Abstract
Metabolic rewiring is considered as a primary feature of cancer. Malignant cells reprogram metabolism pathway in response to various intrinsic and extrinsic drawback to fuel cell survival and growth. Among the complex metabolic pathways, pyrimidine biosynthesis is conserved in all living organism and is necessary to maintain cellular fundamental function (i.e. DNA and RNA biosynthesis). A wealth of evidence has demonstrated that dysfunction of pyrimidine metabolism is closely related to cancer progression and numerous drugs targeting pyrimidine metabolism have been approved for multiple types of cancer. However, the non-negligible side effects and limited efficacy warrants a better strategy for negating pyrimidine metabolism in cancer. In recent years, increased studies have evidenced the interplay of oncogenic signaling and pyrimidine synthesis in tumorigenesis. Here, we review the recent conceptual advances on pyrimidine metabolism, especially dihydroorotate dehydrogenase (DHODH), in the framework of precision oncology medicine and prospect how this would guide the development of new drug precisely targeting the pyrimidine metabolism in cancer.
Introduction of Pyrimidine Metabolism Pathway
In mammal, pyrimidine can be produced through de novo synthesis pathway taking amino acids as substrates or salvage pathway by uptake of the circulating pyrimidines in the bloodstream. Generally, salvage pathway is the main pyrimidines sources for resting or fully differentiated cells, while the de novo pathway is necessary for high-proliferating cells to meet the boosted requirement of pyrimidines. Uridine 5′-monophosphate (UMP) is the first production of de novo pyrimidine pathway and is further converted to other pyrimidine nucleosides for synthesis of DNA and RNA. On the other hand, pyrimidine synthesis is also implicated in other metabolic pathways. Cytidine triphosphate (CTP) acts as a shuttle for PtdCho through cytidine diphosphate (CDP)-choline pathway (1). Uridine-5′-triphosphate (UTP) participates in the formation of UDP-N-acetylglucosamine (UDP-GlcNAc), UDP-glucose, UDP-galactose, and UDP-glucuronic acid (2). Owing to the critical functions of pyrimidine in cell proliferation and survival, the disorder of pyrimidine metabolism has been considered as a vital driver in tumor initiation and progression (3, 4).
Pyrimidine Biosynthesis Through De Novo Pathways
Pyrimidine de novo biosynthesis pathway is initiated by a trifunctional enzyme CAD (carbamoyl phosphate synthetase (CPS), aspartate carbamoyltransferase (ATC) and dihydroorotase) (Figure 1) (5). The CPS leads the first reaction using glutamine, bicarbonate, and ATP to produce carbamoyl phosphate, which is the first committed step in pyrimidine de novo synthesis metabolic flux (6). The second step is catalyzed by the ATC domain via converting aspartate and carbamoyl phosphate to carbamoyl aspartate (7). The dihydroorotase domain, a Zn metalloenzyme locating between CPS and ATC domains, hydrolyzes carbamoyl aspartate to dihydroorotate (DHO) (8). CAD, governing the initiation of pyrimidine de novo synthesis pathway (9–11), is under precise control in cells (12, 13).
Figure 1
Dihydroorotate dehydrogenase (DHODH), a ubiquitous flavoprotein (flavin mononucleotide, FMN) located in the mitochondrial inner membrane, converses DHO to orotic acid (14). DHODH is a unique enzyme that could perform this conversion in cells, representing the second rate-limiting step in pyrimidine de novo synthesis pathway. Additionally, DHODH transfers two electrons to coenzyme Q (CoQ) via ubiquinone and directly couples to the mitochondrial respiratory chain and oxygen consumption (15–18).
Subsequently, uridine monophosphate synthetase (UMPS), a bifunctional protein, transforms orotic acid into uridine monophosphate (UMP) through two catalytic reactions. The N-terminal domain of UMPS converts orotic acid into orotidylate (OMP) taking phospho-α-dribosyl-1-pyrophosphate (PRPP) as co-substrate, while C-terminal converts OMP into UMP (19).
In general, the de novo pyrimidine flux is low in resting or fully matured cells, where the demand of pyrimidines is mostly satisfied through the salvage pathway. Nevertheless, the high pyrimidine flux is indispensable in cancer cells in order to meet their increased need of nucleic acid and other cellular components (4, 20). Besides, owing to the bifunction of DHODH linking the pyrimidine de novo synthesis pathway with mitochondrial respiratory chain and oxygen consumption, this pathway becomes a pacemaker for cell growth and proliferation under limited oxygen tension. Consequently, pyrimidine de novo synthesis represents a promising therapeutic target in delaying cancer progression (15, 21–25).
Pyrimidine Biosynthesis Through Salvage Pathways
Although de novo pathway generally provides competent pyrimidine for cells growth and development, energetic expensiveness of this pathway restricts its application in cells (26). Cells at rest can meet their pyrimidine requirement via salvage pathway using intracellular nucleic acid degradation product or extracellular nucleoside pool in the blood stream (Figure 1) (27).
Intracellularly, uridine/cytidine kinase (UCK) participates in the recycling of cytidine and uridine to CMP and UMP, respectively. CMP is then converted to CTP by cytidine monophosphate kinase (CMPK) and nucleoside-diphosphate kinase (NDPK). For reusing extracellular nucleoside, their availabilities between cells and external environment are primarily mediated by specific nucleosides transport channels and pumps (28, 29). Specifically, nucleoside transport in mammals is comprised of two major gene families, SLC28 (SLC28A1, SLC28A2, SLC28A3) for encoding cation dependent concentrative nucleosides transporters (CNTs) and SLC29 (SLC29A1, SLC29A2, SLC29A3, SLC29A4) for encoding energy independent equilibrate nucleoside transporters (ENTs) (28, 29). These nucleosides transporters differ both in concentration of cation coupling and permeant selectivity. Also, targeting pyrimidine salvage pathway was considered as a valuable approach for reducing pyrimidines accessibility in cancer cells (30–36).
Biological Role of Pyrimidines
As an non-negligible substrate for DNA and RNA biosynthesis, UMP can be phosphorylated to generate produce UDP and UTP (37). UDP is catalyzed into deoxy-UMP (dUMP), which is the substrate to produce deoxyribonucleotides dTMP and dTTP through thymidylate synthase (TS). CTP synthetase (CTPS) converts UTP into CTP using glutamine as amine donor (12). Overall, UMP can supply all kinds of pyrimidine deoxyribonucleotides (dTTP, dCTP) and ribonucleotides (CTP, UTP) for DNA and RNA biosynthesis to sustain transmission of genetic information in cell proliferation (3, 38).
Beyond DNA and RNA biosynthesis, CTP also participates in phospholipids biosynthesis in cells. Phosphatidylcholine (PC) is a primary component of phospholipid among biological membranes as well as a source of lipid-like second messengers (39), indicating that PC is a vital substance in cell metabolism and growth. CDP-choline generation is a rate-limiting step for the PC biosynthesis in mammals, which is produced by choline phosphate cytidylyltransferase (CCT) using CTP and choline phosphate (40). Thus, intracellular CTP pool is critical for the generation of PC and the subsequent phospholipid biosynthesis.
During energy metabolism, glucose-derived glucose-1-phosphate combined with UTP to produce UDP-glucose, which is further catalyzed to UDP-glucuronic acid through UDP-glucose 6-dehydrogenase (UGDH) (41, 42). UDP-glucuronic acid is a requisite precursor for the glycosaminoglycans, a component of proteoglycans for sustaining electrolyte as well as fluid balance in the extracellular matrix.
On the other hand, UTP also takes part in the glycosylation modification of proteins, which has a wide range of function and shows essential role for life even at the single cell level (43). In this case, UTP combines with N-acetylglucosamine (GlcNAc) derived from hexosamine biosynthesis (HBP) pathway involving four enzymes converting fructose 6-phosphate to UDP-GlcNAc, to generate UDP-GlcNAc. UDP-GlcNAc is a necessary substrate for O-linked glycosylation (O-GlcNAcylation), a frequent and essential post-translation modification of protein in linking glycosidic of saccharides to proteins in Golgi apparatus (44). During this process, the O-GlcNAc transferase (OGT) takes UDP-GlcNAc as a substrate to install GlcNAc to the hydroxyl oxygen of serine or threonine residues of proteins (45), while O-GlcNAcase (OGA) catalyzes the removal of O-GlcNAc from modified proteins (46, 47).
Together, these evidences demonstrate that pyrimidine metabolism cooperates with various cell metabolism and signaling pathways, demonstrating the decisive effects of pyrimidine metabolism in determining cell fates including proliferation, differentiation, apoptosis, and metastasis.
Oncogenes Reprogram Pyrimidine Metabolism in Cancers
Dysfunction of Pyrimidine Metabolism in Cancers
In cancer, dysfunctional pyrimidine de novo synthesis not only supplies high-level of nucleotides, but also affects other metabolism pathways and cellular signaling (Figure 2). For instance, the essential characters of CTP in phospholipid synthesis, UDP in glucose metabolism and UTP in protein glycosylation hint the influence of pyrimidine synthesis in tumorigenesis. The high requirement of membrane components in active cells sets the indispensable of phosphatidylcholine obtained from CTP and choline in membrane biogenesis. It is reported that phospholipid synthesis pathway was highly relied on the cellular CTP level, which modulates the mitochondrial phospholipid composition upon altered abundance of ether lipids (18). Moreover, CTP also takes part in signaling transmission, enzymatic processes of membrane as well as cell homeostasis (48–51).
Figure 2
Glycosaminoglycan, derived from UDP and glucose, resides in the extracellular aiming to provide mechanical support for cells, involving in the cell migration, adhesion, motility and wound healing (52). The elevated glycosaminoglycan concentration is identified in multiple types of cancers, such as breast cancer, brain tumor, glioblastoma, and lung cancer (53–56). During the glycosaminoglycan biosynthesis, UGDH, catalyzing conversion of UDP-glucose to UDP-glucuronate, is recognized as the rate-limiting enzyme and has been identified as a potential anti-cancer target (55, 57–60). While, the precursor contents of glycosaminoglycan were rarely investigated. Recently, Xiongjun Wang et al. found that the activation of UGDH by epidermal growth factor receptor (EGFR) attenuates the UDP-glucose-mediated disruption of the interaction of HuR and SNAI1 mRNA, which promotes lung metastasis through initiating the epithelial-mesenchymal transmission (54).
UTP is critical for protein glycosylation by serving as a glycosylation substrate sub-fraction. It is reported that protein glycosylation level is generally elevated in various malignancies and has been identified as a hallmark of cancer (61). As an abundant post-translational modification of protein, glycosylation takes part in sustaining cancer cell growth and proliferation (62–69). O-GlcNAc modification is important for VEGF levels, and disordered glycosylation of VEGFR could regulate its interaction with galectins, which are associated with both angiogenesis and tumor metastasis (70, 71). The O-GlcNAc was also reported to be implicated in the regulation of oncogenic gain-of-function of mutant p53 (72, 73). Additionally, Kevin Qian et al. demonstrated that ERK signaling pathway profoundly affected O-GlcNAc homeostasis via OGA-mediated OGT transcription in pancreatic ductal adenocarcinoma (PDAC), laying the profound role of O-GlcNAc signaling in cancers (74). Decreased O-GlcNAc levels elevates the cell-cycle regulator p27Kip1 and reduces FoxM1 as well as its downstream target genes expression in breast cancer (67). Moreover, Christina M. Ferrer et al. uncovered that O-GlcNAcylation regulates HIF-1α proteasomal degradation via a α-ketoglutarate-dependent manner (75). The pentose phosphate pathway (PPP) is critical for rapidly proliferational cells through maintaining cellular redox homoeostasis and the upregulation of PPP has been appeared in various cancer types (76, 77). The activity of G6PD, the rate-limiting enzyme of PPP, was reported to be regulated by O-linked glycosylation of Ser 84, which significantly elevated PPP metabolism flux in human lung cancer cells related to hypoxia pressure regulation (78). Besides, O-GlcNAcylation inhibits phosphofructokinase 1 (PFK1) activity in response to hypoxia, redirecting glucose flux through PPP to confer growth advantage to lung cancer cells (79).
The chronic inflammatory response plays an essential role in cancer development and abundant evidences shown that glycosylation can modulate multiple key mediators involved in the inflammatory process. It is reported that O-linked glycosylation can upregulate the transcription level of NF-κB (80) and pro-inflammatory factor COX2 (81). Furthermore, the protein glycosylation level of COX2 is associated with the therapeutic efficiency of its inhibitors (82). On the other hand, the pro-inflammatory cytokines in turn balance glycan composition of cells via increasing glycosyltransferases expression and promote cancer-related antigens biosynthesis in pancreatic cancer as well as gastric cancer (83, 84).
Published studies have suggested that cellular O-glycosylation level is highly associated with the activities of OGT or OGA and the level of substrate UDP-GlcNAc (85–88). However, the direct and detailed role of pyrimidine synthesis in the regulation of O-glycosylation is not well established. Thus, the specific mechanism of pyrimidine synthesis in regulating O-glycosylation needed to be further explored.
Additionally, pyrimidine synthesis also participates in tumor metastasis. Pharmacological inhibition of DHODH was reported to reduce liver metastasis in colorectal cancer (89) and small cell lung cancer model (88). Siddiqui, A. et al. reported that thymidylate synthase (TS) was reported to be increased in cancer cells with a mesenchymal phenotype (90). Depletion of TS significantly decreased the expression of ZEB1, a powerful epithelial-mesenchymal transition (EMT) driver, and impeded the migration of cancer cells. Moreover, TS maintains the de-differentiated state of triple negative breast cancers (91) and drives the EMT phenotypes in NSCLC (92). These results further demonstrated the potential association of pyrimidine metabolism to the malignant process of cancer.
Oncogene Reprograms Pyrimidine Metabolism in Cancers
Generally, tumorigenesis is accompanied by enhanced metabolic reprogramming for satisfying cells survival and proliferation in front of their internal and external surrounding microenvironment changes. It is well known that cancer cells intend to remodeling metabolism to increase pyrimidine de novo synthesis flux to support vigorous growth. Here, we regularly review the influence of various oncogenic signaling pathways on gene expression and post-translational modifications in pyrimidine metabolism in cancers.
Oncogenic Signaling Pathways Modulate Gene Transcription in Pyrimidine Metabolism
Oncoprotein c-Myc is critical for cell proliferation, differentiation, apoptosis, and cell metabolism via regulating the expression of hundreds of target genes (93–95). It was reported that c-Myc upregulated the level of CAD by binding to the highly conserved palindromic E box sequence downstream of transcriptional site of CAD promoter (96–98) (Figure 3). Besides, c-Myc is also implicated in the transcription regulation of UMPS and CTPS (99). On the other hand, our studies and others have revealed that intervention of pyrimidine synthesis by targeting DHODH resulted in decreased protein stability and transcription activity of c-Myc in AML and melanoma (100, 101). These studies highlight the complex interplay between c-Myc oncogenic signaling and pyrimidine metabolic reprogramming in cancers (24, 99, 102, 103).
Figure 3
Hypoxia-inducible factor (HIF-1α) was identified to bind to CAD promoter and repress its transcription of in vivo, triggering the inhibition of pyrimidine de novo synthesis and subsequently cell-cycle arrest (104, 105). In addition, specificity protein 1 (Sp-1) interacts with estrogen receptor α (ERα) physically and results in transcriptional activation of estrogen-responsive genes expression including CAD (106–110).
Oncogenic Signaling Pathways Affect Post-Translation Modification of Pyrimidine Metabolism Enzyme
Post-translation modification of pyrimidine pathway is also regulated by various oncogenic signaling pathways. As a hexamer of a 243 KD polypeptide, CAD folds into three functional domains autonomously (111), including CPS, aspartate ATC and dihydroorotase. The activity of CPS domains, governing the flux of pyrimidine de novo synthesis, can be reversible inhibited by UTP and allosterically activated by PRPP. Lee M. Graves et al. described that EGFR-mitogen-activated protein (MAP) kinase cascade promotes cell proliferation via phosphorylation of CAD at Thr456 (Figure 3) (112). This allosteric regulation abolishes the feedback inhibition of UTP as well as endow more sensitive to PRPP, which increases the flux of pyrimidine de novo synthesis (113). While, protein kinase A (PKA) induced phosphorylation of Ser1406 elevated UTP inhibition and decreased sensitivity upon PRPP, consequently decreased CAD activity and pyrimidine flux (114). Additionally, both MAPK and PKA complexes with CAD stably and the steric interference by the bound kinase will influence each other (115). Thus, the reciprocal phosphorylation of CAD is under the precise control of PKA and MAPK signaling cascades, which provides the elegant mechanism to regulate pyrimidine de novo synthesis.
The mammalian target of rapamycin complex 1 (mTORC1) is a Ser-Thr kinase sensing growth signals to modulate cell metabolism and proliferation (116). PI3K-PTEN-mTORC1 signaling pathway has been shown to phosphorylate Ser1859 of CAD via downstream ribosomal protein S6 kinase 1 (S6K1) and promoted CAD oligomerization, facilitating the concerted action of three enzymatic domains (6, 111) and stimulating pyrimidine de novo synthesis (10, 117). Karina N. et al. found that loss of sirtuin 3 (SIRT3), a tumor suppressor, also hyperactivates mTORC1-CAD axis and hence increases de novo pyrimidine synthesis (118), however the specific mechanism is still to be explored. Furthermore, LKB1 suppressed CPS1 transcriptional level through AMPK, which is known to inhibit mTOR and KRAS. LKB1 mutation speeds the dependence on pyrimidine synthesis in lung cancer cells (13). In addition, loss of argininosuccinate synthase (ASS1) increases CAD activity via mTOR-S6K1 signaling pathway and facilitates pyrimidine synthesis to support proliferation (7).
Targeting Pyrimidine Metabolism Vulnerability in Oncogene-Driven Cancers
A plethora of studies have showed that oncogene-driven cancers exhibit higher level of pyrimidine de novo pathway, pointing out the opportunity of precision cancer therapy by targeting the pyrimidine dependence (Table 1). In recent years, DHODH has emerged as a promising synthetic lethal target for various oncogenic events, including BRAF (V600E) mutation (101), PTEN deficiency (23) (22), and RAS mutation (119) (Figure 3).
Table 1
| Drug | Structure | Target | Disease |
|---|---|---|---|
| Teriflunomide | 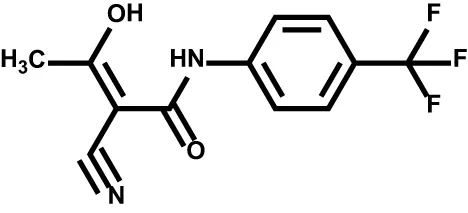 | DHODH | Multiple sclerosis |
| Leflunomide | 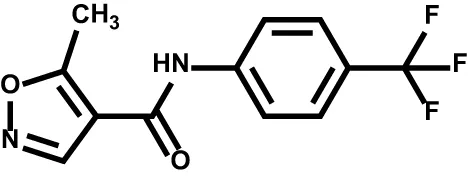 | DHODH | Rheumatoid arthritis |
| Fluorouracil | 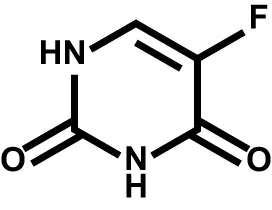 | Thymidylate synthase; DNA; RNA | Multiple solid tumors |
| Floxuridine | 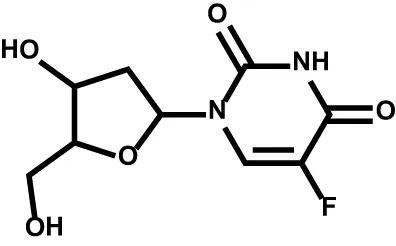 | Thymidylate synthase | Metastatic hepatic cancer Stage 4 gastrointestinal adenocarcinoma |
| Gemcitabine | 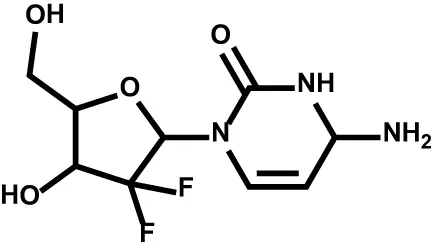 | DNA; RRM1; TYMS; CMPK1 | Non-small cell lung cancer Pancreatic cancer Bladder cancer Breast cancer |
Small molecular inhibitors of pyrimidine metabolic pathway approved by FDA.
In BRAF (V600E) mutant melanoma cells, DHODH modulates transcriptional elongation and its inhibition completely abrogated cell growth, providing the potential therapeutic strategy through combination of DHODH and BRAF (V600E) inhibitors (101). PTEN, one of the commonly mutated tumor suppressor, is an important negative regulator of oncogenic PI3K signaling (120). It is reported that DHODH inhibition caused inherent defects in DNA repair and accumulated DNA damage contributes to synthetic lethality in PTEN mutant TNBC cells (23). Besides, Kristin K. Brown et al. characterized adaptive metabolic reprogramming of pyrimidine de novo synthesis as an early issue promoting chemotherapy resistance in TNBC treatment. Thus, targeting DHODH-driven pyrimidine synthesis seems to be a feasible strategy in overcoming chemotherapy resistance in TNBC clinically (22).
KRAS mutation is one of the most common mutation in human cancer. Previous studies have demonstrated that KRAS mutants induce pyrimidine metabolism reprogramming in multiple malignancies types (121, 122). Malvika Koundinya et al. have reported that KRAS mutant cells seems to exhibit high sensitivity to DHODH inhibitors (119). Decreased expression of mutant KRAS has been shown to downregulate the genes transcriptional level in pyrimidine pathways (123, 124). KRAS mutation activates classic MAPK pathway, which results in MYC upregulation and transcriptional level of the non-oxidative PPP gene ribose 5-phosphate isomerase A (RPIA), promoting the nucleotide biosynthesis (123). Notably, interference with nucleotide biosynthesis resulted in glutamine deprivation in KRAS-driven cancer cells and thus lead to S-phase cell cycle arrest and DNA replication stress (125). These evidences fully define the increase pyrimidine synthesis flux in KRAS mutant cancer cell. On the other hand, although KRAS mutant cells exhibits decreased oxidative phosphorylation (126), the mitochondrial electron transport via DHODH in sustaining hyperpolarized mitochondrial membrane potential and pro-survival (127) could explain for the increased sensitivity of KRAS mutant cells to DHODH inhibition.
In addition, p53 also plays a pivotal role in regulating cellular metabolism (128–132). For instance, Irem Kaymak et al. stated that p53 deficiency activates the mevalonate pathway through SREBP2 and speeds ubiquinone synthesis which is crucial to the DHODH catalyzed pyrimidine synthesis (133). Combination of DHODH and CHK1 inhibitors demonstrate synergistically effect in p53 dysfunctional cancer cells through inducing aberrant cell cycle and massive cell death (134). While, the potential association between p53 and DHODH should to be further elaborated.
In addition, cancer cells with electronic transmission chain (ETC) deficiency displayed the dysfunction of DHODH due to the lack of complex III. Restoration of ETC recovers DHODH activity and cell growth (15, 17). These results suggested the indispensable role of DHODH-driven pyrimidine synthesis in linking ETC and tumorigenesis. Furthermore, David B. Sykes et al. described that DHODH inhibitors significantly decreased levels of leukemia-initiating cells, and improved survival of leukemia-bearing mice. This pioneering study defined DHODH as a metabolic regulator of differentiation in multiple subtypes of acute myeloid leukemia (AML) and DHODH inhibition emerged as a potential strategy for overcoming differentiation blockade in treating AML (135). Of note, multiple DHODH inhibitors were found to promote AML cell differentiation and launched to clinical research in AML treatment (Table 2) (100, 136–139).
Table 2
| Drug | Structure | Disease | Phase |
|---|---|---|---|
| Brequinar | 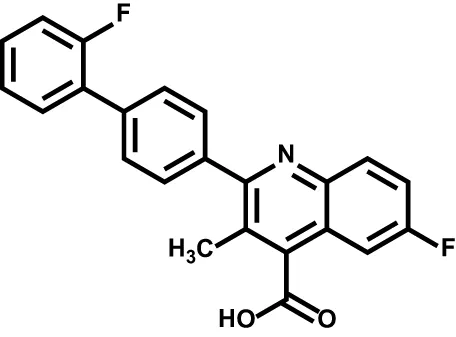 | Acute myeloid leukemia | Phase II |
| BAY2402234 | 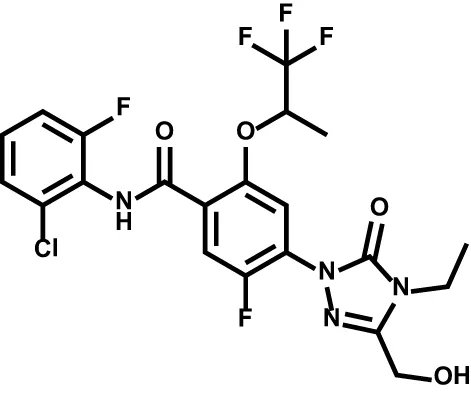 | Leukemia | Phase Ia |
| ASLAN003 | 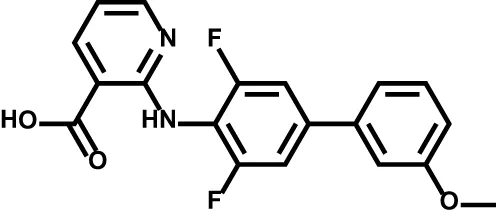 | Acute myeloid leukemia | Phase II |
| PTC299 | 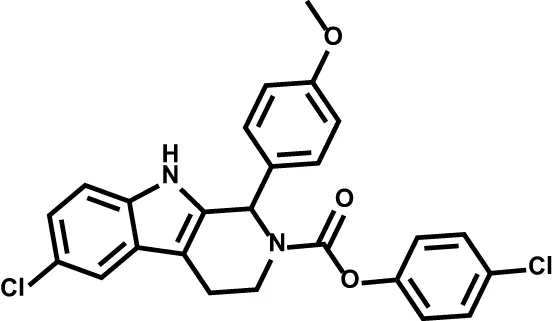 | Acute myeloid leukemia | Phase I |
| AG-636 | Unavailable | Lymphoma | Phase I |
| Leflunomide | 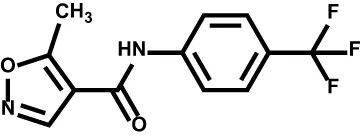 | Relapsing multiple sclerosis COVID-19 | Phase II |
| ML390 |  | Acute myeloid leukemia | – |
| Isobavachalcone | 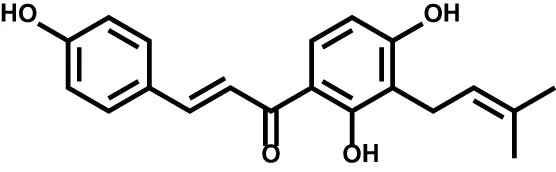 | Acute myeloid leukemia | – |
| Piperine | 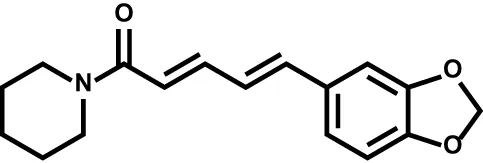 | Multiple sclerosis | – |
Representative small-molecular inhibitors of DHODH.
clinical trial has been terminated due to the lack of sufficient clinical benefit.
Bioinformatics Analysis of Pyrimidine Metabolism and Signaling in Cancers
We then turn to investigate the gene expression profiling of pyrimidine metabolizing enzymes in cancer genomics. We first score the pyrimidine metabolism pathway in 31 TCGA cancers compared with corresponding normal tissues in GEPIA (140). As shown in Figure 4A, more than 80% cancers demonstrate significantly increased expression of pyrimidine metabolism related genes compared to normal tissues, which indicates the closing correlation of pyrimidine metabolism in tumor progression. Specifically, AML exhibits the highest score.
Figure 4
In order to further examine the importance of pyrimidine metabolism in AML, we then analyzed the cellular responses in the presence of various shRNA in AML cell lines from Cancer Cell Line Encyclopedia (CCLE) (141). As shown in Figure 4B, knock down of genes involved in pyrimidine metabolism generally decrease the cell viability, suggesting the indispensable role of pyrimidine pathway in the development of AML. Specifically, the most negative shRNA score occurs in AML is DHODH, suggesting the significant cell proliferation inhibition treated with shRNA. DHODH catalyzes pyrimidine de novo pathway simultaneously transferring electrons to mitochondrial respiration chain. Depletion of DHODH significantly disturbs tumor formation with complete functional oxidative phosphorylation. It is reported that DHODH-driven pyrimidine synthesis rather than mitochondrial ATP synthase connects respiration to tumorigenesis (15). Furthermore, DHODH inhibition restructures the ETC upon respirasome assembly and activity owing to the modified phospholipid synthesis dependent on CTP level. Hence, the dual function of DHODH provides the plausible explanation for the prominent effect of shDHODH in AML. Moreover, survival analysis from GEPIA database indicated that higher expression of DHODH was closely associated with shorter disease-free survival (DFS) (Figure 4C). These oncology genomics data suggest that DHODH plays a decisive role in the cell survival and proliferation of leukemia.
We then analyzed the potential proteins directly interacting with DHODH using STRING v11.0 and clustered these enzymes through KEGG pathways (142). As shown in Figure 4D, in addition to two key enzymes involved in pyrimidine de novo pathway, CAD and UMPS, enzymes related with many other cellular metabolism pathways are associated with DHODH, including purine metabolism, one carbon poll by folate, antifolate resistance as well some hall markers in various diseases especially in neurodegenerative diseases. These data suggest that pyrimidine metabolism is paired with other cellular metabolisms and the balance and crosstalk of these metabolism pathway support the cell proliferation and growth. Moreover, patients with high DHODH expression had much worse prognosis than these with lower level, especially in AML. Hence, detecting the expression level of DHODH may provide a promising approach in early diagnosis or predict outcomes in clinical. Taken together, cancer genomic analysis revealed a unique clue for precisely targeting of pyrimidine metabolism pathway in cancer.
Emerging Therapeutics Targeting Pyrimidine Metabolism in Cancers
CAD Inhibitors
PALA
N-(phosphonacetyl)-l-aspartate (PALA) is a potent inhibitor of de novo pyrimidine pathway targeting aspartate transcarbamylase domain of CAD (143). PALA had failed in a phase II trial as single agent owing to the non-ideal efficacy as well as serious side effect due to nonspecifically causing DNA damage in normal cells (144). Additionally, PALA was reported to targeting other metabolic enzymes, such as carbonic anhydrase IV (145). Therefore, more potent, and specific CAD inhibitor is needed to reevaluate the feasibility of targeting CAD.
On the other hand, PALA had showed synergistic anticancer effect in drug combination therapy. For example, Kensler et al. have shown that treatment with drug combination of PALA and acivicin resulted in >80% inhibition of Lewis lung carcinoma tumor growth, and 50% increases in life span (146). Besides, PALA showed synergistical cytotoxic effects with Fluorouracil (5-FU) across most human colon cancer cell lines (147).
DHODH Inhibitors
Leflunomide/Teriflunomide
Leflunomide is an effective DHODH inhibitor that has gained FDA approval for the treatment of rheumatoid arthritis and psoriatic arthritis (148). Leflunomide inhibits DHODH by binding to the ubiquinone binding channel and prevents the production of the orotic acid (149). Teriflunomide (also known as A771726), an active metabolite of leflunomide, was approved by FDA for the treatment of relapsing multiple sclerosis (MS) (150, 151).
In preclinical models, leflunomide and teriflunomide showed anti-proliferative effects against cancer cells from various cancer types through targeting DHODH-driven nucleotide pools accessibility (152, 153). Synergy between leflunomide/teriflunomide and genotoxic chemotherapy agents (doxorubicin, cisplatin, etoposide, and topotecan) was observed by exacerbating DNA damage and overwhelming the DNA damage response (154). In recent years, leflunomide was advanced into clinical trials of solid tumor and hematologic tumor: metastatic TNBC (NCT03709446) and Plasma Cell Myeloma (NCT04370483).
Brequinar
Brequinar is a potent and specific inhibitor of DHODH, originally developed by DuPont Pharmaceuticals (DUP 785; NSC 368390) (155) with an IC50 of ~20 nM upon DHODH in vitro and has a in vivo half-life of ~12 hr. Brequinar has been advanced into early clinical trials of patients with advanced solid tumor malignancies, whereas serious toxic effects limit its clinical development (156–159). In 2016, a pioneering study from David B. Sykes group have revealed the excellent therapeutic benefit and safety of brequinar in treating leukemia using an improved dosage regimen in preclinical models. Inspired by these evidences, DHODH targeting by brequinar was recently re-evaluated towards AML in clinical (NCT03760666).
Other Synthetic DHODH Inhibitors in Clinical
Recently, Bayer AG have reported a highly potent and selective DHODH inhibitor BAY 2402234, which exhibits strong differentiation therapy potential in preclinical leukemia models. However, the clinical trial has been terminated due to the lack of sufficient clinical benefit (NCT 03404726) (137). ASLAN003, a DHODH inhibitor developed by ASLAN Pharmaceuticals (Singapore, SG), was initial designed for the treatment of autoimmune disease. Subsequent studies have shown that ASLAN003 can also induce leukemia cell differentiation in vitro and in vivo, providing a rational basis for clinical application of ASLAN003 in leukemia (NCT 03451084) (136). PTC299, an inhibitor of VEGFA mRNA translation, was subsequently identified as a DHODH inhibitor. The unique dual-mechanism of PTC299 may provide a promising therapeutic opportunity for patients with refractory cancers (NCT 03761069) (138).
Natural Product-Derived Inhibitors
Natural products represent a critical and valuable source of lead compounds and drugs. Currently, only few natural-occurring compounds have been identified as DHODH inhibitor. We previously reported that isobavachalcone (IBC), derived from Traditional Chinese Medicine Psoralea corylifolia, inhibits DHODH with high affinity. IBC dramatically triggers apoptosis as well as overcomes differentiation blockade of multiple AML cell lines. Additionally, the combination of IBC and adriamycin effectively prolong mouse survival in a mouse model of AML (160). On the other hand, using a panel of biochemical assays and structural biology approach, we identified that piperine, a main bioactive constituent of black pepper, as a potent inhibitor of DHODH. We characterized that piperine impairs T cell proliferation as well as reduced inflammation in MOG-induced EAE mouse model with lessened myelin and bold-brain barrier (BBB) destruction by targeting DHODH (161). Collectively, these natural products-derived DHODH inhibitors may represent promising treatment strategy across various diseases.
UMPs Inhibitors
Pyrazofurin
Pyrazofurin was found to inhibit the orotidine 5′-monophosphate decarboxylase activity of UMPS as a nucleoside analogue in the last century with high efficacy especially in acute myelogenous leukemia patients (162). Nevertheless, the resistance to pyrazofurin appeared early in clinical (163–165).
Other Inhibitors
Fluorouracil/Floxuridine/Gemcitabine
Fluorouracil is an analogue of uracil with a fluorine atom at the C-5 position in place of hydrogen, which mimicked uracil incorporating into DNA and RNA, producing intracellularly several active metabolites: fluorodeoxyuridine monophosphate (FdUMP), fluorodeoxyuridine triphosphate (FdUTP) and fluorouridine triphosphate (FUTP) (166). Fluorouracil is reported to bind the deoxyribonucleotide of the FdUMP and the folate cofactor, N5–10-methylenetetrahydrofolate, to thymidylate synthase (TS) to form a covalently bound ternary complex. Up to now, 5-FU has been approved for treating multiple solid tumors including breast, pancreas, stomach, head and neck, and colorectal cancers (167).
Floxuridine is a pyrimidine analogue used as an antineoplastic antimetabolite that is metabolized to fluorouracil, usually as a continuous hepatic arterial infusion to treat hepatic metastases from colon cancer (168). Unlike 5-fluorouracil (5-FU), floxuridine is specifically incorporated into DNA, not into RNA. Inhibition of cell proliferation resulted by floxuridine is 10- to 100-fold higher than that of 5-FU (169, 170).
Gemcitabine is a deoxycytidine analogue, a pyrimidine antimetabolite related to cytarabine. Gemcitabine is converted into phosphate metabolites, which are incorporated in the metabolism of pyrimidine bases and disturb DNA synthesis. Gemcitabine exhibits cell phase specificity, primarily killing cells undergoing DNA synthesis (S-phase) and blocking the progression of cells through the G1/S-phase boundary. In clinical, gemcitabine has been approved to treat various cancers including non-small-cell lung cancer, pancreatic cancer and gallbladder cancer (171–173).
Drugs Resistance and Potential Combination Therapy in the Future
A wealth of clinical studies has demonstrated the strong tolerance and toxicity of multiple inhibitors of pyrimidine pathway, highlighting the urgent need of improved strategy targeting pyrimidine metabolism. Herein, we mainly summary the emerging role of DHODH inhibition in drugs resistance and combination therapy.
DHODH inhibition can sensitize cancer cells to conventional chemotherapy and overcomes corresponding resistance mechanisms by targeting metabolic dependencies. It is reported that pretreatment with leflunomide induced pyrimidine depletion in TNBC cells and overcame doxorubicin resistance (22) and brequinar was reported to increased cell sensitivity to TRAIL therapy (174). Pyrimidine depletion induced by leflunomide may result in a higher incorporation of chemotherapeutics gemcitabine (175).
KRAS mutant driven cancer cell lines exhibit synthetic lethal vulnerability towards pyrimidine biosynthesis (119). For instance, DHODH inhibitors affect energy metabolism as well as glutamine levels in KRAS mutant cell lines with high sensibility (176). As the potential ability to increase p53 synthesis, DHODH inhibition could combine the inhibitors of p53 degradation and enhance the antitumor effect (177). Therefore, DHODH inhibitors combining traditional chemotherapeutic as well as targeted drugs may increase effectiveness and control unwanted adverse side effects in clinical. Nevertheless, the design of efficient combination projects across multiple cancers types remained to be undeniably challenging.
Conclusion and Prospects
Metabolic dysregulation has been identified as an emerging hallmark of cancer. A wealth of evidence has demonstrated that metabolic dependencies and phenotypes can evolve in cancer progresses from premalignant lesions to clinically evident cancers to metastasis malignancies. Therefore, understanding sophisticated cancer metabolism and identifying liabilities will expedite the development of new therapeutics to treat human cancer.
Pyrimidine is a basic and indispensable substrate for nucleic acids, phospholipid, glucose metabolism, and protein glycosylation. Recent works in pyrimidine metabolism intended to focus on assessing the interplay of metabolic phenotypes and intrinsic genetic alternation in cancer. It was identified that KRAS mutant, PTEN deficiency as well as p53 deficiency cells exhibits increased pyrimidine de novo synthesis flux. This dependence on pyrimidine pathway leads to the synthetic lethal target of pyrimidine synthesis in these cells, suggesting the hardwired metabolic vulnerabilities in different gain-of-function mutant cancers. The crosstalk between pyrimidine pathway and other metabolic signaling generates a greater understanding for metabolic heterogeneity and devises approaches to targeting for clinical therapy. Hence, the discovery of treatments targeting precise pyrimidine metabolism could be done once the underlying metabolic difference is recognized in individual patients.
Considering the crucial role of DHODH in rapidly proliferating cells (like lymphocytes), pharmacological targeting DHODH has revealed ideal effect in inflammatory disease, and autoimmune disease. Importantly, increasing evidence have demonstrated that DHODH closely correlates with various oncogenic signaling pathways in many cancers as discussed above. In clinical, DHODH is actively explored as a target in AML differentiation therapy with several inhibitors advanced in multiple clinical trials. Notably, the development of innovative biomarkers of targeting DHODH in AML is critical for precision or personalized medicine. On the other hand, combination therapy of DHODH inhibitors and conventional chemotherapeutics/targeted agents may offer superior clinical strategies for refractory/resistant AML.
Besides, it is reported that DHODH regulates β-catenin pathway through interacting with NH2 terminal of β-catenin directly independent on catalyzed activity (178), suggesting the multi-function properties of DHODH protein. Thus, more diverse, and precise targeting strategies, such as proteolysis-targeting chimera (PROTAC) and molecular glue, are needed to negate the pro-tumor activity of DHODH.
In summary, pyrimidine metabolic remodeling facilitates tumor progression and introduces metabolic vulnerability that can be intervened to treat cancer. It should be noted that cancer metabolism is heterogeneous and flexible, and classical “one-size-fits-all” treatment may fail to achieve satisfactory clinical benefit. With the development of cancer genomics, proteomics, and metabolomics, we prospect that the detailed characterization of the interplay between cellular metabolism and oncogenic signaling will facilitate the development of mechanism-driven precision cancer medicine.
Funding
This work was funded by the National Natural Science Foundation of China (81973362, 81972828), Shanghai Committee of Science and Technology (18431900500, 19ZR1473500), and Open Research Project of Key Laboratory of High-Incidence-Tumor Prevention & Treatment (Guangxi Medical University), Ministry of Education.
Statements
Author contributions
WL and JH contributed to conception and design of the review. WW wrote the first draft of the manuscript. JC and HM performed the statistical analysis. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
RenYLiuQLiuHZhouXZhangYCaiM. Engineering Substrate and Energy Metabolism for Living Cell Production of Cytidine-5′-Diphosphocholine. Biotechnol Bioeng (2020) 117:1426–35. doi: 10.1002/bit.27291
2
Vander HeidenMGDeBerardinisRJ. Understanding the Intersections Between Metabolism and Cancer Biology. Cell (2017) 168:657–69. doi: 10.1016/j.cell.2016.12.039
3
LuengoAGuiDYVander HeidenMG. Targeting Metabolism for Cancer Therapy. Cell Chem Biol (2017) 24:1161–80. doi: 10.1016/j.chembiol.2017.08.028
4
ReavesMLYoungBDHosiosAMXuYFRabinowitzJD. Pyrimidine Homeostasis is Accomplished by Directed Overflow Metabolism. Nature (2013) 500:237–41. doi: 10.1038/nature12445
5
SigoillotFDKotsisDHSerreVSigoillotSMEvansDRGuyHI. Nuclear Localization and Mitogen-Activated Protein Kinase Phosphorylation of the Multifunctional Protein CAD. J Biol Chem (2005) 280:25611–20. doi: 10.1074/jbc.M504581200
6
Moreno-MorcilloMGrande-GarciaARuiz-RamosADel Cano-OchoaFBoskovicJRamon-MaiquesS. Structural Insight Into the Core of CAD, the Multifunctional Protein Leading De Novo Pyrimidine Biosynthesis. Structure (2017) 25:912–923 e915. doi: 10.1016/j.str.2017.04.012
7
RabinovichSAdlerLYizhakKSarverASilbermanAAgronSet al. Diversion of Aspartate in ASS1-deficient Tumours Fosters De Novo Pyrimidine Synthesis. Nature (2015) 527:379–83. doi: 10.1038/nature15529
8
Grande-GarciaALallousNDiaz-TejadaCRamon-MaiquesS. Structure, Functional Characterization, and Evolution of the Dihydroorotase Domain of Human CAD. Structure (2014) 22:185–98. doi: 10.1016/j.str.2013.10.016
9
EvansDRGuyHI. Mammalian Pyrimidine Biosynthesis: Fresh Insights Into an Ancient Pathway. J Biol Chem (2004) 279:33035–8. doi: 10.1074/jbc.R400007200
10
Ben-SahraIHowellJJAsaraJMManningBD. Stimulation of De Novo Pyrimidine Synthesis by Growth Signaling Through mTOR and S6K1. Science (2013) 339:1323–8. doi: 10.1126/science.1228792
11
del Caño-OchoaFMoreno-MorcilloMRamón-MaiquesS. Cad, A Multienzymatic Protein At the Head of De Novo Pyrimidine Biosynthesis. Subcellular Biochem (2019) 93:505–38. doi: 10.1007/978-3-030-28151-9_17
12
MartinEPalmicNSanquerSLenoirCHauckFMongellazCet al. CTP Synthase 1 Deficiency in Humans Reveals its Central Role in Lymphocyte Proliferation. Nature (2014) 510:288–92. doi: 10.1038/nature13386
13
KimJHuZCaiLLiKChoiEFaubertBet al. CPS1 Maintains Pyrimidine Pools and DNA Synthesis in KRAS/LKB1-mutant Lung Cancer Cells. Nature (2017) 546:168–72. doi: 10.1038/nature22359
14
ReisRAGCalilFAFelicianoPRPinheiroMPNonatoMC. The Dihydroorotate Dehydrogenases: Past and Present. Arch Biochem biophysics (2017) 632:175–91. doi: 10.1016/j.abb.2017.06.019
15
BajzikovaMKovarovaJCoelhoARBoukalovaSOhSRohlenovaKet al. Reactivation of Dihydroorotate Dehydrogenase-Driven Pyrimidine Biosynthesis Restores Tumor Growth of Respiration-Deficient Cancer Cells. Cell Metab (2019) 29:399–416 e310. doi: 10.1016/j.cmet.2018.10.014
16
BoukalovaSHubackovaSMilosevicMEzrovaZNeuzilJRohlenaJet al. Dihydroorotate Dehydrogenase in Oxidative Phosphorylation and Cancer. Biochim Biophys Acta Mol basis Dis (2020) 1866:165759. doi: 10.1016/j.bbadis.2020.165759
17
Martínez-ReyesICardonaLRKongHVasanKMcElroyGSWernerMet al. Mitochondrial Ubiquinol Oxidation is Necessary for Tumour Growth. Nat (2020) 585:288–92. doi: 10.1038/s41586-020-2475-6
18
BennettCFO’MalleyKEPerryEABalsaELatorre-MuroPRileyCLet al. Peroxisomal-Derived Ether Phospholipids Link Nucleotides to Respirasome Assembly. Nat Chem Biol (2021). doi: 10.1038/s41589-021-00772-z
19
CantorJRAbu-RemailehMKanarekNFreinkmanEGaoXLouissaintAJret al. Physiologic Medium Rewires Cellular Metabolism and Reveals Uric Acid as an Endogenous Inhibitor of UMP Synthase. Cell (2017) 169:258–272 e217. doi: 10.1016/j.cell.2017.03.023
20
SykesDB. The Emergence of Dihydroorotate Dehydrogenase (DHODH) as a Therapeutic Target in Acute Myeloid Leukemia. Expert Opin Ther Targets (2018) 22:893–8. doi: 10.1080/14728222.2018.1536748
21
OkesliAKhoslaCBassikMC. Human Pyrimidine Nucleotide Biosynthesis as a Target for Antiviral Chemotherapy. Curr Opin Biotechnol (2017) 48:127–34. doi: 10.1016/j.copbio.2017.03.010
22
BrownKKSpinelliJBAsaraJMTokerA. Adaptive Reprogramming of De Novo Pyrimidine Synthesis Is a Metabolic Vulnerability in Triple-Negative Breast Cancer. Cancer Discovery (2017) 7:391–9. doi: 10.1158/2159-8290.cd-16-0611
23
MathurDStratikopoulosEOzturkSSteinbachNPegnoSSchoenfeldSet al. Pten Regulates Glutamine Flux to Pyrimidine Synthesis and Sensitivity to Dihydroorotate Dehydrogenase Inhibition. Cancer Discovery (2017) 7:380–90. doi: 10.1158/2159-8290.CD-16-0612
24
CunninghamJTMorenoMVLodiARonenSMRuggeroD. Protein and Nucleotide Biosynthesis are Coupled by a Single Rate-Limiting Enzyme, PRPS2, to Drive Cancer. Cell (2014) 157:1088–103. doi: 10.1016/j.cell.2014.03.052
25
MadakJTBankheadACuthbertsonCRShowalterHDNeamatiN. Revisiting the Role of Dihydroorotate Dehydrogenase as a Therapeutic Target for Cancer. Pharmacol Ther (2019) 195:111–31. doi: 10.1016/j.pharmthera.2018.10.012
26
LofflerMFairbanksLDZameitatEMarinakiAMSimmondsHA. Pyrimidine Pathways in Health and Disease. Trends Mol Med (2005) 11:430–7. doi: 10.1016/j.molmed.2005.07.003
27
Pedroza-GarciaJANajera-MartinezMMazubertCAguilera-AlvaradoPDrouin-WahbiJSanchez-NietoSet al. Role of Pyrimidine Salvage Pathway in the Maintenance of Organellar and Nuclear Genome Integrity. Plant J (2019) 97:430–46. doi: 10.1111/tpj.14128
28
YoungJD. The SLC28 (CNT) and SLC29 (ENT) Nucleoside Transporter Families: A 30-Year Collaborative Odyssey. Biochem Soc Trans (2016) 44:869–76. doi: 10.1042/BST20160038
29
Pastor-AngladaMUrtasunNPerez-TorrasS. Intestinal Nucleoside Transporters: Function, Expression, and Regulation. Compr Physiol (2018) 8:1003–17. doi: 10.1002/cphy.c170039
30
KrysDHamannIWuestMWuestF. Effect of Hypoxia on Human Equilibrative Nucleoside Transporters hENT1 and hENT2 in Breast Cancer. FASEB J (2019) 33(12):13837–51. doi: 10.1096/fj.201900870RR
31
JiraskovaLRyskaADuintjer TebbensEJHornychovaHCeckaFStaudFet al. Are ENT1/ENT1, NOTCH3, and Mir-21 Reliable Prognostic Biomarkers in Patients With Resected Pancreatic Adenocarcinoma Treated With Adjuvant Gemcitabine Monotherapy? Cancers (Basel) (2019) 11(11). doi: 10.3390/cancers11111621
32
OkamuraYYasukawaSNarimatsuHBokuNFukutomiAKonishiMet al. Human Equilibrative Nucleoside Transporter-1 Expression is a Predictor in Patients With Resected Pancreatic Cancer Treated With Adjuvant S-1 Chemotherapy. Cancer Sci (2020) 111:548–60. doi: 10.1111/cas.14258
33
WangCBuolamwiniJK. A Novel RNA Variant of Human Concentrative Nucleoside Transporter 1 (hCNT1) That is a Potential Cancer Biomarker. Exp Hematol Oncol (2019) 8:18. doi: 10.1186/s40164-019-0144-y
34
VlachodimouAKonstantinopoulouKAPIJHeitmanLH. Affinity, Binding Kinetics and Functional Characterization of Draflazine Analogues for Human Equilibrative Nucleoside Transporter 1 (SLC29A1). Biochem Pharmacol (2020) 172:113747. doi: 10.1016/j.bcp.2019.113747
35
Molina-ArcasMMoreno-BuenoGCano-SoldadoPHernández-VargasHCasadoFJPalaciosJet al. Human Equilibrative Nucleoside Transporter-1 (hENT1) is Required for the Transcriptomic Response of the Nucleoside-Derived Drug 5′-DFUR in Breast Cancer MCF7 Cells. Biochem Pharmacol (2006) 72:1646–56. doi: 10.1016/j.bcp.2006.07.036
36
MaZLuSSunDBaiMJiangTLinNet al. Roles of Organic Anion Transporter 2 and Equilibrative Nucleoside Transporter 1 in Hepatic Disposition and Antiviral Activity of Entecavir During non-Pregnancy and Pregnancy. Br J Pharmacol (2019) 176:3236–49. doi: 10.1111/bph.14756
37
LiouJ-YDutschmanGELamWJiangZChengY-C. Characterization of Human Ump/Cmp Kinase and Its Phosphorylation of d- and l-Form Deoxycytidine Analogue Monophosphates. Cancer Res (2002) 62:1624.
38
Ben-SahraIHowellJJAsaraJMManningBD. Stimulation of De Novo Pyrimidine Synthesis by Growth Signaling Through mTOR and S6K1. Sci (New York N.Y.) (2013) 339:1323–8. doi: 10.1126/science.1228792
39
ExtonJH. Phosphatidylcholine Breakdown and Signal Transduction. Biochim Biophys Acta (BBA) - Lipids Lipid Metab (1994) 1212:26–42. doi: 10.1016/0005-2760(94)90186-4
40
CornellRBRidgwayND. CTP:Phosphocholine Cytidylyltransferase: Function, Regulation, and Structure of an Amphitropic Enzyme Required for Membrane Biogenesis. Prog Lipid Res (2015) 59:147–71. doi: 10.1016/j.plipres.2015.07.001
41
EggerSChaikuadAKavanaghKLOppermannUNidetzkyB. Structure and Mechanism of Human UDP-glucose 6-Dehydrogenase. J Biol Chem (2011) 286:23877–87. doi: 10.1074/jbc.M111.234682
42
EggerSChaikuadAKavanaghKLOppermannUNidetzkyB. UDP-Glucose Dehydrogenase: Structure and Function of a Potential Drug Target. Biochem Soc Trans (2010) 38:1378–85. doi: 10.1042/bst0381378
43
HartGWHousleyMPSlawsonC. Cycling of O-linked beta-N-acetylglucosamine on Nucleocytoplasmic Proteins. Nature (2007) 446:1017–22. doi: 10.1038/nature05815
44
PinhoSSReisCA. Glycosylation in Cancer: Mechanisms and Clinical Implications. Nat Rev Cancer (2015) 15:540–55. doi: 10.1038/nrc3982
45
<O-GlcNAc-JBC1997-O-Linked GlcNAc Transferase Is a Conserved Nucleocytoplasmic Protein Containing Tetratricopeptide Repeats.
46
ToscaniniMAFavaroloMBGonzalez FlechaFLEbertBRautengartenCBredestonLM. Conserved Glu-47 and Lys-50 Residues are Critical for UDP-N-acetylglucosamine/UMP Antiport Activity of the Mouse Golgi-associated Transporter Slc35a3. J Biol Chem (2019) 294:10042–54. doi: 10.1074/jbc.RA119.008827
47
KreppelLKBlombergMAHartGW. Dynamic Glycosylation of Nuclear and Cytosolic Proteins: CLONING and CHARACTERIZATION of A Unique O-GlcNAc Transferase WITH Multiple TETRATRICOPEPTIDE Repeats. J Biol Chem (1997) 272:9308–15. doi: 10.1074/jbc.272.14.9308
48
FagonePJackowskiS. Phosphatidylcholine and the CDP-choline Cycle. Biochim Biophys Acta (2013) 1831:523–32. doi: 10.1016/j.bbalip.2012.09.009
49
BrewerJWJackowskiS. Upr-Mediated Membrane Biogenesis in B Cells. Biochem Res Int (2012) 2012:738471. doi: 10.1155/2012/738471
50
McMasterCR. Lipid Metabolism and Vesicle Trafficking: More Than Just Greasing the Transport Machinery. Biochem Cell Biol = Biochimie biologie cellulaire (2001) 79:681–92. doi: 10.1139/o01-139
51
SarriESicartALázaro-DiéguezFEgeaG. Phospholipid Synthesis Participates in the Regulation of Diacylglycerol Required for Membrane Trafficking At the Golgi Complex. J Biol Chem (2011) 286:28632–43. doi: 10.1074/jbc.M111.267534
52
ElgundiZPapanicolaouMMajorGCoxTRMelroseJWhitelockJMet al. Cancer Metastasis: The Role of the Extracellular Matrix and the Heparan Sulfate Proteoglycan Perlecan. Front Oncol (2020) 9:1482. doi: 10.3389/fonc.2019.01482
53
AfratisNGialeliCNikitovicDTsegenidisTKarousouETheocharisADet al. Glycosaminoglycans: Key Players in Cancer Cell Biology and Treatment. FEBS J (2012) 279:1177–97. doi: 10.1111/j.1742-4658.2012.08529.x
54
WangXLiuRZhuWChuHYuHWeiPet al. UDP-Glucose Accelerates SNAI1 mRNA Decay and Impairs Lung Cancer Metastasis. Nature (2019) 571:127–31. doi: 10.1038/s41586-019-1340-y
55
OyinladeOWeiSLalBLaterraJZhuHGoodwinCRet al. Targeting UDP-α-D-glucose 6-Dehydrogenase Inhibits Glioblastoma Growth and Migration. Oncogene (2018) 37:2615–29. doi: 10.1038/s41388-018-0138-y
56
GuoJ-YChiuC-HWangM-JLiF-AChenJ-Y. Proteoglycan Serglycin Promotes non-Small Cell Lung Cancer Cell Migration Through the Interaction of its Glycosaminoglycans With CD44. J BioMed Sci (2020) 27:2–2. doi: 10.1186/s12929-019-0600-3
57
VigettiDOriMViolaMGenasettiAKarousouERizziMet al. Molecular Cloning and Characterization of UDP-glucose Dehydrogenase From the Amphibian Xenopus Laevis and its Involvement in Hyaluronan Synthesis. J Biol Chem (2006) 281:8254–63. doi: 10.1074/jbc.M508516200
58
García-GarcíaMJAndersonKV. Essential Role of Glycosaminoglycans in Fgf Signaling During Mouse Gastrulation. Cell (2003) 114:727–37. doi: 10.1016/s0092-8674(03)00715-3
59
GoodwinCRAhmedAKXiaS. Udp-α-D-glucose 6-Dehydrogenase: A Promising Target for Glioblastoma. Oncotarget (2019) 10:1542–3. doi: 10.18632/oncotarget.26670
60
WeiQGalbenusRRazaACernyRLSimpsonMA. Androgen-Stimulated UDP-glucose Dehydrogenase Expression Limits Prostate Androgen Availability Without Impacting Hyaluronan Levels. Cancer Res (2009) 69(6):2332–9. doi: 10.1158/0008-5472.CAN-08-3083
61
KimJLeeHMCaiFKoBYangCLieuELet al. The Hexosamine Biosynthesis Pathway is a Targetable Liability in KRAS/LKB1 Mutant Lung Cancer. Nat Metab (2020) 2(12):1401–12. doi: 10.1038/s42255-020-00316-0
62
ParkS-YYoonS-JFreire-de-LimaLKimJ-HHakomoriS-I. Control of Cell Motility by Interaction of Gangliosides, Tetraspanins, and Epidermal Growth Factor Receptor in A431 Versus KB Epidermoid Tumor Cells. Carbohydr Res (2009) 344:1479–86. doi: 10.1016/j.carres.2009.04.032
63
TodeschiniARDos SantosJNHandaKHakomoriS-I. Ganglioside GM2-Tetraspanin Cd82 Complex Inhibits Met and Its Cross-Talk With Integrins, Providing a Basis for Control of Cell Motility Through Glycosynapse. J Biol Chem (2007) 282:8123–33. doi: 10.1074/jbc.M611407200
64
JulienSBobowskiMSteenackersALe BourhisXDelannoyP. How Do Gangliosides Regulate Rtks Signaling? Cells (2013) 2:751–67. doi: 10.3390/cells2040751
65
TaniguchiNA. Sugar-Coated Switch for Cellular Growth and Arrest. Nat Chem Biol (2007) 3:307–9. doi: 10.1038/nchembio0607-307
66
ItkonenHMMinnerSGuldvikIJSandmannMJTsourlakisMCBergeVet al. O-Glcnac Transferase Integrates Metabolic Pathways to Regulate the Stability of c-MYC in Human Prostate Cancer Cells. Cancer Res (2013) 73(16):5277. doi: 10.1158/0008-5472.CAN-13-0549
67
CaldwellSAJacksonSRShahriariKSLynchTPSethiGWalkerSet al. Nutrient Sensor O-GlcNAc Transferase Regulates Breast Cancer Tumorigenesis Through Targeting of the Oncogenic Transcription Factor Foxm1. Oncogene (2010) 29:2831–42. doi: 10.1038/onc.2010.41
68
BoscherCDennisJWNabiIR. Glycosylation, Galectins and Cellular Signaling. Curr Opin Cell Biol (2011) 23:383–92. doi: 10.1016/j.ceb.2011.05.001
69
LauKSPartridgeEAGrigorianASilvescuCIReinholdVNDemetriouMet al. Complex N-Glycan Number and Degree of Branching Cooperate to Regulate Cell Proliferation and Differentiation. Cell (2007) 129:123–34. doi: 10.1016/j.cell.2007.01.049
70
CrociDOCerlianiJPPintoNAMorosiLGRabinovichGA. Regulatory Role of Glycans in the Control of Hypoxia-Driven Angiogenesis and Sensitivity to Anti-Angiogenic Treatment. Glycobiology (2014) 24:1283–90. doi: 10.1093/glycob/cwu083
71
LynchTPFerrerCMJacksonSRShahriariKSVossellerKReginatoMJ. Critical Role of O-Linked β-N-acetylglucosamine Transferase in Prostate Cancer Invasion, Angiogenesis, and Metastasis. J Biol Chem (2012) 287:11070–81. doi: 10.1074/jbc.M111.302547
72
FardiniYDehennautVLefebvreT. O-Glcnacylation: A New Cancer Hallmark? Front Endocrinol (Lausanne) (2013) 4:99. doi: 10.3389/fendo.2013.00099
73
YangWHKimJENamHWJuJWKimHSKimYSet al. Modification of p53 With O-linked N-Acetylglucosamine Regulates p53 Activity and Stability. Nat Cell Biol (2006) 8:1074–83. doi: 10.1038/ncb1470
74
QianKWangSFuMZhouJSinghJPLiM-Det al. Transcriptional Regulation of O-GlcNAc Homeostasis is Disrupted in Pancreatic Cancer. J Biol Chem (2018) 293:13989–4000. doi: 10.1074/jbc.RA118.004709
75
FerrerCMLynchTPSodiVLFalconeJNSchwabLPPeacockDLet al. O-GlcNAcylation Regulates Cancer Metabolism and Survival Stress Signaling Via Regulation of the HIF-1 Pathway. Mol Cell (2014) 54:820–31. doi: 10.1016/j.molcel.2014.04.026
76
PatraKCHayN. The Pentose Phosphate Pathway and Cancer. Trends Biochem Sci (2014) 39:347–54. doi: 10.1016/j.tibs.2014.06.005
77
JiangPDuWWuM. Regulation of the Pentose Phosphate Pathway in Cancer. Protein Cell (2014) 5:592–602. doi: 10.1007/s13238-014-0082-8
78
RaoXDuanXMaoWLiXLiZLiQet al. O-GlcNAcylation of G6PD Promotes the Pentose Phosphate Pathway and Tumor Growth. Nat Commun (2015) 6:8468–8. doi: 10.1038/ncomms9468
79
YiWClarkPMMasonDEKeenanMCHillCGoddardWA,3et al. Phosphofructokinase 1 Glycosylation Regulates Cell Growth and Metabolism. Sci (New York N.Y.) (2012) 337:975–80. doi: 10.1126/science.1222278
80
RamakrishnanPClarkPMMasonDEPetersECHsieh-WilsonLCBaltimoreD. Activation of the Transcriptional Function of the NF-κb Protein c-Rel by O-GlcNAc Glycosylation. Sci Signal (2013) 6:ra75–5. doi: 10.1126/scisignal.2004097
81
SevignyMBLiC-FAlasMHughes-FulfordM. Glycosylation Regulates Turnover of Cyclooxygenase-2. FEBS Lett (2006) 580:6533–6. doi: 10.1016/j.febslet.2006.10.073
82
SevignyMBGrahamKPonceELouieMCMitchellK. Glycosylation of Human Cyclooxygenase-2 (COX-2) Decreases the Efficacy of Certain COX-2 Inhibitors. Pharmacol Res (2012) 65:445–50. doi: 10.1016/j.phrs.2012.01.001
83
BassagañasSAllendeHCoblerLOrtizMRLlopEde BolósCet al. Inflammatory Cytokines Regulate the Expression of Glycosyltransferases Involved in the Biosynthesis of Tumor-Associated Sialylated Glycans in Pancreatic Cancer Cell Lines. Cytokine (2015) 75:197–206. doi: 10.1016/j.cyto.2015.04.006
84
PadróMMejías-LuqueRCoblerLGarridoMPérez-GarayMPuigSet al. Regulation of Glycosyltransferases and Lewis Antigens Expression by IL-1β and IL-6 in Human Gastric Cancer Cells. Glycoconjugate J (2011) 28:99–110. doi: 10.1007/s10719-011-9327-4
85
TanWJiangPZhangWHuZLinSChenLet al. Posttranscriptional Regulation of De Novo Lipogenesis by Glucose-Induced O-Glcnacylation. Mol Cell (2021) 81(9):1890–904.e7. doi: 10.1016/j.molcel.2021.02.009
86
ChenJDongXChengXZhuQZhangJLiQet al. Ogt Controls Neural Stem/Progenitor Cell Pool and Adult Neurogenesis Through Modulating Notch Signaling. Cell Rep (2021) 34:108905. doi: 10.1016/j.celrep.2021.108905
87
HuangHWuQGuoXHuangTXieXWangLet al. O-GlcNAcylation Promotes the Migratory Ability of Hepatocellular Carcinoma Cells Via Regulating FOXA2 Stability and Transcriptional Activity. J Cell Physiol (2021). doi: 10.1002/jcp.30385
88
XiangJChenCLiuRGouDChangLDengHet al. Gluconeogenic Enzyme PCK1 Deficiency Promotes CHK2 O-GlcNAcylation and Hepatocellular Carcinoma Growth Upon Glucose Deprivation. J Clin Invest (2021) 131. doi: 10.1172/jci144703
89
YamaguchiNWeinbergEMNguyenALibertiMVGoodarziHJanjigianYYet al. PCK1 and DHODH Drive Colorectal Cancer Liver Metastatic Colonization and Hypoxic Growth by Promoting Nucleotide Synthesis. Elife (2019) 8:e52135. doi: 10.7554/eLife.52135
90
SiddiquiAVazakidouMESchwabANapoliFFernandez-MolinaCRapaIet al. Thymidylate Synthase is Functionally Associated With ZEB1 and Contributes to the Epithelial-to-Mesenchymal Transition of Cancer Cells. J Pathol (2017) 242:221–33. doi: 10.1002/path.4897
91
SiddiquiAGollavilliPNSchwabAVazakidouMEErsanPGRamakrishnanMet al. Thymidylate Synthase Maintains the De-Differentiated State of Triple Negative Breast Cancers. Cell Death Differ (2019) 26:2223–36. doi: 10.1038/s41418-019-0289-6
92
SiddiquiMAGollavilliPNRameshVParmaBSchwabAVazakidouMEet al. Thymidylate Synthase Drives the Phenotypes of Epithelial-to-Mesenchymal Transition in non-Small Cell Lung Cancer. Br J Cancer (2021) 124:281–9. doi: 10.1038/s41416-020-01095-x
93
DangCV. MYC on the Path to Cancer. Cell (2012) 149:22–35. doi: 10.1016/j.cell.2012.03.003
94
VitaMHenrikssonM. The Myc Oncoprotein as a Therapeutic Target for Human Cancer. Semin Cancer Biol (2006) 16:318–30. doi: 10.1016/j.semcancer.2006.07.015
95
SoucekLWhitfieldJMartinsCPFinchAJMurphyDJSodirNMet al. Modelling Myc Inhibition as a Cancer Therapy. Nature (2008) 455:679–83. doi: 10.1038/nature07260
96
MiltenbergerRJSukowKAFarnhamPJ. An E-box-mediated Increase in Cad Transcription At the G1/S-phase Boundary is Suppressed by Inhibitory c-Myc Mutants. Mol Cell Biol (1995) 15:2527. doi: 10.1128/MCB.15.5.2527
97
EberhardySRFarnhamPJ. Myc Recruits P-TEFb to Mediate the Final Step in the Transcriptional Activation of the Cad Promoter. J Biol Chem (2002) 277:40156–62. doi: 10.1074/jbc.M207441200
98
GarganoBAmenteSMajelloBLaniaL. P-TEFb is a Crucial Co-Factor for Myc Transactivation. Cell Cycle (2007) 6:2031–7. doi: 10.4161/cc.6.16.4554
99
SatohKYachidaSSugimotoMOshimaMNakagawaTAkamotoSet al. Global Metabolic Reprogramming of Colorectal Cancer Occurs At Adenoma Stage and is Induced by MYC. Proc Natl Acad Sci USA (2017) 114:E7697–706. doi: 10.1073/pnas.1710366114
100
WuDWangWChenWLianFLangLHuangYet al. Pharmacological Inhibition of Dihydroorotate Dehydrogenase Induces Apoptosis and Differentiation in Acute Myeloid Leukemia Cells. Haematologica (2018) 103:1472–83. doi: 10.3324/haematol.2018.188185
101
WhiteRMCechJRatanasirintrawootSLinCYRahlPBBurkeCJet al. DHODH Modulates Transcriptional Elongation in the Neural Crest and Melanoma. Nature (2011) 471:518–22. doi: 10.1038/nature09882
102
MacSMFarnhamPJ. CAD, a c-Myc Target Gene, is Not Deregulated in Burkitt’s Lymphoma Cell Lines. Mol Carcinog (2000) 27:84–96. doi: 10.1002/(SICI)1098-2744(200002)27:2<84::AID-MC4>3.0.CO;2-3
103
DangCVLeAGaoP. MYC-Induced Cancer Cell Energy Metabolism and Therapeutic Opportunities. Clin Cancer Res (2009) 15:6479–83. doi: 10.1158/1078-0432.CCR-09-0889
104
GodaNRyanHEKhadiviBMcNultyWRickertRCJohnsonRS. Hypoxia-Inducible Factor 1alpha is Essential for Cell Cycle Arrest During Hypoxia. Mol Cell Biol (2003) 23:359–69. doi: 10.1128/mcb.23.1.359-369.2003
105
ChenK-FLaiY-YSunHSTsaiS-J. Transcriptional Repression of Human Cad Gene by Hypoxia Inducible Factor-1alpha. Nucleic Acids Res (2005) 33:5190–8. doi: 10.1093/nar/gki839
106
KhanSAbdelrahimMSamudioISafeS. Estrogen Receptor/Sp1 Complexes are Required for Induction of Cad Gene Expression by 17beta-Estradiol in Breast Cancer Cells. Endocrinology (2003) 144:2325–35. doi: 10.1210/en.2002-0149
107
SavilleBWormkeMWangFNguyenTEnmarkEKuiperGet al. Ligand-, Cell-, and Estrogen Receptor Subtype (α/β)-Dependent Activation At GC-rich (Sp1) Promoter Elements. J Biol Chem (2000) 275:5379–87. doi: 10.1074/jbc.275.8.5379
108
TongYTanYZhouCMelmedS. Pituitary Tumor Transforming Gene Interacts With Sp1 to Modulate G1/S Cell Phase Transition. Oncogene (2007) 26:5596–605. doi: 10.1038/sj.onc.1210339
109
LaneANFanTWM. Regulation of Mammalian Nucleotide Metabolism and Biosynthesis. Nucleic Acids Res (2015) 43:2466–85. doi: 10.1093/nar/gkv047
110
AbdelrahimMSamudioISmithRBurghardtRSafeS. Small Inhibitory Rna Duplexes for Sp1 Mrna Block Basal and Estrogen-induced Gene Expression and Cell Cycle Progression in MCF-7 Breast Cancer Cells. J Biol Chem (2002) 277:28815–22. doi: 10.1074/jbc.M203828200
111
LeeLKellyREPastra-LandisSCEvansDR. Oligomeric Structure of the Multifunctional Protein CAD That Initiates Pyrimidine Biosynthesis in Mammalian Cells. Proc Natl Acad Sci USA (1985) 82:6802–6. doi: 10.1073/pnas.82.20.6802
112
SigoillotFDBerkowskiJASigoillotSMKotsisDHGuyHI. Cell Cycle-Dependent Regulation of Pyrimidine Biosynthesis. J Biol Chem (2003) 278:3403–9. doi: 10.1074/jbc.M211078200
113
GravesLMGuyHIKozlowskiPHuangMLazarowskiEPopeRMet al. Regulation of Carbamoyl Phosphate Synthetase by MAP Kinase. Nature (2000) 403:328–32. doi: 10.1038/35002111
114
GaravitoMFNarváez-OrtizHYZimmermannBH. Pyrimidine Metabolism: Dynamic and Versatile Pathways in Pathogens and Cellular Development. J Genet Genomics (2015) 42:195–205. doi: 10.1016/j.jgg.2015.04.004
115
KotsisDHMaskoEMSigoillotFDGregorioRDGuy-EvansHIEvansDR. Protein Kinase A Phosphorylation of the Multifunctional Protein CAD Antagonizes Activation by the MAP Kinase Cascade. Mol Cell Biochem (2007) 301:69–81. doi: 10.1007/s11010-006-9398-x
116
IadevaiaVHuoYZhangZFosterLJProudCG. Roles of the Mammalian Target of Rapamycin, mTOR, in Controlling Ribosome Biogenesis and Protein Synthesis. Biochem Soc Trans (2012) 40:168–72. doi: 10.1042/bst20110682
117
RobitailleAMChristenSShimobayashiMCornuMFavaLLMoesSet al. Quantitative Phosphoproteomics Reveal Mtorc1 Activates De Novo Pyrimidine Synthesis. Science (2013) 339:1320. doi: 10.1126/science.1228771
118
Gonzalez HerreraKNZaganjorEIshikawaYSpinelliJBYoonHLinJRet al. Small-Molecule Screen Identifies De Novo Nucleotide Synthesis as a Vulnerability of Cells Lacking Sirt3. Cell Rep (2018) 22:1945–55. doi: 10.1016/j.celrep.2018.01.076
119
KoundinyaMSudhalterJCourjaudALionneBTouyerGBonnetLet al. Dependence on the Pyrimidine Biosynthetic Enzyme Dhodh Is a Synthetic Lethal Vulnerability in Mutant Kras-Driven Cancers. Cell Chem Biol (2018) 25:705–717 e711. doi: 10.1016/j.chembiol.2018.03.005
120
LiJYenCLiawDPodsypaninaKBoseSWangSIet al. PTEN, a Putative Protein Tyrosine Phosphatase Gene Mutated in Human Brain, Breast, and Prostate Cancer. Science (1997) 275:1943. doi: 10.1126/science.275.5308.1943
121
LevineAJPuzio-KuterAM. The Control of the Metabolic Switch in Cancers by Oncogenes and Tumor Suppressor Genes. Science (2010) 330:1340. doi: 10.1126/science.1193494
122
WiseDRDeBerardinisRJMancusoASayedNZhangX-YPfeifferHKet al. Myc Regulates a Transcriptional Program That Stimulates Mitochondrial Glutaminolysis and Leads to Glutamine Addiction. Proc Natl Acad Sci USA (2008) 105:18782–7. doi: 10.1073/pnas.0810199105
123
Santana-CodinaNRoethAAZhangYYangAMashadovaOAsaraJMet al. Oncogenic KRAS Supports Pancreatic Cancer Through Regulation of Nucleotide Synthesis. Nat Commun (2018) 9:4945. doi: 10.1038/s41467-018-07472-8
124
YingHKimmelmanACLyssiotisCAHuaSChuGCFletcher-SananikoneEet al. Oncogenic Kras Maintains Pancreatic Tumors Through Regulation of Anabolic Glucose Metabolism. Cell (2012) 149:656–70. doi: 10.1016/j.cell.2012.01.058
125
PatelDMenonDBernfeldEMrozVKalanSLoayzaDet al. Aspartate Rescues s-Phase Arrest Caused by Suppression of Glutamine Utilization in KRas-driven Cancer Cells. J Biol Chem (2016) 291:9322–9. doi: 10.1074/jbc.M115.710145
126
WeinbergFHamanakaRWheatonWWWeinbergSJosephJLopezMet al. Mitochondrial Metabolism and ROS Generation are Essential for Kras-mediated Tumorigenicity. Proc Natl Acad Sci USA (2010) 107:8788–93. doi: 10.1073/pnas.1003428107
127
van der WindtGJEvertsBChangCHCurtisJDFreitasTCAmielEet al. Mitochondrial Respiratory Capacity is a Critical Regulator of CD8+ T Cell Memory Development. Immunity (2012) 36:68–78. doi: 10.1016/j.immuni.2011.12.007
128
KawauchiKArakiKTobiumeKTanakaN. p53 Regulates Glucose Metabolism Through an IKK-NF-κb Pathway and Inhibits Cell Transformation. Nat Cell Biol (2008) 10:611–8. doi: 10.1038/ncb1724
129
RosSFlöterJKaymakIDa CostaCHouddaneADubuisSet al. 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 is Essential for p53-null Cancer Cells. Oncogene (2017) 36:3287–99. doi: 10.1038/onc.2016.477
130
BensaadKTsurutaASelakMAVidalMNCNakanoKBartronsRet al. TIGAR, a P53-Inducible Regulator of Glycolysis and Apoptosis. Cell (2006) 126:107–20. doi: 10.1016/j.cell.2006.05.036
131
JiangPDuWWangXMancusoAGaoXWuMet al. p53 Regulates Biosynthesis Through Direct Inactivation of glucose-6-phosphate Dehydrogenase. Nat Cell Biol (2011) 13:310–6. doi: 10.1038/ncb2172
132
HitosugiTZhouLElfSFanJKangH-BSeoJHet al. Phosphoglycerate Mutase 1 Coordinates Glycolysis and Biosynthesis to Promote Tumor Growth. Cancer Cell (2012) 22:585–600. doi: 10.1016/j.ccr.2012.09.020
133
KaymakIMaierCRSchmitzWCampbellADDankworthBAdeCPet al. Mevalonate Pathway Provides Ubiquinone to Maintain Pyrimidine Synthesis and Survival in P53-Deficient Cancer Cells Exposed to Metabolic Stress. Cancer Res (2020) 80:189. doi: 10.1158/0008-5472.CAN-19-0650
134
HubackovaSDavidovaEBoukalovaSKovarovaJBajzikovaMCoelhoAet al. Replication and Ribosomal Stress Induced by Targeting Pyrimidine Synthesis and Cellular Checkpoints Suppress p53-deficient Tumors. Cell Death Dis (2020) 11:110. doi: 10.1038/s41419-020-2224-7
135
SykesDBKfouryYSMercierFEWawerMJLawJMHaynesMKet al. Inhibition of Dihydroorotate Dehydrogenase Overcomes Differentiation Blockade in Acute Myeloid Leukemia. Cell (2016) 167:171–86.e115. doi: 10.1016/j.cell.2016.08.057
136
ZhouJQuahJYNgYChooiJYTohSHLinBet al. ASLAN003, a Potent Dihydroorotate Dehydrogenase Inhibitor for Differentiation of Acute Myeloid Leukemia. Haematologica (2019) 105(9):2286–97. doi: 10.3324/haematol.2019.230482
137
ChristianSMerzCEvansLGradlSSeidelHFribergAet al. The Novel Dihydroorotate Dehydrogenase (DHODH) Inhibitor BAY 2402234 Triggers Differentiation and is Effective in the Treatment of Myeloid Malignancies. Leukemia (2019) 33:2403–15. doi: 10.1038/s41375-019-0461-5
138
CaoLWeetallMTrottaCCintronKMaJKimMJet al. Targeting of Hematologic Malignancies With PTC299, A Novel Potent Inhibitor of Dihydroorotate Dehydrogenase With Favorable Pharmaceutical Properties. Mol Cancer Ther (2019) 18:3–16. doi: 10.1158/1535-7163.MCT-18-0863
139
LewisTASykesDBLawJMMunozBRustiguelJKNonatoMCet al. Development of ML390: A Human DHODH Inhibitor That Induces Differentiation in Acute Myeloid Leukemia. ACS Med Chem Lett (2016) 7:1112–7. doi: 10.1021/acsmedchemlett.6b00316
140
TangZLiCKangBGaoGLiCZhangZet al. GEPIA: A Web Server for Cancer and Normal Gene Expression Profiling and Interactive Analyses. Nucleic Acids Res (2017) 45:W98–W102. doi: 10.1093/nar/gkx247
141
BarretinaJCaponigroGStranskyNVenkatesanKMargolinAAKimSet al. The Cancer Cell Line Encyclopedia Enables Predictive Modelling of Anticancer Drug Sensitivity. Nature (2012) 483:603–7. doi: 10.1038/nature11003
142
SzklarczykDGableALLyonDJungeAWyderSHuerta-CepasJet al. String v11: Protein-Protein Association Networks With Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res (2019) 47:607–d613. doi: 10.1093/nar/gky1131
143
CollinsKDStarkGR. Aspartate Transcarbamylase. Interaction With the Transition State Analogue N-(Phosphonacetyl)-L-Aspartate. J Biol Chem (1971) 246:6599–605. doi: 10.1016/S0021-9258(19)34156-0
144
HastakKPaulRKAgarwalMKThakurVSAminARAgrawalSet al. DNA Synthesis From Unbalanced Nucleotide Pools Causes Limited DNA Damage That Triggers ATR-CHK1-dependent p53 Activation. Proc Natl Acad Sci USA (2008) 105:6314–9. doi: 10.1073/pnas.0802080105
145
WinumJYInnocentiAGagnardVMonteroJLScozzafavaAVulloDet al. Carbonic Anhydrase Inhibitors. Interaction of Isozymes I, Ii, IV, V, and IX With Organic Phosphates and Phosphonates. Bioorganic Med Chem Lett (2005) 15:1683–6. doi: 10.1016/j.bmcl.2005.01.049
146
KenslerTWReckLJCooneyDA. Therapeutic Effects of Acivicin and N-(Phosphonacetyl)-L-Aspartic Acid in a Biochemically Designed Trial Against a N-(Phosphonacetyl)-L-Aspartic Acid-Resistant Variant of the Lewis Lung Carcinoma. Cancer Res (1981) 41:905.
147
WadlerSMaoXBajajRHallamSSchwartzEL. N-(Phosphonacetyl)-L-aspartate Synergistically Enhances the Cytotoxicity of 5-Fluorouracil/interferon-alpha-2a Against Human Colon Cancer Cell Lines. Mol Pharmacol (1993) 44:1070–6.
148
TeschnerSBurstV. Leflunomide: A Drug With a Potential Beyond Rheumatology. Immunotherapy (2010) 2:637–50. doi: 10.2217/imt.10.52
149
RobitailleAMChristenSShimobayashiMCornuMFavaLLMoesSet al. Quantitative Phosphoproteomics Reveal Mtorc1 Activates De Novo Pyrimidine Synthesis. Science (2013) 339:1320–3. doi: 10.1126/science.1228771
150
OhJO’ConnorPW. Teriflunomide in the Treatment of Multiple Sclerosis: Current Evidence and Future Prospects. Ther Adv Neurol Disord (2014) 7:239–52. doi: 10.1177/1756285614546855
151
FragosoYDBrooksJBB. Leflunomide and Teriflunomide: Altering the Metabolism of Pyrimidines for the Treatment of Autoimmune Diseases. Expert Rev Clin Pharmacol (2015) 8:315–20. doi: 10.1586/17512433.2015.1019343
152
HerrmannMLSchleyerbachRKirschbaumBJ. Leflunomide: An Immunomodulatory Drug for the Treatment of Rheumatoid Arthritis and Other Autoimmune Diseases. Immunopharmacology (2000) 47:273–89. doi: 10.1016/s0162-3109(00)00191-0
153
Miret-CasalsLSebastiánDBreaJRico-LeoEMPalacínMFernández-SalgueroPMet al. Identification of New Activators of Mitochondrial Fusion Reveals a Link Between Mitochondrial Morphology and Pyrimidine Metabolism. Cell Chem Biol (2018) 25:268–278.e264. doi: 10.1016/j.chembiol.2017.12.001
154
BrownKKSpinelliJBAsaraJMTokerA. Adaptive Reprogramming of De Novo Pyrimidine Synthesis Is a Metabolic Vulnerability in Triple-Negative Breast Cancer. Cancer Discovery (2017) 7:391–9. doi: 10.1158/2159-8290.CD-16-0611
155
DexterDLHessonDPArdeckyRJRaoGVTippettDLDusakBAet al. Activity of a Novel 4-Quinolinecarboxylic Acid, NSC 368390 [6-Fluoro-2-(2’-Fluoro-1,1’-biphenyl-4-yl)-3-methyl-4-quinolinecarb Oxylic Acid Sodium Salt], Against Experimental Tumors. Cancer Res (1985) 45:5563–8.
156
ArteagaCLBrownTDKuhnJGShenHSO’RourkeTJBeougherKet al. Phase I Clinical and Pharmacokinetic Trial of Brequinar Sodium (Dup 785; NSC 368390). Cancer Res (1989) 49:4648–53.
157
BurrisHARaymondEAwadaAKuhnJGO’RourkeTJBrentzelJet al. Pharmacokinetic and Phase I Studies of Brequinar (DUP 785; NSC 368390) in Combination With Cisplatin in Patients With Advanced Malignancies. Invest New Drugs (1998) 16:19–27. doi: 10.1023/a:1016066529642
158
NoeDARowinskyEKShenHSClarkeBVGrochowLBMcGuireWBet al. Phase I and Pharmacokinetic Study of Brequinar Sodium (NSC 368390). Cancer Res (1990) 50:4595–9.
159
SchwartsmannGDodionPVermorkenJBten Bokkel HuininkWWJoggiJWinogradBet al. Phase I Study of Brequinar Sodium (NSC 368390) in Patients With Solid Malignancies. Cancer Chemother Pharmacol (1990) 25:345–51. doi: 10.1007/bf00686235
160
WuDWangWChenWLianFLangLHuangYet al. Pharmacologic Inhibition of Dihydroorotate Dehydrogenase Induces Apoptosis and Differentiation in Acute Myeloid Leukemia Cells. Haematologica (2018), haematol.2018.188185. doi: 10.3324/haematol.2018.188185
161
LiuZHuQWangWLuSWuDZeSet al. Natural Product Piperine Alleviates Experimental Allergic Encephalomyelitis in Mice by Targeting Dihydroorotate Dehydrogenase. Biochem Pharmacol (2020) 177:114000. doi: 10.1016/j.bcp.2020.114000
162
CadmanECDixDEHandschumacherRE. Clinical, Biological, and Biochemical Effect of Pyrazofurin. Cancer Res (1978) 38:682–8.
163
RingerDPHowellBAEtheredgeJL. Alteration in De Novo Pyrimidine Biosynthesis During Uridine Reversal of Pyrazofurin-Inhibited DNA Synthesis. J Biochem Toxicol (1991) 6:19–27. doi: 10.1002/jbt.2570060104
164
KanalasJJSuttleDP. Amplification of the UMP Synthase Gene and Enzyme Overproduction in Pyrazofurin-Resistant Rat Hepatoma Cells. Molecular Cloning of a cDNA for UMP Synthase. J Biol Chem (1984) 259:1848–53. doi: 10.1016/S0021-9258(17)43486-7
165
SuttleDPStarkGR. Coordinate Overproduction of Orotate Phosphoribosyltransferase and Orotidine-5’-Phosphate Decarboxylase in Hamster Cells Resistant to Pyrazofurin and 6-Azauridine. J Biol Chem (1979) 254:4602–7. doi: 10.1016/S0021-9258(17)30053-4
166
WohlhueterRMMcIvorRSPlagemannPGW. Facilitated Transport of Uracil and 5-Fluorouracil, and Permeation of Orotic Acid Into Cultured Mammalian Cells. J Cell Physiol (1980) 104:309–19. doi: 10.1002/jcp.1041040305
167
MyersCE. The Pharmacology of the Fluoropyrimidines. Pharmacol Rev (1981) 33:1–15.
168
CalabresiPWelchAD. Chemotherapy of Neoplastic Diseases. Annu Rev Med13:147–202. doi: 10.1146/annurev.me.13.020162.001051
169
LaskinJDEvansRMSlocumHKBurkeDHakalaMT. Basis for Natural Variation in Sensitivity to 5-Fluorouracil in Mouse and Human Cells in Culture. Cancer Res (1979) 39:383–90.
170
YamadaMNakagawaHFukushimaMShimizuKHayakawaTIkenakaKet al. In Vitro Study on Intrathecal Use of 5-Fluoro-2’-Deoxyuridine (FdUrd) for Meningeal Dissemination of Malignant Brain Tumors. J Neurooncol (1998) 37:115–21. doi: 10.1023/a:1005869226496
171
ScagliottiGVNovelloS. Gemcitabine (Gemzar®)-Based Induction Chemotherapy in non-Small-Cell Lung Cancer. Lung Cancer (2002) 38:13–9. doi: 10.1016/S0169-5002(02)00352-5
172
SunJRussellCCScarlettCJMcCluskeyA. Small Molecule Inhibitors in Pancreatic Cancer. RSC Med Chem (2020) 11:164–83. doi: 10.1039/c9md00447e
173
BeaulieuCLuiAYusufDAbdelazizZRandolphBBatuyongEet al. A Population-Based Retrospective Study of Biliary Tract Cancers in Alberta, Canada. Curr Oncol (Toronto Ont.) (2021) 28:417–27. doi: 10.3390/curroncol28010044
174
HeTHaapa-PaananenSKaminskyyVOKohonenPFeyVZhivotovskyBet al. Inhibition of the Mitochondrial Pyrimidine Biosynthesis Enzyme Dihydroorotate Dehydrogenase by Doxorubicin and Brequinar Sensitizes Cancer Cells to TRAIL-induced Apoptosis. Oncogene (2014) 33:3538–49. doi: 10.1038/onc.2013.313
175
LuWYaoXOuyangPDongNWuDJiangXet al. Drug Repurposing of Histone Deacetylase Inhibitors That Alleviate Neutrophilic Inflammation in Acute Lung Injury and Idiopathic Pulmonary Fibrosis Via Inhibiting Leukotriene A4 Hydrolase and Blocking Ltb4 Biosynthesis. J Med Chem (2017) 60:1817–28. doi: 10.1021/acs.jmedchem.6b01507
176
KoundinyaMSudhalterJCourjaudALionneBTouyerGBonnetLet al. Dependence on the Pyrimidine Biosynthetic Enzyme Dhodh Is a Synthetic Lethal Vulnerability in Mutant Kras-Driven Cancers. Cell Chem Biol (2018) 25:705–17.e711. doi: 10.1016/j.chembiol.2018.03.005
177
LaddsMvan LeeuwenIMMDrummondCJChuSHealyARPopovaGet al. A DHODH Inhibitor Increases p53 Synthesis and Enhances Tumor Cell Killing by p53 Degradation Blockage. Nat Commun (2018) 9:1107. doi: 10.1038/s41467-018-03441-3
178
QianYLiangXKongPChengYCuiHYanTet al. Elevated DHODH Expression Promotes Cell Proliferation Via Stabilizing β-Catenin in Esophageal Squamous Cell Carcinoma. Cell Death Dis (2020) 11:862. doi: 10.1038/s41419-020-03044-1
Summary
Keywords
metabolic reprogram, pyrimidine metabolism, precision medicine, dihydroorotate dehydrogenase, pyrimidine inhibitor
Citation
Wang W, Cui J, Ma H, Lu W and Huang J (2021) Targeting Pyrimidine Metabolism in the Era of Precision Cancer Medicine. Front. Oncol. 11:684961. doi: 10.3389/fonc.2021.684961
Received
24 March 2021
Accepted
27 April 2021
Published
28 May 2021
Volume
11 - 2021
Edited by
Adriano Venditti, University of Rome Tor Vergata, Italy
Reviewed by
Jakub Rohlena, Academy of Sciences of the Czech Republic (ASCR), Czechia; Parames C. Sil, Bose Institute, India
Updates
Copyright
© 2021 Wang, Cui, Ma, Lu and Huang.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weiqiang Lu, wqlu@bio.ecnu.edu.cn; Jin Huang, huangjin@ecust.edu.cn
This article was submitted to Cancer Metabolism, a section of the journal Frontiers in Oncology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.