 Taida Huang1,2†
Taida Huang1,2† Mo Yang1†Yunxin Zeng1Xiaomin Huang1Nan Wang1
Mo Yang1†Yunxin Zeng1Xiaomin Huang1Nan Wang1 Yun Chen1Peng Li1
Yun Chen1Peng Li1 Jinqiu Yuan1Chun Chen1*
Jinqiu Yuan1Chun Chen1* Brian G. Oliver3,4‡
Brian G. Oliver3,4‡ Chenju Yi1*‡
Chenju Yi1*‡- 1Research Center, The Seventh Affiliated Hospital of Sun Yat-sen University, Shenzhen, China
- 2Faculty of Medicine, School of Biomedical Sciences, The Chinese University of Hong Kong, Hong Kong, China
- 3Faculty of Science, School of Life Sciences, University of Technology Sydney, Sydney, NSW, Australia
- 4Respiratory Cellular and Molecular Biology, Woolcock Institute of Medical Research, Sydney, NSW, Australia
Objectives: Maternal smoking causes fetal underdevelopment and results in births which are small for gestation age due to intrauterine undernutrition, leading to various metabolic disorders in adulthood. Furthermore, postnatal high fat diet (HFD) consumption is also a potent obesogenic factor, which can interact with maternal smoking. In this study, we aimed to determine whether maternal HFD consumption during pregnancy can reverse the adverse impact of maternal smoking and change the response to postnatal HFD consumption.
Methods: Female mice were exposed to cigarette smoke (SE, 2 cigarettes/day) or sham exposed for 5 weeks before mating, with half of the SE dams fed HFD (43% fat, SE+HFD). The same treatment continued throughout gestation and lactation. Male offspring from each maternal group were fed the same HFD or chow after weaning and sacrificed at 13 weeks.
Results: Maternal SE alone increased body weight and fat mass in HFD-fed offspring, while SE+HFD offspring showed the highest energy intake and glucose metabolic disorder in adulthood. In addition, postnatal HFD increased the body weight and aggravated the metabolic disorder caused by maternal SE and SE+HFD.
Conclusions: Maternal HFD consumption could not ameliorate the adverse effect of maternal SE but exaggerate metabolic disorders in adult offspring. Smoking cessation and a healthy diet are needed during pregnancy to optimize the health outcome in the offspring.
What This Paper Adds
What Is Already Known on This Subject
Maternal smoking is one of the known risk factors for the development of obesity in offspring. A number of studies have provided evidence that maternal cigarette smoke exposure (SE) leads to low birth weight and faster weight gain during the suckling period, called catch-up growth. This fast postnatal growth is commonly observed in children, which may lead to obesity during childhood as shown in the weaning offspring in this study.
What Important Gaps in Knowledge Exist on This Topic
Maternal high fat diet (HFD) consumption, on the other hand, can lead to in utero overnutrition. However, it is unclear whether in the setting of maternal SE, HFD consumption may mask the impact of in utero undernutrition caused by maternal SE. Furthermore, we cannot exclude the possibility that postnatal HFD consumption may worsen the regulation of energy consumption in offspring with in utero SE.
What This Paper Adds
Our study demonstrated that maternal SE during pregnancy results in increased adiposity and metabolic disorders if the offspring are exposed to HFD after weaning. The additional exposure to HFD failed to counteract with cigarette SE leading to even more severe metabolic disorders in adult offspring. Therefore, both quitting smoking and maintaining a healthy diet are vital for the healthy future of the offspring.
Key Messages
- Maternal smoking exposure (SE) decreased body weight without affecting the daily energy intake of breeders.
- Maternal HFD consumption could not ameliorate the adverse effect of maternal SE but exaggerate metabolic disorders in adult offspring.
- Postnatal HFD failed to counteract with cigarette SE but further increased the body weight and aggravated the metabolic disorder caused by maternal SE and SE+HFD.
Introduction
Obesity is occurring at alarming rates globally, which is linked to various complications, such as metabolic disorders, diabetes, cardiovascular disease, cancers, osteoarthritis, and reproductive problems (1–3). About 2.1 billion people worldwide are estimated to be overweight or obese, increasing their risk of developing associated insulin resistance and cardiovascular disease, adding to the already enormous cost of obesity-related diseases (3, 4). The consumption of food high in energy and fat is the major driver of the obesity pandemic. However, many obese people may also have had a suboptimal intrauterine environment which may interact with obesity-induced risks.
Maternal smoking is one of the known risk factors for the development of obesity in offspring. Epidemiological investigations revealed that maternal smoking/second-hand cigarette smoke exposure (SE) during pregnancy is also a major cause of intrauterine undernutrition, even the mothers do not necessarily eat less during pregnancy compared with non-smokers (5, 6). This is caused by placental limitation, leading to pre-term birth, low body weight, and reduced head circumference at birth (7–9). However, postnatal catch-up growth results in maternal smoking being associated with an increased risk of obesity in offspring in both childhood and adulthood (10–12). Secondary to this catch-up growth, maternal SE has also been shown to contribute to later metabolic disorders in the offspring, such as glucose intolerance and type 2 diabetes, fatty liver changes, dyslipidemia, and cardiovascular disease (13–17). This has been suggested to be linked to increased eating disorders in such offspring (8, 18). Therefore, the impact of maternal SE offspring is more than just intrauterine undernutrition and fetal growth restriction, with long-lasting effects on one's adulthood.
Maternal high fat diet (HFD) consumption, on the other hand, can lead to in utero overnutrition. However, it is unclear whether in the setting of maternal SE, HFD consumption may mask the impact of in utero undernutrition caused by maternal SE. Furthermore, we can't exclude the possibility that maternal HFD consumption may worsen the regulation of energy consumption in offspring with in utero SE, as both can encourage energy overconsumption in rodent models (19, 20). Maternal nicotine exposure is a strong risk factor for obesogenic overeating in childhood (21). Interestingly, prenatal growth retardation during infancy has been reported in obese mothers who smoked during pregnancy (5). This indicates that HFD consumption might not be able to reverse the intrauterine undernutrition resulted from maternal smoking. In animal studies, maternal HFD consumption during pregnancy also results in increased milk intake during the suckling period, and overfeeding during this period can have additive effects to further induce fat over accumulation resulting in metabolic disorder (22, 23). However, this conclusion is difficult to apply to humans due to the complexity of dietary and smoking behaviors between different individuals, as well as other external factors.
One of the widely studied appetite regulatory networks is in the hypothalamus, consisting of neurons expressing the appetite stimulator neuropeptide Y (NPY) and its counterpart, appetite suppressors α-melanocyte stimulating hormone (α-MSH) coded by proopiomelanocortin (POMC) (24). While maternal SE may induce smoking quitting type of rebound response of NPY in the offspring's brain (6), maternal obesity leads to a heightened response of NPY signaling after fasting (25). However, it is unclear how maternal SE and maternal HFD consumption interact to influence brain appetite regulators in the offspring. Skeletal muscle is a key metabolic organ for glucose metabolism. Both maternal HFD and SE exposure during pregnancy may affect muscle genesis, and thereafter metabolic function (26, 27). Myoblast determination protein 1 (Myod1) promotes transcriptional activation of Myogenin (Myog) during myogenesis (28). It is unknown how they are affected by prenatal and postnatal insults, which were examined in this study.
Furthermore, epidemiological studies have found that HFD consumed early in life is a risk factor for childhood weight gain and later adulthood obesity, accompanied by various metabolic dysfunction (29, 30). Thus, we hypothesized that post-weaning HFD exposure may further exaggerate metabolic disorders caused by maternal SE, whereas, additional HFD exposure in the SE mothers may ameliorate the adverse impact on the metabolic regulators in the offspring. In this study, we exposed the dams to cigarette smoke with/without access to a HFD and also offered the same HFD to half of the litter after weaning. We aimed to examine the interaction between maternal and postnatal environmental factors on the metabolic outcomes in the offspring, including body weight, organ weight, gene expression of metabolic markers in the hypothalamus and metabolic organs.
Materials and Methods
Animals
According to the previous findings on the strain dependence of the response to SE (31), Balb/c mice were used for this study. Female mice breeders (aged 6 weeks) were housed at 20 ± 2°C in sterile micro-isolator cages and maintained on a 12:12 h light/dark cycle (lights on at 06:00 h). They were allowed a week to adapt to their new environment, with ad libitum access to standard rodent chow and water. The study was approved by the Animal Ethics Committee of the San Yet-sun University (number: SYSU-IACUC-2020-B0552).
Modeling of Maternal SE and HFD Feeding
After acclimatization, female breeders were randomly divided into three groups with similar average body weight: sham exposed fed a chow (CHOW+SHAM, representing a healthy control), chow-fed SE (CHOW+SE, representing smokers consuming a balanced diet), and HFD-fed SE (HFD+SE, representing smokers consuming a “junk” diet). For SE, animals were placed inside a perspex chamber (18 L) and exposed to the smoke produced by two cigarettes (Double Happiness; Tar: 8 mg; nicotine: 0.7 mg; CO: 10 mg), twice daily for 5 weeks before mating, during pregnancy and lactation as described in Chan et al. (32). The sham exposed animals were handled identically but were not exposed to cigarette smoke. Mice were fed either standard rodent chow (3.76 kcal/g, 16% energy as fat, 20% as protein, Research Diets, Inc., United States), or a pellet HFD (4.7 kcal/g, 43% energy as fat, 20% as protein, 35% as carbohydrate, Research Diets, Inc., United States. Supplementary Table 1) (33, 34). Body weight and 24-h caloric intake were measured once a week as previously described (14, 35). After 5 weeks of pre-conditioning, females were housed and mated with male mice. The same treatment was continued until pups weaned.
Postnatal Litter Size Adjustment and Post-Weaning HFD Feeding
On day 1 after birth, litters were adjusted to a size of 4–6 animals per litter (sex ratio 1:1) to minimize the impact of milk competition. At the age of 20 days (weaning age), male pups were used in the current study. Half of the male pups from the same litter were given a chow diet, while the other half were given the pellet HFD used in the dams. This further yielded six experimental groups, described as the maternal diet + maternal exposure – offspring diet. The six groups were CHOW+SHAM-CHOW, CHOW+SE-CHOW, HFD+SE-CHOW, CHOW+SHAM-HFD, CHOW+SE-HFD, and HFD+SE-HFD. Body weight and food intake were measured once a week until mice reached 13 weeks of age.
Offspring IP Glucose Tolerance Test (IPGTT)
Intraperitoneal glucose tolerance test (IPGTT) was carried out as previously described (13). Briefly, 11 weeks old male offspring were fasted for 5 h. Blood samples were collected from the tail tip to establish a baseline glucose level (T0), which was measured again at 15, 30, 60, and 90 min after glucose injection (2 g/kg, ip). The area under the curve (AUC) was calculated for each mouse during IPGTT.
Sample Collection
Dams were culled 1 day after pups were weaned and male offspring were culled at 13 weeks. After overnight fasting and deep anesthesia (ketamine/xylazine 180/32 mg/kg), blood was collected by cardiac puncture and blood glucose was measured immediately (Accu-Chek glucose meter, Roche) in the dams. Plasma was stored at −20°C for insulin and triglyceride (TG) measurements. Then animals were killed by decapitation and the hypothalamus was micro-dissected. Brown adipose tissue (BAT), epididymal fat, retroperitoneal (Rp) fat, mesenteric fat was dissected and weighed, as well as organs (liver and heart) and skeletal muscle [soleus, extensor digitorum longus (EDL), and tibialis]. Rp fat, BAT, and skeletal muscle were kept providing further markers of substrate metabolism.
Plasma TG and Insulin Assays
Plasma TG was measured using glycerol standard (equivalent to 0–8.46 mM; G7793, Sigma-Aldrich) and TG reagent (T2449, Sigma-Aldrich) as previously described (35). Briefly, samples and standards were incubated with triglyceride reagent at 37°C for 20 min and read on a microplate reader (51119000, Thermo Scientific) at 490 nm. Plasma insulin concentrations were measured using a commercially available ELISA kit (KA3812, Abnova).
Quantitative Real-Time PCR
Total RNA was isolated using TriZol reagent (15596026, Invitrogen) according to the manufacturer's instructions. The purified total RNA was used as a template to generate first-strand cDNA synthesis kit (RR036A, Takara). TaqMan probe/primers that were pre-optimized and validated by the manufacturer (only probe sequence provided by Thermo Fisher Scientific, USA, Supplementary Table 2) were used for quantitative real-time PCR (StepOnePlus Real-Time PCR System, Applied Biosystem). Markers of appetite regulation, including Npy, Npy Y1 receptor (Npy1r), Pomc, and single minded gene (Sim1), were measured in the hypothalamus. Marker involved in substrate metabolism carnitine palmitoyl-transferase (Cpt1α) and Tnfα were measured in the Rp fat. Thermogenesis markers Uncoupling protein (Ucp1 and Ucp3) were measured in BAT. Expression of muscle metabolic markers PPARγ coactivator (Pgc1α and Pgc1β), Myog and MyoD were measured in the soleus muscle.
Statistical Methods
Results are expressed as mean ± SEM. Data on blood glucose level change during IPGTT was analyzed using one-way analysis of variance (ANOVA) with repeated measures, followed by post-hoc Fisher's Least Significance Difference (LSD) tests. Differences in other parameters in the dams and offspring were analyzed using one-way ANOVA and two-way ANOVA, respectively, followed by post-hoc LSD tests if the data were normally distributed. If not, data were log transformed to achieve normality of distribution before they were analyzed.
Results
SE Decreased Body Weight Without Affecting the Daily Energy Intake of Breeders
Before the start of the experiment, the average body weight of female mice was similar among the three groups. After 5 weeks of treatments, mice exposed to cigarette smoke showed significantly lower body weight than those with sham exposure (p < 0.05, Table 1), whereas HFD feeding increased the body weight of the mice with SE (p < 0.05, Table 1). This effect on body weight persisted until these breeders were sacrificed. Interestingly, SE did not affect the daily caloric intake, however, mice consumed 45.6% more calories if they were fed a HFD while exposed to cigarette smoke.
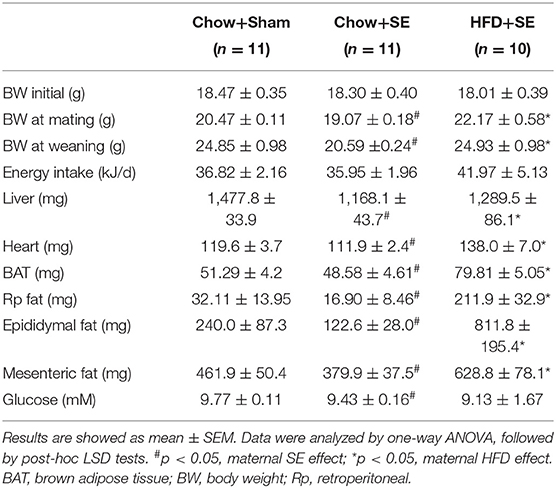
Table 1. Parameters of the dams.
SE reduced liver and fat mass, with reduced blood glucose level (p < 0.05 vs. CHOW+SHAM, Table 1) which was consistent with the literature. HFD+SE dams also had increased liver and heart weight, as well as body fat such as BAT, Rp fat, epididymal fat, and mesenteric fat, while SE markedly reduced the weights of liver, heart, and fat tissues (p < 0.05 vs. CHOW+SE), with some even greater than the control mice, however, the glucose level was further reduced.
Postnatal HFD Increased the Body Weight and Aggravated the Metabolic Disorder Caused by Maternal SE and SE+HFD
At weaning (postnatal day 20), CHOW+SE and HFD+SE pups appeared to have bigger body weights as compared with those from control dams (CHOW+SHAM), indicating that both maternal HFD and SE might raise the risk of obesity in young mice (Table 2).
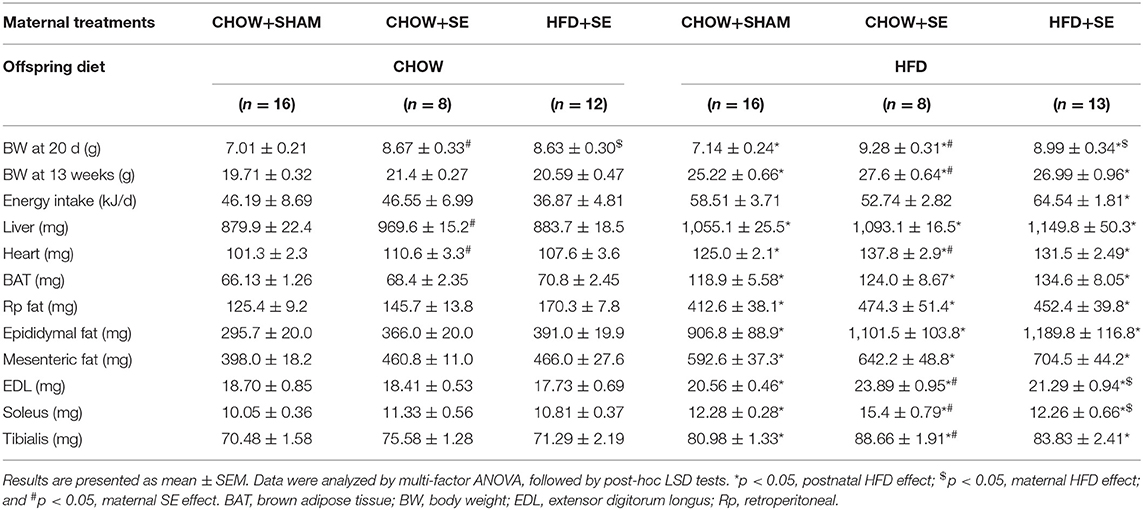
Table 2. Parameters of the male offspring.
At 13 weeks, chow-fed offspring from CHOW+SE and HFD+SE dams showed similar body weight with only larger liver and heart in the SE-CHOW offspring. They also had a similar ability to clear blood glucose during IPGTT (Figures 1A,B), with similar plasma insulin and TG levels among the 3 chow-fed offspring groups (Figures 1C,D).
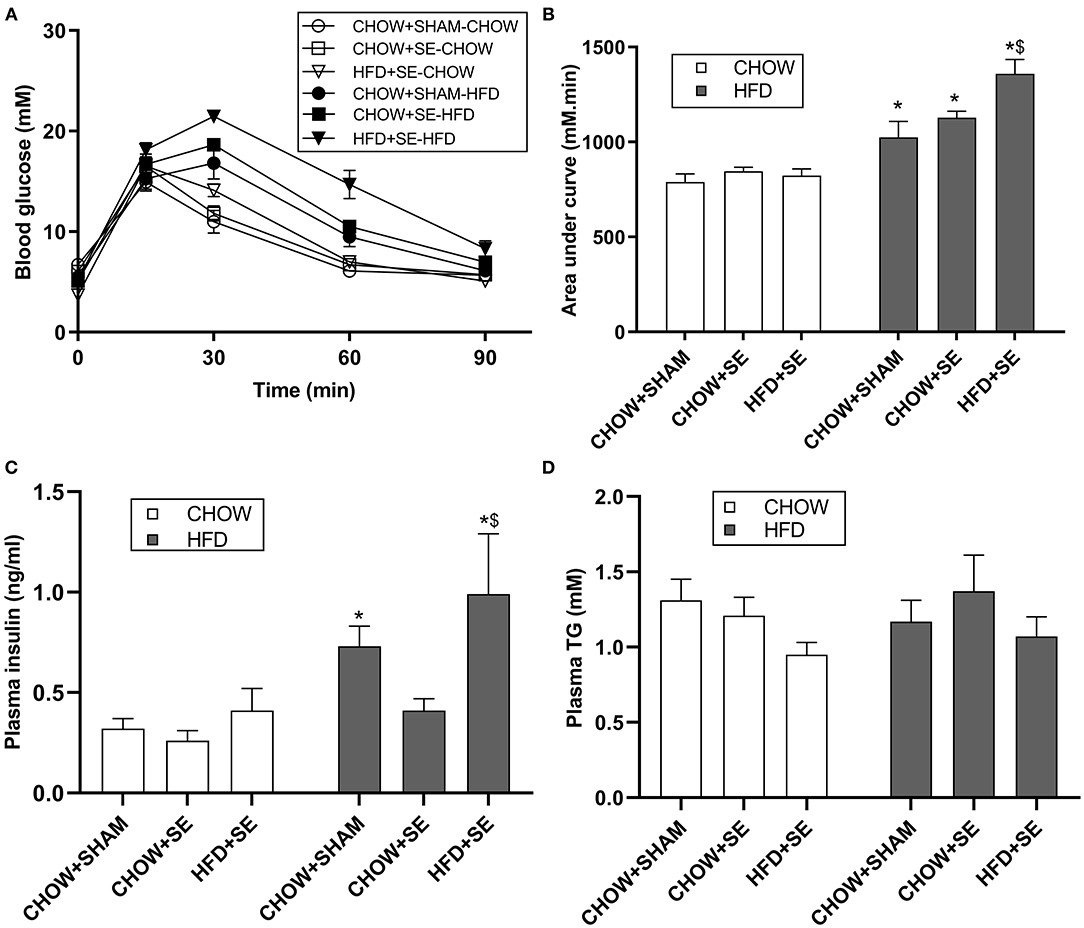
Figure 1. Blood glucose and lipid profile in male offspring. (A) Change in blood glucose levels during an IPGTT at 11 weeks. The area under the curve for panel (A) is shown in panel (B). (C,D) The concentration of insulin (C) and TG (D) in plasma in male offspring at 13 weeks. All statistical results are showed as mean ± SEM. Data were analyzed by two-way ANOVA, followed by post-hoc LSD tests. *p < 0.05 postnatal HFD effect, $p < 0.05 maternal HFD effects.
With postnatal exposure to a HFD, the body weight, body fat (BAT, RP, Epididymal, and Mesenteric fat) and skeletal muscle mass (EDL, soleus, and tibialis), were significantly greater in the offspring treatment. The mice from SE and SE+HFD dams showed a faster growth rate as compared with those from CHOW+SHAM dams (p < 0.05, CHOW+SHAM-HFD vs. CHOW+SE-HFD, CHOW+SE-HFD vs. HFD+SE-HFD, Table 2). A significant increase in energy intake was only observed in the HFD+SE-HFD offspring (Table 2). Moreover, CHOW+SE-HFD offspring showed significantly bigger heart and muscle weights, while HFD+SE-HFD offspring only showed larger muscle weight (EDL and Soleus) (p < 0.05, CHOW+SHAM-HFD vs. CHOW+SE-HFD, CHOW+SE-HFD vs. HFD+SE-HFD, Table 2). HFD+SE-HFD offspring developed more severe glucose intolerance during IPGTT than CHOW+SHAM-HFD and CHOW+SE-HFD offspring (p < 0.05, Figure 1B); however, only CHOW+SHAM-HFD and HFD+SE-HFD offspring showed increased plasma insulin levels (Figure 1C). In addition, plasma TG levels were not affected by postnatal HFD-consumption (Figure 1D).
Thus, maternal intervention, including SE and SE+HFD, resulted in increased body weight at weaning; however, this difference due to maternal programming diminished after consuming a balanced chow diet. While maternal SE increased body weight and heart weight without affecting adiposity when the offspring consumed a HFD, maternal exposure to both SE and HFD significantly increased caloric intake and resulted in the largest fat mass although without statistical significance.
Effects on Hypothalamic Appetite Regulators in Male Offspring
To investigate the effects of the maternal and postnatal HFD consumption on the feeding regulators, we checked the mRNA expression in the hypothalamus. In chow-fed offspring, Pomc mRNA expression was significantly up-regulated in the HFD+SE-CHOW group (p < 0.05 vs. CHOW+SHAM, Figure 2A), whereas Sim 1 was significantly increased in the CHOW+SE-CHOW and HFD+SE-CHOW groups (p < 0.05 vs. CHOW+SHAM, Figure 2B). Moreover, hypothalamic Npy mRNA was only increased in CHOW+SE-CHOW offspring (p < 0.05 vs. CHOW+SHAM, Figure 2C), although the level in the HFD+SE-CHOW group was comparable to the CHOW+SE-CHOW group. However, Npyr1r mRNA was similar among chow-fed offspring.
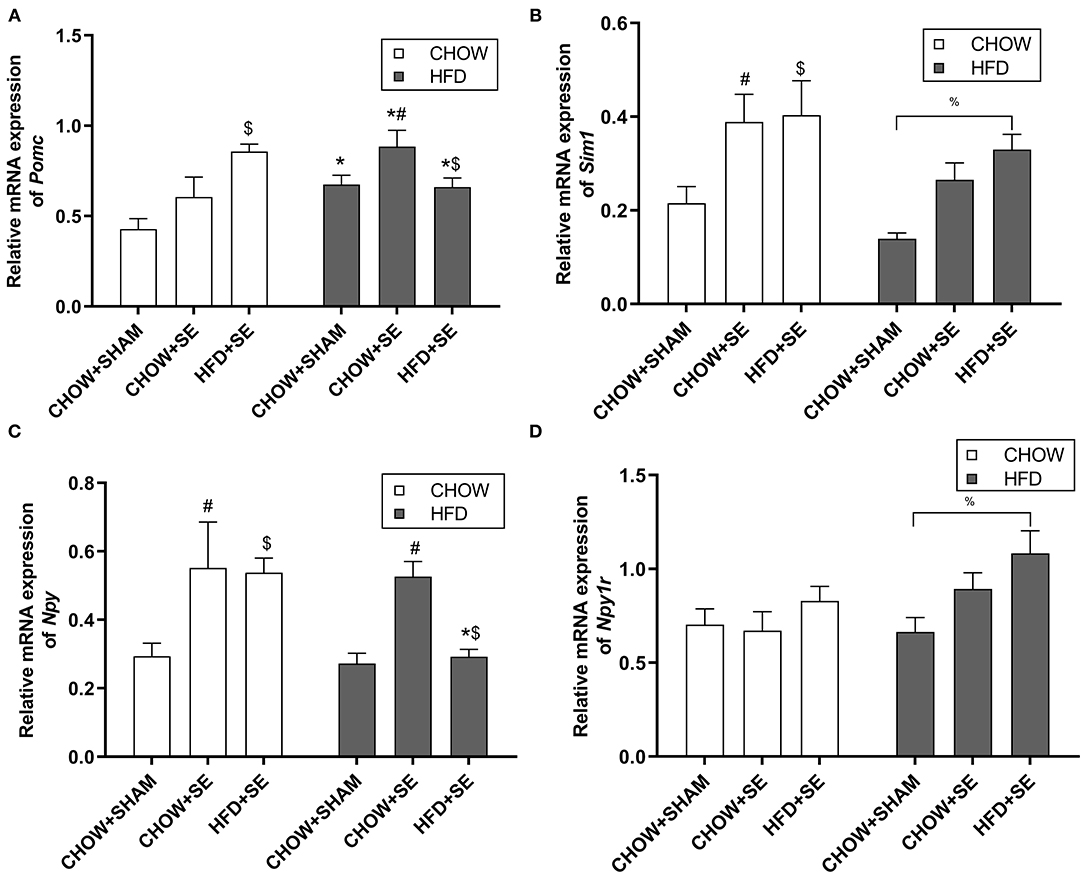
Figure 2. Expression of the energy homeostatic regulator in the hypothalamus. Hypothalamic mRNA expression of Pomc (A), Sim1 (B), Npy (C), and Npy1r (D) in the male offspring at 13 weeks. Results are expressed as mean ± SEM. Data were analyzed by two-way ANOVA, followed by post-hoc LSD tests. *p < 0.05, postnatal HFD effect; $p < 0.05, maternal HFD effect; and #p < 0.05, maternal SE effect. %p < 0.05.
In HFD-fed offspring, Pomc mRNA expression was increased by HFD consumption (p < 0.05 vs. CHOW+SHAM-CHOW, Figure 2A), which was further increased in CHOW+SE-HFD mice (p < 0.05, CHOW+SE-HFD vs. CHOW+SHAM-HFD). There was no difference in Pomc mRNA level between HFD+SE-HFD and CHOW+SE-HFD group. However, Sim 1 was only significantly increased in the HFD+SE-HFD group compared with the CHOW+SHAM-HFD group (p < 0.05, Figure 2B). Npy was significantly upregulated in the CHOW+SE-HFD group (p < 0.05 vs. CHOW+SHAM-HFD, Figure 2C) although similar to its chow-fed littermates, whereas Npyr1r mRNA was significantly upregulated in the HFD+SE-HFD offspring (p < 0.05 vs. CHOW+SHAM-HFD, Figure 2D).
Effects on the Substrate Metabolic in the Fat and Muscle in the Offspring
Cpt1α is the rate-limiting step for fatty acid oxidation in the mitochondrial, while Tnfα is a pro-inflammatory cytokine which plays a key role in insulin resistance. The expression of Cpt1α and Tnfα were no affected by neither maternal nor postnatal interventions (Figures 3A,B). However, the expression of the thermogenesis markers Ucp1 was significantly up-regulated by postnatal HFD consumption in CHOW+SHAM-HFD and HFD+SE-HFD groups compared with their chow-fed littermates (p < 0.05, Figure 3C), while Ucp3 was significantly higher in the HFD+SE-HFD compared with CHOW+SHAM-HFD and CHOW+SE-HFD groups (p < 0.05, Figure 3D).
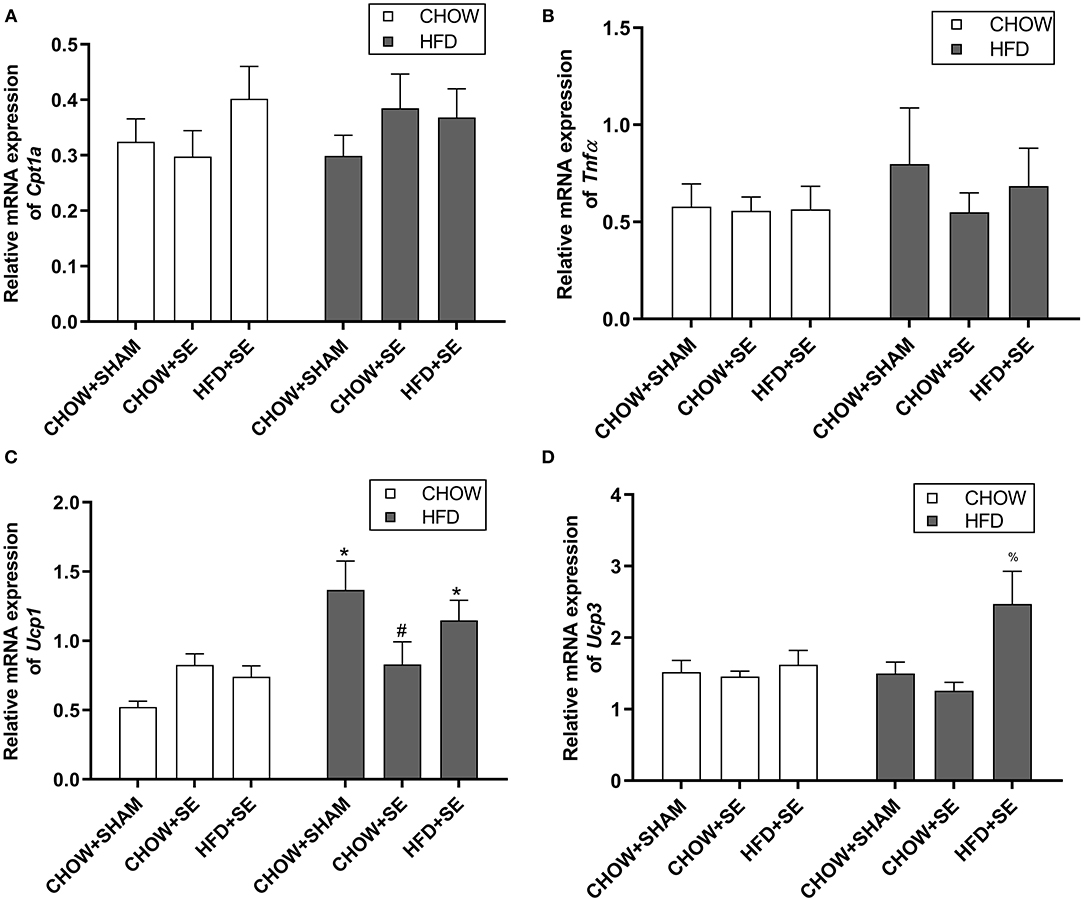
Figure 3. Expression of energy metabolic regulators in fat tissue. mRNA expression of Cpt1α (A), Tnfα (B) in the white fat and, Ucp1 (C), and Ucp3 (D) in the brown fat in the male offspring 13 weeks. Results are expressed as mean ± SEM. Data were analyzed by two-way ANOVA, followed by post-hoc LSD tests. *p < 0.05, postnatal HFD effect and #p < 0.05, maternal SE effect. %p < 0.05 vs. CHOW+SHAM-HFD and CHOW+SE-HFD.
In soleus muscle, we check the expression of mitochondrial biogenesis markers Pgc1α and Pgc1β, which were not significantly affected by maternal programming nor postnatal HFD consumption (Figures 4A,B), albeit a trend decrease in Pgc1β expression in CHOW+SE-HFD and HFD+SE-HFD groups. Furthermore, Myog expression was not different among all groups (Figure 4C); however, Myod1 expression was up-regulated in the HFD+SE-CHOW group (p < 0.05 vs. CHOW+SHAM-CHOW), which was further reduced by postnatal HFD feeding (p < 0.05 vs. HFD+SE-CHOW, Figure 4D).
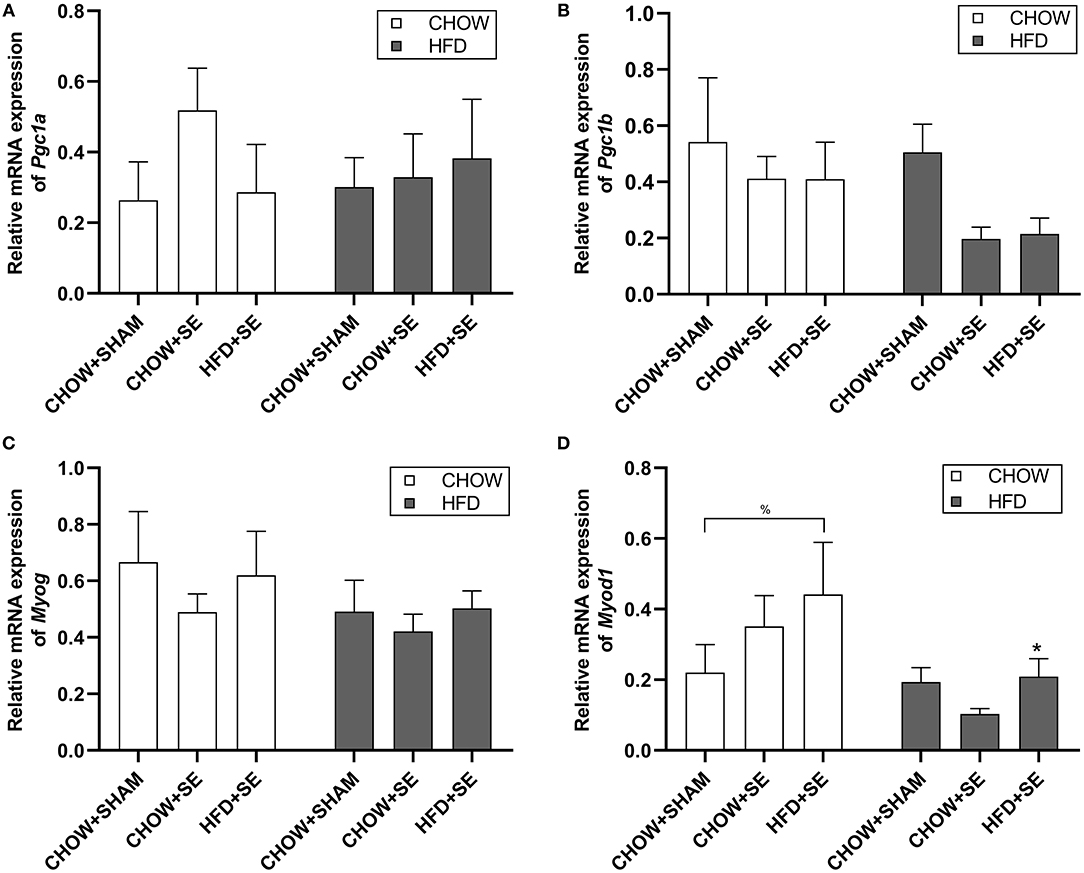
Figure 4. Expression of the metabolic regulator and myogenesis related gene in skeletal muscle tissue. Muscle mRNA expression of Pgc1α (A), Pgc1β (B), Myog (C), and Myod1 (D) in the male offspring at 13 weeks. Results are expressed as mean ± SEM. Data were analyzed by two-way ANOVA, followed by post-hoc LSD tests. *p < 0.05, postnatal HFD effect; %p < 0.05.
Discussion
In this study, we demonstrated that maternal SE exaggerates metabolic disorders if the offspring consume a HFD after weaning. The additional HFD consumption did not ameliorate or reverse the adverse effects due to maternal SE. On the contrary, maternal exposure to both SE and HFD led to the worst outcome in the offspring fed a HFD after weaning.
Exposure to cigarette smoke alone induced significant wasting in the dams, reflected by reduced both fat and lean body mass, consistent with a previous study (13). While additional HFD consumption only significantly increased the body weight at mating, after lactation, the body weight of HFD+SE dams was comparable to the control offspring. However, abdominal fat masses were increased in mice exposed to cigarette smoke at this time point. This is consistent with observations in humans that smoking encourages the development of central obesity albeit smaller body weight (36).
A number of studies have provided evidence that maternal SE led to low birth weight and faster weight gain during the suckling period, called catch-up growth (6, 37, 38). This fast postnatal growth is commonly observed in children, which may lead to obesity during childhood as shown in the weaning offspring in this study (39, 40). Childhood BMI can positively correlate with that in adulthood, which has been well-represented in the offspring with in utero SE. This is not caused by overeating, as the caloric intake was similar between CHOW+SE-HFD and CHOW+SHAM-HFD offspring. Maternal nicotine exposure has been shown to reduce hypothalamic Npy and increase Pomc expression in newborns (6). The increase in both in the CHOW+SE-HFD offspring may reflect a withdrawn rebound, similar to the response in the smokers after quitting. This may be attributed to larger fat mass in CHOW+SE-HFD offspring. There are several homologs of UCPs, including UCP1 and UCP3, which are responsible for mediating thermogenesis and basal metabolic rate in fat (41). Previously study showed that caloric restriction could down-regulate UCP1 to and UCP3 reserve energy expenditure (42, 43). Thus, the HFD leads to an adaptive upregulation of UCPs to increase energy expenditure as shown in CHOW+SHAM-HFD mice. Compared with CHOW+SHAM-HFD offspring, those with intrauterine SE had suppressed thermogenesis marker UCP1 in their BAT, which may impair the adaptive increase in heat production observed in CHOW+SHAM-HFD offspring, resulting in increased fat mass in CHOW+SE-HFD offspring. Glucose intolerance in CHOW+SE-HFD mice may be due to insulin insufficiency, rather than insulin resistance in CHOW+SHAM-HFD, as reflected by plasma insulin levels in these two groups. Increased insulin level is a sign of insulin resistance. HFD has been shown to reduce insulin sensitivity and promote the development of type 2 diabetes (44). Previous studies have shown that maternal nicotine treatment can interrupt β-cell functions in the offspring (45–47).
“Eat for two” is a traditional practice for pregnant women even in the current obesity pandemic. As intrauterine undernutrition is a key to maternal smoking, such practice may be more appealing. However, in this study, we have shown that the combination of maternal HFD and SE further exaggerates the metabolic disorders than maternal SE alone when the offspring were exposed to an obesogenic environment after birth. There are several mechanisms suggested in this study. Firstly, overeating was only observed in HFD+SE-HFD offspring, which may be driven by markedly upregulated Npy1r, the orexigenic receptor for NPY. This increase in activity was not counteracted by the adaptive increase in Sim1 expression which lies downstream of the receptor of the anorexigenic peptide αMSH. Secondly, the heightened glucose intolerance may be a combination of impaired β-cell function due to maternal SE and insulin resistance due to both intrauterine and postnatal HFD exposure. Albeit overeating, the body weight and fat mass in HFD+SE-HFD seem to be controlled. This may be due to more than doubled Ucp3 expression to increase the energy expenditure while energy was over consumed.
During the weight gain by HFD consumption, muscle mass is also increased to support the increased body weight, as shown in HFD-fed mice in this study. The greatest increase in HFD+SE-HFD mice may be proportional to their body weight. Pgc1α controls mitochondrial biogenesis and angiogenesis in skeleton muscle (48–50); whereas, Pgc1β activates an anti-angiogenesis gene program in the skeletal muscle and its overexpression can induce muscle wasting by inhibiting ubiquitin-mediated proteolysis (51, 52). In our present study, the trending decrease in Pgc1β in mice from SE dams can help to explain the increased muscle mass. However, this did not seem to lead to an improvement in muscle metabolic function. Previous studies suggested that Myod1 could promote transcriptional activation to regulate the expression of muscle-specific genes, including Myog, which plays a crucial role in the terminal differentiation during myogenesis (28, 53). In this study, we found significantly altered Myod1 expression but not Myog by maternal HFD+SE. This may be an adaptation to prevent a reduction in muscle mass by maternal SE, which is common in smokers (54). However, the Myod1 changes in offspring from HFD+SE dams was reversed by postnatal HFD consumption. This may be due to myogenesis, as we observed in the other groups fed a HFD. However, further studies are needed to examine the metabolic functions of the muscle as well as mitochondrial function which is beyond the scope of this study.
Whilst there was increased fat mass and unchanged fatty acid metabolic marker Cpt1α, plasma TG was not affected by maternal programming nor postnatal HFD consumption, which might be due to the mouse strain specific. It has previously been shown that with cigarette SE only during lactation, there were decreased proteins levels of Ucp1 and Cpt1 and reduced sympathetic nerve stimulation upon BAT in female adult rat (55). The same was observed with the administration of isolated nicotine through minipumps on the dams at the same period (56). Similarly, in other rat model of SE only during lactation, SE group male adult offspring did not changed insulinemia, despite higher serum glucose levels, suggesting a pancreatic insulin secretory failure (57). Nevertheless, it needs to be noted that none of the above metabolic abnormalities were observed in the chow-fed offspring. This highlights the importance of a healthy diet to prevent the adverse impact of maternal programming on metabolic outcomes in adulthood. As this study was only performed with male mice, it cannot be directly extended to the female offspring, due to the sexual differences during maternal programming as shown by the others (13, 58). In a similar model of SE, females offspring showed increased glucose tolerance by maternal SE, which is consistent with our founding (13). In addition, another study showed that female offspring from the SE dams had lower levels of anorexigenic neuropeptides, Cocaine-, and amphetamine-regulated transcript and MSH, in their brains (59).
In conclusion, this study demonstrated that maternal SE during pregnancy results in increased adiposity and worsened metabolic disorders if the offspring are exposed to HFD after weaning. The additional maternal exposure to HFD interacts with SE which exacerbated metabolic disorders in the male offspring by disrupting metabolic regulators. Therefore, both quitting smoking and maintaining a healthy diet are vital for the healthy future of the offspring.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by Institutional of Animal Care and Committee of San Yet-sun University.
Author Contributions
TH: conceptualization, methodology, visualization, investigation, and writing – original draft. MY and YZ: investigation and editing. XH: investigation. NW: visualization and investigation. YC, PL, and JY: supervision and validation. CC and BO: supervision and review. CY: supervision, writing, review, and editing. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Nature Science Foundation of China (NSFC 81971309), Guangdong Basic and Applied Basic Research Foundation (2019A1515011333), Fundamental Research Funds for the Central Universities (F7201931620002) and the General Project of Shenzhen Science and Technology Innovation Committee (JCYJ20190809161405495).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnut.2021.638576/full#supplementary-material
References
1. Malik VS, Willett WC, Hu FB. Global obesity: trends, risk factors and policy implications. Nat Rev Endocrinol. (2013) 9:13–27. doi: 10.1038/nrendo.2012.199
2. Bayol SA, Farrington SJ, Stickland NC. A maternal 'junk food' diet in pregnancy and lactation promotes an exacerbated taste for 'junk food' and a greater propensity for obesity in rat offspring. Brit J Nutr. (2007) 98:843–51. doi: 10.1017/S0007114507812037
3. Mitchell S, Shaw D. The worldwide epidemic of female obesity. Best Pract Res Cl Ob. (2015) 29:289–99. doi: 10.1016/j.bpobgyn.2014.10.002
4. Di Cesare M, Soric M, Bovet P, Miranda JJ, Bhutta Z, Stevens GA, et al. The epidemiological burden of obesity in childhood: a worldwide epidemic requiring urgent action. Bmc Med. (2019) 17:212. doi: 10.1186/s12916-019-1449-8
5. Haworth JC, Ellestad-Sayed JJ, King J, Dilling LA. Relation of maternal cigarette smoking, obesity, and energy consumption to infant size. Am J Obstet Gynecol. (1980) 138:1185–9. doi: 10.1016/S0002-9378(16)32790-9
6. Grove KL, Sekhon HS, Brogan RS, Keller JA, Smith MS, Spindel ER. Chronic maternal nicotine exposure alters neuronal systems in the arcuate nucleus that regulate feeding behavior in the newborn rhesus macaque. J Clin Endocrinol Metab. (2001) 86:5420–6. doi: 10.1210/jcem.86.11.8033
7. Vardavas CI, Chatzi L, Patelarou E, Plana E, Sarri K, Kafatos A, et al. Smoking and smoking cessation during early pregnancy and its effect on adverse pregnancy outcomes and fetal growth. Eur J Pediatr. (2010) 169:741–48. doi: 10.1007/s00431-009-1107-9
8. Power C, Jefferis BJ. Fetal environment and subsequent obesity: a study of maternal smoking. Int J Epidemiol. (2002) 31:413–9. doi: 10.1093/intjepid/31.2.413
9. Fall CHD, Osmond C, Barker DJP, Clark PMS, Hales CN, Stirling Y, et al. Fetal and infant growth and cardiovascular risk-factors in women. Br Med J. (1995) 310:428–32. doi: 10.1136/bmj.310.6977.428
10. Chen H, Morris MJ. Maternal smoking-A contributor to the obesity epidemic? Obes Res Clin Pract. (2007) 1:155–63. doi: 10.1016/j.orcp.2007.07.004
11. Cunningham J, Dockery DW, Gold DR, Speizer FE. Racial-differences in the association between maternal smoking during pregnancy and lung-function in children. Am J Resp Crit Care. (1995) 152:565–9. doi: 10.1164/ajrccm.152.2.7633708
12. Mendez MA, Torrent M, Ferrer C, Ribas-Fito N, Sunyer J. Maternal smoking very early in pregnancy is related to child overweight at age 5-7 y. Am J Clin Nutr. (2008) 87:1906–13. doi: 10.1093/ajcn/87.6.1906
13. Chen H, Iglesias MA, Caruso V, Morris MJ. Maternal cigarette smoke exposure contributes to glucose intolerance and decreased brain insulin action in mice offspring independent of maternal diet. PLoS One. (2011) 6:e27260. doi: 10.1371/journal.pone.0027260
14. Chen H, Hansen MJ, Jones JE, Vlahos R, Anderson GP, Morris MJ. Detrimental metabolic effects of combining long-term cigarette smoke exposure and high-fat diet in mice. Am J Physiol Endocrinol Metab. (2007) 293:E1564–71. doi: 10.1152/ajpendo.00442.2007
15. Chen H, Hansen MJ, Jones JE, Vlahos R, Bozinovski S, Anderson GP, et al. Cigarette smoke exposure reprograms the hypothalamic neuropeptide Y axis to promote weight loss. Am J Resp Crit Care. (2006) 173:1248–54. doi: 10.1164/rccm.200506-977OC
16. Chen H, Vlahos R, Bozinovski S, Jones J, Anderson GP, Morris MJ. Effect of short-term cigarette smoke exposure on body weight, appetite and brain neuropeptide Y in mice. Neuropsychopharmacol. (2005) 30:713–9. doi: 10.1038/sj.npp.1300597
17. Horta BL, Gigante DP, Nazmi A, Silveira VMF, Oliveira I, Victora CG. Maternal smoking during pregnancy and risk factors for cardiovascular disease in adulthood. Atherosclerosis. (2011) 219:815–20. doi: 10.1016/j.atherosclerosis.2011.08.018
18. Rogers I, Emmett P. The effect of maternal smoking status, educational level and age on food and nutrient intakes in preschool children: results from the Avon Longitudinal Study of Parents and Children. Eur J Clin Nutr. (2003) 57:854–64. doi: 10.1038/sj.ejcn.1601619
19. Sullivan EL, Nousen EK, Chamlou KA. Maternal high fat diet consumption during the perinatal period programs offspring behavior. Physiol Behav. (2014) 123:236–42. doi: 10.1016/j.physbeh.2012.07.014
20. Chen H, Saad S, Sandow SL, Bertrand PP. Cigarette smoking and brain regulation of energy homeostasis. Front Pharmacol. (2012) 3:147. doi: 10.3389/fphar.2012.00147
21. Cummings JR, Gearhardt AN, Miller AL, Hyde LW, Lumeng JC. Maternal nicotine dependence is associated with longitudinal increases in child obesogenic eating behaviors. Pediatr Obes. (2019) 14:e12541. doi: 10.1111/ijpo.12541
22. Chen H, Simar D, Lambert K, Mercier J, Morris MJ. Intrauterine and postnatal overnutrition differentially impact appetite regulators and fuel metabolism. Endocrinology. (2008) 149:5348–56. doi: 10.1210/en.2008-0582
23. Chen H, Simar D, Morris MJ. Maternal obesity impairs brain glucose metabolism and neural response to hyperglycemia in male rat offspring. J Neurochem. (2014) 129:297–303. doi: 10.1111/jnc.12623
24. Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. (2006) 443:289–95. doi: 10.1038/nature05026
25. Chen H, Morris MJ. Differential responses of orexigenic neuropeptides to fasting in offspring of obese mothers. Obesity. (2009) 17:1356–62. doi: 10.1038/oby.2009.56
26. Jagoe RT, Engelen MP. Muscle wasting and changes in muscle protein metabolism in chronic obstructive pulmonary disease. Eur Respir J Suppl. (2003) 46:52s−63s. doi: 10.1183/09031936.03.00004608
27. Samec S, Seydoux J, Dulloo AG. Role of UCP homologs in skeletal-muscles and brown adipose tissue-mediators of thermogenesis or regulators of lipids as fuel substrate. FASEB J. (1998) 12:715–24. doi: 10.1096/fasebj.12.9.715
28. Harada A, Mallappa C, Okada S, Butler JT, Baker SP, Lawrence JB, et al. Spatial re-organization of myogenic regulatory sequences temporally controls gene expression. Nucleic Acids Res. (2015) 43:2008–21. doi: 10.1093/nar/gkv046
29. Ailhaud G, Massiera F, Alessandri JM, Guesnet P. Fatty acid composition as an early determinant of childhood obesity. Genes Nutr. (2007) 2:39–40. doi: 10.1007/s12263-007-0017-6
30. Ailhaud G, Guesnet P. Fatty acid composition of fats is an early determinant of childhood obesity: a short review and an opinion. Obes Rev. (2004) 5:21–6. doi: 10.1111/j.1467-789X.2004.00121.x
31. Vlahos R, Bozinovski S, Jones JE, Powell J, Gras J, Lilja A, et al. Differential protease, innate immunity and NF kappa B induction profiles during lung inflammation induced by sub-chronic cigarette smoke exposure in mice. Am J Physiol Lung Cell Mol Physiol. (2006) 290:L931–45. doi: 10.1152/ajplung.00201.2005
32. Chan YL, Saad S, Machaalani R, Oliver BG, Vissel B, Pollock C, et al. Maternal cigarette smoke exposure worsens neurological outcomes in adolescent offspring with hypoxic-ischemic injury. Front Mol Neurosci. (2017) 10:306. doi: 10.3389/fnmol.2017.00306
33. Nguyen LT, Saad S, Tan Y, Pollock C, Chen H. Maternal high-fat diet induces metabolic stress response disorders in offspring hypothalamus. J Mol Endocrinol. (2017) 59:81–92. doi: 10.1530/JME-17-0056
34. Morris MJ, Chen H, Watts R, Shulkes A, Cameron-Smith D. Brain neuropeptide Y and CCK and peripheral adipokine receptors: temporal response in obesity induced by palatable diet. Int J Obes (Lond). (2008) 32:249–58. doi: 10.1038/sj.ijo.0803716
35. Nguyen LT, Chen H, Mak C, Zaky A, Saad S. SRT1720 attenuates obesity and insulin resistance but not liver damage in the offspring due to maternal and postnatal high-fat diet consumption. Am J Physiol Endocrinol Metab. (2018) 315:E196–E203. doi: 10.1152/ajpendo.00472.2017
36. Chiolero A, Faeh D, Paccaud F, Cornuz J. Consequences of smoking for body weight, body fat distribution, and insulin resistance. Am J Clin Nutr. (2008) 87:801–9. doi: 10.1093/ajcn/87.4.801
37. Mantzoros CS, Varvarigou A, Kaklamani VG, Beratis NG, Flier JS. Effect of birth weight and maternal smoking on cord blood leptin concentrations of full-term and preterm newborns. J Clin Endocrinol Metab. (1997) 82:2856–61. doi: 10.1210/jc.82.9.2856
38. Fried PA, O'Connell CM. A comparison of the effects of prenatal exposure to tobacco, alcohol, cannabis and caffeine on birth size and subsequent growth. Neurotoxicol Teratol. (1987) 9:79–85. doi: 10.1016/0892-0362(87)90082-1
39. Toschke AM, Ehlin AG, von Kries R, Ekbom A, Montgomery SM. Maternal smoking during pregnancy and appetite control in offspring. J Perinat Med. (2003) 31:251–6. doi: 10.1515/JPM.2003.034
40. Oken E, Huh SY, Taveras EM, Rich-Edwards JW, Gillman MW. Associations of maternal prenatal smoking with child adiposity and blood pressure. Obes Res. (2005) 13:2021–8. doi: 10.1038/oby.2005.248
41. Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. (2004) 84:277–359. doi: 10.1152/physrev.00015.2003
42. Sivitz WI, Fink BD, Donohoue PA. Fasting and leptin modulate adipose and muscle uncoupling protein: divergent effects between messenger ribonucleic acid and protein expression. Endocrinology. (1999) 140:1511–9. doi: 10.1210/endo.140.4.6668
43. Valle A, Garcia-Palmer FJ, Oliver J, Roca P. Sex differences in brown adipose tissue thermogenic features during caloric restriction. Cell Physiol Biochem. (2007) 19:195–204. doi: 10.1159/000099207
44. DeFronzo RA. Insulin resistance, lipotoxicity, type 2 diabetes and atherosclerosis: the missing links. The Claude Bernard Lecture 2009. Diabetologia. (2010) 53:1270–87. doi: 10.1007/s00125-010-1684-1
45. Bruin JE, Gerstein HC, Holloway AC. Long-term consequences of fetal and neonatal nicotine exposure: a critical review. Toxicol Sci. (2010) 116:364–74. doi: 10.1093/toxsci/kfq103
46. Bruin JE, Petre MA, Lehman MA, Raha S, Gerstein HC, Morrison KM, et al. Maternal nicotine exposure increases oxidative stress in the offspring. Free Radical Bio Med. (2008) 44:1919–25. doi: 10.1016/j.freeradbiomed.2008.02.010
47. Holloway AC, Lim GE, Petrik JJ, Foster WG, Morrison KM, Gerstein HC. Fetal and neonatal exposure to nicotine in Wistar rats results in increased beta cell apoptosis at birth and postnatal endocrine and metabolic changes associated with type 2 diabetes. Diabetologia. (2005) 48:2661–6. doi: 10.1007/s00125-005-0022-5
48. Lin J, Wu H, Tarr PT, Zhang CY, Wu ZD, Boss O, et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. (2002) 418:797–801. doi: 10.1038/nature00904
49. Arany Z, Foo SY, Ma Y, Ruas JL, Bommi-Reddy A, Girnun G, et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1α. Nature. (2008) 451:1008–12. doi: 10.1038/nature06613
50. Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. (2003) 34:267–73. doi: 10.1038/ng1180
51. Brault JJ, Jespersen JG, Goldberg AL. Peroxisome proliferator-activated receptor gamma coactivator 1α or 1β overexpression inhibits muscle protein degradation, induction of ubiquitin ligases, and disuse atrophy. J Biol Chem. (2010) 285:19460–71. doi: 10.1074/jbc.M110.113092
52. Menconi MJ, Arany ZP, Alamdari N, Aversa Z, Gonnella P, O'Neal P, et al. Sepsis and glucocorticoids downregulate the expression of the nuclear cofactor PGC-1beta in skeletal muscle. Am J Physiol Endocrinol Metab. (2010) 299:E533–43. doi: 10.1152/ajpendo.00596.2009
53. Gerber AN, Klesert TR, Bergstrom DA, Tapscott SJ. Two domains of MyoD mediate transcriptional activation of genes in repressive chromatin: a mechanism for lineage determination in myogenesis. Genes Dev. (1997) 11:436–50. doi: 10.1101/gad.11.4.436
54. Petersen AMW, Magkos F, Atherton P, Selby A, Mittendorfer B. Smoking impairs muscle protein synthesis and increases the expression of myostatin and MAFbx in muscle. Am J Physiol. (2007) 293:843–8. doi: 10.1152/ajpendo.00301.2007
55. Peixoto TC, Moura EG, Oliveira E, Younes-Rapozo V, Soares PN, Rodrigues VST, et al. Neonatal tobacco smoke reduces thermogenesis capacity in brown adipose tissue in adult rats. Braz J Med Biol Res. (2018) 51:e6982. doi: 10.1590/1414-431x20186982
56. Peixoto TC, Moura EG, Soares PN, Bertasso IM, Pietrobon CB, Caramez FAH, et al. Nicotine exposure during breastfeeding reduces sympathetic activity in brown adipose tissue and increases in white adipose tissue in adult rats: sex-related differences. Food Chem Toxicol. (2020) 140:111328. doi: 10.1016/j.fct.2020.111328
57. Santos-Silva AP, Oliveira E, Pinheiro CR, Santana AC, Nascimento-Saba CC, Abreu-Villaca Y, et al. Endocrine effects of tobacco smoke exposure during lactation in weaned and adult male offspring. J Endocrinol. (2013) 218:13–24. doi: 10.1530/JOE-13-0003
58. Jamshed L, Perono GA, Jamshed S, Holloway AC. Early life exposure to nicotine: postnatal metabolic, neurobehavioral and respiratory outcomes and the development of childhood cancers. Toxicol Sci. (2020) 178:3–15. doi: 10.1093/toxsci/kfaa127
59. Peixoto TC, Moura EG, Oliveira E, Younes-Rapozo V, Lisboa PC. Hypothalamic neuropeptides expression and hypothalamic inflammation in adult rats that were exposed to tobacco smoke during breastfeeding: sex-related differences. Neuroscience. (2019) 418:69–81. doi: 10.1016/j.neuroscience.2019.08.006
Keywords: smoke exposure, high fat diet, feeding regulation, glucose intolerance, myogenes
Citation: Huang T, Yang M, Zeng Y, Huang X, Wang N, Chen Y, Li P, Yuan J, Chen C, Oliver BG and Yi C (2021) Maternal High Fat Diet Consumption Exaggerates Metabolic Disorders in Mice With Cigarette-Smoking Induced Intrauterine Undernutrition. Front. Nutr. 8:638576. doi: 10.3389/fnut.2021.638576
Received: 07 December 2020; Accepted: 18 February 2021;
Published: 16 March 2021.
Edited by:
Clare Marie Reynolds, University College Dublin, IrelandReviewed by:
Marcio Alberto Torsoni, State University of Campinas, BrazilEgberto Gaspar Moura, Rio de Janeiro State University, Brazil
Copyright © 2021 Huang, Yang, Zeng, Huang, Wang, Chen, Li, Yuan, Chen, Oliver and Yi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chun Chen, Y2hlbmNodW42OUAxMjYuY29t; Chenju Yi, eWljaGpAbWFpbC5zeXN1LmVkdS5jbg==
†These authors have contributed equally to this work
‡These authors share senior authorship