 Tobias Alexander Kampmann
Tobias Alexander Kampmann David Müller
David Müller Clemens Franz Vorsmann
Clemens Franz Vorsmann Jan Kierfeld
Jan Kierfeld- Physics Department, TU Dortmund University, Dortmund, Germany
We discuss the rejection-free event-chain Monte-Carlo algorithm and several applications to dense soft matter systems. Event-chain Monte-Carlo is an alternative to standard local Markov-chain Monte-Carlo schemes, which are based on detailed balance, for example the well-known Metropolis-Hastings algorithm. Event-chain Monte-Carlo is a Markov chain Monte-Carlo scheme that uses so-called lifting moves to achieve global balance without rejections (maximal global balance). It has been originally developed for hard sphere systems but is applicable to many soft matter systems and particularly suited for dense soft matter systems with hard core interactions, where it gives significant performance gains compared to a local Monte-Carlo simulation. The algorithm can be generalized to deal with soft interactions and with three-particle interactions, as they naturally arise, for example, in bead-spring models of polymers with bending rigidity. We present results for polymer melts, where the event-chain algorithm can be used for an efficient initialization. We then move on to large systems of semiflexible polymers that form bundles by attractive interactions and can serve as model systems for actin filaments in the cytoskeleton. The event chain algorithm shows that these systems form networks of bundles which coarsen similar to a foam. Finally, we present results on liquid crystal systems, where the event-chain algorithm can equilibrate large systems containing additional colloidal disks very efficiently, which reveals the parallel chaining of disks.
1 Event-Chain Monte-Carlo Algorithm
Since its first application to a hard disk system [1], Monte-Carlo (MC) simulations have been applied to virtually all types of models in statistical physics, both on-lattice and off-lattice. MC samples microstates
for a move
The Metropolis rule (1) satisfies detailed balance for the Boltzmann distribution
Typical local MC moves, such as single spin flips in spin systems or single particle moves in off-lattice systems of interacting particles, are often motivated by the actual dynamics of the system. Sampling with local moves can become slow, however, in certain physically relevant situations, most notably, close to a critical point, where large correlated regions exist, or in dense systems, where acceptable moves become rare.
Cluster algorithms deviate from the local Metropolis MC rule and construct MC moves of large non-local clusters, ideally, in a way that the MC move of the cluster is performed rejection-free. For lattice spin systems, the Swendsen-Wang [3] and Wolff [4] algorithms are the most important cluster algorithms with enormous performance gains close to criticality, where they reduce the dynamical exponent governing the critical slowing down in comparison to local MC simulations. These cluster algorithms still fulfill detailed balance but based on a non-trivial trial probability that derives from the cluster construction rules.
The event-chain (EC) MC algorithm introduced by Krauth et al. [5] has been developed to decrease the autocorrelation time in hard disk or sphere systems. It performs rejection-free displacements of several spheres in a single MC move (see Figure 1) and can, therefore, also be classified as a cluster algorithm. The basic idea is to perform a displacement
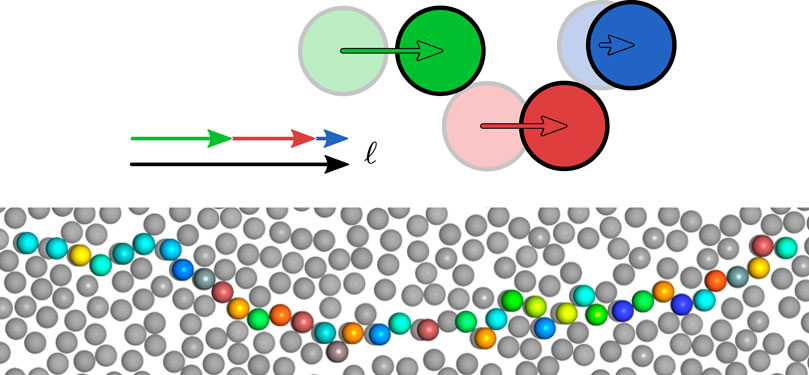
FIGURE 1. The upper scheme illustrates the billiard-type construction of an EC with a total displacement length
In the following, we will give an introduction into the ECMC algorithm starting with the example of simple hard sphere systems. We then discuss how the algorithm realizes the general concept of lifting moves, which is employed in order to achieve global balance. Finally, we review the adaptation and generalization of the ECMC algorithm from athermal systems with hard interactions to systems with soft potentials specified by an interaction energy. Many soft matter systems can be constructed from hard spheres as basic building blocks, eventually with additional interactions, for example spring-like connections to form a polymer chain. We discuss in some detail several applications of the EC algorithm in dense soft matter systems, namely polymer systems, liquid crystal systems as well as mixed systems such as liquid crystal colloids.
1.1 EC Algorithm for Hard Spheres
We consider the conceptually simple system of hard (impenetrable) disks (of diameter σ) in two dimensions in order to introduce the ECMC algorithm. Hard disks are the epitome of a system that is easily described and quickly implemented in a (naive) simulation [1], but exhibits a non-trivial phase transition that has been debated for a long time [6]. The two-dimensional melting phase transition is believed to be a two-step Kosterlitz-Thouless melting via an intermediate hexatic phase [7–9], which is, however, difficult to confirm unambiguously in simulations [10, 11].
In a hard disk system, an EC move is constructed by first selecting a random starting particle, an EC displacement direction, and a total displacement length
The total displacement length
We note that the EC algorithm will be very well suited to simulate dense systems but does not allow to simulate maximally dense, i.e., jammed systems since an EC can not displace any particle if all particles touch each other. In jammed hard disk systems, the mean free path
We will show in the next section that the EC algorithm ensures global balance, regardless of how the EC displacement direction is chosen. If the EC displacement directions are chosen randomly the straight EC algorithm also satisfies detailed balance over many EC events. The most performant version of the straight EC algorithm, however, the so-called xy-version or irreversible straight EC algorithm, breaks detailed balance and uses only displacements along coordinate axes and only in positive direction. Breaking detailed balance but preserving global balance leads to performance gains. Ergodicity must also be ensured, which is done via periodic boundary conditions for the irreversible EC algorithm; moreover, several computations can be simplified for the xy EC algorithm. In Ref. [12], the irreversible EC simulations are roughly 70 times faster than a simulation employing local MC moves. We can obtain similar performance gains in dense soft matter systems.
1.2 Balance Conditions and Lifting for Hard Spheres
The standard Metropolis algorithm (1) employs detailed balance to ensure stationarity of the probability distribution of states by pairwise balancing of probability fluxes between microstates in condition (2). Since circular probability fluxes also fulfill the stationarity requirement for the probability distribution, detailed balance is not a necessary condition and, often, performance can be gained by finding an algorithm that fulfills the weaker condition of global balance,
(where the sums are over all microstates), which is simply the continuity equation for a stationary probability
Global balance algorithms should become particularly performant if maximal global balance is achieved, which means that probability backflow is forbidden leading to the additional constraint
In particular, this implies a rejection-free algorithm, i.e.,
In Figure 2, we show how maximal global balance is achieved in EC algorithms for a hard sphere system. In order to investigate balance conditions, it is often convenient to decompose the EC into infinitesimal moves
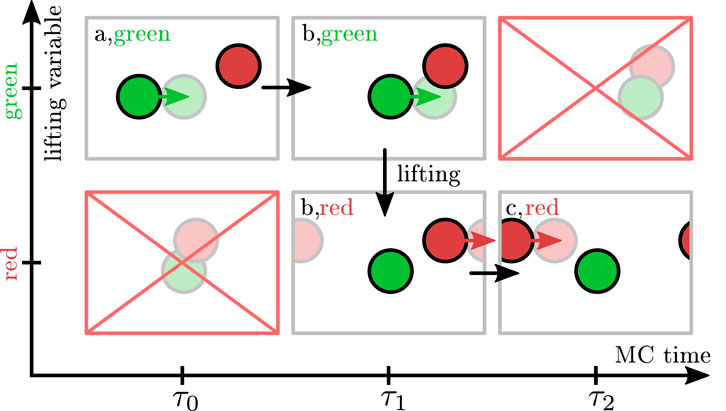
FIGURE 2. Scheme of the lifting formalism for hard spheres. The physical states are labeled with letters (a,b,c) and the lifting variable is indicated by color, i.e., the current active particle is indicated by a colored arrow. The EC displacement direction is to the right. Proposed infinitesimal displacements are indicated by opaque spheres. In case of a physical rejection at MC step
For infinitesimal EC moves, only two spheres can collide at once, and it is sufficient to consider a simplified setting of only two hard spheres (green and red) as shown in the cartoon in Figure 2, where proposed infinitesimal displacements are indicated by opaque spheres. The physical states are labeled with letters (A and b,c), and the lifting variable is indicated by color (red or green particle is active). Physical transitions change the physical states by changing particle positions; this happens by moving the active sphere in the EC displacement direction (i.e., to the right from (a,green) at
In this way, rejections are avoided, and the entire rejection probability flow is redirected to a lifting move probability flow, which ensures maximal global balance if all physical configurations are equally probable, i.e., if the Boltzmann distribution for hard spheres holds:
The total physical inflow
By construction of the EC algorithm, all collisions give rise to lifting such that the lifting flow
Apart from a translation, the states a and c and the forbidden overlapping configurations (upper right and lower left pictures in Figure 2) are identical (please note the periodic boundary conditions).
Therefore, the missing physical inflow
Again, the missing physical inflow
All in all, we obtain
which are exactly the maximal global balance conditions for states (b,green) and (b,red) proving maximal global balance for any EC move on a state space that is extended by the lifting variable denoting the active particle. Global balance holds, regardless of how the EC displacement direction or displacement length is chosen. If the reverse EC displacement directions are offered with the same probability (for example by drawing EC directions completely uniformly in all directions) the resulting EC algorithm also satisfies detailed balance over the course of many EC moves.
For pair interactions between hard spheres the lifting variable is the particle number. We can also introduce hard walls to the system, which represent additional steric one-particle interactions. For such one-particle interactions, the lifting variable will not be the particle number (as there are no interaction partners, the active particle must stay the same) but the EC direction itself. It can be shown that classical reflection of the EC direction upon collision with the hard wall will ensure maximal global balance. Analogously to the forbidden overlapping configurations Figure 2, both the rejected outflow in a collision with a wall and the missing inflow from forbidden configurations are exactly equal to the lifting flow from reflected EC directions.
1.3 Soft Interaction Energies
In many applications other than pure hard core systems, we have to deal with systems containing hard core repulsion alongside with other soft interaction energies
This strategy of handling additional interaction energies by re-introduction of Metropolis sampling and, thus, rejections will surely decrease the efficiency of the simulation. All advantages of the EC algorithm are effectively lost. There is, however, a way to include arbitrary
In a pure hard sphere simulation, a sphere is continuously displaced until it hits another sphere which triggers a lifting event (or the total displacement reaches
Each particle i has a set
In the following, we consider again infinitesimal displacements
We construct a global balance algorithm starting from a Metropolis filter for the acceptance of physical moves based on the energy
It will be important that the factorized Metropolis filter still has the detailed balance property (2). The probability of rejecting the next infinitesimal step
The factorized Metropolis filter can now be used to construct a maximal global balance algorithm on an enlarged state space by redirecting all physical rejection events into lifting events. This is done analogously to the hard sphere case outlined above. If a rejection should take place according to the factorized Metropolis filter, we perform a compensating lifting move instead. We lift to one of the particles that participate in the interaction that caused the rejection. For a pair interaction this already determines the particle to lift to uniquely, for many-body interactions an additional rule is needed to select the particle to lift to properly [15]. Therefore, we first discuss the simpler case of pair interactions and show how we can realize maximal global balance.
1.3.1 Pair Interactions
For states in the enlarged state space, we use the notation
This transition rate is decreased by rejections if a move has
Because only one pair interaction
Lifting moves from
with
and dividing by
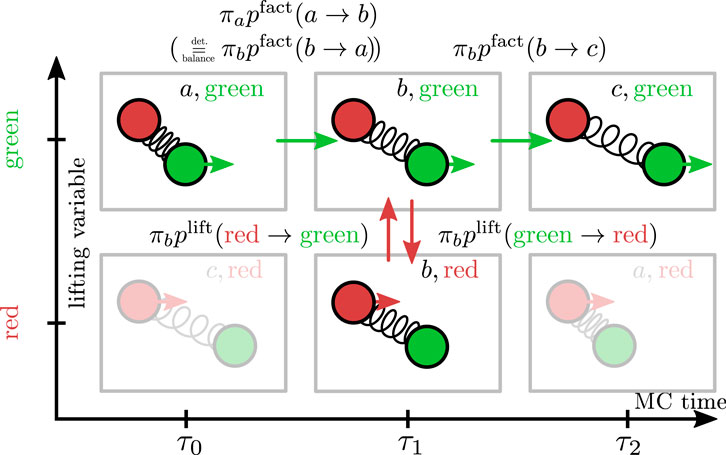
FIGURE 3. Scheme of the lifting formalism for a continuous pair interaction (notation see Figure 2). Using Eq. 5 in the global balance condition (3) for incoming and outgoing currents for state b and dividing by
The last equality holds for a translationally invariant pair interaction, where
which leads to a rejection-free algorithm because the rejection probability of the physical moves (6) is exactly redirected into a lifting probability. We also conclude that
Global balance is established for each move
For an efficient implementation, an event-based approach is chosen [13, 17], in which we determine the distance to the next rejection. This rejection distance corresponds to the distance to the next collision for hard spheres.
The probability
which depends on all uphill energy differences caused by particle j if particle i is moved, see Figure 4. Interpreting
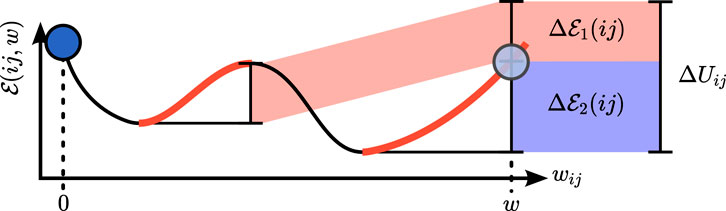
FIGURE 4. Event-driven approach to determine the rejection distance for a pair interaction between i and j. Condition (13) has to be solved for the rejection distance
Transforming the probability distribution Eq. 12, we find that the distribution of the usable (i.e., uphill) energy
for
1.3.2
In many applications, also three-particle interactions occur. This happens, in particular, for extended objects, such as rods or polymers, which can be described by bead-spring models. One prominent example are semiflexible polymer chains with a bending energy. Because the local bending angle involves three neighboring beads in a discrete model, the bending energy is a three-particle intra-polymer interaction in terms of bead positions.
In Ref. [15], the EC algorithm has been generalized to three- and many-particle interactions, thus broadening the range of applicability of rejection-free EC algorithms considerably. For
In order to redirect the rejection probability
as for pairs, see Eq. 11. We need, in addition, a set of conditional lifting probabilities
We first consider the important case of a three-particle interaction (between particles i, j, k), see also Figure 5. Analogously to Eq. 10, the global balance condition (3) applied to flows to state
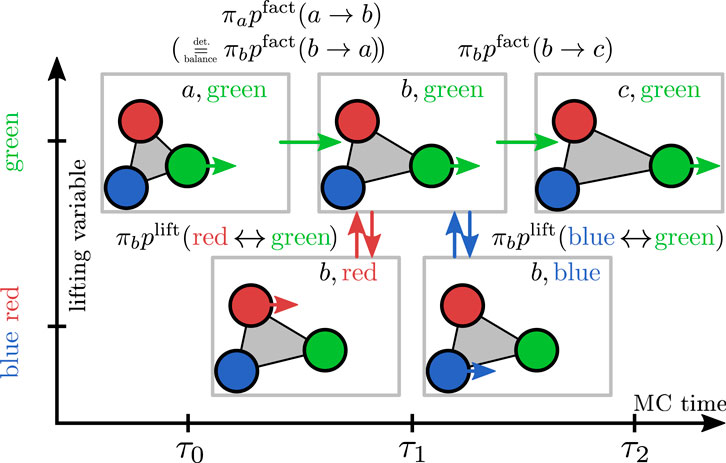
FIGURE 5. Conditional lifting in a system with three-particle interaction. The conditional lifting probabilities can be derived from the balance condition. Analogously to Figure 2 (i.e., using Eq. 5 and dividing by
Inserting (15) this results in the condition
As for pairs, lifting from particle i to j will only take place (
where
For arbitrary
For
Regardless of the number of interacting particles
1.3.3 One-Particle Interactions
Finally, soft external one-particle potentials
for lifting the EC direction to a reflected direction
1.4 ECMC Algorithm
In summary, an ECMC simulation, which we use for soft matter systems, can be structured as follows. Starting from a random particle i in a random EC direction
1. choose a random EC direction
2. calculate the minimal rejection distance
3. if rejection is triggered by a three- or more particle interaction:select
4. move particle i:
5. lift to rejecting particle
6. let
7. if
1.5 Additional ECMC Simulation Features
We want to highlight some useful properties of an ECMC simulation. First, using the factorized Metropolis filter each interaction is independent, making it easy to implement the algorithm in a highly modular manner.
Furthermore, the rejection distance distribution and lifting statistics is intimately linked to the pressure in the system. Without loss of generality, let the EC direction be in x-direction, then the pressure can be expressed by [13].
where
Every configuration visited is weighted properly even if a hard sphere collision just happened. This can be exploited by special MC moves. This is not unconditionally true in a molecular dynamics simulation where, for instance, the statistics is skewed if measurements are taken on particle collisions. Inclusion of special many-particle MC moves can be beneficial for particular systems. For polymer melt simulations, we introduce a bead-swapping move [20], which switches positions of two hard spheres upon collision obeying a standard metropolis filter for the spring energies involved, as explained later.
Many simulation techniques based on single particle moves can benefit from massive parallelization if the simulation system can be efficiently divided into independent pieces. For ECMC algorithms, massive parallelization will not be optimal because of the cluster nature of EC moves [21]. In Ref. [21], a parallelization scheme for the EC algorithm was introduced, and extensive tests for correctness and efficiency were performed for the hard sphere system in two dimensions. For optimal parameters the algorithm achieves a speed up by a factor of roughly the number of cores used compared to a sequential EC simulation. For parallelization we use a spatial partitioning approach into simulation cells. We analyzed the performance gains for the parallel EC algorithm and find the criterion
2 Applications
We now briefly present some soft matter systems where we successfully employed ECMC algorithms, namely a dense polymer melt, a network-forming system of polymers with a short range attraction, hard needle models of liquid crystal and liquid crystal colloids from a mixture of needles and hard spheres. In all of these systems the ECMC algorithm speeds up simulations considerably at high particle densities as compared to standard local MC simulations. The simulations for these soft matter applications have been performed within the highly modular polyeventchain framework, which is particularly suited for soft matter applications and will be presented in detail elsewhere. Jellyfish provides a similar EC framework with a focus on all atom simulations [22, 23].
2.1 Initialization and Simulation of Polymer Melts
The simulation of polymer melts by Molecular Dynamics (MD) or MC simulations is a challenging problem, in particular, for long chains at high densities, where polymers in the melt exhibit slow reptation and entanglement dynamics. For chain molecules of length N the disentanglement time increases
In MC simulations, non-local or collective MC moves can be introduced, for example, chain-topology changing double-bridging moves [37], which speed up equilibration; dynamic properties, however, are no longer realistic if such collective MC moves are employed. Likewise, reptation can also be introduced as explicit additional local MC reptation move [28, 38] (slithering snake moves) to obtain faster equilibration of a polymer melt but reptation dynamics is no longer dynamically realistic then.
In order to apply the above scheme of ECMC simulations to polymer melts, flexible polymers are modeled by a bead-spring model consisting of hard spheres, which are connected by harmonic springs [20]. These are the only additional soft pair interactions in the ECMC scheme for a flexible polymer melt. We choose the bond rest length to equal the hard-sphere diameter σ and a sufficiently large bond stiffness such that bond-crossing becomes inhibited by the impenetrable hard spheres and polymers remain entangled.
ECMC simulations can speed up MC simulations of polymers such that simulation speeds become comparable to MD simulation speeds and such that reptation dynamics can be clearly observed [20]. In each EC move, all beads that would collide successively during a short time interval in a MD simulation are displaced at once. This gives rise to a ECMC dynamics which is effectively very similar to the realistic MD dynamics and statements about polymer dynamics are still possible. In Ref. [20] we compared the ECMC algorithm to MD simulations implemented in LAMMPS regarding the equilibration of polymer melts. We utilized the time, that the positional fluctuation of the most inner bead of each polymer needs to reach the diffusive regime, as gauge to introduce an ad-hoc interpretation of time in the MC simulation (see Table I and Figure 5 in Ref. [20]). This showed that LAMMPS equilibrates more efficiently for moderate chain lengths. For long polymers (
The additional swap-move chanes the topology of entanglements locally. In contrast to the double-bridging move, which changes bonds, the swap move changes topology by changing bead positions. For this purpose we modify the EC move so that the EC does not directly transfer to the next bead upon hard sphere contact but, instead, a swap of the two touching spheres is proposed, see Figure 6. Such an additional swap move is EC specific and allows for a local change of entanglements. An analogous swap move in a standard MC algorithm needs to select pairs of beads such that the swap move has a reasonable acceptance rate (the particles have to be reasonably close). Moreover, the selection rule has to satisfy detailed balance (for example, simply proposing the nearest neighbor for swapping will lead to a violation of detailed balance). Therefore, there is no straightforward analogue of the EC swap move in a standard MC simulation with local moves.
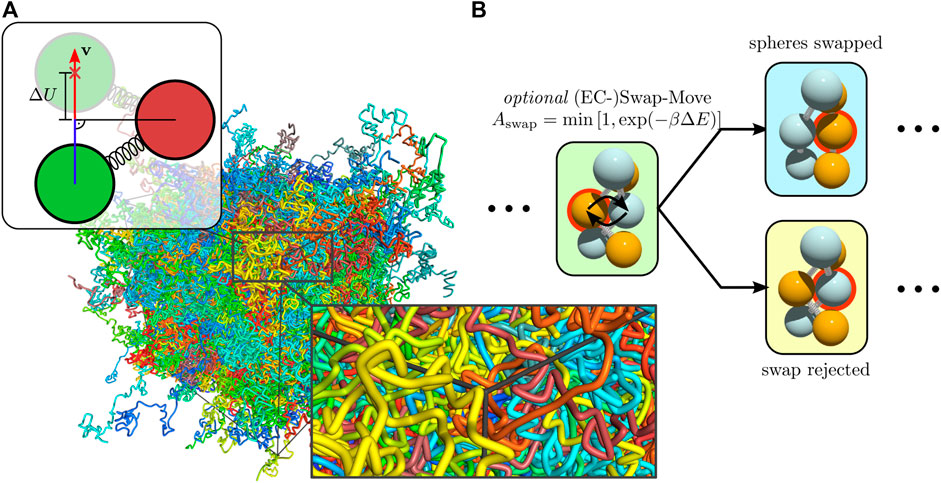
FIGURE 6. The background shows a polymer melt of flexible chains, which are presented as volumetric splines (A) Scheme for the rejection distance for hard sphere chains. If two bonded spheres do not collide, the active sphere can move freely until the minimal distance between both is reached. A usable energy
Here, we want to focus on another important aspect of EC moves in polymer melt simulations. EC moves can also be employed to generate initial configurations that are already representative of equilibrium configurations. This keeps the equilibration or warm-up phase of any subsequent simulation scheme (MC or MD) short. An early strategy to generate initial conditions is to slowly compress a very dilute equilibrated melt, which can take a long time for very dense target systems and becomes more difficult when chain lengths increase [30]. Another method that has been proposed first distributes the chains without interaction (phantom chains) and tries to resolve overlaps in a way which distorts the chain statistics as least as possible [30]. Over the years more sophisticated ways of resolving overlaps have been proposed, for instance a slowly increase of the strongly repulsive excluded volume potential [32]. This procedure is called push-off and the rate at which the potential is increased and the potential range is extended determines to a large extent how much the original statistics of the chains are disturbed. The method can also be applied to more realistic polymer models [39]. Recent work by Moreira et al. [40] shows how the procedure of Auhl et al. can be applied more efficiently and how the equilibration time can be shortened roughly by a factor of 6. An alternative strategy that performs even better for very long chains (again by a factor of
We present an EC-based scheme that is similar to the push-off scheme and takes advantage of the fact that the EC algorithm is well suited to resolve overlaps. To do this, ECs are started in random directions on any sphere that overlaps with at least one other sphere until all overlaps of this sphere are resolved. This procedure ignores spheres that overlap with the active sphere, so that the algorithm itself does not need to be changed. This procedure corresponds to locally rattling the polymer system, and can create enough space around the overlapping bead to insert it.
For relatively dense melts, Flory [44, 45] puts forward the hypothesis that the volume exclusion effect (chain swelling,
This motivates the following scheme to generate pre-equilibrated polymer melts. Since excluded volume interactions between parts of a given chain are more or less screened as Flory suggests, we place phantom/ideal chains into the system in configurations that resemble non-reversal configurations by excluding a certain angular region for backward steps in generating the initial phantom chain configurations as described in Ref. [32]. This captures the structure of the chains on all length scales quite well, but comes with the downside that the asymptotic behavior of the end-to-end distance (the asymptotic Flory ratio
The resulting phantom chain system is then equilibrated for a short time, so that the phantom chains will relax toward ideal chain configurations, i.e., the phantom chains contract starting from the initial non-reversal configurations. Then the hard sphere diameter is increased from
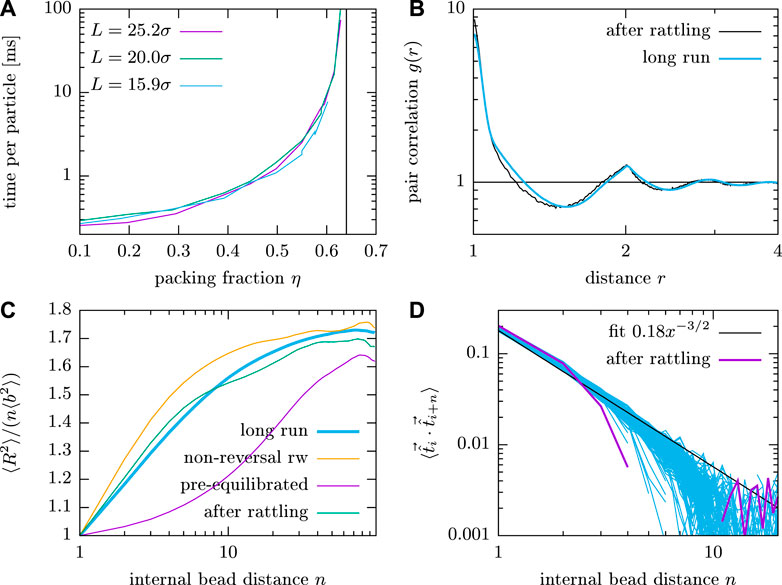
FIGURE 7. (A) Wall time per particle to generate rattled configurations of variable polymer number M with a fixed length of
For this procedure it is vital that the spring constant is quite large. Starting ECs on one particle until overlaps are resolved obviously breaks balance. Presumably, setting the spring constant to a relatively high value (in Figure 7
In general, it is hard to quantitatively compare time scales and thus performance of the EC-based initialization scheme with other schemes from the literature. Hardware development over the years (Moore’s law, number of cores, etc.), underlying polymer models (volume exclusion: hard spheres vs. Lennard-Jones, bonding: harmonic springs vs. FENE vs. tangent hard spheres vs. united atom force fields), criteria of deciding when equilibrium is reached, or simply untested parameter dependencies (regarding system size, chain length, spring constants) can have non-trivial influence, even on an otherwise robust benchmark. Despite these problems to compare quantitatively, the proposed EC-based method seems comparable to other state-of-the-art methods. This has to be checked further in future work.
2.2 Self-Assembly of Filament Bundle Networks
The cytoskeleton is a mechanically important structure which consists of three classes of interacting filamentous proteins (microtubules, filamentous F-actin, intermediate filaments) in animal cells. The different constituents reflect the multi-functionality of the cytoskeleton and the different requirements with respect to force and time scales. F-actin structures in the cell cortex are most important for cell mechanics and cell motility [47, 48]. From the polymer physics point of view, F-actin is a semiflexible polymer since typical contours lengths are of the same order as its persistence length. Therefore, bending rigidity and thermal fluctuations are relevant to understand F-actin structures. Within the cell, F-actin forms locally very dense sub-cellular structures like bundles [49], networks and even networks of bundles to fulfill their biological functionality [47]. In vivo, bundles and also networks are held together by crosslinking proteins [50].
Also minimal cytoskeletal in vitro systems [50], some of which are confined to droplets [51, 52] or in microfluidic chambers [53–55], show formation of bundles and also networks of bundles under the influence of attractive interactions. In vitro, such attractive interactions can not only be induced by crosslinkers but also by counterions and depletion interactions.
The simulation of the cytoskeleton of a cell is a long-standing problem as its physical properties are often governed by the interplay of many long polymers, which are deformable by thermal fluctuations against their bending rigidity and subject to crosslinker-mediated attractive interactions [56]. Theoretical work on crosslinker-mediated bundling of semiflexible polymers [56–59] and the related problem of counterion-mediated binding of semiflexible polyelectrolytes [60] show a discontinuous bundling transition above a threshold concentration of crosslinkers or counterions. The nature of the crosslinkers can influence the mechanical properties of polymer bundles [61].
Simulations of few filaments clearly confirm bundling in a single transition [56]. The emerging bundles of semiflexible polymers (Figure 8) are typically rather densely packed which causes difficulties in equilibrating bundled structures in traditional MC simulations employing local moves. In Ref. [56], MC simulations showed evidence for kinetically arrested states with segregated sub-bundles; in Ref. [62], kinetically arrested bundle networks have been observed for rather small semiflexible polyelectrolyte systems. Further numerical progress requires a faster equilibration of dense bundle structures. Simulations of larger systems consisting of cytoskeletal filaments and crosslinkers [63–65] show different competing phases from isotropic networks to bundles and other aggregates, for example, with bond-orientational order. In Ref. [65], it was also pointed out that the dynamics of bundling and polymerization in combination with entanglement of polymers strongly influence the resulting structure. This raises the question to what extent bundled structures can reach a genuine equilibrium under realistic conditions.
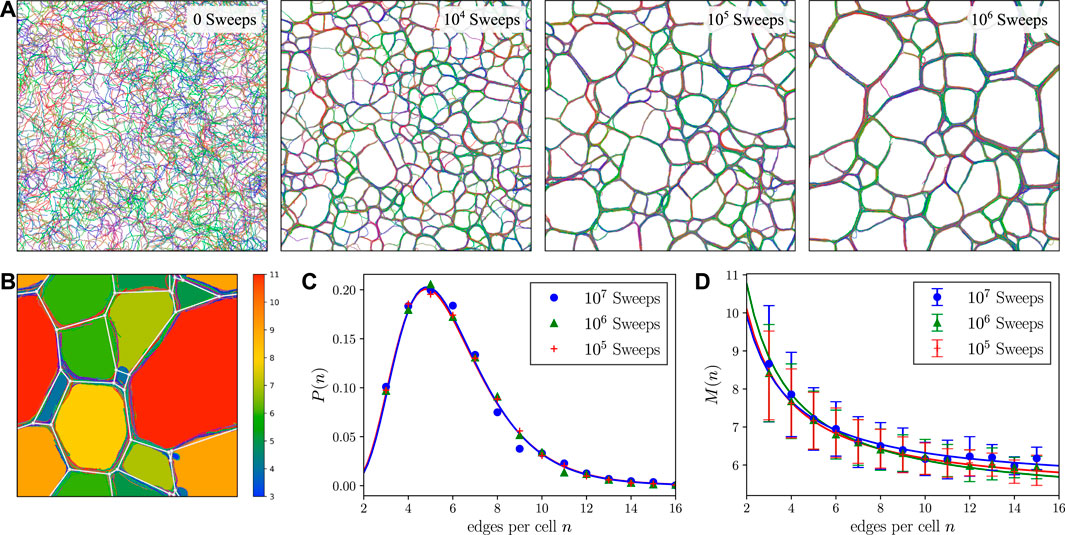
FIGURE 8. (A) A series of snapshots (after 0,
Further numerical progress requires a faster equilibration of dense bundle structures, for which the ECMC algorithm is ideally suited [21]. In comparison to polymer melts, two additional interactions have to be included in this system: (i) Polymers are semiflexible such that we have to include a bending rigidity of each bead-spring polymer, which is an example of a three-particle interaction between three neighboring hard sphere beads along a polymer. (ii) There is a short-range attraction, which mimics a crosslinker- or counterion-mediated attraction between polymers. Therefore, all beads also interact pairwise with a short-ranged attractive square well potential of strength g and range d. We choose a potential strength g well above the critical value for bundling [56, 59], and the range of the attractive square well potential is
We have applied this ECMC algorithm to a system consisting of many2 interacting semiflexible polymers, such as actin filaments, in a flat simulation box in three dimensions. Periodic boundary conditions are used only for the extended direction of the simulation box whereas the spatially short direction is non-periodic. Under the influence of the short-range attraction, we clearly observe formation of densely packed bundles. We also observe that, starting from an isotropic melt-like situation, it is not a single bundle that forms but the system evolves into a structure consisting of a network of bundles.
The simulation does not reach a truly equilibrated stationary state, but the network of bundles keeps evolving by coarsening processes as shown in Figure 8A. This clearly demonstrates that the assembly dynamics plays an important role in structure formation in these systems, as similarly suggested in Ref. [65]. As mentioned above, the ECMC dynamics is effectively very similar to the realistic MD dynamics. Therefore, the observed coarsening dynamics is not an artifact of the MC simulation but a generic property of this multi-filament system. On the time scales of our simulations (up to
Our preliminary results suggest that the bundle networks form structures similar to foams [66], which also continue to coarsen over time, see Figure 8. The dynamics of the coarsening process exhibits further analogies. It can be observed that cells in the bundle network, which enclose a comparatively large area, tend to increase their area at the expense of cells with smaller enclosed area. This behavior is known from foams as a consequence of the von Neumann law for the area growth rate of cells [67] and of the correlation between relative bubble volume and the number of sides [68]. However, the von Neumann law assumes an exchange of the enclosed medium by diffusion through the liquid interfaces and cannot be applied directly to the bundle networks considered here. Other empirical laws found in the context of foams like Aboav-Weaire law [69] for the mean number
While the structure formation in dry foams is driven by the minimization of interfacial energies and diffusion [70], it appears that the bundle networks coarsen by a zipping mechanism [71, 72] which leads – similar to the decrease in interfacial energy in foams – to a decrease of the polymer binding energy (i.e., to more adhesive contacts between polymers). At a fixed amount of polymers this gives rise to a minimization of bundle length, which plays an analogous role to the minimization of surface area in a classical three-dimensional foam. The foam-like bundle networks resemble the structures observed in different in vitro experiments [51, 53], where also polygonal cell structures of the self-assembled networks have been observed.
2.3 Liquid Crystals and Colloidal Suspensions
Composite soft matter systems such as colloidal mixtures containing different colloidal particles, often of different size, represent challenging systems for simulations because their physics are typically governed by effective interactions which arise if microscopic degrees of freedom of one species are integrated out. Effective interactions are essential to characterize stability and potential self-assembly into crystalline phases but hard to access in a microscopic particle-based simulation. The process of integrating out microscopic degrees of freedom corresponds to the numerical evaluation of a potential of mean force between the colloidal species of interest. In order to measure the potential of mean force for one colloidal species accurately, all degrees of freedom must be properly equilibrated.
As an example of such a colloidal mixture we consider a two-dimensional system of needles and colloidal disks. This serves as a simple model system for liquid crystal (LC) colloids, which are colloidal particles suspended in a liquid crystal. Such LC colloids exhibit anisotropic effective interactions between colloidal particles if the LC is in an ordered, e.g., nematic phase [73, 74]. A nematic LC phase forms a rich variety of defect-structures around a spherical inclusion such as Saturn-ring disclination rings or a satellite hedgehog for normal anchoring and boojums for planar anchoring [75–77]. In a dense nematic LC, the effective interactions can be governed both by depletion interactions [78] or by long-range elastic interactions mediated by director field distortions in the nematic hard needles. Because hard needles tend to align tangentially at a hard wall, we expect a quadrupolar elastic interaction, which is characteristic for planar anchoring at the colloidal disk [79–83] but also generic in two dimensions [84]. This interplay has been studied in Ref. [85] employing an ECMC simulation.
The colloidal mixture of hard disks suspended in a nematic host of hard needles is particularly challenging as the hard needle system must be fairly dense to establish a nematic phase. While particle-based simulations exist for dilute rods in the isotropic phase [86, 87], the regime of a nematic host is fairly unexplored up to now and simulations resorted to coarse-graining approaches [83]. So far, only single inclusions [88] or confining geometries [89] have been investigated by particle-based simulations. In order to measure the potential of mean force for larger colloidal disks accurately, all degrees of freedom of the needles but also the relatively slow degrees of freedom of the colloidal disks must be properly equilibrated. This is efficiently achieved by the ECMC algorithm.
The idea for the ECMC simulation of a needle system is to represent needles by their two endpoints, where only one endpoint is moving at a time [15]; the endpoints are connected by an infinitely thin, hard line. For efficient sampling, the distance of the two endpoints, i.e. the length l of the hard needle can fluctuate around its characteristic length
In two dimensions, the needle-needle interaction simplifies effectively to a collision of an endpoint with another needle. The remaining MC move distance is lifted to one of the endpoints of this needle. Therefore, we have a fluid of endpoints with an effective three-particle interaction (two endpoints of a passive needle and the active endpoint). For needles the probability
In a composite colloidal problem the ECMC algorithm also faces the problem of lifting between different species. This can be performed exactly according to the same rules as lifting between a single species. In the presence of additional disks, MC displacement of needle endpoints is also lifted to disks if a needle collides with the disk and vice versa as illustrated in Figure 9A. The example of a needle system also shows that collision detection is often the computational bottleneck of ECMC simulations. Here, we use a sophisticated neighbor list system to achieve high simulation speeds also in the nematic phase. Each particle is confined to a container, which triggers an event when the particle leaves it. Then the neighbor list is updated, which ensures that the neighbor lists are always valid. Particles are added to the neighbor lists of the other particle and vice versa when their containers overlap. This way, for different particles different container shapes can be chosen. For the needles, a very narrow rectangle can be used, which limits the computational effort for calculating the distances to the next collision and makes the simulation significantly more efficient in the nematic phase. In particular, the anisotropy of the needles can be assigned particularly well without sacrificing any flexibility for the bookkeeping of the spheres. Furthermore, we optimize the updating of lists by putting them onto a collision grid.
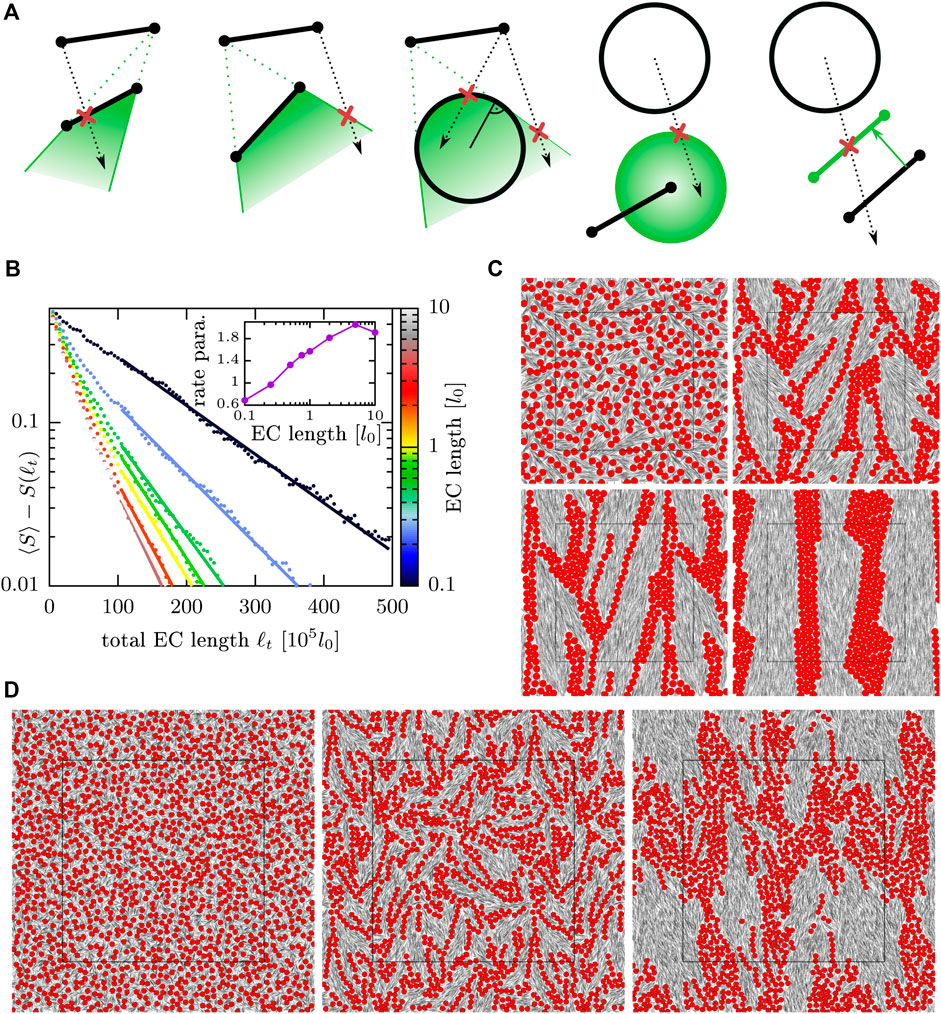
FIGURE 9. (A) List of all geometrical cases where rejections/lifting between needles and disks occur (first two depict needle-needle, the third needle-disk, and the last two ones sphere-needle collisions). If a needle is not hit at one of its endpoints (first and last picture) conditional lifting probabilities
In Figure 9B, we show a detailed benchmarking for a two-dimensional pure needle system. With increasing EC length
In three dimensions, hard needles introduce no excluded volume and therefore lack any phase transitions (we have checked with ECMC simulations not shown here that, independent of the density, the nematic order parameter vanishes for hard needles in equilibrium). In three dimensions hard spherocylinders are a natural extension of needles that introduces a non-vanishing excluded volume. As an additional proof of concept of our ECMC implementation with three-particle lifting, we investigate the series of liquid crystal phase transitions by monitoring the pressure as a function of increasing volume fraction, see Figure 10. We see a sequence of transitions from isotropic to nematic ordering, followed by a transition to smectic order, and ultimately, crystallization in complete agreement with literature results [90].
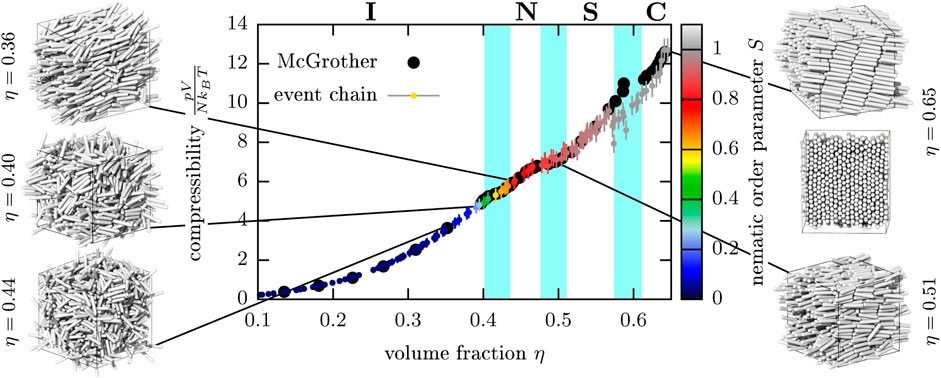
FIGURE 10. Phase diagram of hard spherocylinders (with diameter D and length
For the colloidal mixture of hard disks suspended in a nematic host of hard needles, the ECMC simulation reveals a surprising and robust tendency for parallel chaining of disks along the director axis which seems to contradict the chaining in a
3 Discussion
We presented several applications that demonstrate how ECMC is an effective and fast simulation technique that is particularly suited for dense soft matter systems. Polymer melts, bundles of semiflexible polymers, and the composite system of a liquid crystal colloid are computationally challenging problems, where the ECMC techniques give an improved performance.
We started with an introduction to the algorithm, where we showed how arbitrary interactions between hard spheres can be easily included into the ECMC framework such that a multitude of soft matter systems can be modeled. On this algorithmic side, long-range interactions have not been covered explicitly here but can also be included in an effective manner [91].
Future developments will also address variations of the basic EC moves presented here in order to sample even more efficiently. On a generic collision, one interaction rejects the move and lifting occurs which means the current EC direction has a component parallel to the energy gradient of this specific interaction. One can resample the perpendicular parts of the EC direction in order to avoid reshuffling the EC direction from time to time, i.e., a finite EC length. This procedure has been recently developed and is called forward EC [92]. In specialized settings, the scheme seems very promising with regard to efficient sampling and should also be tested in the context of soft matter systems in the future.
On the application side in the soft matter field, we presented new results for the initialization and pre-equilibration of polymer melts, where a novel rattling algorithm based on ECs can be employed to efficiently remove overlaps in initial configurations. For large systems of semiflexible polymers with a short-range attraction, we demonstrated that ECMC simulations are capable to follow the formation of large networks of bundles and their subsequent foam-like coarsening behavior, see Figure 8. For a two-dimensional hard disk and needle model for LC colloids, the structure formation by parallel chaining of disks could be followed for large systems over large time scales as shown in Figure 9. Here we find that increasing the EC length, i.e., increasing the cluster size in our EC algorithm gives rise to considerable performance gains.
In summary, we addressed compact quasi-zero-dimensional hard sphere particles, one-dimensional hard rods or hard needles, and one-dimensional polymers as constituents in soft matter ECMC simulations. A natural extension for future work is the development of ECMC algorithms for two-dimensional triangulated surfaces with local elastic properties, which are impenetrable for other simulation objects, e.g., hard spheres or polymers. This will allow us to study fluctuating triangulated elastic membranes and, in a compound polymer-membrane system, polymer networks confined to elastic capsules. It was shown that the boundary conditions significantly contribute to the network structure [53]. The elastic properties of a triangulated surface can be described by a set of elastic energies such as the TRBS model [93], where elastic constants like Young’s modulus and Poisson ratio are adjustable and also bending stiffness can be included [94].
The actual implementation will be a very challenging problem. The interaction of a triangle with a point or a hard sphere is an effective 4-particle interaction. The rejection distance is efficiently calculable by the Möller “Trumbore intersection algorithm. To implement non-intersecting surfaces, interactions between two triangles have to be introduced, which can lead to a 6-particle interaction. Therefore, membrane simulations would also represent a further test of the general framework for
Based on the extensive experiences with EC based simulations presented here, we think this simulation technique is suitable to tackle large (biological) systems. In order to verify and evaluate the EC algorithm for triangulated surfaces, we can investigate the crumpling transition [95]. In Ref. [96], the necessary biophysical conditions for the formation of tubular membrane protrusions have been investigated and, it has been suggested that actin filaments polymerizing against a soft membrane are sufficient to form protrusions, which seems a suitable next system to explore the possibilties of EC algorithms for dense soft matter systems.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
TAK conceived research, performed simulations, evaluated data, wrote and revised manuscript, acquired research funding; DM, LPW, CFV performed simulations, evaluated data; JK conceived research, wrote and revised manuscript.
Funding
TAK received support by the Deutsche Forschungsgemeinschaft (DFG) (Grant No. KA 4897/1–1).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
1In our previous work [21] we suggested a steady increase of the hard sphere diameter (similar to the slow push-off) rather than introducing the full diameter at once, but actually implemented the procedure outlined here because of an implementation error. Therefore, the procedure in Ref. [21] was described wrongly, but still produced overlap-free, high quality initial conditions.
2We tried as many as
References
1. Metropolis N, Rosenbluth AW, Rosenbluth MN, Teller AH, Teller E Equation of state calculations by fast computing machines. J Chem Phys (1953) 21:1087–92. doi:10.1063/1.1699114
2. Hastings WK Monte Carlo sampling methods using Markov chains and their applications. Biometrika (1970) 57:97–109. doi:10.1093/biomet/57.1.97
3. Swendsen RH, Wang J Nonuniversal critical dynamics in Monte Carlo simulations. Phys Rev Lett (1987) 58:86–8. doi:10.1103/PhysRevLett.58.86
4. Wolff U Collective Monte Carlo updating for spin systems. Phys Rev Lett (1989) 62:361–4. doi:10.1103/PhysRevLett.62.361
5. Bernard EP, Krauth W, Wilson DB Event-chain Monte Carlo algorithms for hard-sphere systems. Phys Rev E (2009) 80:056704. doi:10.1103/PhysRevE.80.056704
6. Strandburg K Two-dimensional melting. Rev Mod Phys (1988) 60:161–207. doi:10.1103/RevModPhys.60.161
7. Kosterlitz JM, Thouless DJ Ordering, metastability and phase transitions in two-dimensional systems. J Phys C (1973) 6:1181–203. doi:10.1088/0022-3719/6/7/010
8. Halperin BI, Nelson DR Theory of two-dimensional melting. Phys Rev Lett (1978) 41:121–4. doi:10.1103/PhysRevLett.41.121
9. Young AP Melting and the vector Coulomb gas in two dimensions. Phys Rev B (1979) 19:1855–66. doi:10.1103/PhysRevB.19.1855
10. Mak CH Large-scale simulations of the two-dimensional melting of hard disks. Phys Rev E (2006) 73:065104. doi:10.1103/PhysRevE.73.065104
11. Bernard EP, Krauth W Two-step melting in two dimensions: first-order liquid-hexatic transition. Phys Rev Lett (2011) 107:155704. doi:10.1103/PhysRevLett.107.155704
12. Engel M, Anderson JA, Glotzer SC, Isobe M, Bernard EP, Krauth W Hard-disk equation of state: first-order liquid-hexatic transition in two dimensions with three simulation methods. Phys Rev E (2013) 87:042134. doi:10.1103/PhysRevE.87.042134
13. Michel M, Kapfer SC, Krauth W Generalized event-chain Monte Carlo: constructing rejection-free global-balance algorithms from infinitesimal steps. J Chem Phys (2014) 140:054116. doi:10.1063/1.4863991
14. Michel M, Mayer J, Krauth W Event-chain Monte Carlo for classical continuous spin models. EPL (Europhysics Lett (2015) 112. 20003. doi:10.1209/0295-5075/112/20003
15. Harland J, Michel M, Kampmann TA, Kierfeld J Event-chain Monte Carlo algorithms for three- and many-particle interactions. EPL (Europhysics Letters) (2017) 117:30001. doi:10.1209/0295-5075/117/30001
16. Diaconis P, Holmes S, Neal RM Analysis of a nonreversible Markov chain sampler. Ann Appl Probab (2000) 10:726–52. doi:10.1214/aoap/1019487508
17. Peters EAJF, de With G Rejection-free Monte Carlo sampling for general potentials. Phys Rev E (2012) 85:026703. doi:10.1103/PhysRevE.85.026703
18. Nishikawa Y, Michel M, Krauth W, Hukushima K Event-chain algorithm for the Heisenberg model. Phys Rev E (2015) 92:63306. doi:10.1103/PhysRevE.92.063306
19. Gillespie DT Stochastic simulation of chemical kinetics. Annu Rev Phys Chem (2007) 58:35–55. doi:10.1146/annurev.physchem.58.032806.104637
20. Kampmann TA, Boltz HH, Kierfeld J Monte Carlo simulation of dense polymer melts using event chain algorithms. J Chem Phys (2015) 143:044105. doi:10.1063/1.4927084
21. Kampmann TA, Boltz HH, Kierfeld J Parallelized event chain algorithm for dense hard sphere and polymer systems. J Comput Phys (2015) 281:864–75. doi:10.1016/j.jcp.2014.10.059
22. Faulkner MF, Qin L, Maggs AC, Krauth W All-atom computations with irreversible Markov chains. J Chem Phys (2018) 149:064113. doi:10.1063/1.5036638
23. Höllmer P, Qin L, Faulkner MF, Maggs A, Krauth W JeLLyFysh-Version1.0 — a Python application for all-atom event-chain Monte Carlo. Comput Phys Commun (2020) 253:107168. doi:10.1016/j.cpc.2020.107168
25. Curro JG Computer simulation of multiple chain systems—the effect of density on the average chain dimensions. J Chem Phys (1974) 61:1203–7. doi:10.1063/1.1681994
26. Baumgärtner A, Binder K Dynamics of entangled polymer melts: a computer simulation. J Chem Phys (1981) 75:2994–3005. doi:10.1063/1.442391
27. Khalatur P, Pletneva SG, Papulov Y Monte Carlo simulation of multiple chain systems: the concentration dependence of osmotic pressure in two- and three-dimensional spaces. Chem Phys (1984) 83:97–104. doi:10.1016/0301-0104(84)85224-6
28. Haslam AJ, Jackson G, McLeish TCB An investigation of the shape and crossover scaling of flexible tangent hard-sphere polymer chains by Monte Carlo simulation. J Chem Phys (1999) 111:416–28. doi:10.1063/1.479292
29. Rosche M, Winkler RG, Reineker P, Schulz M Topologically induced glass transition in dense polymer systems. J Chem Phys (2000) 112:3051–62. doi:10.1063/1.480880
30. Kremer K, Grest GS Dynamics of entangled linear polymer melts: a molecular—dynamics simulation. J Chem Phys (1990) 92:5057–86. doi:10.1063/1.458541
31. Pütz M, Kremer K, Grest GS What is the entanglement length in a polymer melt?. Europhys Lett (2000) 49:735–41. doi:10.1209/epl/i2000-00212-8
32. Auhl R, Everaers R, Grest GS, Kremer K, Plimpton SJ Equilibration of long chain polymer melts in computer simulations. J Chem Phys (2003) 119:12718–28. doi:10.1063/1.1628670
33. Ramos PM, Karayiannis NC, Laso M Off-lattice simulation algorithms for athermal chain molecules under extreme confinement. J Comput Phys (2018) 375:918–34. doi:10.1016/j.jcp.2018.08.052
34. Hsu HP, Kremer K Static and dynamic properties of large polymer melts in equilibrium. J Chem Phys (2016) 144:154907. doi:10.1063/1.4946033
35. Kremer K Statics and dynamics of polymeric melts: a numerical analysis. Macromolecules (1983) 16:1632–8. doi:10.1021/ma00244a015
36. Paul W, Binder K, Heermann DW, Kremer K Dynamics of polymer solutions and melts. Reptation predictions and scaling of relaxation times. J Chem Phys (1991) 95:7726–40. doi:10.1063/1.461346
37. Karayiannis NC, Mavrantzas VG, Theodorou DN A novel Monte Carlo scheme for the rapid equilibration of atomistic model polymer systems of precisely defined molecular architecture. Phys Rev Lett (2002) 88:105503. doi:10.1103/PhysRevLett.88.105503
38. Wall FT, Mandel F Macromolecular dimensions obtained by an efficient Monte Carlo method without sample attrition. J Chem Phys (1975) 63:4592–5. doi:10.1063/1.431268
39. Sliozberg YR, Kröger M, Chantawansri TL Fast equilibration protocol for million atom systems of highly entangled linear polyethylene chains. J Chem Phys (2016) 144:154901. doi:10.1063/1.4946802
40. Moreira LA, Zhang G, Müller F, Stuehn T, Kremer K Direct equilibration and characterization of polymer melts for computer simulations. Macromol Theor Simulations (2015) 24:419–31. doi:10.1002/mats.201500013
41. Zhang G, Moreira LA, Stuehn T, Daoulas KC, Kremer K Equilibration of high molecular weight polymer melts: a hierarchical strategy. ACS Macro Lett (2014) 3:198–203. doi:10.1021/mz5000015
42. Wittmer JP, Beckrich P, Meyer H, Cavallo A, Johner A, Baschnagel J Intramolecular long-range correlations in polymer melts: the segmental size distribution and its moments. Phys Rev E (2007) 76:011803. doi:10.1103/PhysRevE.76.011803
43. Wittmer JP, Beckrich P, Johner A, Semenov AN, Obukhov SP, Meyer H, et al. Why polymer chains in a melt are not random walks. Europhys Lett (2007) 77:56003. doi:10.1209/0295-5075/77/56003
44. Flory PJ The configuration of real polymer chains. J Chem Phys (1951) 19:1315–6. doi:10.1063/1.1748031
46. Foteinopoulou K, Karayiannis NC, Laso M, Kröger M, Mansfield ML Universal scaling, entanglements, and knots of model chain molecules. Phys Rev Lett (2008) 101:265702. doi:10.1103/PhysRevLett.101.265702
47. Huber F, Schnauß J, Rönicke S, Rauch P, Müller K, Fütterer C, et al. Emergent complexity of the cytoskeleton: from single filaments to tissue. Adv Phys (2013) 62:1–112. doi:10.1080/00018732.2013.771509
48. Blanchoin L, Boujemaa-Paterski R, Sykes C, Plastino J Actin dynamics, architecture, and mechanics in cell motility. Physiol Rev (2014) 94:235–63. doi:10.1152/physrev.00018.2013
49. Bartles JR Parallel actin bundles and their multiple actin-bundling proteins. Curr Opin Cel Biol. (2000) 12:72–8. doi:10.1016/S0955-0674(99)00059-9
50. Lieleg O, Claessens MMAE, Bausch AR Structure and dynamics of cross-linked actin networks. Soft Matter (2010) 6:218–25. doi:10.1039/B912163N
51. Huber F, Strehle D, Käs J Counterion-induced formation of regular actin bundle networks. Soft Matter (2012) 8:931–6. doi:10.1039/C1SM06019H
52. Huber F, Strehle D, Schnauß J, Käs J Formation of regularly spaced networks as a general feature of actin bundle condensation by entropic forces. New J Phys (2015) 17:043029. doi:10.1088/1367-2630/17/4/043029
53. Deshpande S, Pfohl T Hierarchical self-assembly of actin in micro-confinements using microfluidics. Biomicrofluidics (2012) 6:034120. doi:10.1063/1.4752245
54. Deshpande S, Pfohl T Real-time dynamics of emerging actin networks in cell-mimicking compartments. PLoS One (2015) 10:e0116521. doi:10.1371/journal.pone.0116521
55. Strelnikova N, Herren F, Schoenenberger CA, Pfohl T formation of actin networks in microfluidic concentration gradients. Front Mater (2016) 3:1–9. doi:10.3389/fmats.2016.00020
56. Kierfeld J, Kühne T, Lipowsky R Discontinuous unbinding transitions of filament bundles. Phys Rev Lett (2005) 95:038102. doi:10.1103/PhysRevLett.95.038102
57. Kierfeld J, Lipowsky R Unbundling and desorption of semiflexible polymers. Europhys Lett (2003) 62:285–91. doi:10.1209/epl/i2003-00139-0
58. Kierfeld J, Lipowsky R Duality mapping and unbinding transitions of semiflexible and directed polymers. J Phys A Math Gen (2010) 38:L155. doi:10.1088/0305-4470/38/9/L01.–L161
59. Kampmann TA, Boltz HH, Kierfeld J Controlling adsorption of semiflexible polymers on planar and curved substrates. J Chem Phys (2013) 139:034903. doi:10.1063/1.4813021
60. Borukhov I, Bruinsma RF, Gelbart WM, Liu AJ Elastically driven linker aggregation between two semiflexible polyelectrolytes. Phys Rev Lett (2001) 86:2182–5. doi:10.1103/PhysRevLett.86.2182
61. Shabbir H, Dellago C, Hartmann M A high coordination of cross-links is beneficial for the strength of cross-linked fibers. Biomimetics (2019) 4:12. doi:10.3390/biomimetics4010012
62. Stevens MJ Bundle binding in polyelectrolyte solutions. Phys Rev Lett (1999) 82:101–4. doi:10.1103/PhysRevLett.82.101
63. Chelakkot R, Lipowsky R, Gruhn T Self-assembling network and bundle structures in systems of rods and crosslinkers - a Monte Carlo study. Soft Matter (2009) 5:1504–13. doi:10.1039/b808580c
64. Cyron CJ, Müller KW, Schmoller KM, Bausch AR, Wall WA, Bruinsma RF Equilibrium phase diagram of semi-flexible polymer networks with linkers. EPL (Europhysics Lett (2013) 102:38003. doi:10.1209/0295-5075/102/38003
65. Foffano G, Levernier N, Lenz M The dynamics of filament assembly define cytoskeletal network morphology. Nat Commun (2016) 7:13827. doi:10.1038/ncomms13827
66. Ohlenbusch H, Aste T, Dubertret B, Rivier N The topological structure of 2D disordered cellular systems. Eur Phys J B (1998) 2:211–20. doi:10.1007/s100510050242
67. Glazier JA, Weaire D The kinetics of cellular patterns. J Phys Condens Matter (1992) 4:1867–94. doi:10.1088/0953-8984/4/8/004
68. Glazier JA, Gross SP, Stavans J Dynamics of two-dimensional soap froths. Phys Rev A (1987) 36:306–12. doi:10.1103/PhysRevA.36.306
69. Weaire D, Rivier N Soap, cells and statistics—random patterns in two dimensions. Contemp Phys (1984) 25:59–99. doi:10.1080/00107518408210979
71. Kierfeld J, Gutjahr P, Kühne T, Kraikivski P, Lipowsky R Buckling, bundling, and pattern formation: from semi-flexible polymers to assemblies of interacting filaments. J Comput Theor Nanosci (2006) 3:898–911. doi:10.1166/jctn.2006.006
72. Kühne T, Lipowsky R, Kierfeld J Zipping mechanism for force generation by growing filament bundles. EPL (Europhysics Lett (2009) 86:68002. doi:10.1209/0295-5075/86/68002
73. Stark H Physics of colloidal dispersions in nematic liquid crystals. Phys Rep (2001) 351:387–474. doi:10.1016/S0370-1573(00)00144-7
74. Lev BI, Chernyshuk SB, Tomchuk PM, Yokoyama H Symmetry breaking and interaction of colloidal particles in nematic liquid crystals. Phys Rev E (2002) 65:021709. doi:10.1103/PhysRevE.65.021709
75. Terentjev EM Disclination loops, standing alone and around solid particles, in nematic liquid crystals. Phys Rev E (1995) 51:1330–7. doi:10.1103/PhysRevE.51.1330
76. Kuksenok OV, Ruhwandl RW, Shiyanovskii SV, Terentjev EM Director structure around a colloid particle suspended in a nematic liquid crystal. Phys Rev E (1996) 54:5198–203. doi:10.1103/PhysRevE.54.5198
77. Ruhwandl RW, Terentjev EM Monte Carlo simulation of topological defects in the nematic liquid crystal matrix around a spherical colloid particle. Phys Rev E (1997) 56:5561–5. doi:10.1103/PhysRevE.56.5561
78. Lekkerkerker HNW, Tuinier R Colloids and the depletion interaction. in Lecture notes in physics. Berlin: Springer (2011). doi:10.1007/978-94-007-1223-2
79. Ramaswamy S, Nityananda R, Raghunathan VA, Prost J Power-law forces between particles in a nematic. Mol Cryst Liq Cryst (1996) 288:175–80. doi:10.1080/10587259608034594
80. Ruhwandl RW, Terentjev EM Long-range forces and aggregation of colloid particles in a nematic liquid crystal. Phys Rev E (1997) 55:2958–61. doi:10.1103/PhysRevE.55.2958
81. Mozaffari MR, Babadi M, Fukuda J, Ejtehadi MR Interaction of spherical colloidal particles in nematic media with degenerate planar anchoring. Soft Matter (2011) 7:1107–13. doi:10.1039/C0SM00761G
82. Tasinkevych M, Silvestre NM, Telo da Gama MM Liquid crystal boojum-colloids. New J Phys (2012) 14:073030. doi:10.1088/1367-2630/14/7/073030
83. Püschel-Schlotthauer S, Stieger T, Melle M, Mazza MG, Schoen M Coarse-grained treatment of the self-assembly of colloids suspended in a nematic host phase. Soft Matter (2016) 12:469–80. doi:10.1039/C5SM01860A
84. Tasinkevych M, Silvestre NM, Patrício P, Telo da Gama MM Colloidal interactions in two-dimensional nematics. Eur Phys J E (2002) 9:341–7. doi:10.1140/epje/i2002-10087-y
85. Müller D, Kampmann TA, Kierfeld J Chaining of hard disks in nematic needles: particle-based simulation of colloidal interactions in liquid crystals. Sci Rep (2020) 10:12718. doi:10.1038/s41598-020-69544-4
86. Schmidt M Density functional theory for colloidal rod-sphere mixtures. Phys Rev E (2001) 63:050201. doi:10.1103/PhysRevE.63.050201
87. Kim EB, Guzmán O, Grollau S, Abbott NL, de Pablo JJ Interactions between spherical colloids mediated by a liquid crystal: a molecular simulation and mesoscale study. J Chem Phys (2004) 121:1949–61. doi:10.1063/1.1761054
88. Rahimi M, Ramezani-Dakhel H, Zhang R, Ramirez-Hernandez A, Abbott NL, De Pablo JJ Segregation of liquid crystal mixtures in topological defects. Nat Commun (2017) 8:121. doi:10.1038/ncomms15064
89. Gârlea IC, Dammone O, Alvarado J, Notenboom V, Jia Y, Koenderink GH, et al. Colloidal liquid crystals confined to synthetic tactoids. Sci Rep (2019) 9:20391. doi:10.1038/s41598-019-56729-9
90. McGrother SC, Williamson DC, Jackson G A re-examination of the phase diagram of hard spherocylinders. J Chem Phys (1996) 104:6755–71. doi:10.1063/1.471343
91. Kapfer SC, Krauth W Cell-veto Monte Carlo algorithm for long-range systems. Phys Rev E (2016) 94:031302. doi:10.1103/PhysRevE.94.031302
92. Michel M, Durmus A, Sénécal S Forward event-chain Monte Carlo: fast sampling by randomness control in irreversible Markov chains. J Comput Graph Stat (2020) 0:1–14. doi:10.1080/10618600.2020.1750417
93. Delingette H Triangular springs for modeling nonlinear membranes. IEEE Trans Vis Comput Graph (2008) 14:329–41. doi:10.1109/TVCG.2007.70431
94. Tamstorf R, Grinspun E Discrete bending forces and their Jacobians. Graph Models (2013) 75:362–70. doi:10.1016/j.gmod.2013.07.001
95. Kantor Y, Nelson DR Crumpling transition in polymerized membranes. Phys Rev Lett (1987) 58:2774–7. doi:10.1103/PhysRevLett.58.2774
Keywords: Monte Carlo simulations, soft matter physics, hard spheres, liquid crystal colloids, polymer melts, semiflexible polymers, polymer bundle networks, foam
Citation: Kampmann TA, Müller D, Weise LP, Vorsmann CF and Kierfeld J (2021) Event-Chain Monte-Carlo Simulations of Dense Soft Matter Systems. Front. Phys. 9:635886. doi: 10.3389/fphy.2021.635886
Received: 30 November 2020; Accepted: 08 February 2021;
Published: 14 April 2021.
Edited by:
Loukas D. Peristeras, National Centre of Scientific Research Demokritos, GreeceReviewed by:
Nikos Karayiannis, Polytechnic University of Madrid, SpainGeorgios Vogiatzis, National Technical University of Athens, Greece
Copyright © 2021 Kampmann, Müller, Weise, Vorsmann and Kierfeld. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jan Kierfeld, amFuLmtpZXJmZWxkQHR1LWRvcnRtdW5kLmRl