 Huan Zhu1,2
Huan Zhu1,2 Minna Yang1
Minna Yang1 Juan J. Loor3Ahmed Elolimy3Lingyan Li1
Juan J. Loor3Ahmed Elolimy3Lingyan Li1 Chuang Xu1Weidong Wang4Shuxin Yin1Yongli Qu1*
Chuang Xu1Weidong Wang4Shuxin Yin1Yongli Qu1*- 1Heilongjiang Provincial Key Laboratory of Prevention and Control of Bovine Diseases, College of Animal Science and Veterinary Medicine, Heilongjiang Bayi Agricultural University, Daqing, China
- 2College of Science, Heilongjiang Bayi Agricultural University, Daqing, China
- 3Mammalian NutriPhysioGenomics, Department of Animal Sciences and Division of Nutritional Sciences, University of Illinois, Urbana, IL, United States
- 4Heilongjiang Provincial Key Laboratory of Environmental Microbiology and Recycling of Argo-Waste in Cold Region, College of Life Science and Technology, Heilongjiang Bayi Agricultural University, Daqing, China
Hindgut microorganisms in newborn calves play an important role in the development of immunity and metabolism, and optimization of performance. However, knowledge of the extent to which microbiome colonization of the calf intestine is dependent on maternal characteristics is limited. In this study, placenta, umbilical cord, amniotic fluid, colostrum, cow feces, and calf meconium samples were collected from 6 Holstein cow-calf pairs. Microbial composition was analyzed by 16S rRNA gene high-throughput sequencing, and maternal transfer characteristics assessed using SourceTracker based on Gibbs sampling to fit the joint distribution using the mean proportions of each sample with meconium as the “sink” and other sample types as different “sources.” Alpha and beta diversity analyses revealed sample type-specific microbiome features: microbial composition of the placenta, umbilical cord, amniotic fluid, colostrum, and calf feces were similar, but differed from cow feces (p < 0.05). Compared with profiles of meconium vs. placenta, meconium vs. umbilical cord, and meconium vs. colostrum, differences between the meconium and amniotic fluid were most obvious. SourceTracker analysis revealed that 23.8 ± 2.21% of the meconium OTUs matched those of umbilical cord samples, followed by the meconium-placenta pair (15.57 ± 2.2%), meconium-colostrum pair (14.4 ± 1.9%), and meconium-amniotic fluid pair (11.2 ± 1.7%). The matching ratio between meconium and cow feces was the smallest (10.5 ± 1%). Overall, our data indicated that the composition of the meconium microflora was similar compared with multiple maternal sites including umbilical cord, placenta, colostrum, and amniotic fluid. The umbilical cord microflora seemed to contribute the most to colonization of the fecal microflora of calves. Bacteria with digestive functions such as cellulose decomposition and rumen fermentation were mainly transmitted during the maternal transfer process.
Introduction
The newborn hindgut microbiome plays important metabolic and nutritional functions (1), with one of its main roles being the development of the intestinal barrier (2) and the maturation of the innate immune system in early life (3). Available studies in humans, lambs, and foals have focused on understanding the hindgut microbiome when microbial colonization starts (4–6). However, few studies have focused on the calf microbiome (7–9).
Whether the fetal gut and the maternal uterus harbor a microbiome prior to delivery has long been controversial (10). For instance, a number of studies have reported that the uterus is sterile and that mammals are exposed to exogenous microorganisms for the first time only at birth (11, 12). However, in recent years, a microbiota has been detected in amniotic fluid, placenta, and umbilical cord of humans during pregnancy and hindgut of newborn calves prior to colostrum feeding (7, 13, 14). Consequently, these data have given rise to the hypothesis of vertical transmission of the microbiome from mothers to offspring (4, 6). Although the precise source of the newborn gut microbiome is not known with certainty (4), some scholars considered that the newborn meconium arises from intrauterine seeding (15–17). In bovine, Klein-Jöbstl et al. (18) and Alipour et al. (19) evaluated the possibility of colonization of the calf fecal microbiota, and concluded that it was maternally related.
SourceTracker script has been used to evaluate quantitatively the contribution of different maternal microbiome constituents in the meconium of the offspring (4). For instance, He et al. (4) working with humans explored maternal transfer characteristics by comparing the microbiome in meconium with that in various maternal sites and concluded that the microbiome in meconium was inoculated from amniotic fluid, feces, vaginal fluid, and saliva, with the amniotic fluid making the greatest contribution. It is unknown to what extent, if any, similar events occur in livestock species such as dairy calves.
We hypothesized that hindgut flora colonization of newborn dairy calves is strongly influenced by different maternal sources including placenta, umbilical cord, amniotic fluid, colostrum and feces. To address this objective, we screened 6 cow-calf pairs to analyze the contribution of the microbiome in the maternal feces, placenta, amniotic fluid, colostrum, and umbilical cord to the seeding of the meconium microbiome using 16S rRNA sequencing technology and SourceTracker software.
Materials and Methods
Animal care and experimental procedures were approved by the Animal Welfare and Ethics Committee of Heilongjiang Bayi Agriculture University, DaQing, China. Animal care and handling followed the guidelines of the regulations of the Administration of Affairs Concerning Experimental Animals (State Science and Technology Commission of China, 1988).
Experimental Animals
Fifteen 3- to 5-year-old pregnant Holstein cows were procured from a large-scale commercial dairy farm within a 2-week period in December 2020. Selection of cow-calf pairs followed published criteria (20): (1) single calf; (2) calving difficulty score <3; (3) dam's colostrum quality assessed by a bovine colostrometer (HT-113ATC, Hengan Electronic Technology Co., China) of >50 mg/mL of IgG; (4) dam produced at least 3.8 L of good-quality first colostrum; and (5) calf birth weight >36 kg. Related information about cow-calf is shown in Table 1. According to the selection criteria, a total of 6 Holstein cow-calf pairs (two heifers and four steers) were selected for this research. To reduce environmental, management, and seasonal bias, all cows were fed the same diets and calved in a group calving pen without assistance, and sampled by the same experienced veterinarian. Ingredient and nutrient composition of diets and forages are shown in Table 2.

Table 1. Screening information for cows and calves.
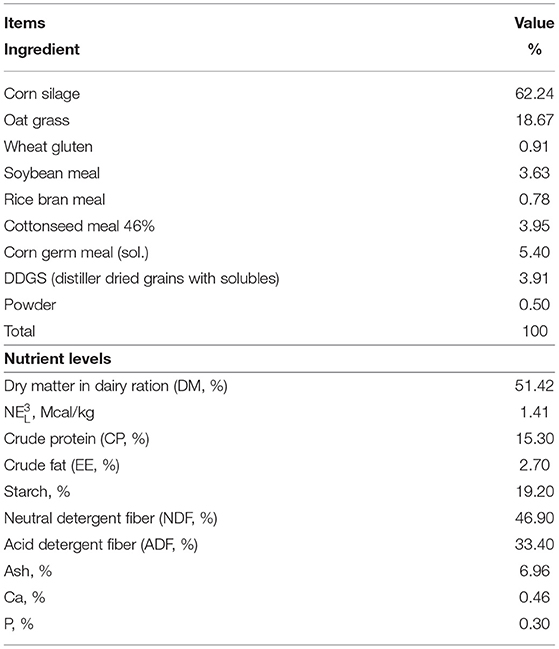
Table 2. Ingredients and chemical composition of dietary treatments of post-perinatal period dairy cow.
Sample Collection
During the second stage of labor when amniotic fluid vesicles were clearly visible and intact, a 60 mL sterile syringe was used to puncture these vesicles wearing sterile surgical gloves to harvest 50 mL of amniotic fluid that were subsequently deposited in two sterile tubes (6). Samples of placenta, umbilical cord, colostrum, cow feces, and meconium were collected aseptically within 1 h after delivery. After the natural delivery of the placenta, veterinarians wearing masks and sterile gloves collected two 1 cm3 slices from different regions of the placenta and umbilical cord using sterile scalpels (15), rinsed with physiologic saline. Once the placenta dropped on the ground, sampling stopped and the cow-calf were removed from the experimental animal. When sampling the umbilical cord, care was taken to avoid collecting at the site where the cord blood vessels pass through to prevent contamination of the sample by blood. During the colostrum collection process, the teats of the cows and surrounding areas were cleaned with sterile water, and then scrubbed with 75% ethanol by veterinarians wearing masks and sterile gloves. The first few drops of colostrum (~5 mL) were discarded, and the colostrum samples (50 mL) were collected into two sterile tubes (21). Considering the non-invasive nature of the sampling, cow feces and calf meconium were all collected from the rectum of the cow and calf, respectively, by veterinarians wearing sterile gloves. Meconium samples were collected before colostrum was fed to calves just after birth. Approximately 20 g were placed into each of two sterile tubes (19). All samples were stored temporarily in liquid nitrogen after collection and transported promptly to a −80°C freezer until analysis.
DNA Extraction
Frozen samples were thawed at room temperature and total DNA extracted from each 1.5 mL sample of colostrum and 0.5 g sample of placenta, umbilical cord, amniotic fluid, cow feces and meconium using a CTAB (modified cetyltrimethylammonium bromide) method (22). The purity and concentration of DNA were assessed by agarose gel electrophoresis. A suitable amount of sample DNA was taken in a centrifuge tube and diluted with sterile water to 1 ng/μL. After extraction, the integrity of the DNA was detected by 1% agarose gel electrophoresis, and the concentration and purity of DNA detected by a NanoDrop 2000 (Thermo Fisher Scientific, United States). The isolated DNA was kept at −20°C until processing.
16S rRNA Amplification and Sequencing
Diluted genomic DNA was used as the template, and the bacterial V4 hypervariable region of 16S rDNA was amplified by PCR using specific primers with barcodes, Phusion® High-Fidelity PCR Master Mix with GC Buffer from New England Biolabs, and a high-efficiency high-fidelity enzyme according to the selection of the sequencing region. The primer pair was 515F (5′-GTGCCAGCMGCCGCGGTAA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′). The PCR products were multiplexed in a single pool in equimolar amounts and then detected by electrophoresis using 2% agarose gel electrophoresis after full mixing. The target bands were recovered using a gel recovery kit provided by Qiagen, and a TruSeq® DNA PCR-Free Sample Preparation Kit was used for amplicon library preparation. All PCR reactions were carried out in 30 μL reactions, 0.2 μM of forward and reverse primers, and about 10 ng template DNA. Thermal cycling consisted of initial denaturation at 98°C for 1 min, followed by 30 cycles of denaturation at 98°C for 10 s, annealing at 50°C for 30 s, and elongation at 72°C for 30 s. Finally 72°C for 5 min. After quantification of the library with a Qubit 2.0 Fluorometer (Thermo Fisher Scientific Inc.) and quantitative PCR, sequencing was conducted using a NovaSeq6000 platform (Illumina, San Diego, CA, United States).
Sequence Analyses
According to the barcode sequence and PCR amplification primer sequence, each set of sample data was separated from the accessory data. After the barcode and primer sequences were trimmed, FLASH software (V1.2.7) (23) was used to assemble reads that were barcode and primer free to obtain the raw tags (24). QIIME software (V1.9.1) (25) was used to filter out low-quality tags, detect sequences by comparison with the species annotation database, and remove chimeras. Lastly, the effective tags were retained for further analysis (26). Uparse software (V7.0.1001) was used to cluster all tags effectively to operational taxonomic units (OTUs) based on 97% identity of the sequences (27). The Mothur method and SSUrRNA database of SILVA132 (28, 29) were used to select and annotate the representative OTUs with the highest frequencies of occurrence for taxonomic information. Alpha and beta diversity were analyzed with QIIME software (V1.9.1). All graphs were drawn with R software (V4.0.3). SourceTracker software was used to predict the likely origin of the meconium microbiome using the maternal microbiome communities as potential “sources” and the meconium microbiome communities as “sink.” LEfSe software was used to perform LDA effect size (LEfSe) analysis, and the default LDA Score filter value was 4.
Statistical Analyses
Data regarding composition of different samples were all analyzed for statistical significance via R software (V4.0.3). Differences between two groups were analyzed using Wilcoxon tests, and Tukey's test and the Wilcoxon test were selected if there were differences among more than two groups, and the confidence level was 0.05. Principal coordinate analysis and Permutational multivariate analysis of variance (PERMANOVA) were performed based on the weighted and unweighted UniFrac distances to evaluate the structural difference in the microbiota between different sample groups.
Results
Alpha Diversity of the Microbiome Community in Meconium and Maternal Samples
Thirty-six examined samples were used as input for NovaSeq6000 to generate 2,260,449 high-quality sequencing reads at the genus level. Shannon, inverse Simpson, and Chao 1 estimator values for genera are also shown in Supplementary Table 1. The Shannon diversity curves leveled off, suggesting that the sequencing depth was enough to capture representative microbial diversity (Figure 1). The Shannon diversity index values varied in different groups (colostrum, 6.93 ± 2.34; meconium, 5.34 ± 2.19; cow feces, 7.76 ± 0.24; umbilical cord, 7.49 ± 0.62; placenta, 8.17 ± 1.29; amniotic fluid, 8.03 ± 1.77). Pairwise comparison by Wilcoxon test on Shannon diversity indexes for meconium and maternal parts (P < 0.05; Table 3). The coverage depth ranking is shown by rank–abundance curves; the OTU curve represented higher microbial diversity and richness (4).
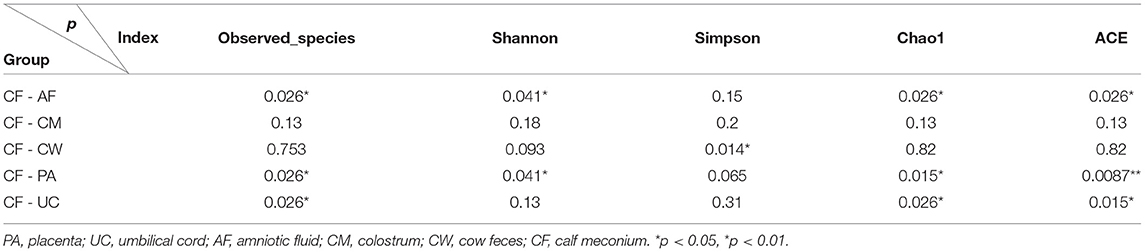
Table 3. Wilcox test of alpha diversity index for meconium and maternal parts.
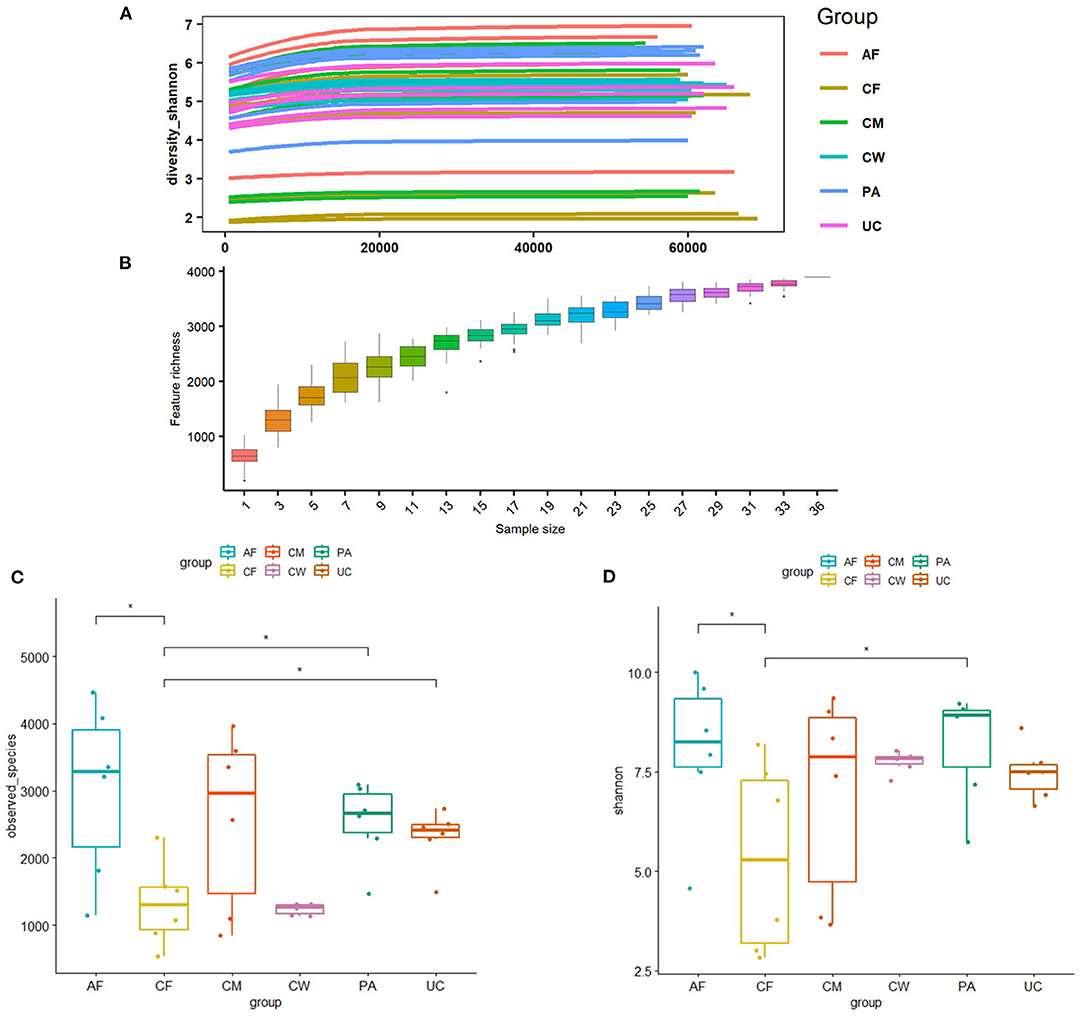
Figure 1. (A) Shannon diversity sparse curves, (B) box plot dilution curves, (C) box plot of the Shannon diversity index, and (D) observed characteristic index of all samples. AF, cow amniotic fluid; CF, calf meconium; CM, cow colostrum; CW, cow feces; PA, cow placenta; UC, cow umbilical cord.
Beta Diversity of the Microbiome Community in Calf Meconium and Maternal Samples
Differences in the microbiome structure among the meconium and different maternal samples were evaluated by PCoA [permutational multivariate analysis of variance (PERMANOVA) by Adonis (Table 4)] and Bray-Curtis dissimilarity (Figure 2). The maternal feces clustered distinctly on the weighted and unweighted UniFrac. The weighted and unweighted UniFrac distance score 3D plot both showed that the meconium samples obviously clustered together (Figures 2A,B). In the weighted bray_curtis_dm matrix, the intergroup distance between meconium-placenta, meconium-umbilical cord, meconium-amniotic fluid, and meconium-colostrum were lower (0.33–0.46), and those between meconium-cow feces were higher (0.74). The differences between cow feces and other groups of samples were also higher (0.62–0.88, Figure 2C).
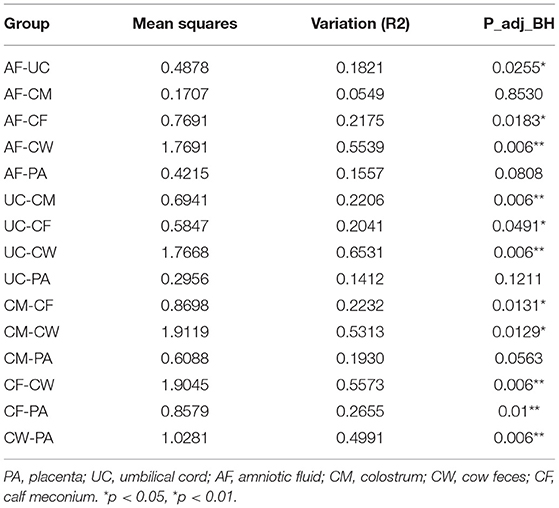
Table 4. PERMANOVA assessment of differences in bacterial community structure between two groups by Adonis.
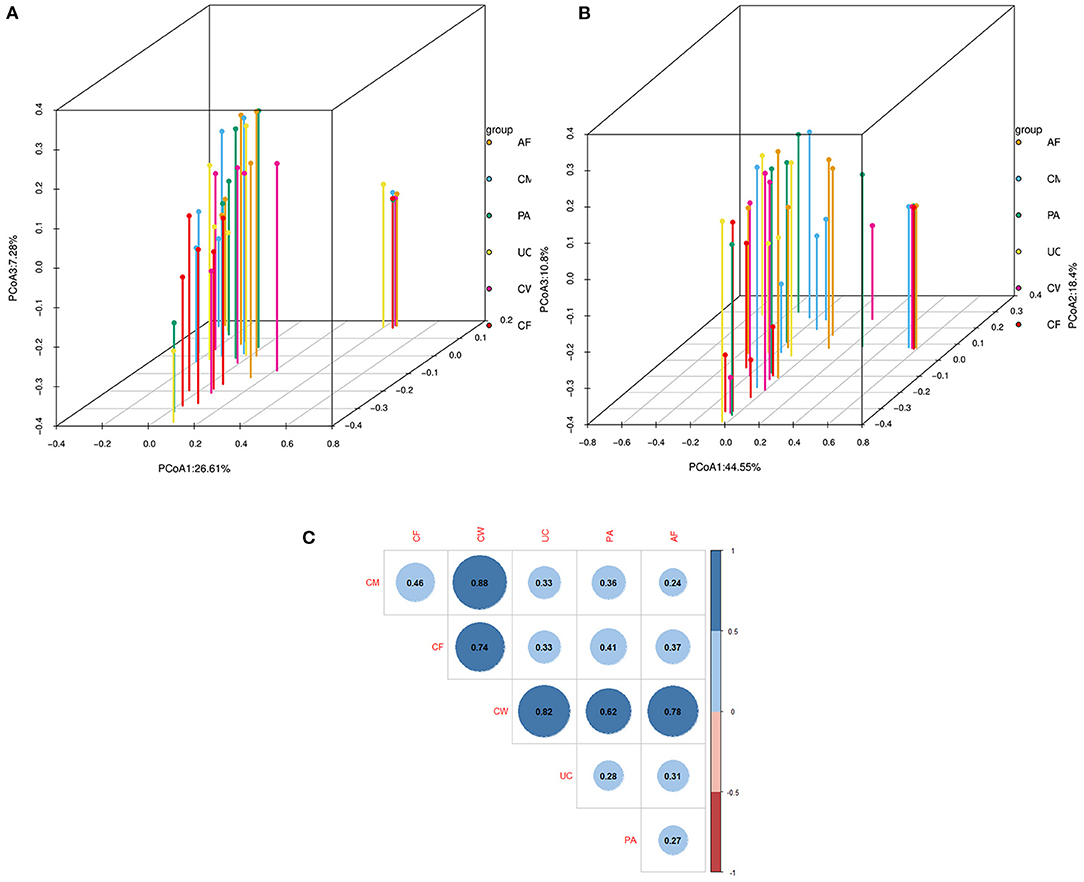
Figure 2. Dissimilarity-based multivariate analyses of microbiome communities of different sample types. Score 3D plots of principal coordinates analysis (PCoA) of different microbiome communities based on (A) weighted and (B) unweighted UniFrac distances. (C) Bray-Curtis dissimilarity matrix calculated based on microbial abundance patterns of operational taxonomic units (OTUs) of different sample types (Blue: positive, red: negative. The larger the absolute value is, the larger the area of the circle). The smaller the value of different degree indexes is, the higher the similarity (i.e., the difference between samples). AF, cow amniotic fluid; CF, calf meconium; CM, cow colostrum; CW, cow feces; PA, cow placenta; UC, cow umbilical cord.
Composition and Difference Analysis of the Microbiome Community in Meconium and Maternal Samples
The relative abundance and clustering characteristics of bacteria at the phylum, family, and genus levels from different sample types are shown in Figure 3. At the phylum level, the relative abundance of Proteobacteria in six parts, Firmicutes in five parts except colostrum, Bacteroidetes in amniotic fluid, placenta and cow feces were all >10%, and Firmicutes in colostrum and Bacteroidetes in colostrum, umbilical cord and calf feces were all close to 10, 9.8, 9.86, 9.76 and 8.94%, respectively (Figure 3A). The top represented bacterial families identified in the placenta, umbilical cord, and amniotic fluid were Pseudomonas, Moraxella, and Ruminococcus (Figure 3B). The colostrum contained mainly Burkholderiaceae, Caulobacteraceae, and Pseudomonadaceae families, and the meconium was dominated by Halomonadaceae, Moraxellaceae, and Pseudomonadaceae families. Lastly, cow feces contained mainly Ruminococcaceae, Rikenellaceae, Lachnospiraceae, and Bacteroidetes families. The microbial composition was dominated by Bacteroidetes, Brevundimonas, Halomonas, Limnobacter, Pseudomonas, and Psychrobacter.
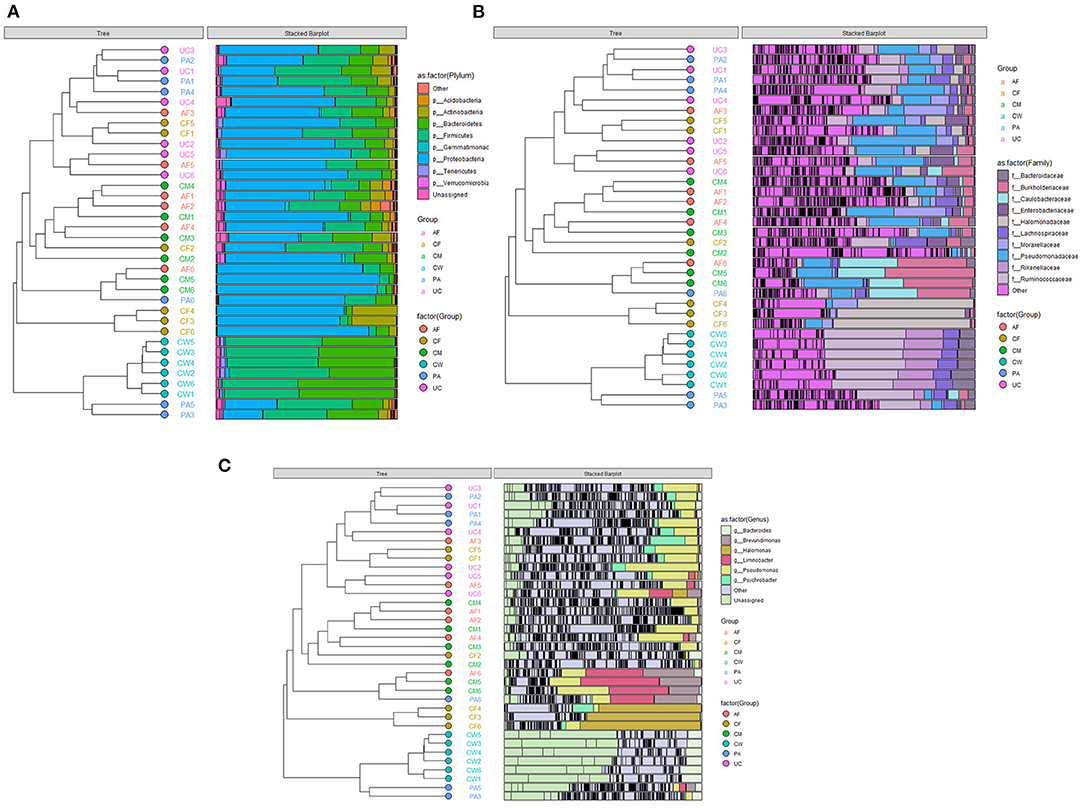
Figure 3. Clustering graph based on Bray-Curtis distance and stacked bar charts showing the microbiome compositions of the six types of samples at the (A) phylum level, (B) family level, and (C) genus level. AF, amniotic fluid; CF, calf meconium; CM, colostrum; CW, cow feces; PA, placenta; UC, umbilical cord.
Sample type-specific OTUs (detected exclusively in one sample type) were identified in all sample groups at the genus level (Figure 4). There were 26, 22, 22, 21, 20, and 4 type-specific OTUs in the placenta, colostrum, amniotic fluid, umbilical cord, meconium, and cow feces, respectively, and 170 OTUs shared by all parts were found (Figure 4). OTUs found exclusively in one type of sample at genus level are shown in Supplementary Table 3.
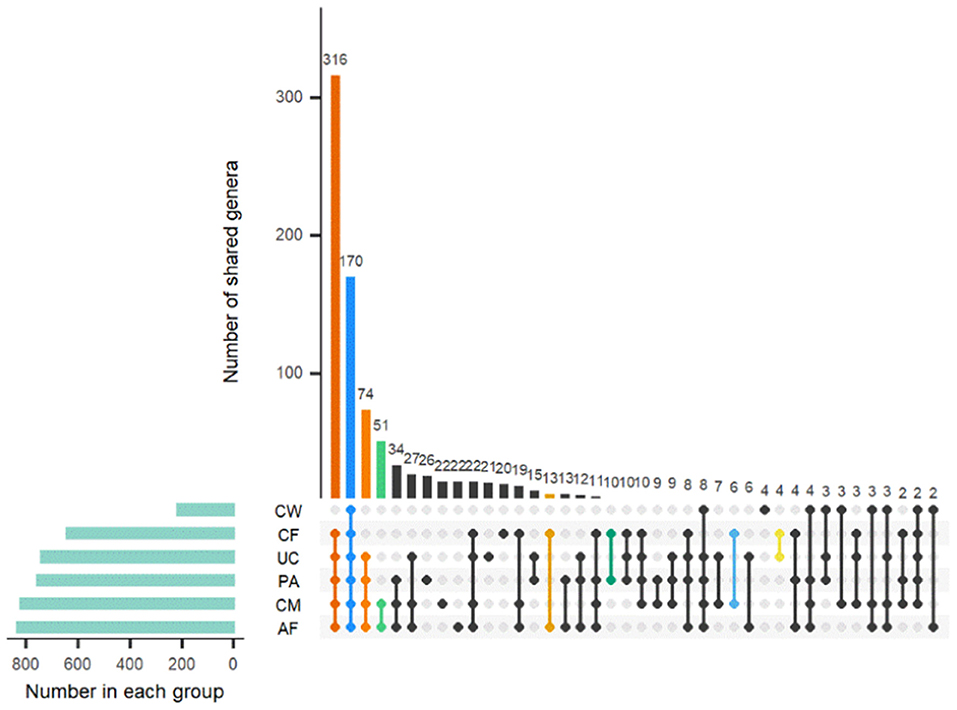
Figure 4. Distribution of OTUs shared among different sample types. AF, amniotic fluid; CF, calf meconium; CM, colostrum; CW, cow feces; PA, placenta; UC, umbilical cord.
LEfSe analysis identified biomarkers with statistically significant differences among different sample types, which can be represented by the LDA score. As shown in Figure 5, 42 microorganisms at different taxonomic levels had LDA scores > 4. The microflora constituents with significant differences in abundance (largest LDA score) between the meconium and other sample types were Gammaproteobacteria, Oceanospirillales, Halomonadaceae, Halomonas, Actinobacteria, unidentified_Actinobacteria, Corynebacteriales, Dietziaceae, Dietzia, unidentified_Enterobacteriaceae, Alteromonadales, Idiomarinaceae, and Aliidiomarina, and Gammaproteobacteria.
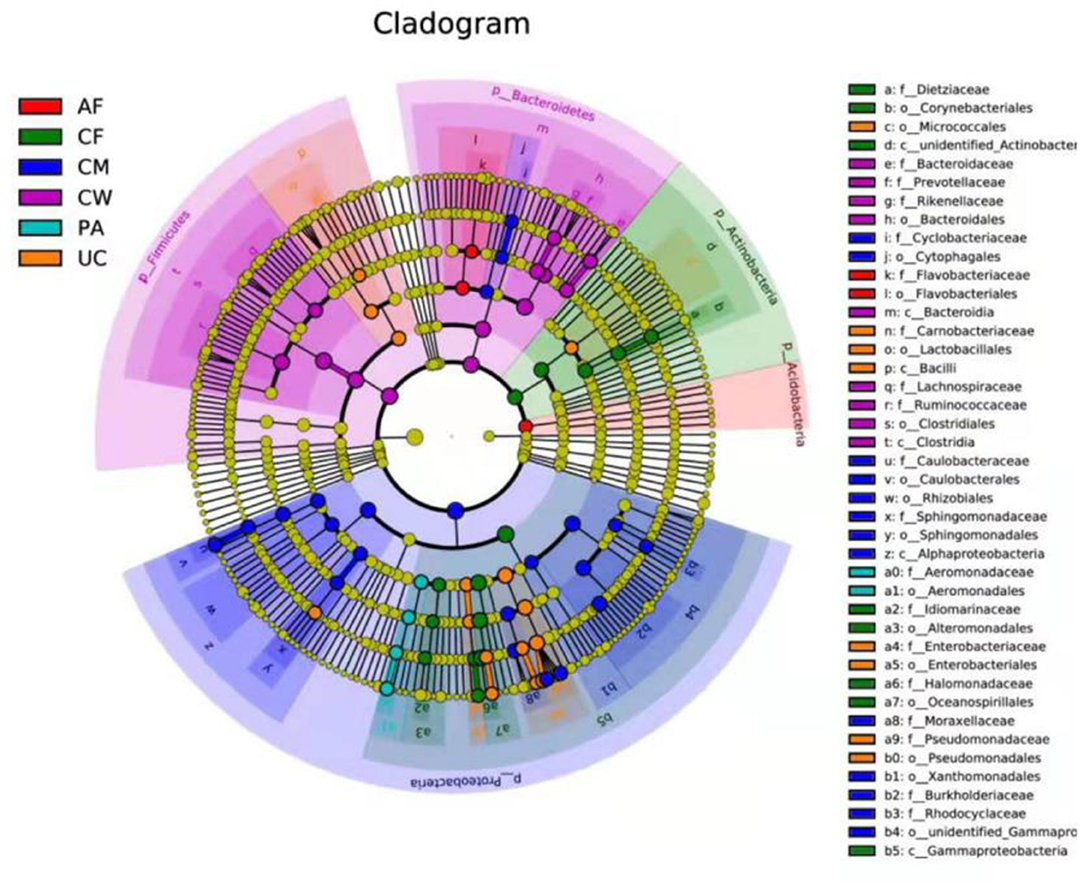
Figure 5. LEfSe-derived phylogenetic trees of the microbiome in all different sample types. AF, amniotic fluid; CF, calf meconium; CM, colostrum; CW, cow feces; PA, placenta; UC, umbilical cord.
Source Tracing Analysis of the Microbiome Community in the Meconium and Different Maternal Sample Types
Overall Source Tracing Analysis of the Microbiome Community in the Meconium and Different Maternal Samples
SourceTracker software predicted the source of microbial communities in the input sample set, with meconium as the “sink” and the other sample types as different “sources.” The matching ratio of the meconium to other sample types was ordered from high to low for the umbilical cord (23.8 ± 2.21%), placenta (15.57 ± 2.2%), colostrum (14.4 ± 1.9%), amniotic fluid (11.2 ± 1.7%), and cow feces (10.5 ± 1%) (Figure 6A).
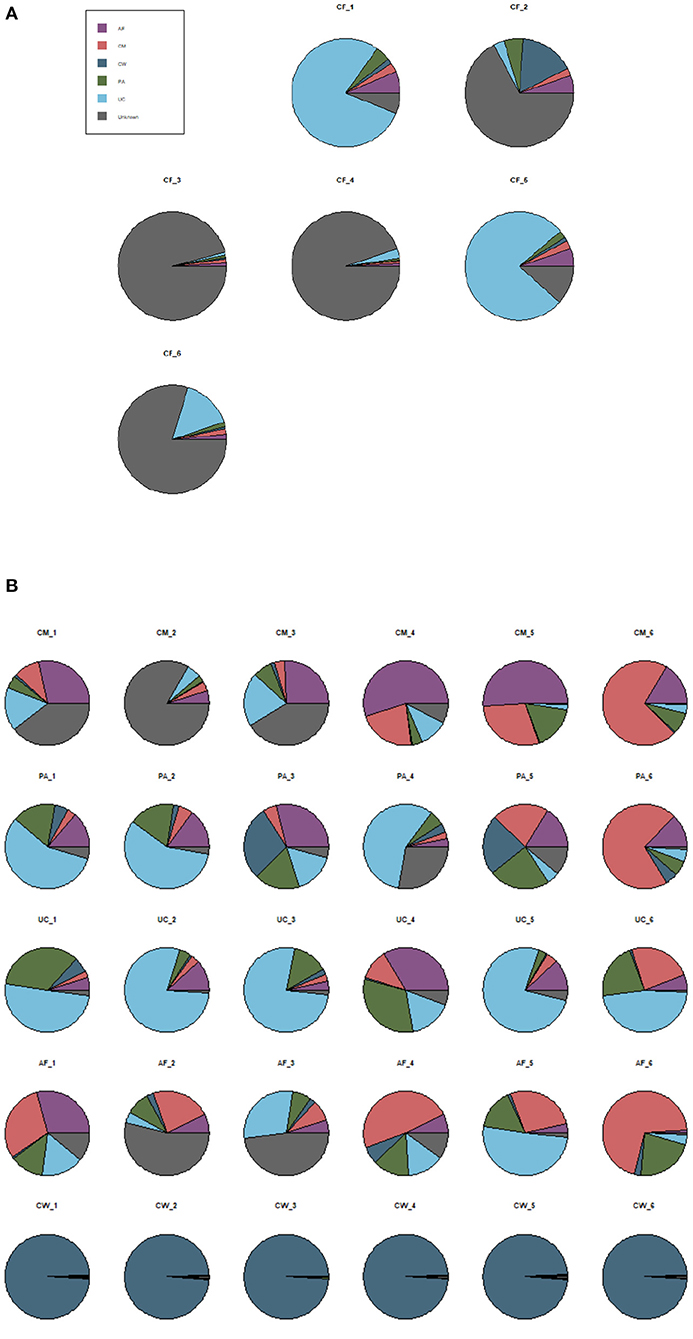
Figure 6. SourceTracker proportion estimates for a subset of “sink” samples and different “source” samples. (A) The matching ratio of microorganisms between meconium and different maternal sample types. (B) The matching ratio among the five “source” environments. AF, amniotic fluid; CF, calf meconium; CM, colostrum; CW, cow feces; PA, placenta; UC, umbilical cord; Unknown, unknown parts.
SourceTracker software was also used to compare the microorganisms in the five sample types as “sources.” Figure 6B shows that, compared with the other sample types (the autologous microorganism structural characteristics were similar to those in other sample types), the specificities were not obvious in the colostrum, placenta, umbilical cord, and amniotic fluid. However, the specificity between the cow feces and the other sample types was significant.
Source Tracing Analysis of the Microbial Community in the Meconium and Different Maternal Sample Types at the Phylum and Genus Levels
SourceTracker software was used to carry out traceability analysis for each cow-calf pair at the phylum and genus levels according to the different floras. This allowed further exploration of the matching ratio of dominant microbiome constituents between the meconium and different maternal sample types. The flora constituents that appeared in the meconium samples of the six calves were selected to ensure the significance of traceability analysis.
After screening, a total of 11 phyla, Cyanobacteria, Deinococcus-Thermus, Verrucomicrobia, Spirochaetes, Tenericutes, Acidobacteria, Proteobacteria, Firmicutes, Bacteroidetes, Actinobacteria, and unidentified_Bacteria, were observed in the maternal transmission process at the phylum level. At the genus level, there were 118 types of major genera in each maternal sample type during the maternal transmission process, which were distributed in Actinobacteria (15 genera), Bacteroidetes (15 genera), Firmicutes (39 genera), Proteobacteria (44 genera), Cyanobacteria (1 genus), Deinococcus-Thermus (1 genus), Verrucomicrobia (1 genus), and unidentified_Bacteria (2 genera). Figure 7 presents the main phyla and genera observed during maternal transmission, and the detailed data can be found in Table 5.
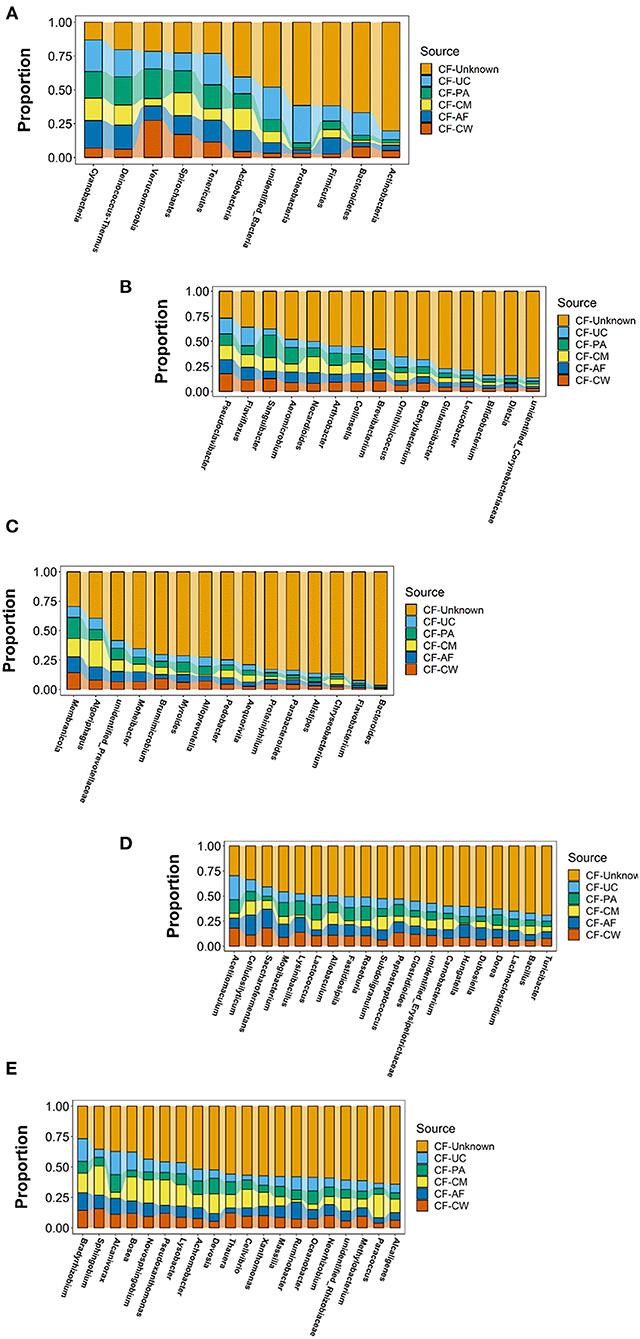
Figure 7. Estimation of the matching ratio of meconium constituents to the main genera in each maternal sample type during maternal transmission. (A) Estimation of the matching proportion of the major microbiome constituents at the phylum level. (B) Estimation of the matching proportion of major microbiome constituents in Actinobacteria. (C) Estimation of the matching proportion of major microbiome constituents in Bacteroidetes. (D) Estimation of the matching proportion of major microbiome constituents in Firmicutes (selection of matching proportion in the top 20). (E) Estimation of the matching proportion of major microbiome constituents in Proteobacteria (selection of matching proportion in the top 20). AF, amniotic fluid; CF, calf meconium; CM, colostrum; CW, cow feces; PA, placenta; UC, umbilical cord; Unknown, unknown parts.
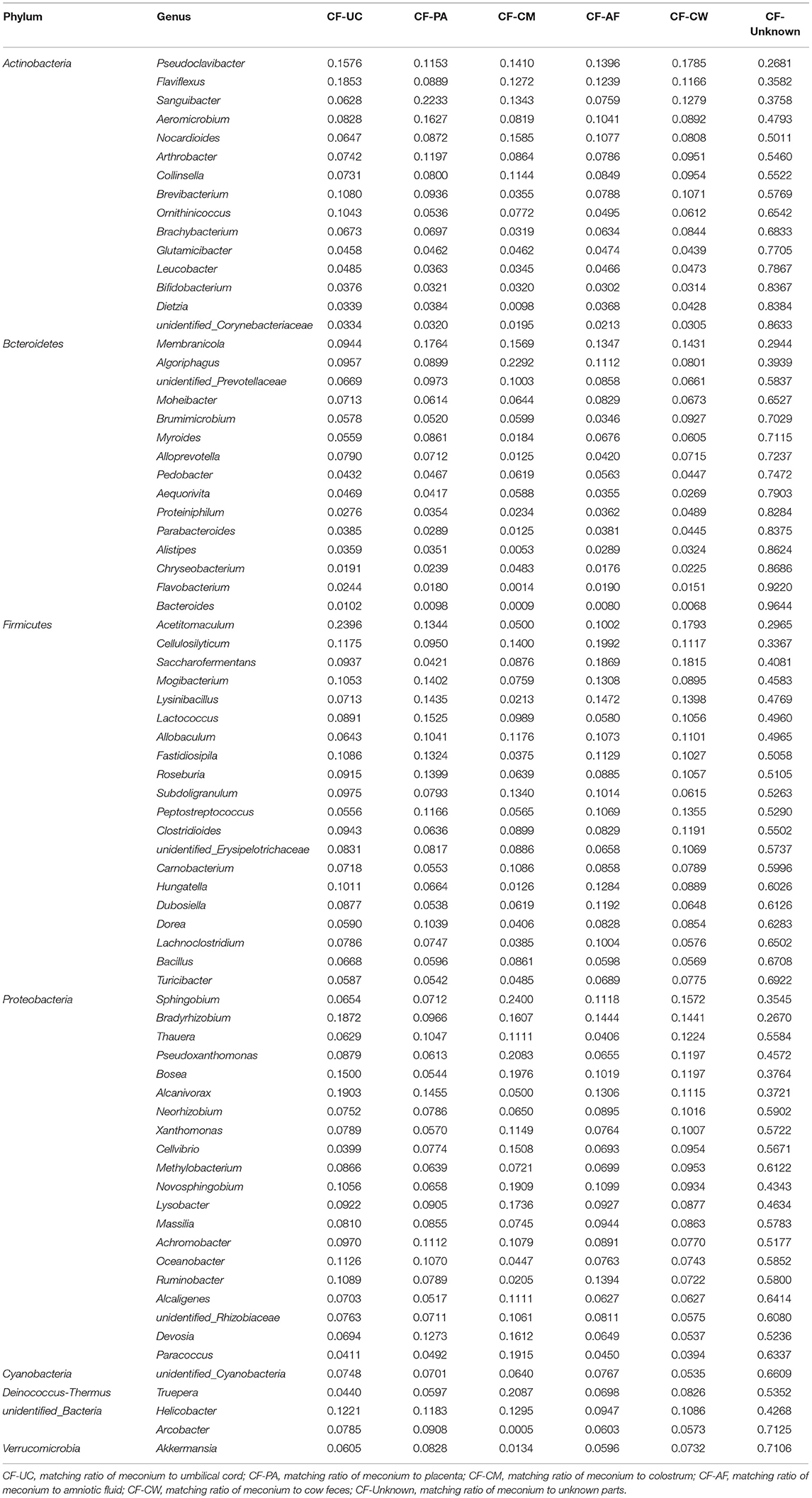
Table 5. Estimation of the matching ratio of meconium to the main genera in each part of their mothers during maternal transmission.
Discussion
Several studies have previously investigated the presence of microorganisms in the prenatal fetal gut and compared the offspring hindgut microbiome with that from different maternal sites such as the vagina, colostrum, and maternal feces (5, 6, 18, 30). Studies used healthy mare-foal, ewe-lamb, and cow-calf pairs as animal models and concluded that the prenatal gut harbored active microorganisms and that the fetal gut microbiome was seeded antenatally (7, 31). Regarding the hindgut microflora of calves, the current literature indicates that microbial communities across hindgut segments differ (32, 33). Thus, by examining the microbiome in meconium and different maternal sample types we were able to explore the possible sources of the hindgut microbiome in newborn calves. To obtain repeat and subsequent samples without euthanasia, a non-invasive and practical method (i.e., meconium examination) was chosen. The meconium microbiome represented the gut microbiome at birth without environmental influences such as feeding.
Meconium is the feces present in the hindgut of the calf before birth and can largely reflect the condition of the fetus' intestinal flora in the mother's womb. Both, the placenta after birth. Amniotic fluid is the only environment for the fetus to survive in the mother's womb and the placenta and umbilical cord are important ways for the mother to transfer nutrients to the fetus and for the fetus to metabolize them. This suggests that the similarity of the early fetal gut flora structure to its environment at the phylum level can reveal important information about the origin of microbial colonization in utero.
The bacterial microbiome of the samples from each site was analyzed using the Illumina Nova sequencing platform. At the phylum level, the dominant microbial components of the placenta, umbilical cord, amniotic fluid, colostrum, meconium, and cow feces included Proteobacteria, Firmicutes, Bacteroidetes, and Actinobacteria.
Proteobacteria are gram-negative bacteria that can produce LPS (lipopolysaccharide) that can enter the blood, reduce the number of hindgut barrier cells, and increase hindgut permeability (34). Bacteroidetes produce butyrate, a product of colonic fermentation with antineoplastic properties, and are beneficial to interactions within the immune system of the host, which can activate T cell-mediated responses (35) and limit the colonization of potentially pathogenic bacteria in the GI tract (35). A previous study found that Proteobacteria, which colonized the intestines of young mice at an early stage, can activate the young mouse's immune system. The high levels of Proteobacteria in meconium in this study may also be related to the construction of the early immune system in calves (36). Thus, the presence of these bacteria in the hindgut of the neonatal calf is advantageous in the context of health (37). Actinobacteria can use carbohydrates to produce lactic acid, which can maintain the acidity of the environment and suppress the growth of pathogenic bacteria in the intestine (38). In addition, microbiota-depleted mice inoculated with Bacteroidetes and Firmicutes reflected that the two phyla both have distinct effects on hindgut immunity by differentially inducing primary and secondary response genes (39).
Similar to previous studies (18, 19, 30), the results in the present study indicated that the core community in meconium was enriched in Halomonadaceae, Pseudomonadaceae, Dietziaceae, Ruminococcaceae, Moraxellaceae, and Enterobacteriaceae. These responses were also similar to those in human studies (40). Accordingly, we speculate that the microbiome in the abovementioned core community is the “pioneer flora” during early life of the offspring. As such, it plays a central role in shaping the community of anaerobic organisms. Results from other studies (19, 30) can also be corroborated by differences in the composition of the microbiome between meconium and cow feces in the present study: compared with those in newborn calves, Proteobacteria levels were reduced in young and adult calves, and Firmicutes and Bacteroides dominated the fecal microbiome (33). These results indicated that the increasing diversity and richness in hindgut microbiome communities as the animal ages are indicative of progressive establishment of a complex microbiome during early life stages (33).
The presence of Pseudomonas, Limnobacter, and Brevundimonas as the most abundant genera in colostrum underscored the key role of colostrum in helping colonize the neonatal gut with microflora with obvious beneficial effects (e.g., probiotic effect) (41). Pseudomonas has been consistently reported to be the dominant microbe in colostrum (42) and raw milk (43). Li et al. (44) concluded that Pseudomonas, Lactococcus, and Acinetobacter were the most common genera, and Hang et al. (45) found that Streptococcus, Acinetobacter, Enterobacter, and Corynebacterium were the dominant microbiota. Although those studies differed from our results, it is possible that differences in experimental approaches and even environment account for most of the discrepancies. For example, Hang et al. (45) squeezed colostrum samples into a non-sterile bucket and collected them directly from the bucket after mixing. Clearly, use of a non-sterile container likely would have contaminated the samples.
Transmission of microbiota from milk to the developing offspring may exert many short- or long- term influences on the physiology of the offspring (46). For example, Lactobacilli in milk include species associated with the hindgut microbiome (47). These microorganisms can produce a large quantity of lactic acid, which can inhibit the growth of pathogenic bacteria (38). Lactic acid can also be converted to butyrate, which maintains the acidity of the environment and suppresses growth of pathogens in the intestine (48). In addition, it has been documented that flora in the maternal gut can reach the mammary gland via intestinal mononuclear cells during late gestation and lactation, also suggesting the existence of bacterial transmission via intestinal-lacteal routes (46). The offspring's intestinal microbiota and its immune evolution are related to milk microbiota, which are derived from the maternal entero-mammary pathway (49). Interestingly, although calves did not have access to the udder at the time of sampling in this experiment, at the phylum level the colostrum flora matched the meconium flora by 14.4%, which was very close to the match between the placental flora and the meconium flora (15.5%). This suggested that colostrum microorganisms have some influence on meconium. DiGiulio et al. (50) suggested that the perinatal transfer of beneficial microorganisms from the maternal gut to the mammary gland via the bloodstream, i.e., the adjustment of the oligo-oligosaccharides, immune factors and microbial communities in milk before delivery helped prepared, so that these prepared “beneficial bacteria” for transmission to the offspring through milk after birth. This may be a specific evolutionary phenomenon.
Quercia et al. (6) concluded that amniotic fluid and intestinal ecosystems can also contribute uniquely to the meconium microbiome community in foals. He et al. (4) studied the association of the microbiome in infant meconium with that in maternal vagina, saliva, amniotic fluid, and feces samples. Their data indicated that the meconium microbiome was seeded from multiple maternal body sites, with amniotic fluid microbiome contributing the most. Thus, vertical transmission of the microbiome from the mother to the offspring may exist. A hypothetical “enteromammary” pathway was proposed in which the selected bacteria in the maternal intestine can access the mammary glands, and dendritic cells and CD18+ cells can take up non-pathogenic bacteria from gut epithelial cells and carry them to other locations (51). The placental microbiome in mice is colonized by invasion and crossing of the endothelial lining (52), a process thought to occur during early vascularization and placentation (15). Dendritic cells from the mare penetrate the host epithelia including hindgut epithelium carrying luminal bacteria or bacterial antigens that are then released into the placenta via the bloodstream (6). Once the amniotic fluid is reached, these microbial factors may have access to the fetal gut and become a part of the meconium ecosystem (53). Various bacteria can also be released into the breast through the blood. Thus, we speculate that the main biological function of microbial factor transfer from the intrauterine region to the fetus may be beneficial to the development of digestive function and the construction of the immune system of newborn calves after delivery.
SourceTracker analysis showed that in the maternal transmission process, cow feces mainly transmitted acid-producing bacteria such as Saccharofermentans, Acetitomaculum, and Pseudoclavibacter. Saccharofermentans are fibrolytic (54) and produce short-chain fatty acids and low-density lipoprotein cholesterol (6) both of which help maintain health and provide energy for the developing intestinal wall (55). Acetitomaculum and Pseudoclavibacter produce mainly acetic acid and butyric acid, respectively (55, 56). Butyrate, as an energy source for host epithelial cells, can regulate growth and the differentiation-related activator protein 1 (AP-1) signaling pathway (57) leading to an increase in the number of immunoregulatory T regulatory (T-reg) cells. These functions may reduce the likelihood of maternal rejection of the fetal allograft (58).
Other groups of bacteria that appear mainly transmitted from cow feces are common in the digestive tract and are associated with nutrition such as the aerobic denitrification bacteria Thauera, Lysinibacillus, and Peptostreptococcus and Novosphingobium, which are core members of the gut flora in the cow (18). Cellulosilyticum, Saccharofermentans, and Ruminobacter, the main cellulose-degrading bacteria (59, 60), and Bradyrhizobium, Mogibacterium, Alcanivorax, Fastidiosipila, Saccharofermentans, and Ruminobacter, are all common bacteria in alimentary canals involved in fiber degradation (59, 61, 62). These are all key microbiome communities transmitted by amniotic fluid. In fact, these bacteria were the main transmitted bacteria that were not only in cow feces and amniotic fluid, but also in placenta and umbilical cord and dominated in the succession that occurred in early life (58). Algoriphagus, Pseudoxanthomonas, Bradyrhizobium, and Novosphingobium were the main cellulose-degrading microbiome constituents (63–65) transmitted via colostrum. In addition, it was reported that the special genus Truepera, which was mainly transmitted by colostrum, was the core flora constituent in bedding used to house cows (66).
Overall, microbial communities of the cow-calf pair encompassed a complex and shared microbiome that likely interacted to maintain health in both cows and calves (40, 67). Although there were differences in microbial community structure among different sample types of dams and offspring, the microbiome involved in cellulose degradation, fermentation, and the common flora in alimentary canals were seeded into the calf via the maternal transmission process and affected the calf's nutrition and the microbial communities existing in the calf intestine.
Some limitations in the present study must be mentioned. First, the number of cow-calf pairs could be considered small for a robust evaluation of maternal transmission. Second, a deviation in PCR results may have occurred in the analysis of low-microbial-biomass samples, and the possibility of contamination of samples cannot be completely excluded. Third, the inherent limits of molecular analyses did not allow for studying whether live or dead bacteria, even microbial debris, were present in the samples collected. Lastly, we identified numerous flora in the meconium, but the origins and timing of the colonization were not investigated, and knowledge about the influence of these microflora on the metabolism and immune function remains limited.
Conclusion
Data provide evidence that the fetal hindgut microbiome of the calf may arise from different maternal parts. The composition of the meconium microflora originated from multiple maternal sites including umbilical cord, placenta, colostrum, and amniotic fluid. Characteristics of the microorganisms in the placenta, umbilical cord, colostrum, and meconium were more obvious than those in amniotic fluid, and differences in the microbial characteristics between meconium and cow feces were the largest. Microflora with digestive functions such as cellulose decomposition and rumen fermentation were highly matched during the maternal transmission process. Overall, the present findings advanced our understanding of the calf gut microbiome and lays a foundation for improving the growth and development of offspring, hindgut health, and lactation potential of calves by intervening in the gut microecology of pregnant cows. Further studies are required to gain an in-depth understanding of the origin, composition, function, dynamics, and colonization time of the calf gut microbiome. Elucidating the effects of the fetal gut microbiome on development, immunity, and health throughout early life will be an important undertaking.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation. The data presented in the study are deposited in the NCBI BioProject dataset repository, and the BioProject ID is PRJNA768139.
Ethics Statement
The animal study was reviewed and approved by Animal Welfare and Ethics Committee of Heilongjiang Bayi Agriculture University.
Author Contributions
HZ, CX, and YQ: conceptualization, methodology, and supervision. MY and SY: experiments and data curation. HZ and LL: writing-original draft preparation and software. JL, AE, and WW: reviewing and editing. All authors read and approved the final version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (Grant Nos. 32072758, U20A2062, and 31902186), the Natural Science Foundation of Heilongjiang Province of China (LH2021C069), and the Scientific Research Starting Foundation for Returned Overseas Chinese Scholars (Grant No. ZRCLG201903).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to acknowledge Licheng Liu, the dairy farm manager, and the workers at Jinao Animal Husbandry Co., Ltd. for their help in collecting experimental data.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnut.2021.736270/full#supplementary-material
References
1. Ren H, Bai H, Su X, Pang J, Li X, Wu S, et al. Decreased amylolytic microbes of the hindgut and increased blood glucose implied improved starch utilization in the small intestine by feeding rumen-protected leucine in dairy calves. J Dairy Sci. (2020) 103:4218–35. doi: 10.3168/jds.2019-17194
2. Vacca I. Microbiota: clostridia protect from gut infections in early life. Nat Rev Microbiol. (2017) 15:321. doi: 10.1038/nrmicro.2017.56
3. Sommer F, Bäckhed F. The gut microbiota–masters of host development and physiology. Nat Rev Microbiol. (2013) 11:227–38. doi: 10.1038/nrmicro2974
4. He Q, Kwok LY, Xi X, Zhong Z, Ma T, Xu H, et al. The meconium microbiota shares more features with the amniotic fluid microbiota than the maternal fecal and vaginal microbiota. Gut Microbes. (2020) 12:1794266. doi: 10.1080/19490976.2020.1794266
5. Bi Y, Tu Y, Zhang N, Wang S, Zhang F, Suen G, et al. Multiomics analysis reveals the presence of a microbiome in the gut of fetal lambs. Gut. (2021) 70:853–64. doi: 10.1136/gutjnl-2020-320951
6. Quercia S, Freccero F, Castagnetti C, Soverini M, Turroni S, Biagi E, et al. Early colonisation and temporal dynamics of the gut microbial ecosystem in Standardbred foals. Equine Vet J. (2019) 51:231–7. doi: 10.1111/evj.12983
7. Elolimy A, Alharthi A, Zeineldin M, Parys C, Helmbrecht A, Loor JJ. Supply of methionine during late-pregnancy alters fecal microbiota and metabolome in neonatal dairy calves without changes in daily feed intake. Front Microbiol. (2019) 10:2159. doi: 10.3389/fmicb.2019.02159
8. Hennessy ML, Indugu N, Vecchiarelli B, Bender J, Pappalardo C, Leibstein M, et al. Temporal changes in the fecal bacterial community in Holstein dairy calves from birth through the transition to a solid diet. PLoS ONE. (2020) 15:e0238882. doi: 10.1371/journal.pone.0238882
9. Song Y, Li F, Fischer-Tlustos AJ, Neves ALA, He Z, Steele MA, et al. Metagenomic analysis revealed the individualized shift in ileal microbiome of neonatal calves in response to delaying the first colostrum feeding. J Dairy Sci. (2021) 104:8783–97. doi: 10.3168/jds.2020-20068
10. Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. (2012) 7402:207–14. doi: 10.1038/nature11234
11. Rehbinder EM, Lødrup Carlsen KC, Staff AC, Angell IL, Landrø L, Hilde K, et al. Is amniotic fluid of women with uncomplicated term pregnancies free of bacteria? Am J Obstet Gynecol. (2018) 219:289.e1–289.e12. doi: 10.1016/j.ajog.2018.05.028
12. de Goffau MC, Lager S, Sovio U, Gaccioli F, Cook E, Peacock SJ, et al. Human placenta has no microbiome but can contain potential pathogens. Nature. (2019) 572:329–34. doi: 10.1038/s41586-019-1451-5
13. Collado MC, Rautava S, Aakko J, Isolauri E, Salminen S. Human gut colonisation may be initiated in utero by distinct microbial communities in the placenta and amniotic fluid. Sci Rep. (2016) 6:23129. doi: 10.1038/srep23129
14. Franasiak JM, Scott RT. Endometrial microbiome. Curr Opin Obstet Gynecol. (2017) 29:146–52. doi: 10.1097/GCO.0000000000000357
15. Aagaard K, Ma J, Antony KM, Ganu R, Petrosino J, Versalovic J. The placenta harbors a unique microbiome. Sci Transl Med. (2014) 237:237ra65. doi: 10.1126/scitranslmed.3008599
16. Urbaniak C, Cummins J, Brackstone M, Macklaim JM, Gloor GB, Baban CK, et al. Microbiota of human breast tissue. Appl Environ Microbiol. (2014) 80:3007–14. doi: 10.1128/AEM.00242-14
17. Stinson LF, Payne MS, Keelan JA. Planting the seed: origins, composition, and postnatal health significance of the fetal gastrointestinal microbiota. Crit Rev Microbiol. (2017) 43:352–69. doi: 10.1080/1040841X.2016.1211088
18. Klein-Jöbstl D, Quijada NM, Dzieciol M, Feldbacher B, Wagner M, Drillich M, et al. Microbiota of newborn calves and their mothers reveals possible transfer routes for newborn calves' gastrointestinal microbiota. PLoS ONE. (2019) 14:e0220554. doi: 10.1371/journal.pone.0220554
19. Alipour MJ, Jalanka J, Pessa-Morikawa T, Kokkonen T, Satokari R, Hynönen U, et al. The composition of the perinatal intestinal microbiota in cattle. Sci Rep. (2018) 8:10437. doi: 10.1038/s41598-018-28733-y
20. Jacometo CB, Zhou Z, Luchini D, Trevisi E, Corrêa MN, Loor JJ. Maternal rumen-protected methionine supplementation and its effect on blood and liver biomarkers of energy metabolism, inflammation, and oxidative stress in neonatal Holstein calves. J Dairy Sci. (2016) 99:6753–63. doi: 10.3168/jds.2016-11018
21. Zhao J, Fan H, Kwok LY, Guo F, Ji R, Ya M, et al. Analyses of physicochemical properties, bacterial microbiota, and lactic acid bacteria of fresh camel milk collected in Inner Mongolia. J Dairy Sci. (2020) 103:106–16. doi: 10.3168/jds.2019-17023
22. Doyle CJ, Gleeson D, O'Toole PW, Cotter PD. Impacts of seasonal housing and teat preparation on raw milk microbiota: a high-throughput sequencing study. Appl Environ Microbiol. (2016) 83:e02694–16. doi: 10.1128/AEM.02694-16
23. Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. (2011) 27:2957–63. doi: 10.1093/bioinformatics/btr507
24. Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, et al. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods. (2013) 10:57–9. doi: 10.1038/nmeth.2276
25. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. (2010) 7:335–6. doi: 10.1038/nmeth.f.303
26. Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: a versatile open source tool for metagenomics. PeerJ. (2016) 4:e2584. doi: 10.7717/peerj.2584
27. Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. (2011) 21:494–504. doi: 10.1101/gr.112730.110
28. Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. (2013) 10:996–8. doi: 10.1038/nmeth.2604
29. Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. (2007) 73:5261–7. doi: 10.1128/AEM.00062-07
30. Yeoman CJ, Ishaq SL, Bichi E, Olivo SK, Lowe J, Aldridge BM. Biogeographical differences in the influence of maternal microbial sources on the early successional development of the bovine neonatal gastrointestinal tract. Sci Rep. (2018) 8:3197. doi: 10.1038/s41598-018-21440-8
31. Elolimy A, Alharthi A, Zeineldin M, Parys C, Loor JJ. Residual feed intake divergence during the preweaning period is associated with unique hindgut microbiome and metabolome profiles in neonatal Holstein heifer calves. J Anim Sci Biotechnol. (2020) 11:13. doi: 10.1186/s40104-019-0406-x
32. Liang G, Malmuthuge N, Bao H, Stothard P, Griebel PJ, Guan le L. Transcriptome analysis reveals regional and temporal differences in mucosal immune system development in the small intestine of neonatal calves. BMC Genomics. (2016) 17:602. doi: 10.1186/s12864-016-2957-y
33. Malmuthuge N, Liang G, Griebel PJ, Guan LL. Taxonomic and functional compositions of the small intestinal microbiome in neonatal calves provide a framework for understanding early life gut health. Appl Environ Microbiol. (2019) 85:e02534–18. doi: 10.1128/AEM.02534-18
34. Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. (2007) 56:1761–72. doi: 10.2337/db06-1491
35. Mazmanian SK, Round JL, Kasper DL. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature. (2008) 453:620–5. doi: 10.1038/nature07008
36. Friedman JE. The maternal microbiome: cause or consequence of obesity risk in the next generation? Obesity. (2017) 25:497–8. doi: 10.1002/oby.21795
37. Kim YS, Milner JA. Dietary modulation of colon cancer risk. J Nutr. (2007) 137:2576S−9S. doi: 10.1093/jn/137.11.2576S
38. Lin PP, Hsieh YM, Tsai CC. Antagonistic activity of Lactobacillus acidophilus RY2 isolated from healthy infancy feces on the growth and adhesion characteristics of enteroaggregative Escherichia coli. Anaerobe. (2009) 15:122–6. doi: 10.1016/j.anaerobe.2009.01.009
39. Brown RL, Larkinson MLY, Clarke TB. Immunological design of commensal communities to treat intestinal infection and inflammation. PLoS Pathog. (2021) 17:e1009191. doi: 10.1371/journal.ppat.1009191
40. Dogra S, Sakwinska O, Soh SE, Ngom-Bru C, Brück WM, Berger B, et al. Rate of establishing the gut microbiota in infancy has consequences for future health. Gut Microbes. (2015) 6:321–5. doi: 10.1080/19490976.2015.1078051
41. Lindner J, Santarelli M, Yamaguishi CT, Soccol CR, Neviani E. Recovery and identification of bovine colostrum microflora using traditional and molecular approaches. Food Technol Biotechnol. (2011) 49:364–8. doi: 10.1007/s12223-011-0057-6
42. Lima SF, Teixeira AGV, Lima FS, Ganda EK, Higgins CH, Oikonomou G, et al. The bovine colostrum microbiome and its association with clinical mastitis. J Dairy Sci. (2017) 100:3031–42. doi: 10.3168/jds.2016-11604
43. Soto Del Rio MLD, Dalmasso A, Civera T, Bottero MT. Characterization of bacterial communities of donkey milk by high-throughput sequencing. Int J Food Microbiol. (2017) 251:67–72. doi: 10.1016/j.ijfoodmicro.2017.03.023
44. Li N, Wang Y, You C, Ren J, Chen W, Zheng H, et al. Variation in raw milk microbiota throughout 12 months and the impact of weather conditions. Sci Rep. (2018) 8:2371. doi: 10.1038/s41598-018-20862-8
45. Hang BPT, Wredle E, Dicksved J. Analysis of the developing gut microbiota in young dairy calves-impact of colostrum microbiota and gut disturbances. Trop Anim Health Prod. (2020) 53:50. doi: 10.1007/s11250-020-02535-9
46. Fernández L, Langa S, Martín V, Maldonado A, Jiménez E, Martín R, et al. The human milk microbiota: origin and potential roles in health and disease. Pharmacol Res. (2013) 69:1–10. doi: 10.1016/j.phrs.2012.09.001
47. Papademas P, Kamilari E, Aspri M, Anagnostopoulos DA, Mousikos P, Kamilaris A, et al. Investigation of donkey milk bacterial diversity by 16S rDNA high-throughput sequencing on a Cyprus donkey farm. J Dairy Sci. (2021) 104:167–78. doi: 10.3168/jds.2020-19242
48. Chang MN, Wei JY, Hao LY, Ma FT, Li HY, Zhao SG, et al. Effects of different types of zinc supplement on the growth, incidence of diarrhea, immune function, and rectal microbiota of newborn dairy calves. J Dairy Sci. (2020) 103:6100–13. doi: 10.3168/jds.2019-17610
49. Addis MF, Tanca A, Uzzau S, Oikonomou G, Bicalho RC, Moroni P. The bovine milk microbiota: insights and perspectives from -omics studies. Mol Biosyst. (2016) 12:2359–72. doi: 10.1039/C6MB00217J
50. DiGiulio DB, Gervasi MT, Romero R, Vaisbuch E, Mazaki-Tovi S, Kusanovic JP, et al. Microbial invasion of the amniotic cavity in pregnancies with small-for-gestational-age fetuses. J Perinat Med. (2010) 38:495–502. doi: 10.1515/jpm.2010.076
51. Rodríguez JM. The origin of human milk bacteria: is there a bacterial entero-mammary pathway during late pregnancy and lactation? Adv Nutr. (2014) 5:779–84. doi: 10.3945/an.114.007229
52. Fardini Y, Chung P, Dumm R, Joshi N, Han YW. Transmission of diverse oral bacteria to murine placenta: evidence for the oral microbiome as a potential source of intrauterine infection. Infect Immun. (2010) 78:1789–96. doi: 10.1128/IAI.01395-09
53. Perez PF, Doré J, Leclerc M, Levenez F, Benyacoub J, Serrant P, et al. Bacterial imprinting of the neonatal immune system: lessons from maternal cells? Pediatrics. (2007) 119:e724–32. doi: 10.1542/peds.2006-1649
54. Chen S, Niu L, Zhang Y. Saccharofermentans acetigenes gen. nov., sp. nov., an anaerobic bacterium isolated from sludge treating brewery wastewater. Int J Syst Evol Microbiol. (2010) 60:2735–8. doi: 10.1099/ijs.0.017590-0
55. Li JH, Yousif MH, Li ZQ, Wu ZH, Li SL, Yang HJ, et al. Effects of antibiotic residues in milk on growth, ruminal fermentation, and microbial community of preweaning dairy calves. J Dairy Sci. (2019) 102:2298–307. doi: 10.3168/jds.2018-15506
56. Luoyizha W, Wu X, Zhang M, Guo X, Li H, Liao X. Compared analysis of microbial diversity in donkey milk from Xinjiang and Shandong of China through high-throughput sequencing. Food Res Int. (2020) 137:109684. doi: 10.1016/j.foodres.2020.109684
57. Nepelska M, Cultrone A, Béguet-Crespel F, Le Roux K, Doré J, Arulampalam V, et al. Butyrate produced by commensal bacteria potentiates phorbol esters induced AP-1 response in human intestinal epithelial cells. PLoS One. (2012) 7:e52869. doi: 10.1371/journal.pone.0052869
58. Blaser MJ, Dominguez-Bello MG. The human microbiome before birth. Cell Host Microbe. (2016) 20:558–60. doi: 10.1016/j.chom.2016.10.014
59. Hu Y, He Y, Gao S, Liao Z, Lai T, Zhou H, et al. The effect of a diet based on rice straw co-fermented with probiotics and enzymes versus a fresh corn Stover-based diet on the rumen bacterial community and metabolites of beef cattle. Sci Rep. (2020) 10:10721. doi: 10.1038/s41598-020-67716-w
60. Hu R, Zou H, Wang H, Wang Z, Wang X, Ma J, et al. Dietary energy levels affect rumen bacterial populations that influence the intramuscular fat fatty acids of fattening yaks (Bos grunniens). Animals. (2020) 10:1474. doi: 10.3390/ani10091474
61. Fang W, Yan D, Wang Q, Huang B, Ren Z, Wang X, et al. Changes in the abundance and community composition of different nitrogen cycling groups in response to fumigation with 1,3-dichloropropene. Sci Total Environ. (2019) 650:44–55. doi: 10.1016/j.scitotenv.2018.08.432
62. Wang L, Liu K, Wang Z, Bai X, Peng Q, Jin L. Bacterial community diversity associated with different utilization efficiencies of nitrogen in the gastrointestinal tract of goats. Front Microbiol. (2019) 10:239. doi: 10.3389/fmicb.2019.00239
63. Wu X, Sun Y, Deng L, Meng Q, Jiang X, Bello A, et al. Insight to key diazotrophic community during composting of dairy manure with biochar and its role in nitrogen transformation. Waste Manag. (2020) 105:190–7. doi: 10.1016/j.wasman.2020.02.010
64. Chorazyczewski AM, Huang IS, Abdulla H, Mayali X, Zimba PV. The influence of bacteria on the growth, lipid production, and extracellular metabolite accumulation by phaeodactylum tricornutum (Bacillariophyceae). J Phycol. (2021) 57:931–40. doi: 10.1111/jpy.13132
65. Li A, Yang Y, Zhang Y, Lv S, Jin T, Li K, et al. Microbiome analysis reveals the alterations in gut microbiota in different intestinal segments of Yimeng black goats. Microb Pathog. (2021) 155:104900. doi: 10.1016/j.micpath.2021.104900
66. Wang JM, Gan XM, Pu FJ, Wang WX, Ma M, Sun LL, et al. Effect of fermentation bed on bacterial growth in the fermentation mattress material and cecum of ducks. Arch Microbiol. (2021) 203:1489–97. doi: 10.1007/s00203-020-02145-x
Keywords: maternal transfer, hindgut microbiome, dairy calf, diversity, SourceTracker
Citation: Zhu H, Yang M, Loor JJ, Elolimy A, Li L, Xu C, Wang W, Yin S and Qu Y (2021) Analysis of Cow-Calf Microbiome Transfer Routes and Microbiome Diversity in the Newborn Holstein Dairy Calf Hindgut. Front. Nutr. 8:736270. doi: 10.3389/fnut.2021.736270
Received: 05 July 2021; Accepted: 27 September 2021;
Published: 25 October 2021.
Edited by:
Yangchun Cao, Northwest A and F University, ChinaReviewed by:
Zhijun Cao, China Agricultural University, ChinaHangshu Xin, Northeast Agricultural University, China
Copyright © 2021 Zhu, Yang, Loor, Elolimy, Li, Xu, Wang, Yin and Qu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongli Qu, eWxxdTAwN0AxMjYuY29t