 Salvador Herrera-Velarde1
Salvador Herrera-Velarde1 José Ramón Villanueva-Valencia2
José Ramón Villanueva-Valencia2 Paola Mendoza-Espinosa3
Paola Mendoza-Espinosa3 Ramón Castañeda-Priego4*
Ramón Castañeda-Priego4*- 1Subdirección de Posgrado e Investigación, Instituto Tecnológico Superior de Xalapa, Tecnológico Nacional de México, Veracruz, Mexico
- 2Department of Experimental Medical Science, Lund University, Lund, Sweden
- 3Tecnologico de Monterrey, The Institute for the Obesity Research, Monterrey, Mexico
- 4Departamento de Ingeniería Física, División de Ciencias e Ingenierías, Universidad de Guanajuato, Guanajuato, Mexico
Conformational changes and stability of interacting double-stranded DNA chains under high hydrostatic pressure in biological systems are striking topics of importance to study several biomolecular phenomena. For example, to unravel the physiological conditions at which life might occur and to ensure the right functionality of the biochemical processes into the cell under extreme thermodynamic conditions. Furthermore, such processes could shed light on the physicochemical properties of the DNA under high confinement and how, through different mechanisms, a virus releases its genome in order to infect a cell and, therefore, to promote the process of viral replication. To achieve a few steps toward this direction, we propose an all-atomistic molecular dynamics approach in the NPT isothermal-isobaric ensemble to account for how the interplay of DNA—DNA interaction, hydrogen bonding, and the hydrostatic pressure modifies both the DNA conformational degrees of freedom and the spatial organization of the DNA chains in the available volume. We consider two interacting double-stranded DNA chains immersed in an explicit aqueous solution, i.e., water and ions. Our preliminary results highlight the role of hydrogen bonding and electrostatic interactions between DNA strands to avoid denaturation and, therefore, to provide mechanical stability for the DNA molecules. However, the structural evolution, whose kinetics depends on the relaxation of the stresses induced by the pressure, indicates that almost in all pressure conditions, the equilibrium configuration corresponds to an alignment of the two double-stranded DNA molecules along their main axis of symmetry; the rearrangement between the two approaching DNA dodecamers does not always correspond to complementary base pairs and becomes a function of the thermodynamic conditions.
1 Introduction
Among the millions of biochemical processes and physicochemical activities involved in the proper functionality of living matter, perhaps one of the most interesting phenomena is the structural organization of double-stranded DNA (dsDNA) chains into some specific type of bio-macromolecules [1–6]. All of them protecting in some manner the genome from external agents. From the atomic composition of dsDNA, it is well-known that this polyelectrolyte has an effective negative charge due to the phosphate groups along the chain [7]. This type of charge distribution causes that the DNA interacts electrostatically with itself and other molecules, such as proteins, enzymes and ions, leading to a complex self-organization of the DNA chains that is linked to their functionality. Thus, the full understanding of the DNA properties under controlled experimental conditions is critical to improve our knowledge of its function and relation to certain biochemical activities.
Under bulk conditions, the pioneering contributions of Rau and Parsegian, who carried out X-ray diffraction experiments for long (length ≫ cross section) and parallel DNA strands, elucidated the effects of osmotic pressure on the interstrand distance and the emergence of the so-called hydration forces [8, 9], as well as the consequence of varying the ionic conditions on the interstrand interactions, which can be switched from repulsive to attractive to promote, for example, DNA condensation, depending on the polyvalency of the buffer [10, 11]. These experiments were carried out under thermodynamic conditions that allowed the interchange of water and ion molecules with the buffer in order to keep an homogeneous chemical potential at several polyethylene glycol concentrations, which established an osmotic pressure that induced the ordering of DNA strands.
In Biology, there are some examples of bio-particles, such as cells, sperm and viruses that, in addition to their complex internal components, have DNA chains with a characteristic length [12]. Notably, this DNA is tightly packed into a small volume, which gives rise to a high DNA packing density, whose structural arrangement is influenced by the temperature, interstrand interaction (mediated by the ionic strength of the buffer) and DNA persistence length (bending stress). Even more, the internal pressure of such biomacromolecules plays a key role in the appropriate function of them as the case, for example, of the lytic life cycle of viruses [13]. For example, in the wild-type bacteriophage lambda and herpes simplex virus type 1 [14, 15], due to the high packing of the viral DNA into the capsid, there is experimental evidence of the huge internal pressure, which is of some tens of atmospheres. This high pressure environment facilitates the rapid ejection of the DNA from the capsid into the host cell, which will be infected to initiate the process of viral replication.
Providing knowledge on the physical properties of DNA at high pressures, either hydrostatic or osmotic, could help in, for example, the development of new technologies, such as antiviral molecules [16], as well as to explain pathological processes that might be associated with cancer [17, 18] and the formation of new vectors for gene therapy [19, 20]. Another highlighting phenomenon is the so-called pressure-driven dsDNA denaturation process, which is mainly related to ionic strength variations [21, 22]. In this case, the increase of ionic concentration causes an osmotic pressure difference, which corresponds to a completely different physical scenario than the Rau and Parsegian’s experiments discussed above. In particular, pressure-driven dsDNA denaturation has been described in terms of a thermal-electrostatic competition, i.e., under certain thermodynamic conditions, thermo-mechanical variations can lead to DNA denaturation [21, 22]; pressure-induced DNA melting is basically achieved when the breaking of the hydrogen bonds between base pairs takes place. This mechanism can also be associated with irreversible damage caused to DNA that might be linked to the onset of cancer [23]. This issue plays a key role in the transcription of genomic information, particularly in the synthesis of other biomolecules.
On the other hand, in recent years, there has been a growing interest in the self-assembly and the thermodynamic behavior of bioparticles, such as lysozyme [24–26] and monoclonal antibodies [27, 28], to investigate the mechanisms that give rise to, for instance, the phase separation and aggregation (or particle clustering); processes that are crucial and may affect the functionality of such biomolecules [29, 30]. Typically, the interaction between bioparticles is mediated by the heterogeneous surface charge distribution [31–33], i.e., proteins and enzymes have regions or specific active sites that are electrically charged to recognize other agents in order to bind them, and lock them or catalyze them, mainly, to complete a specific biochemical function [34–36]. This type of process implies a specific orientation or spatial organization of the bioparticles in the available volume, which should also depend on the thermodynamic conditions. This contribution explores the organization and stability of two double-stranded and electrostatically interacting DNA chains under high hydrostatic pressures, i.e., our interest is focused on the hydrostatic pressure exerted by solvent molecules over DNA chains and its effect on their spatial organization. To this end, we have performed all-atomistic molecular dynamics (MD) simulations of two short DNA chains to provide some preliminary insights into the effects of high pressurization and finite particle size on the self-assembly of DNA chains. The MD simulations include explicitly the presence of water molecules and ions to emulate, as close as possible, the physiological conditions where the DNA is typically embedded. We have also explored hydrostatic pressures that are biologically relevant.
2 Molecular dynamics simulation of DNA strands
The results discussed in this work are based on atomistic MD trajectories for two interacting double-stranded B-DNA dodecamers using the parmbsc1 force field [37–39] in conjunction with the GROMACS software package version 2019 [40]; parmbsc1 can be found in GROMACS as amber99bsc1. The parmbsc1 is a general-purpose atomistic force field that takes into account high-level quantum mechanical data [37] and has been tested over a wide variety of DNA sequences. In a coarse-grained picture, we have simulated two strongly screened like-charged flexible rods, immersed in a salty aqueous medium, whose interaction at the cytosine and guanine ends is of electrostatic nature.
The sequence and starting structure of each oligomer were obtained from the Protein Data Bank with code 1BNA. This DNA structure is typically used as a benchmark for the development of force-fields [37]. Each MD run was performed using two double-stranded B-DNA dodecamers (d(CGCGAATTCGCG)2) initially separated by 3 nm and centered in a cubic box of around 8.5 nm, i.e., 2.1 the linear size of a DNA chain (see Figure 1). Although the cubic simulation box has the computational disadvantage of requiring a greater number of water molecules, as compared to other simulation cells with different geometrical shape, it gives rise to the same spatial freedom in the three spatial directions. Therefore, the dodecamers have the same possibility of translational and rotational motion. In addition, other simulation boxes, such as the rhombic dodecahedron and truncated octahedron, are more suitable for globular biomolecules. Several initial conditions and starting DNA arrangements, i.e., parallel, perpendicular and oblique, were used in order to disregard any effect associated with the initial configuration. Structures were then solvated with the TIP3P water model with a minimum of 1.2 nm buffer solvation layer beyond the solute, neutralized, and 0.5 M excess NaCl was added. A typical simulated system consisted of around 18600 water molecules and a total number of 58000 atoms. Larger systems were also considered to discard finite size effects (data not shown). DNA chains, water and salt molecules are in a closed thermodynamic system, so we are essentially testing the effects of hydrostatic pressure.
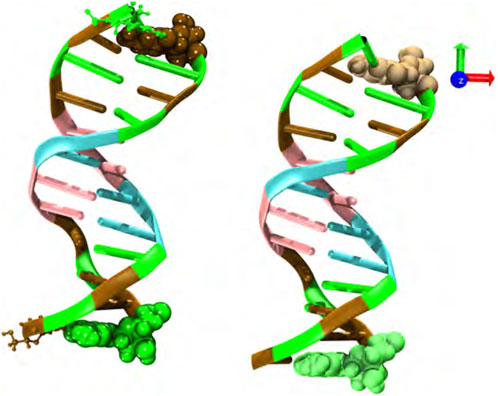
FIGURE 1. Snapshot of the initial configuration; two double-stranded B-DNA dodecamers (d(CGCGAATTCGCG)2) located at the center of the simulation box and initially separated by 3 nm. Water molecules and neutralizing and salt ions are not explicitly shown. Code color: Ochre-cytosine, green-guanine, cyan-adenine and pink-thymine.
MD simulations using the standard leap-frog integrator at the absolute temperature of 300 K and six different hydrostatic pressure values, namely, p = 1, 10, 20, 30, 40, and 50 bar, were carried out using the Verlet cut-off scheme [40]; periodic boundary conditions were applied in all spatial directions. An integration step of 2 fs in conjunction with SHAKE to constrain and fix the X-H bonds to the corresponding default values were considered [41]. The Lennard-Jones and Coulomb interactions were used and computed within a cutoff radius of 1.0 nm, and long-range electrostatic interactions were calculated using the particle mesh Ewald (PME) method [42] with cubic interpolation; GROMACS automatically tunes the load balance by scaling the short-range electrostatic cutoff and grid spacing.
The water-ion-DNA system at 1 bar was optimized using the standard steepest descent energy minimization, followed by a thermalization process during 5 ns using the NVT canonical ensemble. During this equilibration, the temperature was kept constant using the velocity-rescale thermostat [43], with the restriction of fixing the position of heavy atoms to equilibrate the solvent and ions around both DNA molecules. The last configuration of this process was used as the initial configuration in the following steps. After reaching the desired equilibrium temperature, a further pre-equilibrated run for 5 ns in the NPT isobaric-isothermal ensemble was performed using the so-called Berendsen barostat [44] to reach in each case the equilibrium hydrostatic pressure. Subsequently, we have equilibrated each water-ion-DNA system at the targeted hydrostatic pressure for an additional 10 ns period using the Parrinello-Rahman [45] barostat with a 2 ps relaxation time and compressibility of 4.5 × 10−5 bar−1. Finally, a run of 240 ns for each water-ion-DNA system, i.e., p = 1–50 bar, was carried out for gathering statistics and production. We make sure that both DNA chains freely rotate their own size in a timescale shorter than the time window used in the MD simulations. Conformational configurations were saved every 50 ps given a total of 4800 configurations, and visualization snapshots were prepared with the VMD software [46].
3 Results
We now show the results of the MD computer simulations of two short DNA chains immersed in an explicit aqueous solution under different hydrostatic pressures. This work focuses on the evolution of the relative separation between DNA chains. This information is explicitly reported in Figure 2. We show the results that were obtained by considering that both DNA molecules are initially distributed in a parallel configuration and located at a relative distance (measured from the centers of mass (COM)) of 3 nm. However, as mentioned above, we have used other initial configurations and considered larger simulation boxes (keeping fixed the total density) within an extended time window, obtaining practically the same result (data not shown). As seen in the figure, in most pressure conditions, both chains reach an equilibrium co-linear configuration, except for the p = 10 bar case (see snapshots at t = 240 ns). The fact that the two dsDNA chains align along the same main axis is, of course, a consequence of the finite size of the chains. This phenomenology contrasts with the completely different scenario reported for the DNA at the bulk under high osmotic pressure conditions [8, 9], where the osmolyte induced a parallel ordering of the DNA strands. Although hydrostatic and osmotic pressures are not the same, either type can induce a chain-like arrangement of DNA oligomers. Thus, the structural evolution towards the co-linear state clearly depends on the thermodynamic conditions, i.e., hydrostatic pressure. No mechanical instability or denaturation of the DNA molecules is observed. Interestingly, at low and high pressures, the relative distance between chains is the same, about 4 nm, whereas at p = 10 bar, it reaches a value of 3 nm.
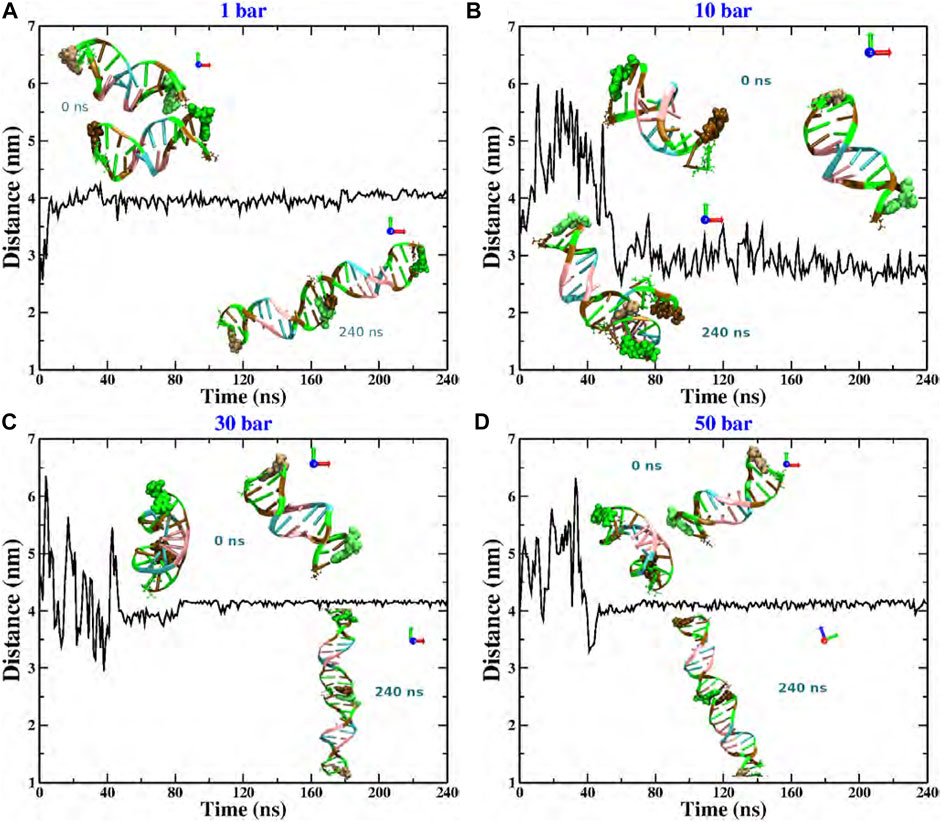
FIGURE 2. Relative distance between the COM of two dodecamers under different hydrostatic pressure conditions: (A) 1 bar, (B) 10 bar, (C) 30 bar, and (D) 50 bar. The snapshots in each case correspond to the initial (upper) and final configurations (bottom). Code color: Ochre-cytosine, green-guanine, cyan-adenine and pink-thymine.
More explicitly, at p = 1 bar (see Figure 2A), the structural reorganization of the DNA molecules occurs monotonically and very fast (at around 10 ns), and the fluctuations around the equilibrium state do not exceed 0.25 nm. This behavior can be understood as follows. Under this pressure condition, the couple of dsDNA molecules start to evolve from a quasi-parallel configuration; however, the electrostatic interactions strongly dominate over the solvent-mediated interaction, which causes the fast rearrangement of chains, looking for the most favorable configuration that minimizes the energy; see Figure 3A. On the other hand, at p = 10 bar (see Figure 2B), the evolution towards the co-linear configuration is not monotonous anymore and, in fact, it is not the predominant one, but, instead, an L-type conformation appears. For this particular hydrostatic pressure condition, one can clearly appreciate that the DNA biomolecules explore an extended configurational landscape and it seems that during an extended time window, they try to be far away from each other. This scenario can be linked to the competition between the electrostatics and the solvent-mediated interaction, i.e., mechanical stresses; the latter enhanced by the pressure; see Figure 3B. Furthermore, the structural relaxation takes a much longer time (
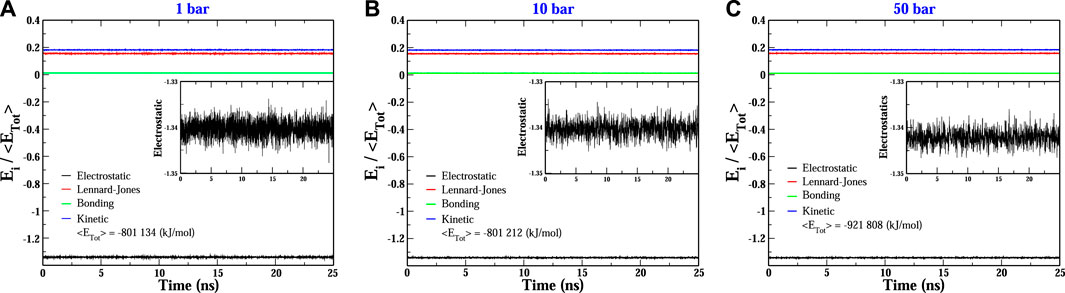
FIGURE 3. Evolution of the main energetic contributions relative to the total energy of the two-dodecamer system under different hydrostatic pressure conditions: (A) 1 bar, (B) 10 bar and (C) 50 bar. Insets show a close view of the electrostatic fluctuations. In all conditions, the electrostatic contribution dominates over the other ones.
It is quite interesting the fact that an increase of one order of magnitude of the pressure value induces a slower structural relaxation. Nevertheless, if the pressure is further increased, Figures 2C, D, which corresponds to pressure values of p = 30 and 50 bar, respectively, the chains are twisted to each other and, at earlier times, they try to explore, again, a rich set of configurations in order to reach the most stable one. The latter being, once again, a co-linear spatial organization whose chain distance fluctuations are much smaller than the previous pressure conditions, describing a persistent equilibrium conformation; see, e.g., Figure 3C. We should point out that the results presented in Figure 2 are very robust in the sense that different initial conditions and several starting DNA configurations were tested and the outcome in all cases was basically the same, even the case p = 10 bar exhibited the same L-type configuration regardless the initial configuration or distribution of dsDNA molecules. In appendix, we discuss in more detail the effect of the initial conditions and the biomolecular recognition on the final configuration state reached by the dsDNA molecules at p = 10 bar.
4 Discussion
The values of the hydrostatic pressure covered in the MD simulations and explored in this work are commonly found in biological environments; larger pressure values, although of academic interest, might not be realistic. Under these conditions, since the density of the whole water-ion-DNA system is basically determined by the water density, as expected, the volume (or dimensions of the simulation box) where the dsDNA molecules are immersed did not change significantly (data not shown), i.e., the water behaves as an incompressible fluid under the thermodynamic conditions here considered. However, it was interesting to note that the relaxation of the internal stresses (produced by the hydrostatic pressure) in the fluid clearly affected the exploration of the configurations that minimize the energy. As discussed in the previous section, except for p = 10 bar, the equilibrium state in the other cases corresponded to a co-linear configuration, which resulted from a delicate interplay of electrostatic interactions and hydrogen bonding between base pairs; the kinetics or speed of the exploration of the configurational states seemed to be mainly determined by both the hydrostatic pressure and molecular recognition, as discussed further below. Other cases at p = 20 and 40 bar were also studied and the co-linear configuration was again the equilibrium state achieved by the dsDNA chains (data not shown). This reflects the generality of the preliminary results reported in this contribution. We should also point out that the fluctuations of the COM could be further elucidated by characterizing the relative dynamics of the two DNA dodecamers, i.e., it is appropriate to determine if those fluctuations are induced, for instance, by hydrodynamic correlations or if they are entirely inherent to the DNA backbone of each dodecamer. This point will be reported elsewhere.
More importantly, we have observed a co-linear arrangement during the different pressure conditions with a non-specific interaction. For example, in the situations at p = 1 and 30 bar, the rearrangement between the DNA dodecamers is generated between terminal cytosine-guanine chains A and B, respectively, but at p = 50 bar it shows that the base pair interaction was between terminal cytosine-cytosine and guanine-guanine of the A and B chains, respectively. The most interesting case was at p = 10 bar, where the pair base interaction is, as already discussed, like an L form. This kind of interaction is due to the structural complementarity or biomolecular recognition, which seemed to result from the pressure exerted on the water-ion-DNA system. However, as displayed in Figure 3, the electrostatic energy became the most important energetic contribution of the water-ion-DNA system, then another aspect that could shed light on the feasibility of the L-shape configuration observed for p = 10 bar is the spatial organization of the ions around the dodecamers. In Figure 4, two snapshots of the water-ion-DNA system are displayed. As illustrated in the figure, the Na+ ions are preferentially distributed on the surface of the DNA chains, indicating that they dominate on the DNA-DNA interaction, as expected. The L-type conformation in dsDNA chains has also been observed in DNA molecules confined between lipid membranes [47]. We should stress out that, however, at all pressure conditions here studied, intrastrand complementarity (G-C) was preserved at the sites of co-linearity of the dsDNAs (see snapshots of Figure 2).

FIGURE 4. Layer of water (red and grey objects) and ions (Na+; blue spheres, and Cl−; cyan spheres) within a cutoff distance of 0.8 nm around the two dodecamers at p = 10 bar and different times, namely, (A) t = 0 ns and (B) t = 100 ns.
During the analysis of the structural evolution of both dsDNA molecules, we also focused on the local structural changes of the typical conformation of B- to non-B-DNA, since it is well-known that they can be induced by changes in the environmental conditions, protein effects, and superhelical tension [48]. These conformational variations directly affect replication, gene expression, recombination, and mutagenesis. Our results indicate that the dodecamers maintained a DNA canonical structure, keeping backbone angles and helical parameters within the expected values for a B-DNA duplex (data not shown). In general, highly charged polyelectrolytes, such as duplex DNA, exhibit significant chain stiffness compared to neutral polymers of similar molecular length due to electrostatic repulsion arising from localized point charges along the molecular boundary. Within our preliminary findings, we have observed a different behavior at p = 10 bar. Under this pressure condition, the water-ion-DNA system reached an equilibrium configuration at a similar time to the other states, but the relative distance between the COM of the dodecamers is 1 nm lower, as a result of the arrangement in L between the dsDNA molecules.
On the other hand, the sugar-phosphate backbone of DNA is hydrophilic. The internal portion of the bases (the rings) has less polar characteristics, although not totally hydrophobic, than the rest of the molecule [49]. The interactions generated between the faces of the rings of the bases of each nucleotide are of the “stacking” type and are highly stable [50, 51]. These aromatic interactions have been proposed to consist of vdW scattering, electrostatic, and hydrophobic forces, but each component’s relative contribution and magnitude still need to be determined [50, 52]. There are three reported representative conformations of pi-stacking interactions with different energy of conformation. These conformations include face-to-face (−1.48 kcal/mol), edge-to-face or T-shaped (−2.46 kcal/mol) and offset (−2.48 kcal/mol) [52]. The offset interaction is present in the interior of the dsDNA chain and on the edge of the co-linear interactions observed in our MD simulations, since it represents the lowest conformation energy. In contrast, a ring-interacting T-shaped conformation seems to be responsible of the L-shaped conformation at p = 10 bar. In this sense, the co-linear conformation generated by the interaction between two ends in the dsDNA sequences is more stable than that created by a single interaction, such as in the L-shaped conformation. Therefore, our hypothesis is that the L-shaped conformation corresponds with a local minimum that is very close to the global one (see appendix). This hypothesis will be tested with further computer simulations.
Although the outcome of this contribution is centered on the very dilute regime at biologically relevant hydrostatic pressures, in contrast, there have been others all-atomistic computer simulation efforts to understand the phase transition of DNA molecules at finite concentration. Most notably, a novel computational method on a multiscale simulation based on AdResS [53, 54] has shown some interesting results at different DNA densities. In particular, it has been reported the influence of the osmotic pressure on the transition of the DNA spatial organization from cholesteric to hexagonal to orthorhombic arrays [55–57], as well as the effect of the ionic strength, concluding that hydration forces are the dominant interaction at high DNA packing, as experimentally highlighted a while ago [10, 11]. This confirms the importance of the explicit inclusion of solvent molecules in order to consider, as much as possible, all fundamental interactions involved in the phase state of any kind of biomacromolecule. Even though the high stress on DNA chains via hydrostatic or osmotic pressures describes different biophysical scenarios, both of them clearly influence the DNA arrangement into the available volume and shed light on our understanding of the thermodynamic conditions that the DNA faces into living matter.
As stated above, we have reported preliminary results that point toward an interesting structural evolution of interacting dsDNA molecules under the influence of hydrostatic pressure. Of course, there are many routes that can be further explored. For example, to carry out a systematic analysis of the pressure-induced self-assembly of a larger number of dsDNA chains, to study longer DNA sequences to mimic the biological conditions and to examine the effect of hydration forces on the structural organization of dsDNA. Work along these lines is currently in progress.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
We would like to acknowledge financial support from CONACyT grants Nos. 237425, 287067 and A1-S-9098, and DAIP-UG 059/2022. JRV-V acknowledges to Mats Paulsson Foundation.
Acknowledgments
Authors also acknowledge computer resources provided by the ITSX.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Charlesworth B, Sniegowski P, Stephan W. The evolutionary dynamics of repetitive DNA in eukaryotes. Nature (1994) 371:215–20. doi:10.1038/371215a0
2. Masai H, Matsumoto S, You Z, Yoshizawa-Sugata N, Oda M. Eukaryotic chromosome DNA replication: Where, when, and how? Annu Rev Biochem (2010) 79:89–130. doi:10.1146/annurev.biochem.052308.103205
3. Bikard D, Loot C, Baharoglu Z, Mazel D. Folded DNA in action: Hairpin formation and biological functions in prokaryotes. Microbiol Mol Biol Rev (2010) 74(4):570–88. doi:10.1128/mmbr.00026-10
4. Rogozin IB, Makarova KS, Natale DA, Spiridonov AN, Tatusov RL, Wolf YI, et al. Congruent evolution of different classes of non-coding DNA in prokaryotic genomes. Nucleic Acids Res (2002) 30(19):4264–71. doi:10.1093/nar/gkf549
5. Shakya A, Park S, Rana N, King JT. Liquid-Liquid phase separation of histone proteins in cells: Role in chromatin organization. Biophys J (2020) 118(3):753–64. doi:10.1016/j.bpj.2019.12.022
6. Levone BR, Lenzken SC, Antonaci M, Maiser A, Rapp A, Conte F, et al. FUS-dependent liquid–liquid phase separation is important for DNA repair initiation. J Cel Biol. (2021) 220(5):e202008030. doi:10.1083/jcb.202008030
7. Dickerson RE. DNA structure from A to Z. Methods Enzymol (1992) 211:67–111. doi:10.1016/0076-6879(92)11007-6
8. Rau DC, Lee B, Parsegian VA. Measurement of the repulsive force between polyelectrolyte molecules in ionic solution: Hydration forces between parallel DNA double helices. Proc Natl Acad Sci USA (1984) 81:2621–5. doi:10.1073/pnas.81.9.2621
9. Parsegian VA, Rand RP, Fuller NL, Rau DC. Osmotic stress for the direct measurement of intermolecular forces. Methods Enzymol (1986) 127:400–16. doi:10.1016/0076-6879(86)27032-9
10. Rau DC, Parsegian VA. Direct measurement of the intermolecular forces between counterion-condensed DNA double helices. Evidence for long range attractive hydration forces. Biophys J (1992) 61(1):246–59. doi:10.1016/s0006-3495(92)81831-3
11. Rau DC, Parsegian VA. Direct measurement of temperature-dependent solvation forces between DNA double helices. Biophys J (1992) 61(1):260–71. doi:10.1016/s0006-3495(92)81832-5
12. Estévez-Torres A, Baigl D. DNA compaction: Fundamentals and applications. Soft Matter (2011) 7(15):6746–56. doi:10.1039/c1sm05373f
13. Gandon S. Why Be temperate: Lessons from bacteriophage λ. Trends Microbiol (2016) 24(5):356–65. doi:10.1016/j.tim.2016.02.008
14. Evilevitch A, Lavelle L, Knobler CM, Raspaund E, Gelbart WM. Osmotic pressure inhibition of DNA ejection from phage. Proc Natl Acad Sci USA (2003) 100(16):9292–5. doi:10.1073/pnas.1233721100
15. Bauer DW, Huffman JB, Homa FL, Evilevitch A. Herpes virus genome, the pressure is on. J Am Chem Soc (2013) 135(30):11216–21. doi:10.1021/ja404008r
16. Brandariz-Nuñez A, Robinson SJ, Evilevitch A. Pressurized DNA state inside herpes capsids—a novel antiviral target. Plos Pathog (2020) 16(7):e1008604. doi:10.1371/journal.ppat.1008604
17. Luo J, Luo Y, Sun J, Zhou Y, Zhang Y, Yang X. Adeno-associated virus-mediated cancer gene therapy: Current status. Cancer Lett (2015) 356:347–56. doi:10.1016/j.canlet.2014.10.045
18. Li C, Bowles D, van Dyke T, Samulski RJ. Adeno-associated virus vectors: Potential applications for cancer gene therapy. Cancer Gene Ther (2005) 12:913–25. doi:10.1038/sj.cgt.7700876
19. Naso MF, Tomkowicz B, Perry WL, Strohl WR. Adeno-associated virus (AAV) as a vector for gene therapy. BioDrugs (2017) 31:317–34. doi:10.1007/s40259-017-0234-5
20. Wright JF. Manufacturing and characterizing AAV-based vectors for use in clinical studies. Gene Ther (2008) 15:840–8. doi:10.1038/gt.2008.65
21. Hernández-Lemus E, Nicasio-Collazo LA, Castañeda-Priego R. Hysteresis in pressure-driven DNA denaturation. PLoS ONE (2012) 7(4):e33789. doi:10.1371/journal.pone.0033789
22. Nicasio-Collazo LA, Delgado-González A, Hernández-Lemus E, Castañeda-Priego R. Counterion accumulation effects on a suspension of DNA molecules: Equation of state and pressure-driven denaturation. J Chem Phys (2017) 146:164902. doi:10.1063/1.4981208
23. Nicasio-Collazo LA, Delgado-González A, Castañeda-Priego R, Hernández-Lemus E. Stress-induced DNA damage: A case study in diffuse large B-cell lymphoma. J R Soc Interf (2014) 11:20140785. doi:10.1098/rsif.2014.0785
24. Hentschel L, Hansen J, Egelhaaf SU, Platten F. The crystallization enthalpy and entropy of protein solutions: Microcalorimetry, van’t hoff determination and linearized Poisson–Boltzmann model of tetragonal lysozyme crystals. Phys Chem Chem Phys (2021) 23:2686–96. doi:10.1039/d0cp06113a
25. Hansen J, Uthayakumar R, Pedersen JS, Egelhaaf SU, Platten F. Interactions in protein solutions close to liquid–liquid phase separation: Ethanol reduces attractions via changes of the dielectric solution properties. Phys Chem Chem Phys (2021) 23:22384–94. doi:10.1039/d1cp03210k
26. Hansen J, Pedersen JN, Pedersen JS, Egelhaaf SU, Platten F. Universal effective interactions of globular proteins close to liquid–liquid phase separation: Corresponding-states behavior reflected in the structure factor. J Chem Phys (2022) 156:244903. doi:10.1063/5.0088601
27. Wang G, Varga Z, Hofmann J, Zarraga IE, Swan JW. Structure and relaxation in solutions of monoclonal antibodies. J Phys Chem B (2018) 122(11):2867–80. doi:10.1021/acs.jpcb.7b11053
28. Hung JJ, Dear BJ, Karouta CA, Chowdhury AA, Godfrin PD, Bollinger JA, et al. Protein-protein interactions of highly concentrated monoclonal antibody solutions via static light scattering and influence on the viscosity. J Phys Chem B (2019) 123:739–55. doi:10.1021/acs.jpcb.8b09527
29. Yearley EJ, Zarraga I, Shire S, Scherer T, Gokarn Y, Wagner N, et al. Small-angle neutron scattering characterization of monoclonal antibody conformations and interactions at high concentrations. Biophys J (2013) 105:720–31. doi:10.1016/j.bpj.2013.06.043
30. Yearley EJ, Godfrin P, Perevozchikova T, Zhang H, Falus P, Porcar L, et al. Observation of small cluster formation in concentrated monoclonal antibody solutions and its implications to solution viscosity. Biophys J (2014) 106:1763–70. doi:10.1016/j.bpj.2014.02.036
31. Raybould MIJ, Marks C, Krawczyk K, Taddese B, Nowak J, Lewis AP, et al. Five computational developability guidelines for therapeutic antibody profiling. Proc Natl Acad Sci USA (2019) 116(10):4025–30. doi:10.1073/pnas.1810576116
32. Davies DR, Padlan EA, Sheriff S. Antibody-antigen complexes. Annu Rev Biochem (1990) 59:439–73. doi:10.1146/annurev.bi.59.070190.002255
33. Stadler AM, Stingaciu L, Radulescu A, Holderer O, Monkenbusch M, Biehl R, et al. Internal nanosecond dynamics in the intrinsically disordered myelin basic protein. J Am Chem Soc (2014) 136:6987–94. doi:10.1021/ja502343b
34. Chaudhri A, Zarraga IE, Kamerzell TJ, Brandt JP, Patapoff TW, Shire SJ, et al. Coarse-grained modeling of the self-association of therapeutic monoclonal antibodies. J Phys Chem B (2012) 116:8045–57. doi:10.1021/jp301140u
35. Chaudhri A, Zarraga IE, Yadav S, Patapoff TW, Shire SJ, Voth GA. The role of amino acid sequence in the self-association of therapeutic monoclonal antibodies: Insights from coarse-grained modeling. J Phys Chem B (2013) 117:1269–79. doi:10.1021/jp3108396
36. Zhang H, Khodadadi S, Fiedler SL, Curtis JE. Role of water and ions on the dynamical transition of RNA. J Phys Chem Lett (2013) 4:3325–9. doi:10.1021/jz401406c
37. Ivani I, Dans PD, Noy A, Perez A, Faustino I, Hospital A, et al. Parmbsc1: A refined force field for DNA simulations. Nat Methods (2016) 13:55–8. doi:10.1038/nmeth.3658
38. Dans PD, Ivani I, Hospital A, Portella G, Gonzalez C, Orozco M. How accurate are accurate force-fields for B-DNA? Nucleic Acids Res (2017) 45(7):4217–30. doi:10.1093/nar/gkw1355
39. Galindo-Murillo R, Robertson JC, Zgarbova M, Sponer J, Otyepka M, Jurecka P, et al. Assessing the current state of amber force field modifications for DNA. J Chem Theor Comput. (2016) 12:4114–27. doi:10.1021/acs.jctc.6b00186
40. Abraham MJ, Murtola T, Schulz R, Pall S, Smith JC, Hess B, et al. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX (2015) 1:19–25. doi:10.1016/j.softx.2015.06.001
41. Ryckaert J-P, Ciccotti G, Berendsen HJ. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J Comput Phys (1977) 23:327–41. doi:10.1016/0021-9991(77)90098-5
42. T Darden DY, Pedersen L. Particle mesh Ewald - an N.log(N) method for Ewald sums in large systems. J Chem Phys (1993) 98:10089–92. doi:10.1063/1.464397
43. Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys (2007) 126:014101. doi:10.1063/1.2408420
44. Berendsen HJC, Postma JPM, Vangunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys (1984) 81:3684–90. doi:10.1063/1.448118
45. Parrinello M, Rahman A. Polymorphic transitions in single crystals: A new molecular dynamics method. J Appl Phys (1981) 52:7182–90. doi:10.1063/1.328693
46. Humphrey W, Dalke A, Schulten K. Vmd - visual molecular dynamics. J Mol Graph (1986) 14:33–8. doi:10.1016/0263-7855(96)00018-5
47. Potaman VN, Sinden RR. Dna: Alternative conformations and Biology. In: Madame curie bioscience database. Germany: Science open (2000–2013). [Internet].
48. Wilton DJ, Ghosh M, Chary KV, Akasaka K, Williamson MP. Structural change in a B-DNA helix with hydrostatic pressure. Nucleic Acids Res (2008) 36:4032–7. doi:10.1093/nar/gkn350
49. Feng B, Sosa RP, Mårtensson AKF, Jiang K, Tong A, Dorfman KD, et al. Hydrophobic catalysis and a potential biological role of DNA unstacking induced by environment effects. Proc Natl Acad Sci U S A (2019) 116:17169–74. doi:10.1073/pnas.1909122116
50. Karabiyik H, Sevinçek R, Karabiyik H. π-Cooperativity effect on the base stacking interactions in DNA: Is there a novel stabilization factor coupled with base pairing H-bonds? Phys Chem Chem Phys (2014) 16:15527–38. doi:10.1039/c4cp00997e
51. Cooper VR, Thonhauser T, Puzder A, Schröder E, Lundqvist BI, Langreth DC. Stacking interactions and the twist of DNA. J Am Chem Soc (2008) 130:1304–8. doi:10.1021/ja0761941
52. Zhao Y, Li J, Gu H, Wei D, Xu YC, Fu W, et al. Conformational preferences of π-π stacking between ligand and protein, analysis derived from crystal structure data geometric preference of π-π interaction. Interdiscip Sci (2015) 7:211–20. doi:10.1007/s12539-015-0263-z
53. Praprotnik M, Delle Site L, Kremer K. Adaptive resolution molecular-dynamics simulation: Changing the degrees of freedom on the fly. J Chem Phys (2005) 123:224106. doi:10.1063/1.2132286
54. Praprotnik M, Delle Site L, Kremer K. Multiscale simulation of soft matter: From scale bridging to adaptive resolution. Annu Rev Phys Chem (2008) 59:545–71. doi:10.1146/annurev.physchem.59.032607.093707
55. Yasar S, Podgornik R, Valle-Orero J, Johnson M, Parsegian VA. Continuity of states between the cholesteric → line hexatic transition and the condensation transition in DNA solutions. Sci Rep (2014) 4:6877. doi:10.1038/srep06877
56. Podgornik R, Zavadlav J, Praprotnik M, 2018. Molecular dynamics simulation of high density DNA arrays. Computation, 6, 3, doi.org/doi:10.3390/computation6010003
57. Zavadlav J, Podgornik R, Praprotnik M, 2017. Order and interactions in DNA arrays: Multiscale molecular dynamics simulation. Sci Rep 7, 4775, doi.org/doi:10.1038/s41598-017-05109-2
58. Singh AK, Wen C, Cheng S, Vinh NQ. Long-range DNA-water interactions. Biophysical J (2021) 120(22):4966–79. doi:10.1016/j.bpj.2021.10.016
59. McDermott ML, Vanselous H, Corcelli SA, Petersen PB. DNA’s chiral spine of hydration. ACS Cent Sci (2017) 3(7):708–14. doi:10.1021/acscentsci.7b00100
60. Shweta H, Sen S, 2018. Dynamics of water and ions around DNA: What is so special about them? J Biosciences, 43(3), 499–518. doi.org/doi:10.1007/s12038-018-9771-4
61. Alexiou TS, Alatas Pv., Tsalikis DG, Mavrantza VG. Conformational and dynamic properties of short DNA minicircles in aqueous solution from atomistic molecular dynamics simulations. Macromolecules (2020) 53(14):5903–18. doi:10.1021/acs.macromol.0c00821
62. Schneider B, Berman HM. Hydration of the DNA bases is local. Biophys J (1995) 69(6):2661–9. doi:10.1016/s0006-3495(95)80136-0
Appendix Analysis of the case p = 10 bar: Effects of the initial conditions
Among the pressure conditions we have examined, the p = 10 bar case revealed a unique feature that was not seen in the other cases. As was discussed in the main text, the L-type shape that the dsDNA strands reached could be linked to a local energy minimum. This non-specific contact among dodecamers could result from a process involving the first hydration layers and the unrestricted mobility of the dsDNA chains in the aqueous medium; see snapshots displayed in Figure 4. It has been discovered that DNA exhibits a hydration sphere, which is thought to be crucial for its structure and biological function [58], as well as water molecules that form a chiral skeleton in the minor groove of DNA, whose biological function is unknown [59]. Several spectroscopic [60] and molecular dynamics [61] methods were used to make these observations. As previous works have shown, DNA-water interactions are crucial for the helical stability and conformational variability of DNA itself. In the same way, they are essential to modulate DNA binds to other biomolecules and ligands [61]. Although we did not directly investigate this process in our work, the data show that it has an electrostatic component that is pressure dependent.
It is interesting to note that in our study, two dodecamers of double-stranded B-DNA (d (CGCGAATTCGCG)2) exhibited remarkable stability in their structure because of end positions of cytosine (ochre) and guanine (green) in their sequence which are well known to be triple-bonded and being stronger than the double-bonded thymine and adenine, which are located in the middle of DNA sequence. This structural conformation of each double-chain makes them stable without any possibility of fracture or denaturation. For p = 10 bar, we specifically noticed an L conformation between the two double chains during the simulations. This configuration is related with the molecular interaction between the thymine in the eighth position of one chain and the guanine in the 12th position of the juxtaposed chain in an unspecific but steady manner, see Figure A1A). Visually speaking, in the simulations, one can notice that this thymine-guanine interaction caused a tiny compaction in the DNA double strand itself. The backbone of both nucleotides, and the interactions between their polar atoms played a role in the interactions between thymine and guanine. Experimental observations show that this kind of interaction occurs in the minor groove, since the shortening of the distance in this region inhibits the establishment of stable hydrogen bonds [62]. This makes it easier for the polar atoms of the nearby nucleotides to be exposed.
Finally, we should highlight that a second MD simulation was produced using as initial condition the L configuration at p = 50 bar to either confirm or disregard the existence of a local minimum at p = 10 bar. The simulations revealed again the development of the co-linear structure after 25 ns; see Figure A1B), indicating that the increase of the hydrostatic pressure likely enables the rehydration of the thymine-guanine interaction site and the rearrangement of the two duplexes into a co-linear conformation (Figure 2D). The collinear conformation is produced by non-specific interactions between interphase nucleotides. They can be formed between bases of the same type or crossing, it has been noted. The interaction is between purine and pyrimidine bases in the cases for p = 1 bar and p = 30 bar, while in p = 20 bar and p = 50 bar, it is between identical bases.
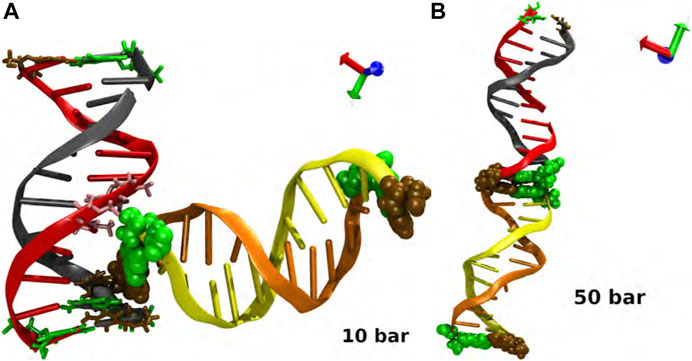
FIGURE A1. Interaction and biomolecular recognition between pairs of DNA at (A) p = 10 bar and (B) p = 50 bar. The snapshots in each case corresponds to a closed view of the intermolecular interactions. Code color: Ochre-cytosine, green-guanine, cyan-adenine and pink-thymine. Color of the DNA chains is just for a better visualization.
Keywords: DNA, denaturation, stability, molecular dynamics, pressure-driven processes
Citation: Herrera-Velarde S, Villanueva-Valencia JR, Mendoza-Espinosa P and Castañeda-Priego R (2023) Stability and structural evolution of double-stranded DNA molecules under high pressures: A molecular dynamics study. Front. Phys. 11:1076787. doi: 10.3389/fphy.2023.1076787
Received: 21 October 2022; Accepted: 18 January 2023;
Published: 06 February 2023.
Edited by:
Leonardo Dagdug, Metropolitan Autonomous University, MexicoReviewed by:
Rudolf Podgornik, University of Chinese Academy of Sciences, ChinaEdward John Sambriski, Delaware Valley University, United States
Copyright © 2023 Herrera-Velarde, Villanueva-Valencia, Mendoza-Espinosa and Castañeda-Priego. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ramón Castañeda-Priego, cmFtb25jcEBmaXNpY2EudWd0by5teA==