic form Na3SiH7 is recoverable to ambient pressure. Computation suggests the accessibility of further sodium hydridosilicate along the composition line NamSiH(4+m) (m = 1–3), i.e., Na2SiH6 and NaSiH5. The structures of phases NamSiH(4+m) constitute octahedral SiH62− complexes which are condensed for m = 1 and occur as isolated species for m = 2, 3. Interestingly, previously predicted superionic Na2SiH6 with a P¯3m1 structure has not been observed during our investigations. It may be speculated that this composition disappears from the convex hull once the most stable structure for NaSiH5 has been identified. The same may hold true for Na3SiH7 at ambient pressure. The composition NaSiH5 is expected to yield a manifold of enthalpically close-lying polymorphic structures based on corner- and edge-condensed SiH62− octahedra. In order to access NaSiH5 structures experimentally, the rather inert elemental Si should be replaced with a more active reactant, such as the Zintl phase NaSi. In addition to a higher reactivity, calculated reaction enthalpies with respect to NaSi (i.e., NaSi + 5/2H2) are about 0.4 eV/Z lower than with respect to NaH + Si + 2H2.
Data availability statement
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Author contributions
KS, HK, and UH contributed to the conception and design of the study. OV and SS performed the computational part. SB, DD, and WC assisted in the implementation of experiments and the evaluation of data. KS and UH wrote the first draft of the manuscript. OV and SS wrote sections of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the Swedish Research Council (VR) through project 2019-06063 and the Bundesministerium fuer Bildung und Forschung (BMBF)—German Federal Ministry of Education and Research (Grant No. 05K20OLA awarded to HK) and the Deutsche Forschungsgemeinschaft (Grant No. 277832266 awarded to HK). The computations were enabled by resources provided by the National Academic Infrastructure for Supercomputing in Sweden (NAISS) at the National Supercomputer Center (NSC) and Center for High Performance Computing (PDC), partially funded by VR through Grant Agreement No. 2022-06725. SS acknowledges the support from VR (Project No. 2019-05551), the Swedish Government Strategic Research Area in Materials Science on Functional Materials at Linköping University (Faculty Grant SFO-Mat-LiU No. 2009-00971), the Knut and Alice Wallenberg Foundation, and the ERC (synergy grant FASTCORR project 854843).
Acknowledgments
The ESRF is thanked for allocating the beamtime CH-5986 and the provision of experimental facilities. The authors also thank Harald Müller for assistance with the Chemistry Laboratory facilities at ESRF. They are grateful to Per Mistenius for skillfully manufacturing the miniature press dies used for sample preparation. Stefan Sonntag and Robert Farla are thanked for assistance and resources needed to perform calibration experiments at P61B-LVP, PETRA III, DESY.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2023.1251774/full#supplementary-material
References
Blöchl, P. E. (1994). Projector augmented-wave method. Phys. Rev. B 50, 17953–17979. doi:10.1103/PhysRevB.50.17953
Bogdanov, E. V., Pogoreltsev, E. I., Gorev, M. V., Kartashev, A. V., Laptash, N. M., and Flerov, I. N. (2019). Heat capacity, thermal expansion and sensitivity to hydrostatic pressure of (NH4)3SiF7 at successive structural phase transitions. J. Solid State Chem. 276, 152–158. doi:10.1016/j.jssc.2019.04.029
Deadmore, D. L., and Bradley, W. F. (1962). The crystal structure of K3SiF7. Acta Cryst. 15, 186–189. doi:10.1107/S0365110X62000493
Guignard, J., and Crichton, W. A. (2015). The large volume press facility at ID06 beamline of the European synchrotron radiation facility as a High Pressure-High Temperature deformation apparatus. Rev. Sci. Instrum. 86, 085112. doi:10.1063/1.4928151
Hofmann, B., and Hoppe, R. (1979). Zur Kenntnis des (NH4)3SiF7-Typs. Neue Metallfluoride A3MF7 mit M = Si, Ti, Cr, Mn, Ni und A = Rb. Cs. Z. Anorg. Allg. Chem. 458, 151–162. doi:10.1002/zaac.19794580121
Hammersley, A. P. (2016). FIT2D: a multi-purpose data reduction, analysis and visualization program. J. Appl. Crystallogr. 49, 646–652. doi:10.1107/S1600576716000455
Hellman, O., Abrikosov, I. A., and Simak, S. I. (2011). Lattice dynamics of anharmonic solids from first principles. Phys. Rev. B 84, 180301. doi:10.1103/PhysRevB.84.180301
Hellman, O., Steneteg, P., Abrikosov, I. A., and Simak, S. I. (2013). Temperature dependent effective potential method for accurate free energy calculations of solids. Phys. Rev. B 87, 104111. doi:10.1103/PhysRevB.87.104111
Hohenberg, P., and Kohn, W. (1964). Inhomogeneous electron gas. Phys. Rev. 136, B864–B871. doi:10.1103/PhysRev.136.B864
Huang, S., Gao, L., and Yu, F. (2020). K2.64Cs0.36SiF7: a new fluorosilicate with a trans-perovskite structure. New J. Chem. 44, 2727–2732. doi:10.1039/C9NJ06037E
Kohn, W., and Sham, L. J. (1965). Self-consistent equations including exchange and correlation effects. Phys. Rev. 140, A1133–A1138. doi:10.1103/PhysRev.140.A1133
Kresse, G., and Furthmüller, J. (1996). Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186. doi:10.1103/PhysRevB.54.11169
Kresse, G., and Hafner, J. (1993). Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561. doi:10.1103/PhysRevB.47.558
Kvashnin, A. G., Tantardini, C., Zakaryan, H. A., Kvashnina, Y. A., and Oganov, A. R. (2020). Computational search for new W–Mo–B compounds. Chem. Mater. 32, 7028–7035. doi:10.1021/acs.chemmater.0c02440
Le Bail, A., Duroy, H., and Fourquet, J. L. (1988). Ab-initio structure determination of LiSbWO6 by X-ray powder diffraction. Mater. Res. Bull. 23, 447–452. doi:10.1016/0025-5408(88)90019-0
Liang, T., Zhang, Z., Feng, X., Jia, H., Pickard, C. J., Redfern, S. A. T., et al. (2020). Ternary hypervalent silicon hydrides via lithium at high pressure. Phys. Rev. Mater. 4, 113607. doi:10.1103/PhysRevMaterials.4.113607
Liang, T., Zhang, Z., Yu, H., Cui, T., Feng, X., Pickard, C. J., et al. (2021). Pressure-induced superionicity of H – in hypervalent sodium silicon hydrides. J. Phys. Chem. Lett. 12, 7166–7172. doi:10.1021/acs.jpclett.1c01809
Lyakhov, A. O., Oganov, A. R., Stokes, H. T., and Zhu, Q. (2013). New developments in evolutionary structure prediction algorithm USPEX. Comput. Phys. Commun. 184, 1172–1182. doi:10.1016/j.cpc.2012.12.009
Matsui, M., Higo, Y., Okamoto, Y., Irifune, T., and Funakoshi, K.-I. (2012). Simultaneous sound velocity and density measurements of NaCl at high temperatures and pressures: application as a primary pressure standard. Am. Mineral. 97, 1670–1675. doi:10.2138/am.2012.4136
Mel’nikova, S. V., Molokeev, M. S., Laptash, N. M., and Misyul, S. V. (2016). A non-typical sequence of phase transitions in (NH4)3GeF7: optical and structural characterization. Dalton Trans. 45, 5321–5327. doi:10.1039/C5DT04907E
Mel’nikova, S. V., Molokeev, M. S., Laptash, N. M., Pogoreltsev, E. I., Misyul, S. V., and Flerov, I. N. (2017). Sequence of phase transitions in (NH4)3SiF7. Dalton Trans. 46, 2609–2617. doi:10.1039/C6DT04874A
Mel’nikova, S. V., Pogoreltsev, E. I., Flerov, I. N., and Laptash, N. M. (2014). Unusual sequence of phase transitions in (NH4)3TiF7 detected by optic and calorimetric studies. J. Fluor. Chem. 165, 14–19. doi:10.1016/j.jfluchem.2014.05.016
Molokeev, M., Misjul, S. V., Flerov, I. N., and Laptash, N. M. (2014). Reconstructive phase transition in (NH4)3TiF7 accompanied by the ordering of TiF6 octahedra. Acta Cryst. B 70, 924–931. doi:10.1107/S2052520614021192
Monkhorst, H. J., and Pack, J. D. (1976). Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192. doi:10.1103/PhysRevB.13.5188
Nylén, J., Sato, T., Soignard, E., Yarger, J. L., Stoyanov, E., and Häussermann, U. (2009). Thermal decomposition of ammonia borane at high pressures. J. Chem. Phys. 131, 104506. doi:10.1063/1.3230973
Oganov, A. R., and Glass, C. W. (2006). Crystal structure prediction using ab initio evolutionary techniques: principles and applications. J. Chem. Phys. 124, 244704. doi:10.1063/1.2210932
Oganov, A. R., Lyakhov, A. O., and Valle, M. (2011). How evolutionary crystal structure prediction works—And why. Acc. Chem. Res. 44, 227–237. doi:10.1021/ar1001318
Perdew, J. P., Burke, K., and Ernzerhof, M. (1997). Errata: generalized gradient approximation made simple [phys Rev Lett 77, 1396 (1996)]. Phys. Rev. Lett. 78, 1396. doi:10.1103/PhysRevLett.78.1396
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868. doi:10.1103/PhysRevLett.77.3865
Petříček, V., Dušek, M., and Palatinus, L. (2014). Crystallographic computing system JANA2006: general features. Z. Krist. - Cryst. Mater. 229, 345–352. doi:10.1515/zkri-2014-1737
Pickard, C. J., and Needs, R. J. (2011). Ab initio random structure searching. J. Phys. Condens. Matter 23, 053201. doi:10.1088/0953-8984/23/5/053201
Pickard, C. J., and Needs, R. J. (2006). High-pressure phases of silane. Phys. Rev. Lett. 97, 045504. doi:10.1103/PhysRevLett.97.045504
Pickard, C. J., and Needs, R. J. (2007). Structure of phase III of solid hydrogen. Nat. Phys. 3, 473–476. doi:10.1038/nphys625
Pogoreltsev, E. I., Flerov, I. N., Kartashev, A. V., Bogdanov, E. V., and Laptash, N. M. (2014). Heat capacity, entropy, dielectric properties and T–p phase diagram of (NH4)3TiF7. J. Fluor. Chem. 168, 247–250. doi:10.1016/j.jfluchem.2014.10.016
Puhakainen, K., Benson, D., Nylén, J., Konar, S., Stoyanov, E., Leinenweber, K., et al. (2012). Hypervalent octahedral SiH62− species from high-pressure synthesis. Angew. Chem. Int. Ed. 51, 3156–3160. doi:10.1002/anie.201108713
Rietveld, H. M. (1969). A profile refinement method for nuclear and magnetic structures. J. Appl. Crystallogr. 2, 65–71. doi:10.1107/S0021889869006558
Sato, T., Takagi, S., Sørby, M. H., Deledda, S., Hauback, B. C., and Orimo, S. (2018). Crystal structural determination of SrAlD5 with corner-sharing AlD6 octahedron chains by X-ray and neutron diffraction. Crystals 8, 89. doi:10.3390/cryst8020089
Shirley, R. (2004). Crysfire 2004: An interactive powder indexing support system, 41 Guildford Park Avenue. Surrey, UK: Guildford.
Shlyapnikov, I. M., Goreshnik, E. A., and Mazej, Z. (2018). Increasing structural dimensionality of alkali metal fluoridotitanates(IV). Inorg. Chem. 57, 1976–1987. doi:10.1021/acs.inorgchem.7b02890
Spektor, K., Crichton, W. A., Filippov, S., Klarbring, J., Simak, S. I., Fischer, A., et al. (2020a). Na–Ni–H phase formation at high pressures and high temperatures: hydrido complexes [NiH5]3–versus the perovskite NaNiH3. ACS Omega 5, 8730–8743. doi:10.1021/acsomega.0c00239
Spektor, K., Crichton, W. A., Filippov, S., Simak, S. I., Fischer, A., and Häussermann, U. (2020b). Na3FeH7 and Na3CoH6: hydrogen-rich first-row transition metal hydrides from high pressure synthesis. Inorg. Chem. 59, 16467–16473. doi:10.1021/acs.inorgchem.0c02294
Togo, A., and Tanaka, I. (2015). First principles phonon calculations in materials science. Scr. Mater. 108, 1–5. doi:10.1016/j.scriptamat.2015.07.021
Vekilova, O. Y., Beyer, D. C., Bhat, S., Farla, R., Baran, V., Simak, S. I., et al. (2023). Formation and polymorphism of semiconducting K2SiH6 and Strategy for metallization. Inorg. Chem. 62, 8093–8100. doi:10.1021/acs.inorgchem.2c04370
Weidenthaler, C., Frankcombe, T. J., and Felderhoff, M. (2006). First crystal structure studies of CaAlH5. Inorg. Chem. 45, 3849–3851. doi:10.1021/ic0602042
Wu, S., Li, B., Chen, Z., Hou, Y., Bai, Y., Hao, X., et al. (2022). Phase transitions and superconductivity in ternary hydride Li2SiH6 at high pressures. J. Appl. Phys. 131, 065901. doi:10.1063/5.0080880
Xie, H., Liang, T., Cui, T., Feng, X., Song, H., Li, D., et al. (2022). Structural diversity and hydrogen storage properties in the system K–Si–H. Phys. Chem. Chem. Phys. 24, 13033–13039. doi:10.1039/D2CP00298A
Zhang, P., Sun, Y., Li, X., Lv, J., and Liu, H. (2020). Structure and superconductivity in compressed Li-Si-H compounds: density functional theory calculations. Phys. Rev. B 102, 184103. doi:10.1103/PhysRevB.102.184103
Keywords: hydridosilicate, gigapascal hydrogenation, multi-anvil techniques, crystal structure prediction, hypervalency
Citation: Spektor K, Kohlmann H, Druzhbin D, Crichton WA, Bhat S, Simak SI, Vekilova OY and Häussermann U (2023) Hypervalent hydridosilicate in the Na–Si–H system. Front. Chem. 11:1251774. doi: 10.3389/fchem.2023.1251774
Received: 02 July 2023; Accepted: 17 August 2023;
Published: 08 September 2023.
Edited by:
Gunter Heymann, University of Innsbruck, AustriaReviewed by:
Julien Haines, Centre National de la Recherche Scientifique (CNRS), FranceChristian Tantardini, UiT The Arctic University of Norway, Norway
Copyright © 2023 Spektor, Kohlmann, Druzhbin, Crichton, Bhat, Simak, Vekilova and Häussermann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kristina Spektor, a3Jpc3RpbmEuc3Bla3RvckBkZXN5LmRl; Ulrich Häussermann, dWxyaWNoLmhhdXNzZXJtYW5uQG1tay5zdS5zZQ==