 Yin-Huai Chen1†§
Yin-Huai Chen1†§ Charlotte E. Cheadle1,2§Louise V. Rice1,2¶
Charlotte E. Cheadle1,2§Louise V. Rice1,2¶ Paul E. Pfeffer1,2‡
Paul E. Pfeffer1,2‡ Sarah Dimeloe1‡
Sarah Dimeloe1‡ Atul Gupta1,3‡Andrew Bush4‡
Atul Gupta1,3‡Andrew Bush4‡ Bibek Gooptu1,5,6
Bibek Gooptu1,5,6 Catherine M. Hawrylowicz1,2*†
Catherine M. Hawrylowicz1,2*†- 1Peter Gorer Department of Immunobiology (Formerly Asthma, Allergy and Lung Biology), School of Immunology and Microbial Sciences, King's College London, London, United Kingdom
- 2Medical Research Council and Asthma UK Centre for Allergic Mechanisms of Asthma, Guy's Hospital, King's College London, London, United Kingdom
- 3National Heart and Lung Institute, Royal Brompton & Harefield National Health Service Foundation Trust, London, United Kingdom
- 4Centre for Paediatrics and Child Health, National Heart and Lung Institute, Imperial College, Royal Brompton Hospital, London, United Kingdom
- 5National Institute for Health Research Leicester Biomedical Research Centre-Respiratory and Leicester Institute of Structural & Chemical Biology, University of Leicester, Leicester, United Kingdom
- 6London Alpha-1 Antitrypsin Deficiency Service, Royal Free Hospital, London, United Kingdom
Background: Vitamin D upregulates anti-inflammatory and antimicrobial pathways that promote respiratory health. Vitamin D synthesis is initiated following skin exposure to sunlight, however nutritional supplementation can be required to address deficiency, for example during the winter months or due to cultural constraints. We recently reported that 1α,25-dihydroxyvitamin D3 (1,25(OH)2D3) treatment induced alpha-1 antitrypsin (AAT) expression in CD4+, but not CD8+ T cells, with evidence supporting an immunoregulatory role.
Research Question: To understand the relationship between vitamin D, lung AAT levels and T lymphocytes further we investigated whether TGF-β is required as a co-factor for 1,25(OH)2D3-induced upregulation of AAT by vitamin D in CD8+ T cells in vitro and correlated circulating vitamin D levels with lung AAT levels in vivo.
Results: 1,25(OH)2D3 in combination with TGF-β1 increased AAT expression by CD8+ T cells, as well as VDR and RXRα gene expression, which may partly explain the requirement for TGF-β. CD4+ T cells may also require autocrine stimulation with TGF-β as a co-factor since 1,25(OH)2D3 was associated with increased TGF-β bioactivity and neutralisation of TGF-β partially abrogated 1,25(OH)2D3-induced SERPINA1 gene expression. Neither CD4+ nor CD8+ T cells responded to the circulating vitamin D precursor, 25-hydroxyvitamin D3 for induction of SERPINA1, suggesting that local generation of 1,25(OH)2D3 is required. Transcriptional gene profiling studies previously demonstrated that human bronchial epithelial cells rapidly increased TGF-β2 gene expression in response to 1,25(OH)2D3. Here, human epithelial cells responded to precursor 25(OH)D3 to increase bioactive TGF-β synthesis. CD8+ T cells responded comparably to TGF-β1 and TGF-β2 to increase 1,25(OH)2D3-induced AAT. However, CD8+ T cells from adults with AAT-deficiency, homozygous for the Z allele of SERPINA1, were unable to mount this response. AAT levels in the airways of children with asthma and controls correlated with circulating 25(OH)D3.
Conclusions: Vitamin D increases AAT expression in human T cells and this response is impaired in T cells from individuals homozygous for the Z allele of SERPINA1 in a clinic population. Furthermore, a correlation between circulating vitamin D and airway AAT is reported. We propose that vitamin D-induced AAT contributes to local immunomodulation and airway health effects previously attributed to vitamin D.
Introduction
Vitamin D is increasingly recognised as an important factor in regulating immunity and respiratory health, and vitamin D deficiency is associated with several immune-mediated airway diseases, including asthma and chronic obstructive pulmonary disease (COPD) (1–3). This is attributed to its diverse immuno-modulatory functions including the capacity to promote anti-microbial pathways, suppress inflammatory responses while enhancing immuno-modulatory functions such as anti-inflammatory IL-10 production (4, 5).
Alpha-1 antitrypsin (AAT) plays a critical role in protecting lung parenchymal tissue from direct elastinolytic degradation and pro-inflammatory effects of serine proteases, notably neutrophil elastase (6). It is predominantly synthesized in the liver by hepatocytes and is secreted into the plasma as the most abundant circulating antiprotease. The metastable structure of native AAT and other members of the serine protease inhibitor (serpin) superfamily provides the potential for stabilizing conformational change (7). This is utilized in the functional mechanism of protease inhibition, but also renders AAT vulnerable to point mutations, as seen in the Glu342Lys (Z) variant. Within hepatocytes, that are the major source of systemic AAT, this triggers misfolding in the endoplasmic reticulum, aberrant conformational change and stabilizing intermolecular linkage to form AAT polymers (8). The results are a deficiency of circulating functional AAT, and retention of misfolded and polymerized AAT within hepatocytes associated with pro-inflammatory and pro-fibrotic gain-of-function (9). This combination predisposes Z variant homozygotes (denoted Pi*ZZ) to severe and early-onset COPD and liver disease (hepatitis, cirrhosis, and hepatocellular carcinoma) (10). The lung disease seems predominantly driven by loss-of-function mechanisms though extracellular polymers encourage neutrophil chemotaxis and activity and are found in the circulation and lung (11).
AAT displays multiple immuno-regulatory functions including inhibition of neutrophil chemotaxis (12), induction of IL-1Ra by macrophages (13) and the development of tolerogenic dendritic cells and their production of IL-10 (14, 15). We have recently shown the requirement of AAT in the 1,25(OH)2D3-mediated induction of IL-10 in CD4+ T cells (16). In addition to immunomodulatory effects mediated by antiprotease function, immunomodulatory effects of AAT that are independent of antiprotease function are now recognized (17). The absence of these effects may further exacerbate the pro-inflammatory state in lungs of individuals with AAT deficiency.
Cells within the lungs e.g., alveolar epithelial cells, monocytes and neutrophils are capable of secreting AAT to supplement liver-derived AAT in the airways (18–20). We have demonstrated that the active form of vitamin D, 1,25(OH)2D3, upregulates expression of SERPINA1, the gene encoding the serine protease inhibitor AAT, and AAT protein secretion by human primary CD4+ T cells (16). Conversely this was not seen for monocytes. Moreover, vitamin D reduces secretion of matrix metalloproteinase (MMP-)9 that degrades AAT (21, 22). These data suggest that vitamin D availability may boost AAT levels within the airway microenvironment and so represent an additional mechanism by which vitamin D protects the airways.
Our published data demonstrate that 1,25(OH)2D3 alone does not upregulate AAT in human CD8+ T cells (16). Given the proposed role of CD8+ T cells in chronic airway conditions such as COPD (23), we investigated whether a co-factor that acts in concert with 1,25(OH)2D3 may induce AAT synthesis by human CD8+ T cells. TGF-β was investigated, since previous studies reported the capacity of hepatoma cell lines and human lung epithelial cell lines to produce AAT in response to TGF-β (24, 25). Furthermore, TGF-β and vitamin D are reported to act co-operatively. Independent studies report, for example, that TGF-β increases gene and protein expression for VDR (26) and the enzyme CYP27B1, which converts precursor vitamin D, 25(OH)D, to the active moiety, 1,25(OH)2D3 (27, 28). Furthermore, 1,25(OH)2D3 in combination with TGF-β increases the frequency of CD4+ Foxp3+ Treg cells (29). These data provide the rationale for the present study to investigate whether 1,25(OH)2D3 and TGF-β interact with each other for enhanced effects in human CD8+ T cells.
Materials and Methods
Subject Characteristics
Healthy donors (“Human peripheral blood cell laboratory studies to investigate the role of immune and inflammatory pathways in respiratory disease” by the local research ethics committee, REC reference: 14/LO/1699) were recruited and provided full written informed consent with full Research Ethics Committee approval. COPD patients without AAT deficiency (Chest clinic, Guy's Hospital, 14/LO/1699), and PiZZ-AAT deficient patients with or without COPD (Targeting dysfunctional mechanisms in alpha-1 antitrypsin deficiency, The Royal Free Alpha-1 clinic, REC reference: 13/LO/1085) were recruited and provided full written informed consent with full Research Ethics Committee approval, including anonymization of donors to researchers. The study in a pediatric asthma cohort and control children undergoing diagnostic bronchoscopy for non-asthma related purposes was approved by the Royal Brompton and Harefield Research Ethics Committee (09/H07008/48) which has been previously described Gupta et al. (30). Informed consent was obtained from parents and age-appropriate assent from children. Serum levels of 25-hydroxyvitamin D were measured as previously described by Gupta et al. (30). All work adhered strictly to institutional safety guidelines and procedures. Table 1 provides data on the number of individuals studied throughout, and figure legends identify number of individual donors in each experimental series.
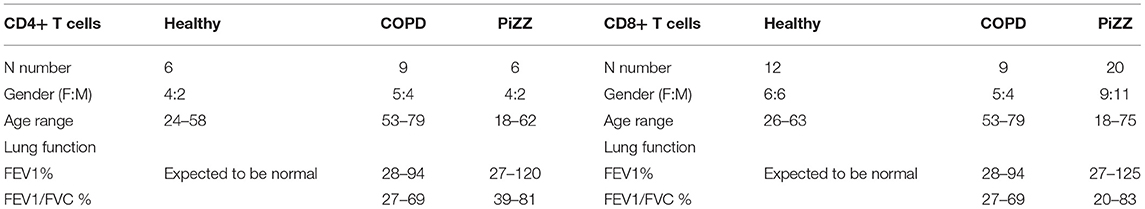
Table 1. Clinical parameters of healthy control, COPD, and PiZZ patients.
Cell Isolation and Culture
Human peripheral blood mononuclear cells (PBMCs) were isolated, as previously described Xystrakis et al. (31). CD4+ and CD8+ T cells were isolated using Dynabeads CD4 or CD8 positive selection kits (Invitrogen, Paisley, UK). Cells (1 × 106/ml) were cultured in RPMI 1640 medium supplemented with 10% FCS, 2 mM L-glutamine, and 50 μg/ml gentamicin, and stimulated with plate-bound anti-CD3 (1 μg/ml; OKT3) plus 50 IU/ml recombinant human IL-2 (Eurocetus, UK) in the presence or absence of 1,25(OH)2D3 (BIOMOL research labs, UK), TGF-β1 (R&D Systems, UK), TGF-β2 (R&D Systems) or anti-TGF-β (R&D Systems) at indicated concentrations for 7 days. NIST SRM1648a Urban Particulate Matter (National Institute of Standards & Technology, USA) is an urban total particulate matter reference material with mean particle diameter 5.85 μm, collected in the USA. NIST was prepared as previously described by Pfeffer et al. (32).
HBEC Isolation and Culture Conditions
Primary human bronchial epithelial cells (HBECs) were acquired and maintained as previously described Pfeffer et al. (33). HBECs were stimulated with 50 μg SRM1648a, a standard reference urban particulate matter, NIST, in the absence or presence of 10, 100, or 1,000 nM 25(OH)D3 for 24 h. Lysed cell monolayers were collected for assessment by qRT-PCR. HBECS were also stimulated with 25(OH)D3 (1 μM) for 48 h and culture supernatant collected for TGF-β bioassay.
qRT-PCR
RNA was extracted from cell pellets using the Rneasy Mini kit (Qiagen, Crawley UK) according to the manufacturer's instructions. Nanodrop ND-1000 spectrophotometer was used to quantify RNA. 250 ng of RNA was reverse transcribed into cDNA. Quantitative real-time PCR was performed, as previously described Urry et al. (34) in triplicates by using an Applied Biosystems 7900 HT system and FAM-labeled Assay-on-Demand reagent sets for SERPINA1 (Hs01097800_m1), VDR (Hs01045846_m1), RXRα (Hs01067640_m1). Quantitative real-time PCR reactions were multiplexed with VIC-labeled 18S primers and probes (Hs99999901_s1) as an endogenous control and analysed with SDS software, version 2.1 (Applied Biosystems, Foster City, Calif), according to 2−ΔCt (× 106).
AAT ELISA
The AAT ELISA employed a commercial polyclonal rabbit anti-human AAT primary capture antibody (Dako, UK) and a biotin-conjugated rabbit anti-human AAT secondary detection antibody [in-house purified, previously described in Dimeloe et al. (16)]. Standards were serially diluted in RPMI medium plus 10% foetal calf serum, by serial 1:2 dilutions from the top standard of 200 ng/ml using human plasma-purified AAT (Sigma-Aldrich, UK). Streptavidin-alkaline phosphatase (Sigma-Aldrich, UK) and 4-Nitrophenyl phosphate disodium salt hexahydrate (1 mg/ml; Sigma-Aldrich, UK) were used to detect AAT. Absorbance was measured at 405 nm on an Anthos HTII plate reader (Anthos, UK) using Softmax pro software and quantified using GraphPad Prism version 6 (GraphPad software Inc., USA). The lower limit of detection was 0.32 ng/ml.
TGF-β Bioassay
TGF-β bioactivity was measured using mink lung epithelial cell lines (MLECs) transfected with plasminogen activator inhibitor-1, a TGF-β target gene, fused with the firefly luciferase construct (kindly donated by Professor Daniel Rifkin, New York University). Briefly, 3 × 105 MLECs/ml were incubated for 14 h at 37°C in 5% CO2 with cell culture supernatants or standards. Culture media was aspirated, washed, cells lysed and luciferase activity determined using Firefly Luciferase Assay Kit (Biotium, USA) and measured by luminescence (1450 MicroBeta TriLux; PerkinElmer, USA) in accordance with the manufacturer's instructions. Light emitted corresponds to the amount of bioactive TGF-β. Serum free RPMI was used for these experiments.
Data Analysis
Data are shown as mean ± standard error of mean (SEM) unless otherwise indicated.
Data analysis was performed using Graphpad Prism version 6.00 for Mac OS X (Graphpad software Inc., USA). Statistical test used as in relevant figure legend.
Results
Exogenous TGF-β1 and 1,25(OH)2D3 Act Cooperatively to Enhance SERPINA1/AAT in CD8+ T Cells
Human CD4+ T cells, but not CD8+ T cells, treated with 1,25(OH)2D3 alone increase SERPINA1 gene expression and AAT protein secretion (16). The requirement for TGF-β as a cofactor to promote synthesis of AAT by CD8+ T cells was investigated. Peripheral blood CD4+ and CD8+ T cells were stimulated with anti CD3/IL-2 in the presence of 10 nM or 100 nM 1,25(OH)2D3, with or without the addition of TGF-β1 over a broad range titration (0.02–20 ng/ml) (Figure 1). After 7 days in culture both 10 and 100 nM 1,25(OH)2D3 significantly increased SERPINA1 transcription and 100 nM 1,25(OH)2D3 significantly upregulated AAT secretion in CD4+ T cells, but not in CD8+ T cell cultures. Addition of 2 or 20 ng/ml TGF-β1 together with the higher concentration of 1,25(OH)2D3 significantly increased both SERPINA1 and AAT expression in CD8+ T cell cultures to levels similar to those observed in CD4+ T cells treated with the same concentration of 1,25(OH)2D3. However, the addition of TGF-β1 alone had no effect, whilst TGF-β1 with 1,25(OH)2D3 had no additional effect in CD4+ T cell cultures. The physiological concentration of TGF-β has been reported to be up to 4 ng/ml in human plasma (35), we therefore have selected 2 ng/ml to proceed in the study.
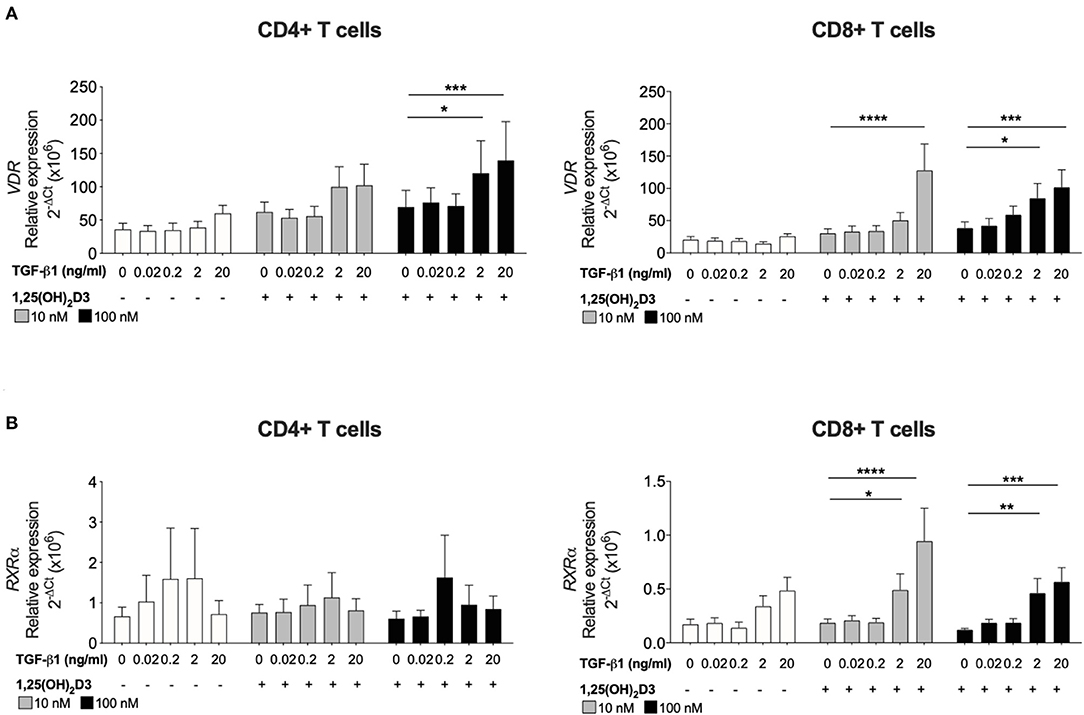
Figure 1. TGF-β1 together with 1,25(OH)2D3 upregulates VDR and RXRα gene expression by CD8+ T cells. CD4+ and CD8+ T cells (n = 6) were stimulated and cultured for 7 days in the absence or presence of 10 or 100 nM 1,25(OH)2D3, with or without TGF-β1 (0.02, 0.2, 2, 20 ng/ml). Gene expression of (A) VDR and (B) RXRα gene expression was quantified by qRT-PCR. Data are expressed as mean ± SEM and statistical analysis employed a 2-way ANOVA with Bonferroni post-tests with *p < 0.05, ****p < 0.0001.
TGF-β1 and 1,25(OH)2D3 Act Together to Upregulate VDR and RXRα Expression in CD8+ T Cells
To investigate how TGF-β1 modulates the response of CD8+ T cells to 1,25(OH)2D3, the effect of TGF-β1 on VDR and RXRα was assessed since the VDR-RXRα complex binds 1,25(OH)2D3 and is essential for subsequent vitamin D-mediated effects. Addition of 1,25(OH)2D3 alone to cultures of CD4+ or CD8+ T cells had no effect on the gene expression of either receptor. Inclusion of TGF-β1 at higher doses (2 and 20 ng/ml) significantly enhanced 1,25(OH)2D3-induced (100 nM) VDR in both CD4+ and CD8+ T cells (Figure 2A). Expression of RXRα was not significantly affected in CD4+ T cells, the combination of 1,25(OH)2D3 and TGF-β1 significantly enhanced RXRα expression in CD8+ T cell cultures (Figure 2B).
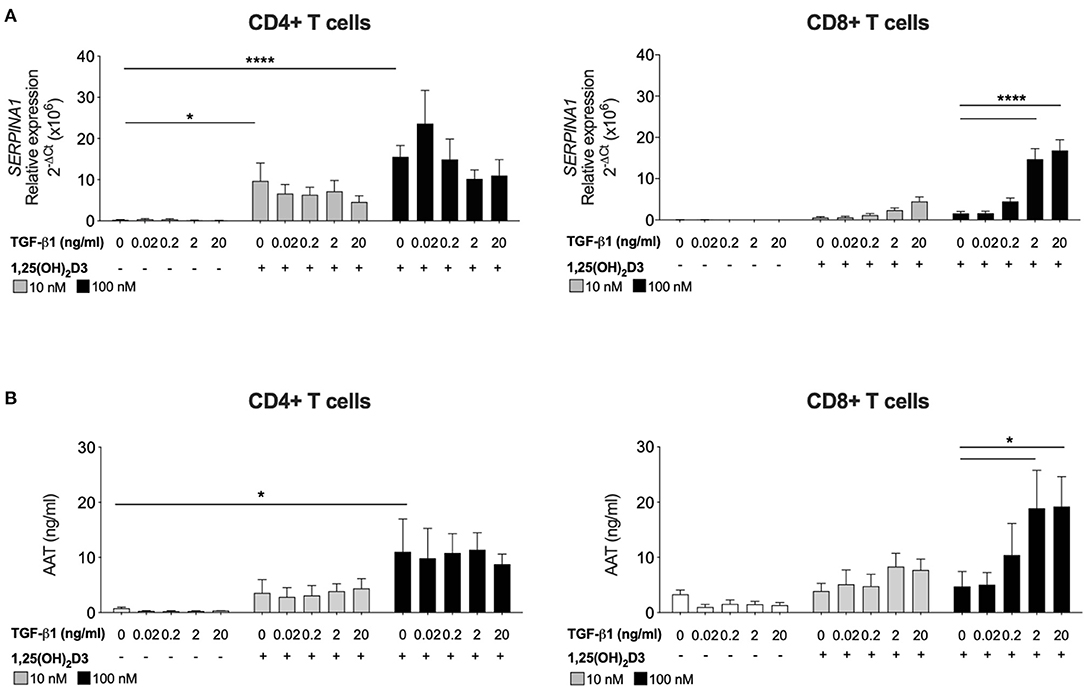
Figure 2. CD8+ T cells require exogenous TGF-β1 to enhance 1,25(OH)2D3-induced SERPINA1/AAT. CD4+ and CD8+ T cells (n = 6) were stimulated and cultured for 7 days in the absence or presence of 10 or 100 nM 1,25(OH)2D3, with or without TGF-β1 (0.02, 0.2, 2, 20 ng/ml). (A) Gene expression of SERPINA1 was quantified by qRT-PCR. (B) Culture supernatants were assessed for AAT protein secreted by ELISA. Data are expressed as mean ± SEM and statistical analysis employed a 2-way ANOVA with Bonferroni post-tests with *p < 0.05, ****p < 0.0001.
Requirement for Endogenous or Exogenous TGF-β in 1,25(OH)2D3-Mediated Regulation of SERPINA1
Since CD4+ T cells did not show the same requirement for exogenous TGF-β as CD8 + T cells for SERPINA1/AAT induction, the issue of whether this was due to their greater endogenous production of bioactive TGF-β in comparison to CD8+ T cells was examined. Bioactive TGF-β secretion was quantified in parallel cultures of CD4+ and CD8+ T cells, using mink lung epithelial cells that were transfected with a TGF-β target gene plasminogen activator inhibitor 1 and a luciferase assay. In the absence of 1,25(OH)2D3, there was no difference in bioactive TGF-β levels between CD4+ and CD8+ T cells at any time point. However, bioactive TGF-β was elevated in supernatants harvested from CD4+ T cells after 5 and 7 days of culture with 100 nM 1,25(OH)2D3. No effect was observed in CD8+ T cells (Figure 3A).
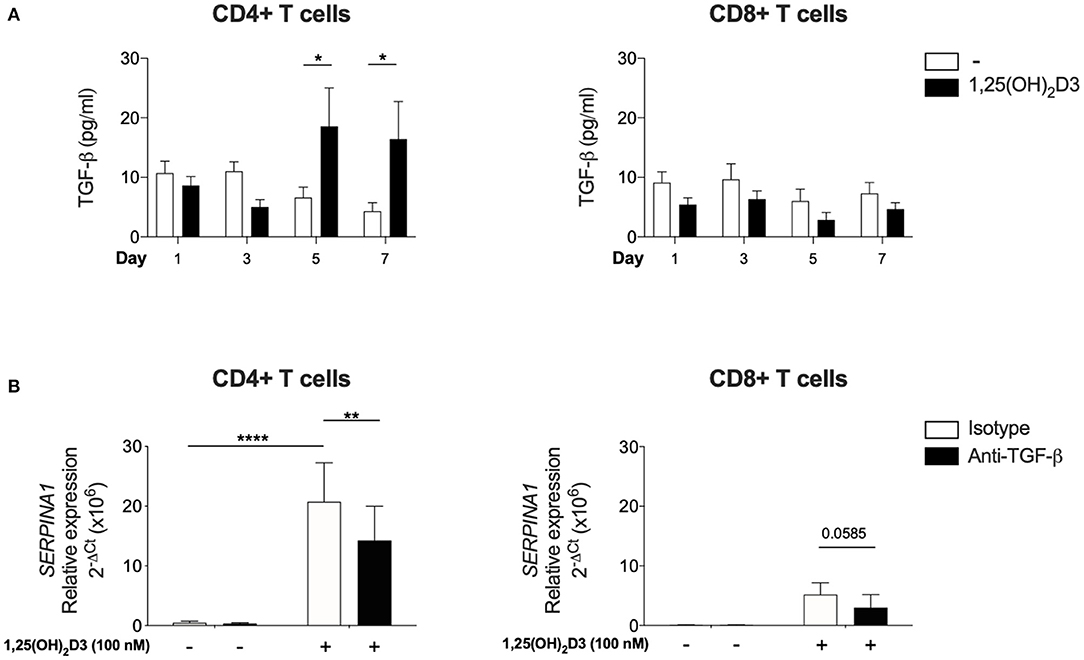
Figure 3. Significant upregulation of bioactive TGF-β is observed with 1,25(OH)2D3 treatment in CD4+ T cells and 1,25(OH)2D3 induction of SERPINA1/AAT is partially inhibited by anti-TGF-β. CD4+ and CD8+ T cells (n = 6) were stimulated and cultured in the absence or presence of 100 nM 1,25(OH)2D3. (A) Supernatant from indicated time points were harvested and TGF-β bioactivity was measured using transfected mink epithelial lung cells and luciferase activity (luminescence, as relative light unit corresponds to TGF-β bioactivity), and converted to pg/ml. (B) Cells were cultured with 100 nM 1,25(OH)2D3 in the presence of anti-TGF-β or an isotype control for 7 days. 1,25(OH)2D3-mediated induction of SERPINA1 expression was assessed by qRT-PCR. Data are expressed as mean ± SEM and statistical analysis employed a 2-way ANOVA with Bonferroni post-tests with *p < 0.05, **p < 0.01, ****p < 0.0001.
In a complementary approach, CD4+ and CD8+ T cells were cultured in the presence of neutralising TGF-β or isotype control antibodies to assess the role of endogenous, autocrine TGF-β in controlling SERPINA1 expression. Anti-TGF-β significantly down-regulated 1,25(OH)2D3-mediated SERPINA1 expression in CD4+ T cell cultures, but in parallel CD8+ T cell cultures, 1,25(OH)2D3 did not significantly increase SERPINA1, and neutralization of TGF-β did not markedly modify this response (Figure 3B). However, modulation of AAT protein expression by neutralization of TGF-β did not achieve statistical significance (data not shown). Together, these observations indicate that the 1,25(OH)2D3-mediated induction of SERPINA1 in both CD4 and CD8 T cells is at least partially TGF-β dependent. CD4+ T cells appear capable of autocrine priming with TGF-β whilst CD8+ T cells require exogenous addition of this co-factor.
Human T Cells Respond to 1,25(OH)2D3 but Not Its Circulating Precursor 25(OH)D3
The studies presented here and in our earlier publication (16) have assessed the capacity of active vitamin D (1,25(OH)2D3) to enhance SERPINA1/AAT production by T cells. Vitamin D circulates predominantly in the precursor form 25(OH)D3, which has a far longer half-life and is found at ~1,000-fold higher concentrations. CYP27B1, the Vitamin D 1-alpha-hydroxylase enzyme which converts 25(OH)D3 to 1,25(OH)2D3, is expressed by CD4+ T cells providing the potential for local modulation of vitamin D activity. However, the degree to which these cells are able to generate physiologically relevant concentrations of the active form remains controversial (36, 37). CD4+ and CD8+ T cells were therefore stimulated in the presence of 25(OH)D3 or 1,25(OH)2D3, with or without 2 ng/ml TGF-β. Upregulation of SERPINA1 was observed in the presence of 1,25(OH)2D3 in the CD4+ T cell cultures, with much lower levels seen in the CD8+ T cells. Neither cell type responded to 25(OH)D3 for upregulation of SERPINA1 (Figure 4A), suggesting that T cells may require 1,25(OH)2D3 generated locally by other cell types to upregulate SERPINA1 in vivo.
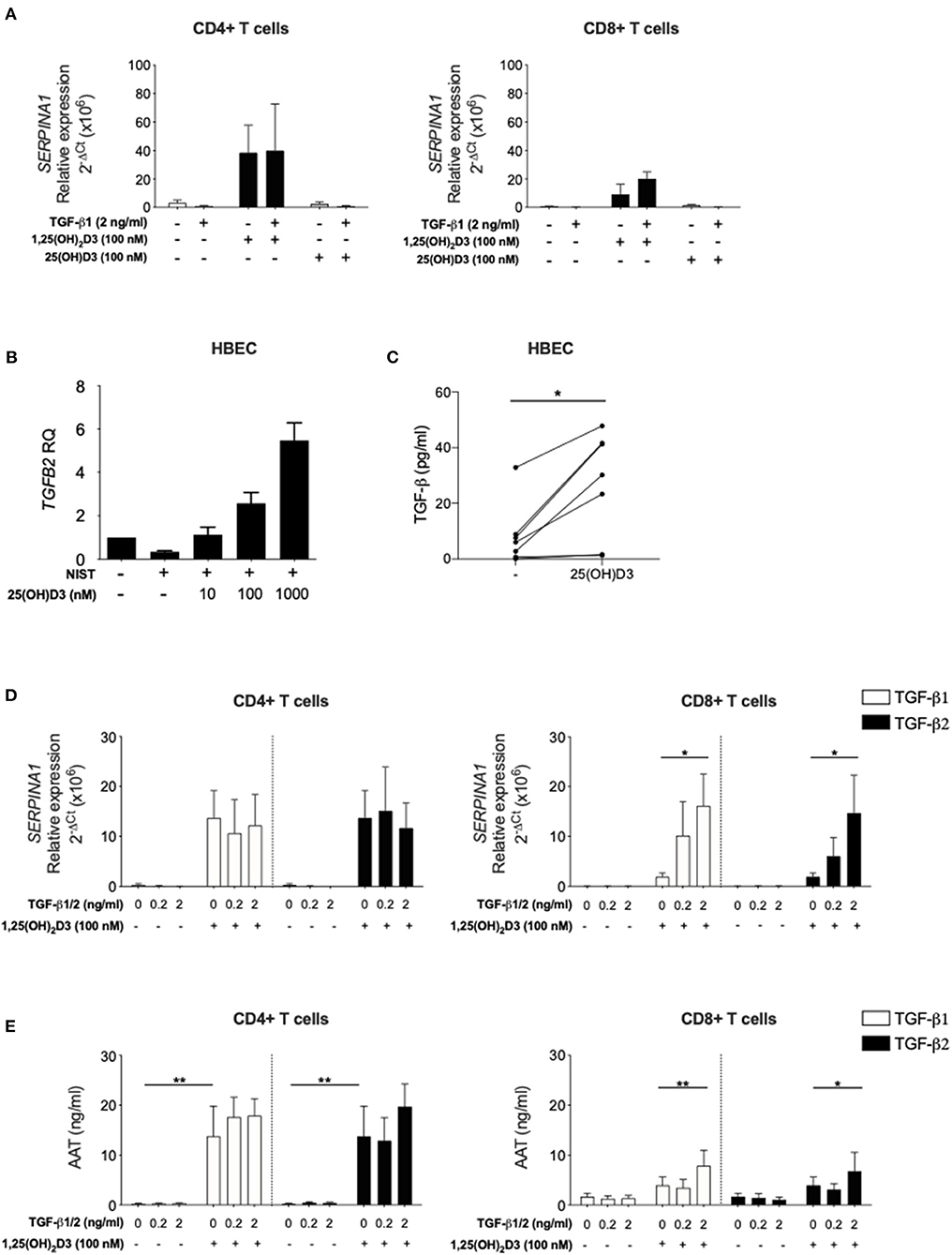
Figure 4. Inactive vitamin D does not induce SERPINA1 expression in T cells, but does induce production of bioactive TGF-β in HBECs. Both TGF-β isoforms upregulate 1,25(OH)2D3-mediated SERPINA1/AAT induction in CD8+ T cells. (A) CD4+ and CD8+ T cells were isolated from peripheral blood of healthy donors (n = 4) and cultured for 7 days in the absence or presence of either 100 nM 1,25(OH)2D3 or 100 nM 25(OH)D, with or without 2 ng/ml TGF-β1. Cell pellets were harvested and assessed for SERPINA1 expression. (B) Human bronchial epithelial cells (HBEC, n = 5) were analysed for TGFB2 gene expression following 24 h stimulation with NIST and different concentrations of 25(OH)D3 (10, 100, and 1,000 nM). (C) HBEC (n = 7) were assessed for their ability to produce bioactive TGF-β in response to 48 h 1 μM 25(OH)D3. Cells were cultured for 7 days in the presence or absence of 100 nM 1,25(OH)2D3 with or without TGF-β1 or TGF-β2 (0.2, 2 ng/ml). (D) Cell pellets were assessed for SERPINA1 gene expression (n = 3–6). (E) Culture supernatants were assessed for AAT protein secretion by ELISA (n = 6). Data are expressed as mean ± SEM and statistical analysis employed a 2-way ANOVA with Bonferroni post-tests with *p < 0.05, **p < 0.01.
Human Bronchial Epithelial Cells May Provide a Local Source of Active Vitamin D to Control T Cell Responses
Human bronchial epithelial cells (HBECs) are known to respond to both 25(OH)D3 and 1,25(OH)2D3 (32, 38) and are considered an important source of local mediators that activate immune cells in the airways (32, 39). In a recent study using transcriptional gene profiling we reported that human primary HBECs stimulated with 1,25(OH)2D3 increased TGFB2 gene expression (32). Here, this observation was extended to demonstrate that primary HBECs increase TGFB2 expression upon 25(OH)D3 stimulation in a concentration-dependent manner, when activated in vitro by exposure to NIST, a standard reference source of total urban particulate matter (SRM-1648a) which has been previously described (32) (Figure 4B). NIST was used as a relevant environmental stimulus of HBEC with previously observed effect on induction of inflammatory pathways (32). Increased bioactive TGF-β was evident at 48 h in HBEC culture supernatants in response to 1 μM 25(OH)D3 in culture (Figure 4C).
In order to support the concept that HBEC derived bioactive TGF-β may enable T cell responsiveness to 1,25(OH)2D3 for upregulation of SERPINA1/AAT, the capacity of TGF-β1 and TGF-β2 isoforms to enhance this response in CD8+ T cells was assessed. CD4+ and CD8+ T cells were stimulated in culture with 0 or 100 nM 1,25(OH)2D3 in the absence or presence of TGF-β1 or TGF-β2. Both TGF-β isoforms in the presence, but not the absence of 1,25(OH)2D3, significantly increased SERPINA1 expression and AAT production in CD8+ T cell cultures. As previously observed, CD4+ T cells responded to 1,25(OH)2D3 with no further effect upon addition of either TGF-β preparation (Figures 4D,E).
Correlation Between Serum Vitamin D and Bronchoalveolar Lavage AAT Concentrations in Children With Asthma
Evidence to support the relevance of these experimental observations was explored in 2 distinct patient populations (Figure 5). At least one independent study reports a link between serum vitamin D levels and AAT levels in autoimmune disease, in this case type 2 diabetes (40). We previously described a clinically well-defined cohort of children with asthma and control children undergoing diagnostic bronchoscopies for non-asthma related investigations, in which lower vitamin D levels were associated with worse asthma control and lung function (30). Using archived serum and bronchoalveolar lavage from this cohort here we observed a significant positive correlation between serum 25(OH)D3 levels and AAT in the bronchoalveolar lavage but not the serum in this paediatric severe asthma cohort (Figure 5A). These findings suggested that circulating vitamin D availability may contribute to control AAT synthesis locally in the airways, at least in part through effects on T cells.
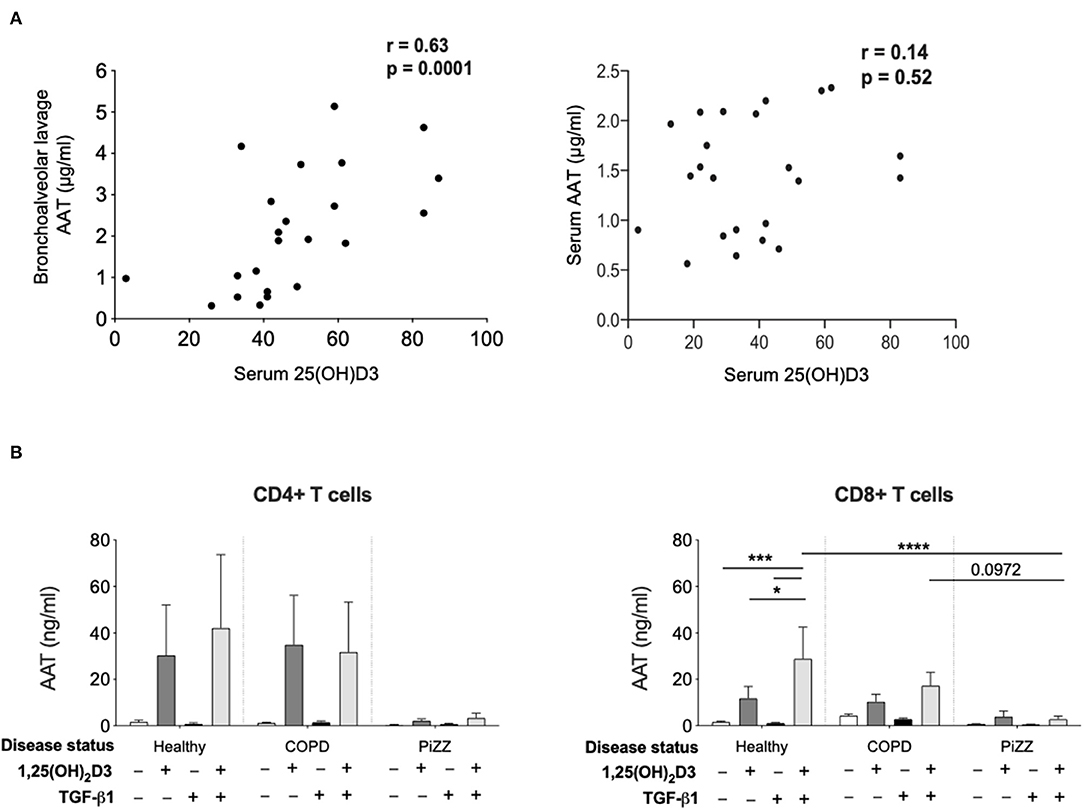
Figure 5. Patient studies to assess in vivo relevance. (A) Serum concentration of 25(OH)D3, as well as bronchoalveolar lavage (left) and serum (right) concentration of AAT were assessed in a paediatric severe asthma cohort and in paediatric controls undergoing bronchoscopy. Correlations were assessed using Spearman's rank correlation. (B) CD4+ and CD8+ T cells were isolated from the peripheral blood of healthy donors (CD4+ n = 6, CD8+ n = 12), non-AAT deficient COPD (n = 9) and PiZZ patients (CD4+ n = 6, CD8+ n = 20) and cultured for 7 days in the absence (–) or presence of 1,25(OH)2D3 (100 nM) with or without TGF-β1 (2 ng/ml) before culture supernatants were assessed for AAT protein secretion by ELISA. Data are expressed as mean ± SEM and statistical analysis employed a 2-way ANOVA with Bonferroni multiple comparison test with *p < 0.05, ***p < 0.001, ****p < 0.0001.
Studies in Adults at Risk of COPD
Hepatocytes synthesise very high levels (1–2 g per day) of AAT, which is secreted into the circulation (41). Patients who are homozygous for the Z variant mutation (PiZZ genotype) in SERPINA1, produce AAT protein carrying a Glu342Lys substitution. This causes it to misfold, polymerise and aggregate within the hepatocyte such that only around 15% of normal levels of AAT protein are secreted, resulting in a similar circulating deficiency (9). These individuals represent up to 95% of the clinically diagnosed cases of AAT deficiency and are at greatly increased risk of early onset COPD. Whilst the situation in hepatocytes appears well-characterised, whether other cell types that produce lower levels of AAT will retain similar proportions of the synthesised protein as intracellular aggregates and demonstrate similarly profound secretion defects in the PiZZ context is not known. We therefore compared the levels of AAT secretion by CD4+ and CD8+ T cells treated with 1,25(OH)2D3 in cohorts of healthy controls, patients with COPD, and PiZZ AAT deficient individuals (Figure 5B). Clinical parameters for each group are summarized in Table 1. The AAT response was impaired in PiZZ CD4+ and CD8+ T cells (with or without TGF-β1). Simultaneous addition of 1,25(OH)2D3 and TGF-β1 significantly enhanced AAT secretion in healthy CD8+ T cells, and COPD CD8+ T cells showed a similar, albeit reduced trend for AAT secretion. However, PiZZ CD8+ T cells failed to secrete AAT in response to any of the treatments. Similar response profiles were observed in CD4+ cells between cohorts, but with greater variability. Overall, these observations strongly support defective secretion of AAT in PiZZ T cells, in response to stimuli that are able to increase AAT in T cells from healthy donors, consistent with the nature of the PiZZ defect in AAT secretion from hepatocytes (9). This may contribute to impaired local immunomodulation around T cells in PiZZ AAT deficiency.
Discussion
A delicate balance between proteases and anti-proteases contributes to homeostasis in the lungs and several mediators, including vitamin D and AAT, contribute to this. This study highlights the following findings: (i) CD8+ T cells, in addition to CD4+ T cells, represent a cellular source for 1,25(OH)2D3-induced AAT in the airway microenvironment, (ii) TGF-β acts as a co-factor in this vitamin D controlled regulatory axis and (iii) this axis is defective in PiZZ patients with severe AAT deficiency. Based on this and our previous findings (16) we propose that T cell-derived AAT is likely to function as an intermediate for immunomodulatory roles of vitamin D in the airway (1, 5). The significant correlation between circulating levels of vitamin D with airway AAT levels in a pediatric cohort further supports a functional relationship between these two mediators in vivo. Our in vitro studies indicate this will be mediated by 1,25(OH)2D3 whilst implicating at least one further mediator upstream of the AAT response that we have not identified in our work to date. This work therefore provides an important starting point for further work to elucidate clear cascades of regulation and so better understand the relationship between vitamin D and AAT.
Hepatocytes are recognised as the major source of circulating AAT secreting 1–2 g/d into the circulation, however there is also good evidence for local synthesis, including in the airways (42, 43). The lower, but still significant levels of AAT secreted by T cells suggest that T cell derived AAT may act locally and have immunomodulatory functions, which have been reviewed elsewhere (44). In brief, AAT acts on structural and innate cells to increase anti-inflammatory mediators such as IL-1Ra, IL-10, to inhibit pro-inflammatory cytokine release, inhibit neutrophil chemotaxis and degranulation, and promote tolerogenic dendritic cells. In the adaptive arm, AAT increases the frequency of Foxp3 Treg cells and T cell synthesis of anti-inflammatory IL-10, whilst inhibiting pro-inflammatory mediators as well as B cell proliferation and autoantibody production. Many of these data arise from animal models where AAT is reported to control autoimmune disease, help to prevent transplant rejection and elastase-induced emphysema (45–48). Importantly, human intervention studies that build on these data are now emerging [reviewed by Song (49)]. Two recent examples report concomitant clinical and parallel immune readouts. AAT suppressed experimental graft vs. host disease (GVHD) by downmodulating inflammation, and AAT infusion was subsequently studied for treatment of steroid-resistant acute GVHD in humans. Magenau et al. reported a durable clinical response in refractory acute GVHD and that AAT administration appeared safe, with low rates of infection and with increasing ratios of T regulatory to effector T cells (50). Campos et al. (51) used double dose AAT infusion in AAT-deficient individuals to restore circulating AAT levels within the normal range, as well as to reduce serine protease activity and numerous inflammatory mediators in bronchoalveolar lavage. Additionally, findings from gene therapy studies demonstrate that low level secretion of AAT is sufficient to induce Treg responses in the local microenvironment, favouring toleration of the associated adeno-associated virus capsid (52).
Many of the immunoregulatory functions ascribed to AAT are also regulated by vitamin D. Observational studies and clinical trials indicate that vitamin D incrementally improves airway health in established chronic respiratory disease, especially in individuals who are profoundly vitamin D deficient (2, 53, 54). The capacity of vitamin D to induce antimicrobial functions, to enhance clinical and immune responses to corticosteroids, to promote epithelial barrier integrity as well as to augment multiple pathways linked to peripheral tolerance are all proposed to underpin these clinical effects (1, 5, 55). It seems probable that vitamin D and AAT demonstrate both common and independent immunoregulatory activities that contribute to airway health, however the interplay between these mediators requires further investigation.
We propose a potential role for vitamin D induced AAT synthesis by human T cells which is discussed in the context of local immune regulation in the airways. Circulating levels of 25-hydroxyvitamin D, the accepted measure of vitamin D status, correlated with AAT levels in the airways in a pediatric asthma cohort and matched non-asthmatic controls provides some support for this concept. However, it is highly unlikely that T cell-derived AAT alone accounts for this correlation. In our earlier publication we found no evidence that 1,25(OH)2D3 increased SERPINA1 mRNA and/or AAT protein levels in monocytes, respiratory epithelial cells or primary hepatocytes (16). However, the capacity of vitamin D to control other pathways that regulate protease:antiprotease balance may also be relevant. This could include the previously reported downregulation by vitamin D of MMP-9 (21) which is known to inactivate AAT (56), that may further enhance the correlation between vitamin D and AAT levels in the lung.
The ChIP-Atlas database suggests there are no VDR binding sites within SERPINA1 implying that indirect and/or non-binding effects of vitamin D may explain the association reported here. The current study does not define the mechanisms by which this may occur. However, links between circulating measures of vitamin D status and AAT levels reported here, in a study of type 2 diabetes patients (40), and suggested by a study in COVID-19 patients (57) support such an association.
Vitamin D has been shown to upregulate TGF-β levels both here and in independent studies, including in T cells and bronchial epithelial cells (32, 58). On the other hand, TGF-β has been shown to enhance VDR, with data showing that TGF-β1 increases VDR expression via SMAD3/4 in human granulosa-lutein cells (26). We speculate that the capacity of TGF-β to both independently increase AAT synthesis in hepatoma and bronchial epithelial cell lines (24, 25) as well as its capacity to enhance the response to vitamin D through increased expression of VDR and RXRα, as described here in T cells, might also contribute to the observed association between airway AAT and circulating vitamin D. Similar effects of TGF-β on VDR gene and protein expression have been reported elsewhere (26). These studies support co-operation between TGF-β1 and vitamin D to drive target gene expression. The interaction of vitamin D and TGF-β to induce an immunomodulatory response via increased local levels of AAT may contribute to the anti-inflammatory repertoire within the pleiotropic potential of TGF-β. Whether TGF-β acts to enhance the response to vitamin D by other cell types and/or other functions of interest remains to be investigated, but the latter at least is suggested by other studies in human T cells (29). As far as we are aware, whether vitamin D supplementation regulates local AAT levels in healthy or patient populations is still unknown but these data are awaited with interest.
PiZZ patients' hepatocytes synthesise normal levels of AAT mRNA and nascent polypeptide, but are unable to secrete signficant amounts of AAT into the circulation (15% of the normal circulating levels) and reduced levels are also evident in the airways (59). We previously demonstrated a loss of correlation of SERPINA1 mRNA levels with an immunomodulatory readout, IL-10 gene expression, in PiZZ patient CD4+ T cells compared with those from controls (16). We attributed this to defective secretion of Z variant AAT protein from these cells. In hepatocytes the sequestration of Z AAT into retained polymeric species is related to the high levels of translation of the aggregation-prone variant. Our data presented here, despite the limitation of comparatively low patient numbers, confirm significantly impaired secretion of AAT is also observed in PiZZ T cells, that generally express far lower levels of AAT protein than the hepatocytes, relative to those from healthy donors. This could not be overcome by vitamin D in the presence or absence of TGF-β.
AAT deficiency such as that associated with the PiZZ mutation, is linked to an inherited increase in the risk of COPD and severe emphysema. This finding led to the paradigm that an imbalance between antiproteases and neutrophil-derived proteases contributes to the pathogenesis of emphysema (10). However, aberrant immune activation in AAT deficiency linked to lung disease also exists, with increasing support for a parallel role of anti-inflammatory properties of AAT that are lost in AAT deficiency. For example Baraldo et al. describe adaptive immune activation in T and B lymphocytes together with a marked increase in lymphoid follicles as a prominent feature in AAT deficiency (60). Similarly AAT-deficient individuals demonstrate increased circulating levels of IL-17A (61) and increased pro-inflammatory mediator profiles in the airways (44, 51).
Bronchial epithelial cells line the airways and are amongst the first cells to be activated by exogenous stimuli such as microbes, particulate matter air pollution, allergens, and other inflammatory stimuli. They are thought to play a pivotal role in controlling immune responses in the airways through actions on dendritic cells and other cells (62). Taken together our data support the responsiveness of lung T cell and airway epithelial cells to 1,25(OH)2D3 as drivers of the correlation of systemic 25(OH)D3 with lung AAT levels observed in vivo. HBECs express CYP27B1 and therefore are able to convert precursor vitamin D to the bioactive 1,25(OH)2D3 (38). Our finding in HBECs further supports this by demonstrating increased bioactive TGF-β synthesis. We therefore propose a model of T cell-epithelial cell interactions in which bronchial epithelial cells may generate bioactive 1,25(OH)2D3 and TGF-β the latter potentially supplemented by bioactive TGF-β production in CD4+ T cells, to promote anti-inflammatory functions in T cells (Figure 6). We previously proposed a T cell-epithelial cell interaction whereby 1,25(OH)2D3 enhances the production of soluble ST2, the decoy receptor that antagonizes IL-33 signaling in HBECs (33). These findings together suggest a physiological supportive network between epithelial and T cells of potential benefit in the control of chronic airway disease.
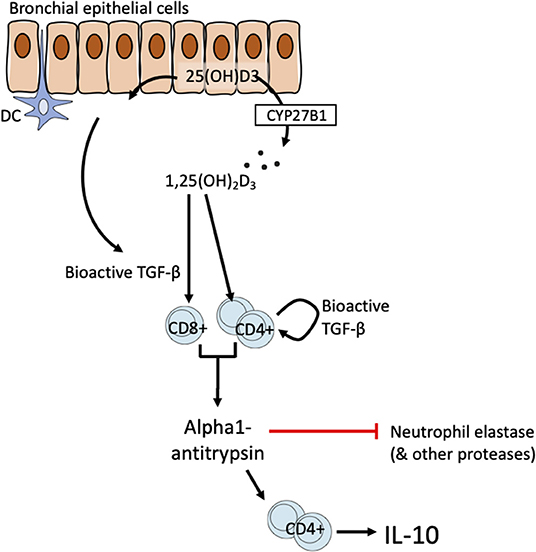
Figure 6. Schematic of proposed T cell-epithelial cell interaction on the vitamin D-TGF-β-AAT axis.
For simplicity the model proposed here highlights HBECs as a likely source of TGF-β. However, a wide range of cells in the airways may also contribute including fibroblasts, smooth muscle cells and inflammatory cells. Defining the most relevant celluar source of TGF-β, and this may not reflect a single cellular source, would likely require a conditional animal knockout model to address this. The precise cellular sources of TGF-β in the microenvironment are not central to the model. TGF-β (in all isoforms) is synthesized as an inactive precursor (a latency-associated protein bound to bioactive TGF-β) that is secreted into the matrix in complex with latent TGF-β binding protein. Activation e.g., mediated by integrins, stretch, and/or proteolysis is then required for the generation of the bioactive moiety (41).
Several limitations of our findings raise questions for future research. For example, whilst we demonstrate the capacity of vitamin D to increase AAT in human T cells, the lack of an obvious vitamin D response element within SERPINA1 suggests that intermediates in this pathway and the regulation by which this effect occurs remain to be clarified. It will also be useful to define the molecular pathways involved in local generation of the active vitamin D metabolite, 1,25(OH)2D3, and TGF-β required for AAT induction. Whilst a correlation between circulating 25-hydroxyvitamin D and airway AAT levels in a pediatric cohort exists, it seems unlikely that this is due solely to vitamin D induction of T cell-derived AAT and the contribution of other vitamin D regulated pathways e.g., downregulation of MMP-9, remain to be fully defined. Furthermore, these data would be complemented by evidence that vitamin D supplementation of vitamin D deficient individuals increases AAT levels in vivo. Finally, defining complementary, distinct and/or overlapping anti-inflammatory effects of vitamin D and AAT in patients will facilitate mechanistic and future clinical studies.
Conclusions and Future Research Directions of This Work
In summary, our study demonstrates that vitamin D increases AAT synthesis in human T cells, via a TGF-dependent mechanism, a pathway that is impaired in individuals with a genetic mutation (PiZZ) that results in decreased levels of AAT secretion. We propose that signals from adjacent airway cells, support vitamin D-mediated induction of AAT in situ in human lung. Some immunoregulatory properties of AAT and vitamin D overlap such as the induction of IL-10 and increase in Foxp3-Treg frequency, leading us to propose that the role of T cell-derived AAT is most likely to be immunomodulatory and may represent an intermediate of some immunodulatory functions of vitamin D. Notably, both oral vitamin D supplementation and AAT infusion are under investigation for their capacity to inhibit inflammation in a range of human immune-mediated pathologies. For AAT these data are still limited and only now emerging (49), whilst for vitamin D these data are complicated by enormous variability in part related to variable study designs (1, 4). Nevertheless, both appear to demonstrate good safety profiles and are well-tolerated, although differences in the cost of a common oral vitamin D supplement available over the counter vs. GMP-grade manufacture of AAT, are likely to be large. Comparison of biological and immune readouts of ongoing trials may provide insight into the most pertinent, overlapping and distinct effects of the two mediators. Sole AAT augmentation therapy is currently being studied, but the potential therapeutic implication of using vitamin D supplementation as an adjuvant/combination therapy to overcome any limitations faced by sole AAT therapy may be of future interest and precedent exists for the use of vitamin D as an adjunct treatment (31, 53).
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Ethics Statement
The studies involving human participants were reviewed and approved by Healthy donors (Human peripheral blood cell laboratory studies to investigate the role of immune and inflammatory pathways in respiratory disease by the local research ethics committee, REC reference: 14/LO/1699) were recruited and provided full written informed consent with full Research Ethics Committee approval. COPD patients without AAT deficiency (Chest clinic, Guy's Hospital, 14/LO/1699), and PiZZ-AAT deficient patients with or without COPD (Targeting dysfunctional mechanisms in alpha-1 antitrypsin deficiency, The Royal Free Alpha-1 clinic, REC reference: 13/LO/1085) were recruited and provided full written informed consent with full Research Ethics Committee approval. The study in a pediatric asthma cohort and control children undergoing diagnostic bronchoscopy for non-asthma related purposes was approved by the Royal Brompton and Harefield Research Ethics Committee (09/H07008/48) which has been previously described (14). Informed consent was obtained from parents and age-appropriate assent from children. Serum levels of 25-hydroxyvitamin D were measured as previously described (14). Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
Author Contributions
CH, BG, and Y-HC designed the study. BG recruited and screened participants. Y-HC performed functional experiments, supported by LR, SD, PP, and CC and supervised by CH and BG. SD, AG, and AB were responsible for studies in the pediatric cohort. CC, Y-HC, and CH wrote the manuscript, which all authors revised and approved. All authors contributed to the article and approved the submitted version.
Funding
CC, LR, and SD were all MRC-funded Ph.D. students through the MRC and Asthma UK Centre for Allergic Mechanisms in Asthma (G1000758). Y-HC, LR, and CC with CH were further supported through Pilot and Feasibility grants from the Alpha 1 Foundation. This research also received support from the National Institute for Health Research (NIHR) Clinical Research Facility at Guy's & St Thomas' NHS Foundation Trust and NIHR Biomedical Research Centre based at Guy's and St Thomas' NHS Foundation Trust and King's College London. BG was supported by the MRC (UK), and the NIHR Leicester Biomedical Research Centre-Respiratory.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We wish to dedicate this paper to our colleague and co-author, LR, who sadly passed away in November 2019. She is greatly missed as both a loyal colleague and friend and as a gifted scientist of great integrity.
We thank the families and blood donors for participation in this study. We thank Professor Daniel Rifkin, New York University, for sharing mink lung epithelial cell lines (MLECs). We thank Professor Sejal Saglani, Imperial College London and Professor John Hurst and his colleagues, Royal Free Hospital London, London Alpha-1 Antitrypsin Deficiency Service, for facilitating studies with paediatric and AAT-deficient patients, respectively.
References
1. Pfeffer PE, Hawrylowicz CM. Vitamin D in Asthma: mechanisms of action and considerations for clinical trials. Chest. (2018) 153:1229–39. doi: 10.1016/j.chest.2017.09.005
2. Martineau AR, James WY, Hooper RL, Barnes NC, Jolliffe DA, Greiller CL, et al. Vitamin D3 supplementation in patients with chronic obstructive pulmonary disease (ViDiCO): a multicentre, double-blind, randomised controlled trial. Lancet Respir Med. (2015) 3:120–30. doi: 10.1016/S2213-2600(14)70255-3
3. Martineau AR, Jolliffe DA, Hooper RL, Greenberg L, Aloia JF, Bergman P, et al. Vitamin D supplementation to prevent acute respiratory tract infections: systematic review and meta-analysis of individual participant data. BMJ. (2017) 356:i6583. doi: 10.1136/bmj.i6583
4. Pfeffer PE, Mann EH, Hornsby E, Chambers ES, Chen YH, Rice L, et al. Vitamin D influences asthmatic pathology through its action on diverse immunological pathways. Ann Am Thorac Soc. (2014) 11:S314–21. doi: 10.1513/AnnalsATS.201405-204AW
5. Bishop E, Ismailova A, Dimeloe SK, Hewison M, White JH. Vitamin D and immune regulation: antibacterial, antiviral, anti-inflammatory. JBMR Plus. (2020) 5:e10405. doi: 10.1002/jbm4.10405
6. Gooptu B, Ekeowa UI, Lomas DA. Mechanisms of emphysema in alpha1-antitrypsin deficiency: molecular and cellular insights. Eur Respir J. (2009) 34:475–88. doi: 10.1183/09031936.00096508
7. Huntington JA, Read RJ, Carrell RW. Structure of a serpin-protease complex shows inhibition by deformation. Nature. (2000) 407:923–6. doi: 10.1038/35038119
8. Faull SV, Elliston ELK, Gooptu B, Jagger AM, Aldobiyan I, Redzej A, et al. The structural basis for Z alpha1-antitrypsin polymerization in the liver. Sci Adv. (2020) 6:eabc1370. doi: 10.1126/sciadv.abc1370
9. Lomas DA, Evans DL, Finch JT, Carrell RW. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature. (1992) 357:605–7. doi: 10.1038/357605a0
10. Gooptu B, Dickens JA, Lomas DA. The molecular and cellular pathology of alpha(1)-antitrypsin deficiency. Trends Mol Med. (2014) 20:116–27. doi: 10.1016/j.molmed.2013.10.007
11. Mahadeva R, Atkinson C, Li Z, Stewart S, Janciauskiene S, Kelley DG, et al. Polymers of Z alpha1-antitrypsin co-localize with neutrophils in emphysematous alveoli and are chemotactic in vivo. Am J Pathol. (2005) 166:377–86. doi: 10.1016/S0002-9440(10)62261-4
12. Bergin DA, Hurley K, McElvaney NG, Reeves EP. Alpha-1 antitrypsin: a potent anti-inflammatory and potential novel therapeutic agent. Arch Immunol Ther Exp. (2012) 60:81–97. doi: 10.1007/s00005-012-0162-5
13. Abecassis A, Schuster R, Shahaf G, Ozeri E, Green R, Ochayon DE, et al. alpha1-antitrypsin increases interleukin-1 receptor antagonist production during pancreatic islet graft transplantation. Cell Mol Immunol. (2014) 11:377–86. doi: 10.1038/cmi.2014.17
14. Elshikha AS, Lu Y, Chen MJ, Akbar M, Zeumer L, Ritter A, et al. Alpha 1 antitrypsin inhibits dendritic cell activation and attenuates nephritis in a mouse model of lupus. PLoS ONE. (2016) 11:e0156583. doi: 10.1371/journal.pone.0156583
15. Ozeri E, Mizrahi M, Shahaf G, Lewis EC. alpha-1 antitrypsin promotes semimature, IL-10-producing and readily migrating tolerogenic dendritic cells. J Immunol. (2012) 189:146–53. doi: 10.4049/jimmunol.1101340
16. Dimeloe S, Rice LV, Chen H, Cheadle C, Raynes J, Pfeffer P, et al. Vitamin D (1,25(OH)2D3) induces alpha-1-antitrypsin synthesis by CD4(+) T cells, which is required for 1,25(OH)2D3-driven IL-10. J Steroid Biochem Mol Biol. (2019) 189:1–9. doi: 10.1016/j.jsbmb.2019.01.014
17. Jonigk D, Al-Omari M, Maegel L, Muller M, Izykowski N, Hong J, et al. Anti-inflammatory and immunomodulatory properties of alpha1-antitrypsin without inhibition of elastase. Proc Natl Acad Sci USA. (2013) 110:15007–12. doi: 10.1073/pnas.1309648110
18. Carroll TP, Greene CM, O'Connor CA, Nolan AM, O'Neill SJ, McElvaney NG. Evidence for unfolded protein response activation in monocytes from individuals with alpha-1 antitrypsin deficiency. J Immunol. (2010) 184:4538–46. doi: 10.4049/jimmunol.0802864
19. Cohen AB. Interrelationships between the human alveolar macrophage and alpha-1-antitrypsin. J Clin Invest. (1973) 52:2793–9. doi: 10.1172/JCI107475
20. Venembre P, Boutten A, Seta N, Dehoux MS, Crestani B, Aubier M, et al. Secretion of alpha 1-antitrypsin by alveolar epithelial cells. FEBS Lett. (1994) 346:171–4. doi: 10.1016/0014-5793(94)80695-0
21. Coussens A, Timms PM, Boucher BJ, Venton TR, Ashcroft AT, Skolimowska KH, et al. 1alpha,25-dihydroxyvitamin D3 inhibits matrix metalloproteinases induced by Mycobacterium tuberculosis infection. Immunology. (2009) 127:539–48. doi: 10.1111/j.1365-2567.2008.03024.x
22. Lacraz S, Isler P, Vey E, Welgus HG, Dayer JM. Direct contact between T lymphocytes and monocytes is a major pathway for induction of metalloproteinase expression. J Biol Chem. (1994) 269:22027–33. doi: 10.1016/S0021-9258(17)31750-7
23. Freeman CM, Han MK, Martinez FJ, Murray S, Liu LX, Chensue SW, et al. Cytotoxic potential of lung CD8(+) T cells increases with chronic obstructive pulmonary disease severity and with in vitro stimulation by IL-18 or IL-15. J Immunol. (2010) 184:6504–13. doi: 10.4049/jimmunol.1000006
24. Boutten A, Venembre P, Seta N, Hamelin J, Aubier M, Durand G, et al. Oncostatin M is a potent stimulator of alpha1-antitrypsin secretion in lung epithelial cells: modulation by transforming growth factor-beta and interferon-gamma. Am J Respir Cell Mol Biol. (1998) 18:511–20. doi: 10.1165/ajrcmb.18.4.2772
25. Mackiewicz A, Ganapathi MK, Schultz D, Brabenec A, Weinstein J, Kelley MF, et al. Transforming growth factor beta 1 regulates production of acute-phase proteins. Proc Natl Acad Sci USA. (1990) 87:1491–5. doi: 10.1073/pnas.87.4.1491
26. Wang F, Chang HM, Yi Y, Lin YM, Li H, Leung PCK. TGF-beta1 promotes vitamin D-induced prostaglandin E2 synthesis by upregulating vitamin D receptor expression in human granulosa-lutein cells. Am J Physiol Endocrinol Metab. (2020) 318:E710–22. doi: 10.1152/ajpendo.00361.2019
27. Schauber J, Dorschner RA, Coda AB, Buchau AS, Liu PT, Kiken D, et al. Injury enhances TLR2 function and antimicrobial peptide expression through a vitamin D-dependent mechanism. J Clin Invest. (2007) 117:803–11. doi: 10.1172/JCI30142
28. Chun RF, Liu PT, Modlin RL, Adams JS, Hewison M. Impact of vitamin D on immune function: lessons learned from genome-wide analysis. Front Physiol. (2014) 5:151. doi: 10.3389/fphys.2014.00151
29. Chambers ES, Suwannasaen D, Mann EH, Urry Z, Richards DF, Lertmemongkolchai G, et al. 1alpha,25-dihydroxyvitamin D3 in combination with transforming growth factor-beta increases the frequency of Foxp3(+) regulatory T cells through preferential expansion and usage of interleukin-2. Immunology. (2014) 143:52–60. doi: 10.1111/imm.12289
30. Gupta A, Sjoukes A, Richards D, Banya W, Hawrylowicz C, Bush A, et al. Relationship between serum vitamin D, disease severity, and airway remodeling in children with asthma. Am J Respir Crit Care Med. (2011) 184:1342–9. doi: 10.1164/rccm.201107-1239OC
31. Xystrakis E, Kusumakar S, Boswell S, Peek E, Urry Z, Richards DF, et al. Reversing the defective induction of IL-10-secreting regulatory T cells in glucocorticoid-resistant asthma patients. J Clin Invest. (2006) 116:146–55. doi: 10.1172/JCI21759
32. Pfeffer PE, Lu H, Mann EH, Chen YH, Ho TR, Cousins DJ, et al. Effects of vitamin D on inflammatory and oxidative stress responses of human bronchial epithelial cells exposed to particulate matter. PLoS ONE. (2018) 13:e0200040. doi: 10.1371/journal.pone.0200040
33. Pfeffer PE, Chen YH, Woszczek G, Matthews NC, Chevretton E, Gupta A, et al. Vitamin D enhances production of soluble ST2, inhibiting the action of IL-33. J Allergy Clin Immunol. (2015) 135:824–7.e3. doi: 10.1016/j.jaci.2014.09.044
34. Urry Z, Xystrakis E, Richards DF, McDonald J, Sattar Z, Cousins DJ, et al. Ligation of TLR9 induced on human IL-10-secreting Tregs by 1alpha,25-dihydroxyvitamin D3 abrogates regulatory function. J Clin Invest. (2009) 119:387–98. doi: 10.1172/JCI32354
35. Wakefield LM, Letterio JJ, Chen T, Danielpour D, Allison RS, Pai LH, et al. Transforming growth factor-beta1 circulates in normal human plasma and is unchanged in advanced metastatic breast cancer. Clin Cancer Res. (1995) 1:129–36.
36. Baeke F, Korf H, Overbergh L, van Etten E, Verstuyf A, Gysemans C, et al. Human T lymphocytes are direct targets of 1,25-dihydroxyvitamin D3 in the immune system. J Steroid Biochem Mol Biol. (2010) 121:221–7. doi: 10.1016/j.jsbmb.2010.03.037
37. Kongsbak M, von Essen MR, Boding L, Levring TB, Schjerling P, Lauritsen JP, et al. Vitamin D up-regulates the vitamin D receptor by protecting it from proteasomal degradation in human CD4+ T cells. PLoS ONE. (2014) 9:e96695. doi: 10.1371/journal.pone.0096695
38. Hansdottir S, Monick MM, Hinde SL, Lovan N, Look DC, Hunninghake GW. Respiratory epithelial cells convert inactive vitamin D to its active form: potential effects on host defense. J Immunol. (2008) 181:7090–9. doi: 10.4049/jimmunol.181.10.7090
39. Hammad H, Lambrecht BN. Dendritic cells and epithelial cells: linking innate and adaptive immunity in asthma. Nat Rev Immunol. (2008) 8:193–204. doi: 10.1038/nri2275
40. Lindley VM, Bhusal K, Huning L, Levine SN, Jain SK. Reduced 25(OH) Vitamin D association with lower alpha-1-antitrypsin blood levels in type 2 diabetic patients. J Am Coll Nutr. (2020) 40:98–103. doi: 10.1080/07315724.2020.1740629
41. Travis J, Salvesen GS. Human plasma proteinase inhibitors. Annu Rev Biochem. (1983) 52:655–709. doi: 10.1146/annurev.bi.52.070183.003255
42. Yuan ZA, Soprano KJ, Kueppers F. Alpha-1 antitrypsin response of stimulated alveolar macrophages. J Cell Biochem. (1992) 49:410–6. doi: 10.1002/jcb.240490411
43. Cichy J, Potempa J, Travis J. Biosynthesis of alpha1-proteinase inhibitor by human lung-derived epithelial cells. J Biol Chem. (1997) 272:8250–5. doi: 10.1074/jbc.272.13.8250
44. Ehlers MR. Immune-modulating effects of alpha-1 antitrypsin. Biol Chem. (2014) 395:1187–93. doi: 10.1515/hsz-2014-0161
45. Kurimoto E, Miyahara N, Kanehiro A, Waseda K, Taniguchi A, Ikeda G, et al. IL-17A is essential to the development of elastase-induced pulmonary inflammation and emphysema in mice. Respir Res. (2013) 14:5. doi: 10.1186/1465-9921-14-5
46. Lewis EC, Mizrahi M, Toledano M, Defelice N, Wright JL, Churg A, et al. alpha1-Antitrypsin monotherapy induces immune tolerance during islet allograft transplantation in mice. Proc Natl Acad Sci USA. (2008) 105:16236–41. doi: 10.1073/pnas.0807627105
47. Tawara I, Sun Y, Lewis EC, Toubai T, Evers R, Nieves E, et al. Alpha-1-antitrypsin monotherapy reduces graft-versus-host disease after experimental allogeneic bone marrow transplantation. Proc Natl Acad Sci USA. (2012) 109:564–9. doi: 10.1073/pnas.1117665109
48. Subramanian S, Shahaf G, Ozeri E, Miller LM, Vandenbark AA, Lewis EC, et al. Sustained expression of circulating human alpha-1 antitrypsin reduces inflammation, increases CD4+FoxP3+ Treg cell population and prevents signs of experimental autoimmune encephalomyelitis in mice. Metab Brain Dis. (2011) 26:107–13. doi: 10.1007/s11011-011-9239-9
49. Song S. Alpha-1 antitrypsin therapy for autoimmune disorders. Chronic Obstr Pulm Dis. (2018) 5:289–301. doi: 10.15326/jcopdf.5.4.2018.0131
50. Magenau JM, Goldstein SC, Peltier D, Soiffer RJ, Braun T, Pawarode A, et al. alpha1-Antitrypsin infusion for treatment of steroid-resistant acute graft-versus-host disease. Blood. (2018) 131:1372–9. doi: 10.1182/blood-2017-11-815746
51. Campos MA, Geraghty P, Holt G, Mendes E, Newby PR, Ma S, et al. The biological effects of double-dose alpha-1 antitrypsin augmentation therapy. A pilot clinical trial. Am J Respir Crit Care Med. (2019) 200:318–26. doi: 10.1164/rccm.201901-0010OC
52. Mueller C, Chulay JD, Trapnell BC, Humphries M, Carey B, Sandhaus RA, et al. Human treg responses allow sustained recombinant adeno-associated virus-mediated transgene expression. J Clin Invest. (2013) 123:5310–8. doi: 10.1172/JCI70314
53. Martineau AR, Cates CJ, Urashima M, Jensen M, Griffiths AP, Nurmatov U, et al. Vitamin D for the management of asthma. Cochrane Database Syst Rev. (2016) 9:CD011511. doi: 10.1002/14651858.CD011511.pub2
54. Martineau AR, Jolliffe DA, Greenberg L, Aloia JF, Bergman P, Dubnov-Raz G, et al. Vitamin D supplementation to prevent acute respiratory infections: individual participant data meta-analysis. Health Technol Assess. (2019) 23:1–44. doi: 10.3310/hta23020
55. Charoenngam N, Holick MF. Immunologic effects of vitamin D on human health and disease. Nutrients. (2020) 12:2097. doi: 10.3390/nu12072097
56. Liu Z, Zhou X, Shapiro SD, Shipley JM, Twining SS, Diaz LA, et al. The serpin alpha1-proteinase inhibitor is a critical substrate for gelatinase B/MMP-9 in vivo. Cell. (2000) 102:647–55. doi: 10.1016/S0092-8674(00)00087-8
57. Shimi G, Zand H. Association of alpha-1-antitrypsin deficiency with vitamin D status: who is most at risk of getting severe COVID-19? Inflamm Res. (2021) 70:375–7. doi: 10.1007/s00011-021-01456-z
58. Mahon BD, Gordon SA, Cruz J, Cosman F, Cantorna MT. Cytokine profile in patients with multiple sclerosis following vitamin D supplementation. J Neuroimmunol. (2003) 134:128–32. doi: 10.1016/S0165-5728(02)00396-X
59. Lomas DA, Mahadeva R. Alpha1-antitrypsin polymerization and the serpinopathies: pathobiology and prospects for therapy. J Clin Invest. (2002) 110:1585–90. doi: 10.1172/JCI0216782
60. Baraldo S, Turato G, Lunardi F, Bazzan E, Schiavon M, Ferrarotti I, et al. Immune activation in alpha1-antitrypsin-deficiency emphysema. beyond the protease-antiprotease paradigm. Am J Respir Crit Care Med. (2015) 191:402–9. doi: 10.1164/rccm.201403-0529OC
61. Pervakova MY, Mazing AV, Lapin SV, Tkachenko OY, Budkova AI, Surkova EA, et al. High serum level of IL-17 in patients with chronic obstructive pulmonary disease and the alpha-1 antitrypsin PiZ allele. Pulm Med. (2020) 2020:9738032. doi: 10.1155/2020/9738032
Keywords: vitamin D, T cell, alpha-1 antitrypsin (AAT), anti-inflammatory, HBEC, airway disease
Citation: Chen Y-H, Cheadle CE, Rice LV, Pfeffer PE, Dimeloe S, Gupta A, Bush A, Gooptu B and Hawrylowicz CM (2021) The Induction of Alpha-1 Antitrypsin by Vitamin D in Human T Cells Is TGF-β Dependent: A Proposed Anti-inflammatory Role in Airway Disease. Front. Nutr. 8:667203. doi: 10.3389/fnut.2021.667203
Received: 12 February 2021; Accepted: 09 July 2021;
Published: 12 August 2021.
Edited by:
Willem Van Eden, Utrecht University, NetherlandsReviewed by:
Michael Roth, University Hospital of Basel, SwitzerlandEmma Derbyshire, Nutritional Insight Limited, United Kingdom
Copyright © 2021 Chen, Cheadle, Rice, Pfeffer, Dimeloe, Gupta, Bush, Gooptu and Hawrylowicz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Catherine M. Hawrylowicz, Y2F0aGVyaW5lLmhhd3J5bG93aWN6QGtjbC5hYy51aw==
†ORCID: Catherine Hawrylowicz orcid.org/0000-0002-2337-7463
Yin-Huai Chen orcid.org/0000-0002-8890-691X
‡Present address: Paul Pfeffer, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, London, United Kingdom
Sarah Dimeloe, Institute of Immunology and Immunotherapy, Institute of Metabolism and Systems Research, College of Medical and Dental Sciences, University of Birmingham, Birmingham, United Kingdom
Atul Gupta, Department of Paediatric Respiratory Medicine, King's College hospital, London, United Kingdom
Andrew Bush, Department of Paediatric Respiratory Medicine, King's College Hospital, London, United Kingdom
§These authors have contributed equally to this work and share first authorship
¶Deceased