 Karla Lienau
Karla Lienau C. A. Triana
C. A. Triana Lukas Reith
Lukas Reith Sebastian Siol2
Sebastian Siol2 Greta R. Patzke
Greta R. Patzke- 1Department of Chemistry, University of Zurich, Zurich, Switzerland
- 2Empa—Swiss Federal Laboratories for Materials Science and Technology, Dübendorf, Switzerland
Water oxidation is the bottleneck reaction for overall water splitting as a direct and promising strategy toward clean fuels. However, the development of robust and affordable heterogeneous water oxidation catalysts remains challenging, especially with respect to the wide parameter space of synthesis and resulting material properties. Oxide catalysts performance in particular has been shown to depend on both synthetic routes and applied catalytic test methods. We here focus on spinel-type Co3O4 as a representative case for an in-depth study of the influence of rather subtle synthetic parameter variations on the catalytic performance. To this end, a series of Co3O4 samples was prepared via time-saving and tunable microwave-hydrothermal synthesis, while systematically varying a single parameter at a time. The resulting spinel-type catalysts were characterized with respect to key materials properties, including crystallinity, oxidation state and surface area using a wide range of analytical methods, such as PXRD, Raman/IR, XAS and XPS spectroscopy. Their water oxidation activity in electrocatalytic and chemical oxidation setups was then compared and correlated with the obtained catalyst properties. Both water oxidation methods displayed related trends concerning favorable synthetic parameters, namely higher activity for lower synthesis temperatures, lower precursor concentrations, addition of hydrogen peroxide and shorter ramping and reaction times, respectively. In addition to the surface area, structural features such as disorder were found to be influential for the water oxidation activity. The results prove that synthetic parameter screening is essential for optimal catalytic performance, given the complexity of the underlying performance-properties relationships.
Introduction
Water splitting is a promising renewable energy approach due to the superior storage options of fuels compared to electricity generated by solar cells. After decades of research into water splitting, the optimal catalytic systems are yet to be found, not only in terms of efficiency but also considering stability and applicability (Chu et al., 2017). To this end, the water oxidation half reaction with its four electron transfer steps remains the bottleneck of the process. Heterogeneous water oxidation catalysts (WOCs) are the most obvious choice for future technical applications due to their superior stability and scale-up possibilities compared to molecular catalysts (Najafpour et al., 2016; Li et al., 2017). A widely used heterogeneous catalyst is spinel-type Co3O4 due to its several advantages, such as long time durability and lower costs compared to noble metal materials (Artero et al., 2011; Wang et al., 2016a; Zhao et al., 2017). Furthermore, Co3O4 is promising for many other applications, such as data storage, sensor applications or battery electrodes as well as for other heterogeneous catalysis processes (Wang et al., 2004, 2015; Li et al., 2005; Luo et al., 2014; Iosub and Stahl, 2015; Zhou et al., 2015; Gao et al., 2018). Heterogeneous WOCs optimization is a complex process that does not only focus on the type of catalyst, but also on other influential parameters, including crystallinity, morphology, size, exposed facets, surface vs. bulk oxidation states and other material properties (Najafpour et al., 2012; Zhao et al., 2017). In order to derive clear design guidelines for heterogeneous WOCs, the investigation of their reaction mechanisms is indispensable. However, this remains a challenging task and requires sophisticated in situ analyses. In a recent representative study on Co3O4 WOCs by Zhang et al. (2014a), the HO–Co2(μ-O/OH)2-OH edge site cobalt motif, i.e., two adjacent Co ions coupled via an oxygen bridge, was shown to be essential for efficient catalysis.
In search of empirical guidelines for catalyst optimization, a number of different synthesis methods have been applied in previous studies to obtain Co3O4 in various particle sizes and morphologies (Grzelczak et al., 2013; Rosen et al., 2014; Zhang et al., 2014a,b, 2017, 2018; Bergmann et al., 2015; Chua et al., 2016; Wang et al., 2016b; Cordeiro and Carvalho, 2018; Liu et al., 2018; Wei et al., 2018; Zhou et al., 2018). We recently conducted a screening investigation where we compared spinel-type Co3O4 samples prepared by a broad spectrum of different synthesis methods with respect to their characteristics and their photo-, electro-, and chemical water oxidation activity (Reith et al., 2019). Indeed we found that the applied synthetic method exerted a major influence on the catalytic water oxidation. Furthermore, considerable deviations between the activity trends for the three applied water oxidation methods were observed. Whereas a strong correlation between the activity and the surface area, crystallinity and disorder, respectively, of the corresponding materials was found for chemical oxidation, electrocatalytic water oxidation was barely influenced by these parameters. This finding agrees with the previously observed in situ formation of cobalt oxyhydroxide in electrocatalytic water oxidation as a general intermediate observed in previous works (Bergmann et al., 2015; Tung et al., 2015).
In the present study, we conduct an in-depth investigation of the influence of microwave-hydrothermal synthesis parameters on the WOC activity of Co3O4 nanocubes, thereby narrowing the parameter range further down in search of optimization guidelines. Microwave synthesis, which is already well-established as a versatile option in organic synthesis, attracts increasing interest in inorganic synthesis, especially for different oxide materials (Bilecka et al., 2009; Bilecka and Niederberger, 2010; Hilaire et al., 2014; Kuzmanoski et al., 2015; Dong et al., 2016; Wang et al., 2016b). Microwaves as non-ionizing and long-wavelength electromagnetic radiation enable high penetration depths and fast heating, thus rendering syntheses economically attractive by decreasing reaction times and temperatures. Due to optimization of the microwave reactor conditions, such as shape, constant motion of the reactor vessels and stirring, thermal gradients and “hot spots” can be largely prevented leading to uniform growth and dimensions of the oxide particles (Nüchter et al., 2004). This renders microwave synthesis a crucial option for well-defined particle fabrication in nanoscience and technology (Hilaire et al., 2014). However, little is still known about the growth mechanisms of metal oxides under the influence of microwaves (Moura et al., 2010; Koziej et al., 2013; Zeng et al., 2013). Therefore, we here provide a broader insight into the crucial influence of a set of different synthesis parameters.
In the case of Co3O4 we recently demonstrated that the growth mechanism can play a key role in the resulting water oxidation activity (Reith et al., 2018). Therefore, we here started from a well-defined microwave protocol for microwave-assisted spinel-type Co3O4 and systematically studied the influence of the synthetic parameters on the properties and catalytic performance of the emerging materials to demonstrate the importance of these parameters in microwave-hydrothermal WOC synthesis (Conrad et al., 2010, 2012, 2013). To this end, the Co3O4 spinel samples were characterized with a wide range of analytical methods and subsequently compared with respect to their chemical and electrocatalytic water oxidation activity. The results of this study shed detailed and practical light on our recent work by illustrating that a given Co3O4 catalyst cannot be simply compared to another specimen without taking the preparative history into account (Reith et al., 2019).
Results and Discussions
In order to screen their influence on the WOC activity of cobalt oxide nanoparticles, the selected standard microwave hydrothermal method was varied with respect to the following parameters: temperature, precursor concentration, amount of hydrogen peroxide added to the synthesis mixture, the ramping and holding times of the synthesis and the stirring speed (see Table 1, cf. experimental section and Table 2 for details). Samples were labeled with respect to the single varied parameter.
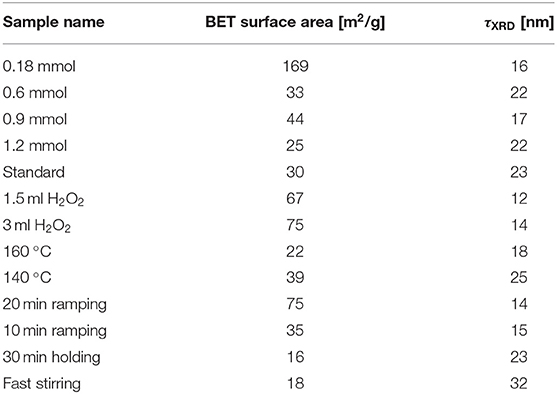
Table 1. Surface area measured by N2-sorption and determined by BET analysis together with crystallite sizes determined from FWHM values of PXRD peaks using the Scherrer equation.
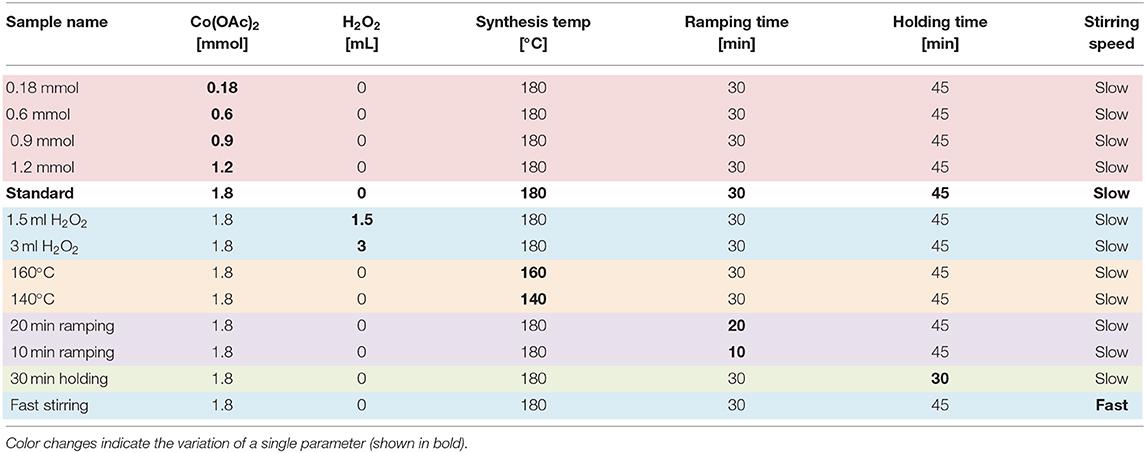
Table 2. Microwave synthesis parameters of the investigated spinel-type Co3O4 samples.
Powder X-ray diffraction (PXRD) patterns show the formation of a phase pure spinel structure [space group Fdm (No.: 227)] for all samples (Figure 1A). Co3O4 crystallizes in the normal spinel structure, i.e., the Co2+ ions occupy one eighth of the tetrahedral voids of cubic close packed oxygen anions while Co3+ ions occupy half of the octahedral voids. The general structural model is in accordance with the obtained Raman spectra shown in Figure 1B, where five phonon excitations can be observed: the A1g, Eg, and the three F2g modes are Raman active (Hadjiev et al., 1988). The symmetric Co-O stretching vibration of the octahedrally coordinated cobalt centers appearing at 693–685 cm−1 is assigned to the A1g mode (Gawali et al., 2018). The Co-O stretching vibration of tetrahedrally coordinated cobalt centers is attributed to the mode at 196–193 cm−1. The two remaining F2g modes and the Eg mode are located at 621–618 cm−1, 523–519 cm−1, and 484–480 cm−1, respectively. The relative shifts of the Raman peaks as well as the different peak broadening indicate a difference in the short-range order of the synthesized Co3O4 spinel samples, analogous to the peak broadening observed in the PXRD patterns. Only very small shifts of the Raman peak centers of A1g and modes are observed for the investigated samples (Figure S1), indicating the presence of very similar sample structures.
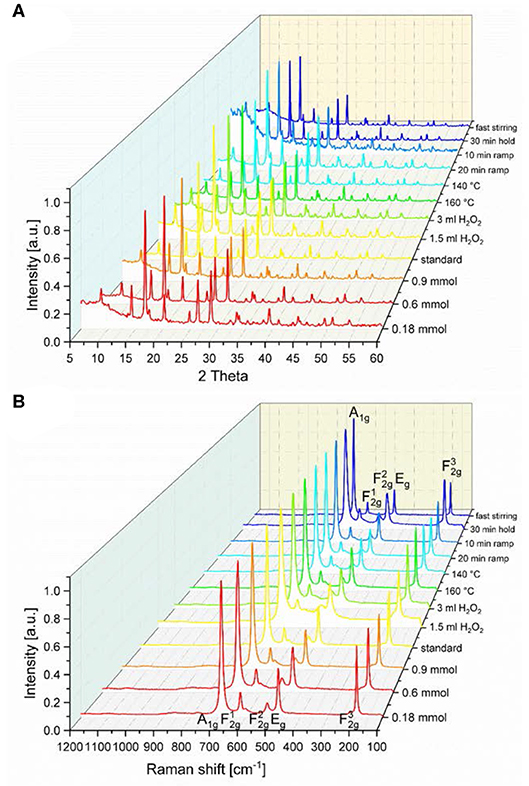
Figure 1. (A) PXRD patterns and (B) Raman spectra of the different spinel-type Co3O4 samples obtained from parameter variations of the standard microwave protocol.
Even though the Co3O4 spinel oxides basically show the same structure, the Bragg reflections of the different samples (Figure 1A) exhibit slightly different full width half maxima (FWHM), from which the crystallite domain size was calculated by means of the Scherrer equation (Scherrer, 1918; Patterson, 1939). The so calculated crystallite sizes τXRD are compared in Table 1. Broader reflections than obtained from the standard synthesis procedure were observed for the materials synthesized with the lowest precursor concentration, with addition of H2O2 and with more rapid heating. These parameter variations resulted in a lower crystallite size for lower concentrations (τXRD ≤ 22 nm), higher amount of H2O2 (τXRD ≤ 14 nm), and shorter ramping (τXRD ≤ 15 nm), compared to the standard procedure with τXRD = 23 nm. Interestingly, the temperature, the stirring speed and the reaction time (holding time) did not exert a significant impact on the FWHM of the Bragg reflections. The values of the surface area obtained from Brunauer-Emmet-Teller (BET) analyses (Table 1) moderately correlate with the crystallite sizes obtained from Scherrer analysis, but appear generally quite low taking the crystallite sizes into account. This may indicate that the obtained Co3O4 particles were agglomerated to a large extent.
TEM images (Figure 2) show only moderately aggregated particles, but this might be due to the different sample preparation for TEM and BET analysis. Whereas for BET measurements, the pestled powder is heated under vacuum to remove adsorbed gases, TEM samples are crushed in a mortar, followed by homogeneous dispersion in ethanol. Furthermore, it is obvious from TEM images that variation of the synthesis parameters exerts a notable influence on the resulting morphologies. Especially shorter ramping times (Figures 2D,G) and the addition of hydrogen peroxide (Figures 2J,K) result in much smaller particles with diameters around 10–20 nm, whereas particle sizes of 30–40 nm were obtained from the standard synthesis. HR-TEM and SEM images are provided in (Figures S2, S3).

Figure 2. TEM images of the spinel samples emerging from parameter variations of the standard microwave protocol (F): (A) and (B) synthesis at 140 and 160 °C, (C) 30 min holding time, (D) and (G) 10 and 20 min ramping time, (E) and (H) 0.9 and 0.18 mmol cobalt precursor, (I) faster stirring speed, (J) and (K) with addition of 1.5 and 3 mL H2O2 (30 wt%). For all images, the same magnification of 50 k was applied (scale bar = 100 nm).
Deeper insight into the atomic short-range order of the different synthesized Co3O4 oxides was obtained through extended X-ray absorption fine structure (EXAFS) analyses. Figure 3 shows the fitting of the Fourier-Transform FT|k3χ(k)| of the experimental Co K-edge EXAFS spectra k3χ(k), for oxides synthesized at different H2O2 concentrations of 3 mL and 1.5 mL, low temperatures of 140 °C and 160 °C and at a lower Co(OAc)2 concentration of 1.2 mmol. Calculated main values for interatomic distances, atomic coordination numbers (N), and Debye-Waller factors (σ2) are given in Table S1. The first peak in the FT|k3χ(k)| spectra at r ≈ 1.55 Å, arising from backscattering of neighboring O atoms, relates to Co2+ and Co3+ cations in tetrahedral {CoO4} and octahedral {CoO6} coordination with oxygen atoms at interatomic distances r ≈ 1.914 Å and r ≈ 1.899 Å, respectively. Those two shells, however, are too close to be resolved in the FT|k3χ(k)| spectra, and hence they convolute to a main Co-O shell with a main interatomic distance Co-O ≈ 1.913 Å and main atomic coordination number N = 5.333. The second and third peaks at r ≈ 2.49 Å and r ≈ 2.95 Å due to backscattering of neighboring Co atoms, relate to the CoOct-CoOct ≈ 2.856 Å [N = 4] and CoTet-CoOct ≈ 3.365 Å [N = 8] coordination shells, respectively. The fourth peak at r ≈ 4.70 Å relates to higher-order Co-O(-Co) coordination shells (Figure 3A and Table S1). The position of the coordination peaks in the FT|k3χ(k)| spectra and of their corresponding Wavelet-Transform (WT) spectra shown in Figure 3B does not change among the different Co3O4 oxides. However, both the FT|k3χ(k)| and WT spectra exhibit a decrease in the magnitude of the Co-O, CoOct-CoOct, CoTet-CoOct and higher Co-Co(-O) coordination peaks. The relative amplitude decay in the FT|k3χ(k)| and WT spectra is correlated with a decrease in the coordination number (N), or an increase in the mean-square disorder parameter σ2, which arises from static structural disorder from crystal defects due to slightly different interatomic distances in the same coordination shell. Therefore, the weakened relative amplitudes of high coordination peaks in the FT|k3χ(k)| and WT spectra offer an indication of the extent of crystalline short-to-medium-range order around the cobalt centers.
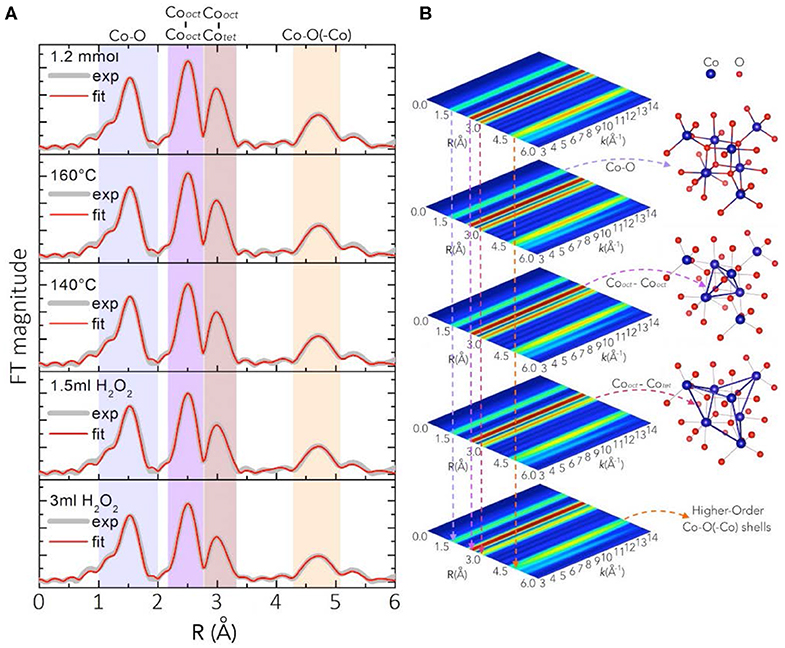
Figure 3. (A) Fitting (red) of the FT|k3χ(k)| spectra (gray) for Co3O4 oxides synthesized in the presence of different H2O2 amounts (3 and 1.5 mL), low temperatures of 140 and 160 °C, and a lower Co(OAc)2 concentration of 1.2 mmol (phase uncorrected). The 2D contour plots in (B) are the WT of the k3χ(k) spectra and their shaded regions highlight the decrease of the peak intensity due to increases in the local disorder σ2 of the Co-O, CoOct-CoOct, CoTet-CoOct and higher Co-Co(-O) coordination shells.
The Co3O4 oxides obtained from the standard synthesis, and those synthetized at 30 min holding and at 10 min ramping time, respectively, display FT|k3χ(k)| spectra similar to those of Co3O4 synthesized at lower Co(OAc)2 concentrations of 0.9 and 1.2 mmol (Figure S4). As shown in Figures 3A,B, Figure S4 and Table S1, the interatomic distances and the atomic coordination numbers N of those oxides remain the same, showing that their atomic short-range order is very similar regardless of the variations in the synthetic parameters, and that they exhibit higher crystallinity among the series of as-synthesized Co3O4 oxides. While the main interatomic distances and N-values for Co3O4 oxides synthesized at lower temperatures of 160 and 140 °C remain quite the same, those oxides show an increase in the mean-square static disorder σ2 (Table S1). This suggests that these oxides have slightly increased local disorder, pointing to the synthesis temperature as a crucial parameter for controlling the local order and the degree of crystallization of the bulk spinel structure. The static local-disorder σ2 increases more remarkably for Co3O4 oxides synthesized at H2O2 concentrations of 1.5 and 3 mL (Figures 3A,B, Figure S4 and Table S1), which indicates increased local disorder and lower degrees of crystallinity in those oxides. The latter may arise from their formation process: when adding H2O2 to the initial suspension (Co(OAc)2·4H2O + 1.5 mL H2O + 25% NH3), some divalent Co2+ cations are replaced by trivalent Co3+ species. The anions in suspension are then intercalated into the interlayer space to compensate the extra positional charge inferred by Co3+ cations, thus leading to the formation of a hydrotalcite-type cobalt compound (Yang et al., 2007). Thereafter, upon hydrothermal treatment at 180 °C, this intermediate hydrotalcite-cobalt compound is converted into spinel-type Co3O4 (Amiri et al., 2011). This structural transformation could induce increased local disorder in the oxides synthesized in the presence of H2O2 as oxidant. This observation agrees with the results from PXRD and Raman spectra, where Co3O4 oxides synthesized at H2O2 concentrations of 1.5 and 3 mL show broadened diffraction and Raman peaks due to the local structural dispersion of the Co and O atoms in the spinel structure (Figure 1).
X-ray absorption near edge structure (XANES) spectra were recorded to obtain further insight into the electronic properties and main oxidation states of Co in the synthesized oxides. Figure 4A shows the XANES spectra for oxides synthesized at H2O2 quantities of 3 and 1.5 mL, low temperatures of 140 and 160 °C and Co(OAc)2 concentrations of 1.2 mmol vs. those of the reference compounds CoIIO and LiCoIIIO2. Co3O4 oxides obtained from standard synthesis, and those synthesized at 30 min holding and at 10 min ramping time displayed XANES spectra similar to those of Co3O4 synthesized at lower Co(OAc)2 concentrations of 0.9 and 1.2 mmol (Figure S5). The Co K-edge absorption edge energy of the different Co3O4 oxides is located at ≈ 7719.64–7719.82 eV (Figures 4A,B), suggesting that the oxidation state of Co is quite the same for all the synthesized Co3O4 oxides. From the linear dependence of the Co K-edge position at the energy corresponding to μ(E) ≈ 0.5 of the normalized XANES spectra of Co3O4 and reference oxides CoIIO, LiCoIIIO2, the main oxidation state of Co was calculated as 2.65 (Figure 4C). This agrees with the main cobalt oxidation state of a normal Co3O4 spinel structure with 8 Co2+ cations located in tetrahedral and 16 Co3+ cations located in octahedral sites, respectively. The slight changes in the white line intensity at ≈ 7729.57 eV (Figure 4A), further indicates the existence of local disorder and a slightly different density of unoccupied Co d-states for Co3O4 oxides synthesized with H2O2 concentrations of 1.5 and 3 mL. This, as previously discussed, could be due to the structural transformations induced when using H2O2 as oxidant, and to some extent be caused by a charge imbalance due to the interaction of Co2+-Co3+ species (Dau et al., 2003).
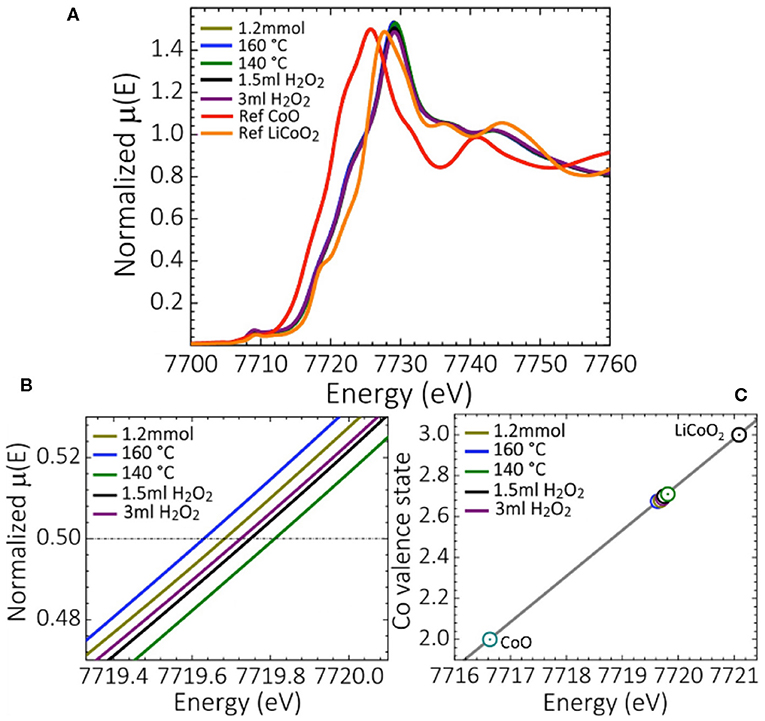
Figure 4. (A) XANES spectra of selected synthesized Co3O4 oxides, and reference compounds CoIIO and LiCoIIIO2, (B) zoom of the Co absorption K-edge of the normalized XANES spectra, and (C) the average Co valence state determined from the XANES spectra.
X-ray photoelectron spectroscopy was conducted on the standard, 3 mL H2O2, 140 °C and 0.18 mmol samples, the latter corresponding to the three synthesis conditions deviating most widely from the standard protocol. The Co 2p spectra are shown in Figure 5B with similar binding energies for all samples of ≈ 780 eV for the Co 2p3/2 and ≈ 795 eV for the Co 2p1/2 peak, which corresponds to literature values (Linstrom, 1997). The spectra of the reference compounds CoIIO and LiCoIIIO2 are shown in the same graph, indicating an increase in surface oxidation state in the order 0.18 mmol < standard ≈ 3 mL H2O2 < 140 °C, which is in accordance with the XANES data shown in Figures 4A–C. The oxidation states were determined from the intensity of the shake-up satellite at 785–786 eV (inset in Figure 5B), which originates exclusively from Co2+, since the binding energies of Co2+ and Co3+ are too close to be distinguished (Yang et al., 2010). For this reason, a Wagner plot containing the kinetic energies of the Co L3VV Auger electrons vs. the corresponding binding energies of the Co 2p3/2 core level photoelectrons is shown in Figure 5A. Wagner plots facilitate chemical state analysis, by illustrating shifts in photoelectron lines and X-ray excited Auger electron lines as well as the modified Auger parameter (AP) which is defined as the sum of the Auger electron kinetic energy and the corresponding core-level photoelectron binding energy (Wagner, 1972). Due to its insensitivity to static charging and variations in the energy scale calibration the AP is particularly useful to compare spectra of insulating samples with literature data and spectra acquired with different instruments. Reference values for the modified AP for CoO, Co and Co3O4 are indicated in Figure 5A as diagonal blue lines (Linstrom, 1997). The APs of the as-synthesized Co3O4 samples are in agreement with previous values reported in literature, while only the parameter of the sample synthesized at low temperature (140 °C) is somewhat lower but still within the error range.
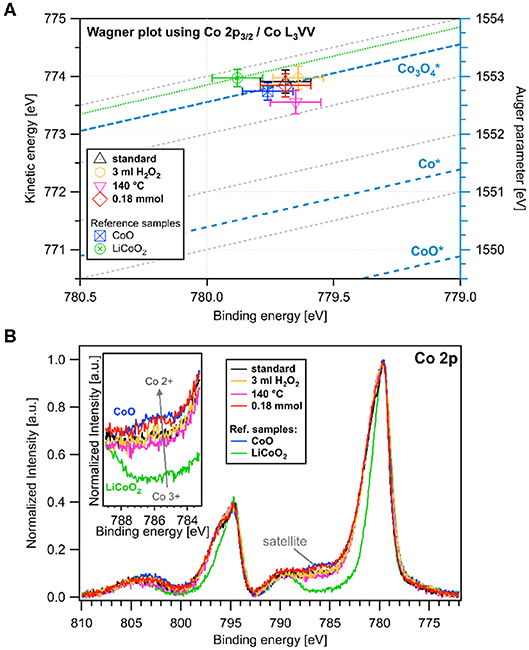
Figure 5. (A) Wagner plot of the four most different samples with the kinetic energy of the Co L3VV Auger electron and Co 2p3/2 core level binding energy with Co*, CoO*, and Co3 references obtained from NIST database.50 (B) XPS spectra of the Co 2p peaks of the same samples and of CoIIO and LiCoIIIO2 reference compounds (inset: satellite of the Co 2p3/2 peak).
The obtained materials were further compared with respect to their water oxidation activity. Water oxidation tests were performed using two different methods, namely chemical and electrocatalytic water oxidation. In the first method, the water oxidation activity was assessed using cerium ammonium nitrate (CAN) which is a standard oxidant for water oxidation with a redox potential of 1.75 V vs. NHE (Patterson, 1939). The amount of formed oxygen was quantified with a luminescent dissolved oxygen electrode (Figure S6) and is shown as a function of the catalyst amount in Figure 6. The dark red bar at the beginning of each group represents the reference yield obtained from Co3O4 synthesized with standard parameters. Obviously, any change of synthesis parameters in the standard protocol leads to a better oxygen evolution. For the first three parameters, i.e., temperature, H2O2 amount and Co-precursor concentration, the oxygen concentration shows an upward trend whereas for the ramping time an optimum was obtained for 20 min. With shorter reaction (holding) time and faster stirring speed the activity was only marginally increased.
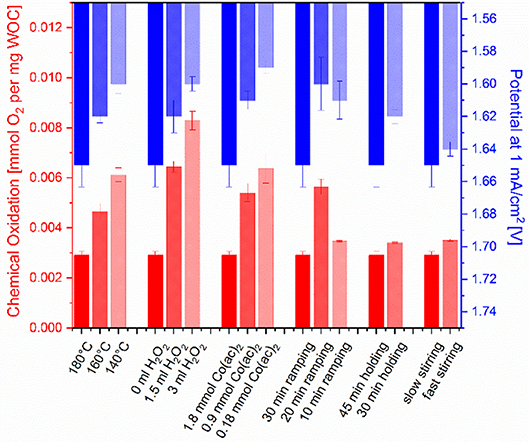
Figure 6. Water oxidation activity (chemical: red, electrocatalytic: blue) of the spinel samples synthesized by varying a single parameter of the standard synthesis method (dark red bar at the left of each triad). Chemical oxidation was assessed with 146 mM cerium(IV) ammonium nitrate (CAN) and the electrocatalytic activity was compared by the potentials vs. RHE at 1 mA/cm2 in 1 M KOH.
Electrocatalytic measurements were performed with a standard three electrode setup using Ag/AgCl as reference and Pt as counter electrode in alkaline environment. The use of Pt counter electrodes was recently under debate since it was shown that metal traces may dissolve in the electrolyte and influence the activity of water reduction catalysts (Cherevko et al., 2014; Chen et al., 2017; Wei et al., 2019). However, this is not applicable for the present water oxidation system, because Pt does not produce any Faradaic current in this region (see Figure S11) and the measurements were performed at pH 14 where Pt dissolution is minimized (Wei et al., 2019). Furthermore, the goal of this study is an internal comparison between spinel-type Co3O4 catalysts emerging from tuned synthetic protocols with standard electrochemical measurement techniques, rather than the discussion of absolute performance values. To this end, stepwise chronoamperometry from 0.45 V to 0.65 V vs. Ag/AgCl was performed. To obtain the bare Faradaic current without diffusion current, the current density value after 5 min holding at the same potential was considered and plotted against the applied potential (Figure S7). Electrochemical impedance spectroscopy (EIS) measurements, showing lower resistivity for Co3O4 synthesized at 140 °C compared to Co3O4 synthesized at 180 °C (standard synthesis) and long-term chronoamperometry measurements are provided in Figures S12, S13 and Table S2.
To facilitate comparison between the samples, the potential at 1 mA/cm2 current density is shown at the left of each triad in Figure 6 (dark blue bars). Compared to our abovementioned study where the influence of substantially different synthetic methods on the electrochemical activity was rather marginal, the present variations in electrocatalytic performance after changing a single synthetic parameter are somewhat higher and display a trend. In contrast, parameter-dependent chemical water oxidation activity differences were slightly smaller compared to the previous study covering a wider range of methods (Figure S8) (Reith et al., 2019). Notably, closely related trends for water oxidation activity were obtained by the two different test methods. Both chemical and electrochemical activity could be increased by either shorter synthesis times, addition of hydrogen peroxide or lower precursor concentrations. The optimal ramping time was found to be 20 min. Shorter synthesis holding times had a more productive influence on the electrocatalytic performance than on the chemical oxidation activity, and stirring speed was the least important parameter in both assays.
The observed water oxidation activities do not correlate exclusively with the surface area or crystallite size of the Co3O4 samples, as seen from comparison with calculated crystallite sizes and BET-determined surface areas (Figure 7). The errors in BET and τXRD are in the range of 1-5 % (0.65 m2g−1 for BET, determined by fourfold measurement of commercial Co3O4 with similar crystallite size to most of the here investigated samples and 0.2–1 nm for τXRD, determined from error propagation from PXRD peak FWHM determination). Furthermore, no correlation to any determined material parameter could be found for surface normalized activities (Figure S9). In light of the key role of the HO-Co2(μ-O/OH)2-OH edge site on Co3O4 assisted water oxidation revealed by Zhang et al. (2014a), we tentatively plotted non normalized chemical water oxidation activities vs. the bond length and disorder of CoOct-CoOct as determined by EXAFS fitting (cf. Figure 7 is below). Error estimations for the disorder parameter σ2 and bond lengths are given in Table S1 as significant decimal digits. Other than for the other structural parameters (Co-O and CoOct-CoTet, see Table S1 and Figure S10), a trend was about to emerge here, but more precise data are required to establish a truly significant correlation. In comparison, other materials characteristics such as oxidation state and crystallinity did not display any clear correlations with the water oxidation activity (Figure S10). As an overall trend, however, smaller particles with larger surface area as well as lower sample crystallinity with higher disorder result in higher water oxidation activities.
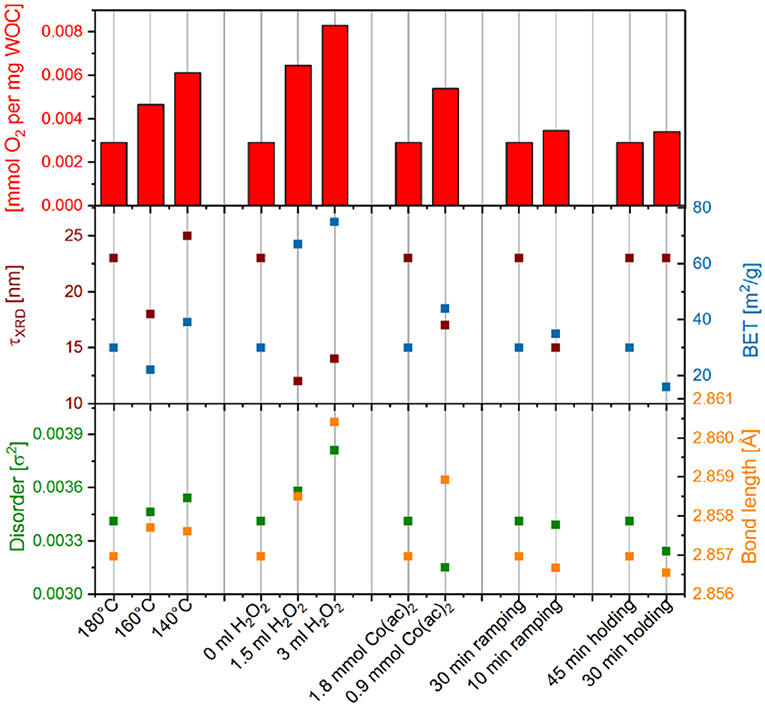
Figure 7. Chemical water oxidation activities of the different Co3O4 samples (top) compared with their respective crystallite size τXRD and BET surface area (middle), as well as, to their mean-square disorder parameter σ2 and CoOct-CoOct bond length determined from EXAFS fitting (bottom).
Conclusions
The present study demonstrates new insights into the importance of synthetic parameters in microwave-hydrothermal synthesis. This synthetic approach is a tunable and rapid method for the production of nanoscale spinel-type Co3O4 water oxidation catalysts, therefore holding great potential for industrial applications. Starting from a standard microwave protocol, the influence of six key synthetic parameters on the properties of the resulting cobalt oxide samples was first screened. In particular, shorter ramping times and addition of hydrogen peroxide were shown to reduce the crystallite and particle size. The crystallinity of the Co3O4 catalysts was decreased most significantly by hydrogen peroxide addition and by lower synthesis temperatures. Therefore, hydrogen peroxide addition was found to be the most influential synthesis parameter, even though the bulk cobalt oxidation state of the emerging spinels was not notably influenced as shown by XANES investigations.
The obtained Co3O4 spinels were characterized with respect to their synthesis parameters using a wide range of analytical methods, including PXRD, BET, XAS, XPS, and Raman spectroscopy as well as electron microscopy. Next, they were compared with respect to their chemical and electrocatalytic water oxidation activity. Significant changes in the respective water oxidation activities were observed as a result of the synthetic parameter variations. In particular, H2O2 addition during synthesis yielded much more active catalysts. Furthermore, lower synthesis temperature, cobalt precursor concentrations and shorter ramping times exerted a beneficial influence on the oxygen evolution performance. Stirring speed and holding time were found to be the least impactful parameters.
Correlations of the observed activity changes with the detailed materials parameters of the Co3O4 spinels indicate that not only the surface area and the crystallite size, but possibly also structural parameters, such as disorder and CoOct-CoOct bond distances as present in the surface edge-site motif determine the water oxidation activity. These results illustrate the key importance of synthetic parameter tuning in heterogeneous catalyst production. Therefore, systematic explorations of the synthetic parameter space along with insight into the underlying mechanisms of catalyst formation and activity are indispensable for informed and efficient design. Future strategies may thus combine rapid machine learning parameter screening with well-designed in situ monitoring experiments.
Experimental
Synthetic Procedure
All Co3O4 samples were obtained from microwave-hydrothermal synthesis. Co(CH3COO)2·4H2O (Sigma-Aldrich, reagent grade) was dissolved in 15 mL water and the pH was adjusted to 11 by addition of aqueous ammonia (25% NH3, Merck, GR for analysis). The dispersion was poured into a 95 mL CEM Omni Teflon vessel equipped with a stirring bar and sealed accordingly. Always, two reactions were conducted simultaneously and temperature and pressure were monitored for the reaction in the reference vessel. The reaction was temperature controlled, i.e., the microwave power was adjusted according to the reference vessel temperature. The reaction mixture was heated to 180 °C in 30 min (referred to as ramping time) and kept at this temperature for 45 min (referred to as holding time), followed by a cooling period of ~30 min, followed by separation of the product by centrifugation. The samples were air-dried at 80 °C prior to further characterization. The parameters were varied as summarized in Table 2 and the samples were labeled accordingly. The standard precursor concentration of 1.8 mmol was adapted from our previous studies in order to guarantee optimal salt loads and filling degrees for the microwave-hydrothermal system to ensure homogeneous and reproducible reaction conditions which warrant comparability to preceding results.
Catalytic Measurements
Electrocatalytic measurements were carried out in a standard three-electrode setup with an Ag/AgCl (Sigma-Aldrich glass reference electrode Ag/AgCl with 3 M KCl) as reference- and a Pt-foil as counter electrode. The working electrode was prepared by first dispersing the WOC in water (2 mg in 100 mL) and 40 mL of this dispersion were drop casted on 1 cm2 FTO (Aldrich, ~7 Ω/sq). After 30 min drying at 80 °C, Nafion solution (10 mL, 1wt-% diluted from 5 wt-%, Nafion™ perfluorinated resin solution, Sigma-Aldrich) was drop-casted on the surface to fix the WOC. For the measurements, 1 M KOH was used as electrolyte. A Bio-Logic SAS SP-150 potentiostat was used. The standard measurement protocol consisted of three steps: (1) Cyclic voltammetry (CV) was measured from 0 to 0.7 V vs. Ag/AgCl with 0.005 mV step size. For data evaluation, only the second cycles were compared to each other. (2) Stepwise chronoamperometry increasing the potential from 0.45 V to 0.65 V in 0.01 V steps and holding for 5 min. The stabilized current at the end of each step was used for further evaluation to eliminate diffusion currents. (3) Chronoamperometry in which a potential of 0.6 V vs. Ag/AgCl was applied for 2 h for stability testing. For EIS and long-term chronoamperometry studies in 1.0 M KOH, 2 mg of the obtained cobalt oxide powders were dispersed in 100 μL of 2% Nafion (ethanol solution) and sonicated for 15 min. A total of 40 μL of the suspension was drop-cast on the carbon cloth (Toray Carbon Paper, TGP-H-60, 1 cm2) and kept at 60 °C for 15 min. Measurement details are provided in the Supplementary Material.
To evaluate the chemical water oxidation activity of the catalysts, the following standard CAN method was applied: Prior to addition of WOC, CAN (2 g) was dissolved in milli-Q water (40 mL) and the solution was degassed with argon. After subsequent addition of WOC (2 mg) the oxygen evolution was recorded for 45 min while stirring the solution, using a luminescent dissolved oxygen sensor (LDO). For data evaluation, only the peak concentration was used. A Hach HQ40d multimeter with LDO 101 sensor was used for these measurements.
Materials and Methods
The Co3O4 spinels were characterized by powder X-ray diffraction (PXRD) using a STOE STADI P diffractometer in transmission mode (Ge monochromator, Mo Kα1 radiation). The surface area was measured using Brunauer–Emmett–Teller (BET) on a Quadrasorb SI machine in N2-adsorption mode. Samples were degassed at 100 °C overnight under vacuum prior to the measurements. Raman spectra were measured on a Renishaw inVia Qontor confocal Raman microscope with a diode laser (785 nm). SEM images were taken with a Zeiss SUPRA 50VP SEM equipped with a Schottky field emitter and an in lens secondary electron (SE) detector. An acceleration voltage of 10 kV and a working distance of 5.4–6 mm were applied. TEM images were acquired using a JEOL JEM-1400 Plus microscope. The camera was a JEOL CCD Ruby (8 M pixel) and the electron beam source a LaB6 crystal operated at 120 kV. HR-TEM images were obtained from a FEI Tecnai F30 FEG device, equipped with a CCD Gatan 794 MultiScan Camera and a Schottky emitter (300 kV). X-ray photoelectron spectroscopy (XPS) was conducted on a Physical Electronics (PHI) Quantum 2000 spectrometer. The monochromatic Al-Kα radiation was generated from an electron beam (15 kV, 35.8 W). For energy scale calibration, Au and Cu reference samples were used. The measurements were carried out at 10−8 mbar, with an electron take off angle of 45° and a pass-energy of 23.5 eV. A low energy electron source was used for charge compensation throughout the measurements. The alignment of the acquired spectra was performed using the main (CC) component of the C1s core level emission. The modified Auger parameter was calculated by adding the kinetic energy of the CoL3VV and Co 2p3/2 binding energies.
X-ray absorption spectroscopy (XAS) spectra were recorded at the Co K-edge on solid powder samples dispersed in cellulose. Measurements of the synthesized Co3O4 samples and reference oxides CoIIO and LiCoIIIO2 were carried out at the SuperXAS beamline at the Swiss Light Source (SLS), Paul Scherrer Institute (PSI) Villigen, Switzerland. Spectra were recorded using quick-scanning (QEXAFS) acquisition data protocols using a three-ionization chamber configuration in transmission mode and a 5-element Silicon Drift Detector. For energy calibration, the spectrum of a metal Co foil was measured simultaneously at the second ionization chamber. The X-ray beam was collimated using a Si coated mirror and the energy was scanned using a channel-cut Si[111] monochromator. A Rh coating toroidal mirror was used after the monochromator to focus the incident X-rays with a spot size of 400 × 200 μm2 on the samples and a photon flux of 5.0 × 1011 photons/s. The measured EXAFS spectra k3χ(k) were extracted by standard data reduction, absorption edge energy calibration and background subtraction as implemented in ATHENA (Ravel and Newville, 2005). The spectra were reduced into the range Δk ≈ 3–14 Å−1 and Fourier transform to FT|k3χ(k)| into the real-space interval of ΔR ≈ 0–6 Å. To calculate main values for the interatomic distances, atomic coordination numbers (N), and Debye-Waller factors (σ2) non-linear least-squares fitting of the FT|k3χ(k)| spectra was carried out with ARTEMIS using atomic clusters of Co3O4 (ICSD code 27498), generated with ATOMS as implemented in IFEFFIT (Ravel and Newville, 2005). The amplitudes and phases shift for single and multiple scattering paths were calculated using FEFF6 (Ankudinov et al., 1998).
Samples with low signal to noise ratio due to low theoretical yields from downscaled precursor concentrations were not included into the SI for the sake of clarity.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
KL and GP conceived the research. KL prepared the samples, carried out PXRD, SEM, HR-TEM, chemical, and electrocatalytic measurements and data analyses. KL and LR carried out Raman and TEM measurements. KL and CT performed XAS experiments and data analyses. SS performed XPS experiments and analyses. KL, CT, SS, and GP wrote the manuscript. All authors have given approval to the final version of the manuscript.
Funding
The authors are grateful to the University of Zurich and the UZH Research Priority Program Solar Light to Chemical Energy Conversion (URPP LightChEC) for financial support. GP acknowledges funding from the Swiss National Science Foundation (Sinergia Grant No. CRSII2_160801/1). SS thanks the COST project IZCNZ0-174856 C16.0075, in the COST Action MP1407 (e-MINDS) for financial support.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Dr. Zbynek Novotny for scientific support with XPS analyses, and Dr. Maarten Nachtegaal and Dr. Olga Safonova for support and access to the SuperXAS beamline to carry out XAS experiments at PSI. We thank S. Esmael Balaghi for conducting and evaluating the ESI and long-term chronoamperometry measurements. Microscopic analyses were done at the Scientific Center for Optical and Electron Microscopy (ScopeM) at ETH Zurich and at the Center for Microscopy and Image Analysis at UZH.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2020.00473/full#supplementary-material
References
Amiri, S. E. H., Vaezi, M. R., and Kandjani, A. E. (2011). A comparison between hydrothermally prepared Co3O4 via H2O2 assisted and calcination methods. J. Ceram. Process. Res. 327–331.
Ankudinov, A. L., Ravel, B., Rehr, J. J., and Conradson, S. D. (1998). Real-space multiple-scattering calculation and interpretation of x-ray-absorption near-edge structure. Phys. Rev. B 58, 7565–7576. doi: 10.1103/PhysRevB.58.7565
Artero, V., Chavarot-Kerlidou, M., and Fontecave, M. (2011). Splitting water with cobalt. Angew. Chem. 50, 7238–7266. doi: 10.1002/anie.201007987
Bergmann, A., Martinez-Moreno, E., Teschner, D., Chernev, P., Gliech, M., Araújo, J. F., et al. (2015). Reversible amorphization and the catalytically active state of crystalline Co3O4 during oxygen evolution. Nat. Commun. 6:8625. doi: 10.1038/ncomms9625
Bilecka, I., Elser, P., and Niederberger, M. (2009). Kinetic and thermodynamic aspects in the microwave-assisted synthesis of ZnO nanoparticles in benzyl alcohol. ACS Nano 3, 467–477. doi: 10.1021/nn800842b
Bilecka, I., and Niederberger, M. (2010). Microwave chemistry for inorganic nanomaterials synthesis. Nanoscale 2:1358. doi: 10.1039/b9nr00377k
Chen, R., Yang, C., Cai, W., Wang, H.-Y., Miao, J., Zhang, L., et al. (2017). Use of platinum as the counter electrode to study the activity of nonprecious metal catalysts for the hydrogen evolution reaction. ACS Energy Lett. 2, 1070–1075. doi: 10.1021/acsenergylett.7b00219
Cherevko, S., Zeradjanin, A. R., Keeley, G. P., and Mayrhofer, K. J. J. (2014). A comparative study on gold and platinum dissolution in acidic and alkaline media. J. Electrochem. Soc. 161, H822–H830. doi: 10.1149/2.0881412jes
Chu, S., Li, W., Yan, Y., Hamann, T., Shih, I., Wang, D., et al. (2017). Roadmap on solar water splitting: current status and future prospects. Nano Futures 1:22001. doi: 10.1088/2399-1984/aa88a1
Chua, C. S., Ansovini, D., Lee, C. J. J., Teng, Y. T., Ong, L. T., Chi, D., et al. (2016). The effect of crystallinity on photocatalytic performance of Co3O4 water-splitting cocatalysts. Phys. Chem. Chem. Phys. 18, 5172–5178. doi: 10.1039/C5CP07589K
Conrad, F., Bauer, M., Weyeneth, S., Zhou, Y., Hametner, K., Günther, D., et al. (2013). Hierarchically structured copper gallium spinels through microwave hydrothermal methods. Solid State Sci. 24, 125–132. doi: 10.1016/j.solidstatesciences.2013.06.016
Conrad, F., Massue, C., Kühl, S., Kunkes, E., Girgsdies, F., Kasatkin, I., et al. (2012). Microwave-hydrothermal synthesis and characterization of nanostructured copper substituted ZnM2O4 (M = Al, Ga) spinels as precursors for thermally stable Cu catalysts. Nanoscale 4, 2018–2028. doi: 10.1039/c2nr11804a
Conrad, F., Zhou, Y., Yulikov, M., Hametner, K., Weyeneth, S., Jeschke, G., et al. (2010). Microwave-hydrothermal synthesis of nanostructured zinc-copper gallates. Eur. J. Inorg. Chem. 2010, 2036–2043. doi: 10.1002/ejic.200901169
Cordeiro, P. V. O., and Carvalho, N. M. F. (2018). Water oxidation reaction catalyzed by Co3O4 treated with organic compounds. Ind. Eng. Chem. Res. 57, 11259–11264. doi: 10.1021/acs.iecr.8b01962
Dau, H., Liebisch, P., and Haumann, M. (2003). X-ray absorption spectroscopy to analyze nuclear geometry and electronic structure of biological metal centers-potential and questions examined with special focus on the tetra-nuclear manganese complex of oxygenic photosynthesis. Anal. Bioanal. Chem. 376, 562–583. doi: 10.1007/s00216-003-1982-2
Dong, H., Kuzmanoski, A., Wehner, T., Müller-Buschbaum, K., and Feldmann, C. (2016). Microwave-assisted polyol synthesis of water dispersible red-emitting Eu3+-modified carbon dots. Materials 10, 1–10. doi: 10.3390/ma10010025
Gao, R., Yang, Z., Zheng, L., Gu, L., Liu, L., Lee, Y., et al. (2018). Enhancing the catalytic activity of Co3O4 for Li–O2 batteries through the synergy of surface/interface/doping engineering. ACS Catal. 8, 1955–1963. doi: 10.1021/acscatal.7b03566
Gawali, S. R., Gandhi, A. C., Gaikwad, S. S., Pant, J., Chan, T.-S., Cheng, C.-L., et al. (2018). Role of cobalt cations in short range antiferromagnetic Co3O4 nanoparticles: a thermal treatment approach to affecting phonon and magnetic properties. Sci. Rep. 8:249. doi: 10.1038/s41598-017-18563-9
Grzelczak, M., Zhang, J., Pfrommer, J., Hartmann, J., Driess, M., Antonietti, M., et al. (2013). Electro- and photochemical water oxidation on ligand-free Co3O4 nanoparticles with tunable sizes. ACS Catal. 3, 383–388. doi: 10.1021/cs3007523
Hadjiev, V. G., Iliev, M. N., and Vergilov, I. V. (1988). The Raman spectra of Co3O4. J. Phys. C: Solid State Phys. 199–201. doi: 10.1088/0022-3719/21/7/007
Hilaire, S., Süess, M. J., Kränzlin, N., Bienkowski, K., Solarska, R., Augustynski, J., et al. (2014). Microwave-assisted nonaqueous synthesis of WO3 nanoparticles for crystallographically oriented photoanodes for water splitting. J. Mater. Chem. A 2, 20530–20537. doi: 10.1039/C4TA04793A
Iosub, A. V., and Stahl, S. S. (2015). Catalytic aerobic dehydrogenation of nitrogen heterocycles using heterogeneous cobalt oxide supported on nitrogen-doped carbon. Org. Lett. 17, 4404–4407. doi: 10.1021/acs.orglett.5b01790
Koziej, D., Floryan, C., Sperling, R. A., Ehrlicher, A. J., Issadore, D., Westervelt, R., et al. (2013). Microwave dielectric heating of non-aqueous droplets in a microfluidic device for nanoparticle synthesis. Nanoscale 5, 5468–5475. doi: 10.1039/c3nr00500c
Kuzmanoski, A., Pankratov, V., and Feldmann, C. (2015). Microwave-assisted ionic-liquid-based synthesis of highly crystalline CaMoO4:RE3+ (RE = Tb, Sm, Eu) and Y2Mo4O15:Eu3+ nanoparticles. Solid State Sci. 41, 56–62. doi: 10.1016/j.solidstatesciences.2015.02.005
Li, J., Güttinger, R., Moré, R., Song, F., Wan, W., and Patzke, G. R. (2017). Frontiers of water oxidation: the quest for true catalysts. Chem. Soc. Rev. 46, 6124–6147. doi: 10.1039/C7CS00306D
Li, W. Y., Xu, L. N., and Chen, J. (2005). Co3O4 Nanomaterials in lithium-ion batteries and gas sensors. Adv. Funct. Mater. 15, 851–857. doi: 10.1002/adfm.200400429
Linstrom, P. (1997). NIST Chemistry WebBook. Washington, DC: National Institute of Standards and Technology, © 1997.
Liu, Q., Chen, Z., Yan, Z., Wang, Y., Wang, E., Wang, S., et al. (2018). Crystal-plane-dependent activity of spinel Co3O4 towards water splitting and the oxygen reduction reaction. ChemElectroChem 5, 1080–1086. doi: 10.1002/celc.201701302
Luo, Y., Kong, D., Luo, J., Wang, Y., Zhang, D., Qiu, K., et al. (2014). Seed-assisted synthesis of Co3O4@α-Fe2O3 core–shell nanoneedle arrays for lithium-ion battery anode with high capacity. RSC Adv. 4:13241. doi: 10.1039/c3ra47189f
Moura, A. P., Cavalcante, L. S., Sczancoski, J. C., Stroppa, D. G., Paris, E. C., Ramirez, A. J., et al. (2010). Structure and growth mechanism of CuO plates obtained by microwave-hydrothermal without surfactants. Adv. Powder Technol. 21, 197–202. doi: 10.1016/j.apt.2009.11.007
Najafpour, M. M., Pashaei, B., and Nayeri, S. (2012). Nano-sized layered aluminium or zinc-manganese oxides as efficient water oxidizing catalysts. Dalton Trans. 41, 7134–7140. doi: 10.1039/c2dt30353a
Najafpour, M. M., Renger, G., Hołynska, M., Moghaddam, A. N., Aro, E.-M., Carpentier, R., et al. (2016). Manganese compounds as water-oxidizing catalysts: from the natural water-oxidizing complex to nanosized manganese oxide structures. Chem. Rev. 116, 2886–2936. doi: 10.1021/acs.chemrev.5b00340
Nüchter, M., Ondruschka, B., Bonrath, W., and Gum, A. (2004). Microwave assisted synthesis – a critical technology overview. Green Chem. 6, 128–141. doi: 10.1039/B310502D
Patterson, A. L. (1939). The scherrer formula for X-ray particle size determination. Phys. Rev. 56, 978–982. doi: 10.1103/PhysRev.56.978
Ravel, B., and Newville, M. (2005). ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537–541. doi: 10.1107/S0909049505012719
Reith, L., Lienau, K., Cook, D. S., Moré, R., Walton, R. I., and Patzke, G. R. (2018). Monitoring the hydrothermal growth of cobalt spinel water oxidation catalysts: from preparative history to catalytic activity. Chem. Eur. J. 24, 18424–18435. doi: 10.1002/chem.201801565
Reith, L., Lienau, K., Triana, C. A., Siol, S., and Patzke, G. R. (2019). Preparative history vs driving force in water oxidation catalysis: parameter space studies of cobalt spinels. ACS Omega 4, 15444–15456. doi: 10.1021/acsomega.9b01677
Rosen, J., Hutchings, G. S., and Jiao, F. (2014). Synthesis, structure, and photocatalytic properties of ordered mesoporous metal-doped Co3O4. J. Catal. 310, 2–9. doi: 10.1016/j.jcat.2013.05.003
Scherrer, P. (1918). “Bestimmung der Größe und der inneren Struktur von Kolloidteilchen mittels Röntgenstrahlen,” in Nachrichten von der Gesellschaft der Wissenschaften zu Göttingen, Mathematisch-Physikalische Klasse 98–100.
Tung, C.-W., Hsu, Y.-Y., Shen, Y.-P., Zheng, Y., Chan, T.-S., Sheu, H.-S., et al. (2015). Reversible adapting layer produces robust single-crystal electrocatalyst for oxygen evolution. Nat. Commun. 6:8106. doi: 10.1038/ncomms9106
Wagner, C. D. (1972). Auger lines in x-ray photoelectron spectrometry. Anal. Chem. 44, 967–973. doi: 10.1021/ac60314a015
Wang, D., Yu, Y., He, H., Wang, J., Zhou, W., and Abruña, H. D. (2015). Template-free synthesis of hollow-structured Co3O4 nanoparticles as high-performance anodes for lithium-ion batteries. ACS Nano 9, 1775–1781. doi: 10.1021/nn506624g
Wang, H.-Y., Hung, S.-F., Chen, H.-Y., Chan, T.-S., Chen, H. M., and Liu, B. (2016b). In operando identification of geometrical-site-dependent water oxidation activity of spinel Co3O4. J. Am. Chem. Soc. 138, 36–39. doi: 10.1021/jacs.5b10525
Wang, J., Cui, W., Liu, Q., Xing, Z., Asiri, A. M., and Sun, X. (2016a). Recent progress in cobalt-based heterogeneous catalysts for electrochemical water splitting. Adv. Mater. 28, 215–230. doi: 10.1002/adma.201502696
Wang, R. M., Liu, C. M., Zhang, H. Z., Chen, C. P., Guo, L., Xu, H. B., et al. (2004). Porous nanotubes of Co3O4: synthesis, characterization, and magnetic properties. Appl. Phys. Lett. 85, 2080–2082. doi: 10.1063/1.1789577
Wei, C., Rao, R. R., Peng, J., Huang, B., Stephens, I. E. L., Risch, M., et al. (2019). Recommended practices and benchmark activity for hydrogen and oxygen electrocatalysis in water splitting and fuel cells. Adv. Mater. 31:e1806296. doi: 10.1002/adma.201806296
Wei, R., Fang, M., Dong, G., Lan, C., Shu, L., Zhang, H., et al. (2018). High-index faceted porous Co3O4 nanosheets with oxygen vacancies for highly efficient water oxidation. ACS Appl. Mater. Inter. 10, 7079–7086. doi: 10.1021/acsami.7b18208
Yang, J., Liu, H., Martens, W. N., and Frost, R. L. (2010). Synthesis and characterization of cobalt hydroxide, cobalt oxyhydroxide, and cobalt oxide nanodiscs. J. Phys. Chem. C 114, 111–119. doi: 10.1021/jp908548f
Yang, Y.-P., Liu, R.-S., Huang, K.-L., Wang, L.-P., Liu, S.-Q., and Zeng, W.-W. (2007). Preparation and electrochemical performance of nanosized Co3O4 via hydrothermal method. Trans. Nonferrous Met. Soc. China 17, 1334–1338. doi: 10.1016/S1003-6326(07)60272-6
Zeng, G., Caputo, R., Carriazo, D., Luo, L., and Niederberger, M. (2013). Tailoring two polymorphs of LiFePO4 by efficient microwave-assisted synthesis: a combined experimental and theoretical study. Chem. Mater. 25, 3399–3407. doi: 10.1021/cm400995g
Zhang, G., Yang, J., Wang, H., Chen, H., Yang, J., and Pan, F. (2017). Co3O4−δ quantum dots as a highly efficient oxygen evolution reaction catalyst for water splitting. ACS Appl. Mater. Inter. 9, 16159–16167. doi: 10.1021/acsami.7b01591
Zhang, M., de Respinis, M., and Frei, H. (2014a). Time-resolved observations of water oxidation intermediates on a cobalt oxide nanoparticle catalyst. Nat. Chem. 6, 362–367. doi: 10.1038/nchem.1874
Zhang, N., Shi, J., Mao, S. S., and Guo, L. (2014b). Co3O4 quantum dots: Reverse micelle synthesis and visible-light-driven photocatalytic overall water splitting. Chem. Comm. 50, 2002–2004. doi: 10.1039/c3cc48026g
Zhang, N., Wang, Y., Hao, Y.-C., Ni, Y.-M., Su, X., Yin, A.-X., et al. (2018). Ultrathin cobalt oxide nanostructures with morphology-dependent electrocatalytic oxygen evolution activity. Nanoscale 10, 20313–20320. doi: 10.1039/C8NR05337E
Zhao, Q., Yan, Z., Chen, C., and Chen, J. (2017). Spinels: controlled preparation, oxygen reduction/evolution reaction application, and beyond. Chem. Rev. 117, 10121–10211. doi: 10.1021/acs.chemrev.7b00051
Zhou, J., Li, J., Zhang, L., Song, S., Wang, Y., Lin, X., et al. (2018). Highly active surface structure in nanosized spinel cobalt-based oxides for electrocatalytic water splitting. J. Phys. Chem. C 122, 14447–14458. doi: 10.1021/acs.jpcc.8b00407
Keywords: Co3O4 spinel, microwave-hydrothermal synthesis, water oxidation, electrocatalysis, synthesis parameters
Citation: Lienau K, Triana CA, Reith L, Siol S and Patzke GR (2020) Microwave-Hydrothermal Tuning of Spinel-Type Co3O4 Water Oxidation Catalysts. Front. Chem. 8:473. doi: 10.3389/fchem.2020.00473
Received: 30 October 2019; Accepted: 06 May 2020;
Published: 09 June 2020.
Edited by:
Ying Jiang, Cranfield University, United KingdomReviewed by:
Gabriella Di Carlo, Italian National Research Council, ItalyZhao Yan, Jiangsu University, China
Copyright © 2020 Lienau, Triana, Reith, Siol and Patzke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Greta R. Patzke, Z3JldGEucGF0emtlQGNoZW0udXpoLmNo