 Ge Sun1
Ge Sun1 Longfei Mao2
Longfei Mao2 Wenjing Deng1
Wenjing Deng1 Shuxiang Xu1
Shuxiang Xu1 Jie Zhao2Jianxue Yang3,4Kaitai Yao1*
Jie Zhao2Jianxue Yang3,4Kaitai Yao1* Miaomiao Yuan5*
Miaomiao Yuan5* Wei Li2*
Wei Li2*- 1Cancer Research Institute, School of Basic Medical Sciences, Southern Medical University, Guangzhou, China
- 2Henan Engineering Research Center of Chiral Hydroxyl Pharmaceutical, School of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang, China
- 3Department of Neurology, The First Affiliated Hospital of Henan University of Science and Technology, Luoyang, China
- 4School of Nursing, Henan University of Science and Technology, Luoyang, China
- 5The Eighth Affiliated Hospital, Sun Yat-sen University, Shenzhen, China
Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) are emerging at the vanguard of therapy for non-small-cell lung cancer (NSCLC) patients with EGFR-activating mutations. However, the increasing therapeutic resistance caused by novel mutations or activated bypass pathways has impaired their performance. In this study, we link one of the commercial EGFR-TKIs, Erlotinib, to different azide compounds to synthesize a novel class of 1,2,3-triazole ring-containing Erlotinib derivatives. We discovered that several new compounds show robust antiproliferation activity against diverse NSCLC cells in vitro including PC-9, H460, H1975 and A549. Two of the most potent compounds, e4 and e12 have been found to be more efficient than Erlotinib in all NSCLC cell lines except PC-9. They significantly induce apoptosis and cell cycle arrest in PC-9 and H460 cells. The antitumor efficacy of compound e4 in vivo is close to that of Erlotinib in a PC-9 xenograft mouse model. Most Erlotinib-1,2,3-triazole compounds exhibit moderate to good inhibitory activities toward wild-type EGFR as indicated by enzyme-linked immunosorbent assay (ELISA), and the EGFR phosphorylation was inhibited in H460 and PC-9 cells exposed to e4 or e12. These data suggest that these Erlotinib-1,2,3-triazole compounds are suitable candidates for use against NSCLC and more unknown mechanisms merit further investigation.
Introduction
It is estimated that 2.09 million new cases of lung cancer (11.6% of the total cases) were diagnosed and 1.76 million deaths were caused by lung cancer (18.4% of the total cancer deaths) globally in 2018 (Bray et al., 2018). Non-small-cell lung cancer (NSCLC) accounts for 85% of all the new cases of lung cancer, and is classified as adenocarcinoma (gland-forming), squamous cell carcinoma and large-cell carcinoma according to the histological distinctions (Travis et al., 2013; Gridelli et al., 2015). Lung cancer remains the most common malignant tumor and the leading cause of cancer deaths worldwide. While surgery is recommended for early-stage NSCLC patients, molecular targeted therapy has become the most important treatment for advanced NSCLC patients, up to 69% of which have a potentially actionable molecular target (Hirsch et al., 2017). The development of targeted therapy has significantly improved the survival of NSCLC patients over the past 2 decades.
Activating mutations in the ATP binding pocket of EGFR and excessive activation of the EGFR signaling pathway are observed in approximately 15–20% of NSCLC patients (Lynch et al., 2004; Paez et al., 2004), and many EGFR tyrosine kinase inhibitors (EGFR-TKIs) have been developed as the first-line therapy for NSCLC, including the first- and second-generation EGFR- TKIs Gefitinib (Figure 1A) (Muhsin et al., 2003), Erlotinib (Figure 1B) (Schettino et al., 2008) and Afatinib (Figure 1C) (Dungo and Keating, 2013), and the third-generation EGFR-TKI Osimertinib (Figure 1D) (Cross et al., 2014). EGFR-TKI inhibits the tyrosine kinase by competing with ATP for the ATP-binding site, thereby suppressing the activation and transduction of downstream signal through RAS-RAF-MEK-ERK and RAS-PI3K-PTEN-AKT-mTOR (Herbst and Bunn, 2003; Kan et al., 2010). Erlotinib is one of the earliest EGFR-TKIs, and it has been shown to be effective in NSCLC patients with or without EGFR-activating mutations, but it is even more potent in patient with EGFR activating mutations (Tsao et al., 2005; Qi et al., 2012; Kobayashi and Hagiwara, 2013). Unfortunately, just like the other first- and second-generation EGFR-TKIs, it has failed large numbers of patients who have eventually develop resistance after administration. More than 60% of the patients develop T790M mutations within the ATP-binding pocket, which prevents drug binding (Sequist et al., 2011). Although Osimertinib is the only inhibitor approved to target the T790M mutation, most third-generation EGFR-TKIs designed to overcome T790M resistance have not shown themselves superior to Osimertinib with respect to antitumor efficacy. It is necessary to identify more competent agents against NSCLC that could work quickly and effectively.
FIGURE 1. Structures of EGFR-TKIs for NSCLC therapy. (A) Gefitinib. (B) Erlotinib. (C) Afatinib. (D) Osimertinib.
In recent years, many researchers focused on synthesizing more potent and safer quinazoline-derived EGFR inhibitors with the aim of overcoming limitations of currently marketed drugs (Bhatia et al., 2020). For example, Elkamhawy and others (Elkamhawy et al., 2015a) designed a novel series of eighteen 6-anilinoquinazolines as inhibitors of EGFR/HER-2. The authors concluded that “11” was a promising candidate through studying the structural activity relationship of the compounds along with their molecular docking studies. In addition, they also reported new quinazoline-urea based compounds with potent cytotoxic activities against TMZ-resistant glioblastoma multiforme (GBM) cells, which may be associated with plausible binding modes in translocator protein 18 kDa (TSPO). Overall, these studies proved that novel quinazoline-based compounds are promising candidates for cancer therapy possessing potency and various activity (Elkamhawy et al., 2015b).
1,2,3-triazoles are prevailing motifs that exist in different compounds with particular bioactivities, such as antitumor, antimicrobial, and antidiabetic effects (Maddili et al., 2018; Safavi et al., 2018; Saeedi et al., 2019). In this study, we aimed to design and synthesize a novel class of 1,2,3-triazole ring-containing Erlotinib derivatives with EGFR tyrosine kinase inhibitory activities and improved antitumor activities against NSCLC.
Results
Chemistry
The strategy for the preparation of the target compounds is illustrated in Scheme 1. Erlotinib was obtained after reaction of 4-chloro-6,7-bis (methoxy ethoxy) quinazolinone and 3-aminophenylacetylene. And the target compounds e1–e20 were obtained via click reaction of Erlotinib with different azido compounds. The reaction conditions were mild and easy to control. The structures of the key intermediates and all target compounds were confirmed by nuclear magnetic resonance (1H NMR and 13C NMR) and high-resolution mass spectrometry (HRMS) (Supplementary Figures S1–20).
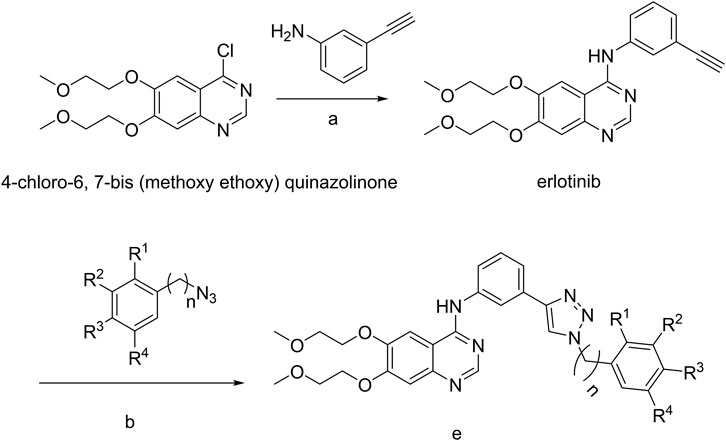
SCHEME 1. a, 85°C, isopropanol; b, copper sulfate pentahydrate, sodium ascorbate; e, Erlotinib derivatives.
In Vitro Antitumor Activity Study
To probe the therapeutic potential of all the synthesized derivatives, CCK-8 (Cell Counting Kit-8) assay was performed to evaluate the in vitro antiproliferative activities of all the new compounds against four human NSCLC cell lines: H460 and A549 express wild type EGFR (Helfrich et al., 2006); PC-9 is sensitive to Erlotinib and characterized by expressing EGFR exon 19 deletion mutation (ΔE746-A750) (Chmielecki et al., 2011); H1975 harbored the EGFR-activating mutations L858R but also the T790M mutation, which is associated with Erlotinib resistance (Helfrich et al., 2006).
Antiproliferation Assay
The antiproliferative effects of the new compounds and Erlotinib were evaluated by CCK-8 assay. As the data shown in Table 1, PC-9 cells were still most sensitive to Erlotinib with an IC50 value of 0.21 μΜ. However, compared to Erlotinib, some compounds including e4, e6, e9, e12 and e16 presented lower but also exceptional inhibitory efficacy against PC-9 with IC50 values of 4.38, 3.27, 7.63, 6.36 and 7.71 μΜ. Besides, for the other three kinds of NSCLC cells, e4, e9, and e12 showed more potent antiproliferative activities than Erlotinib with IC50 values less than 10 μΜ.
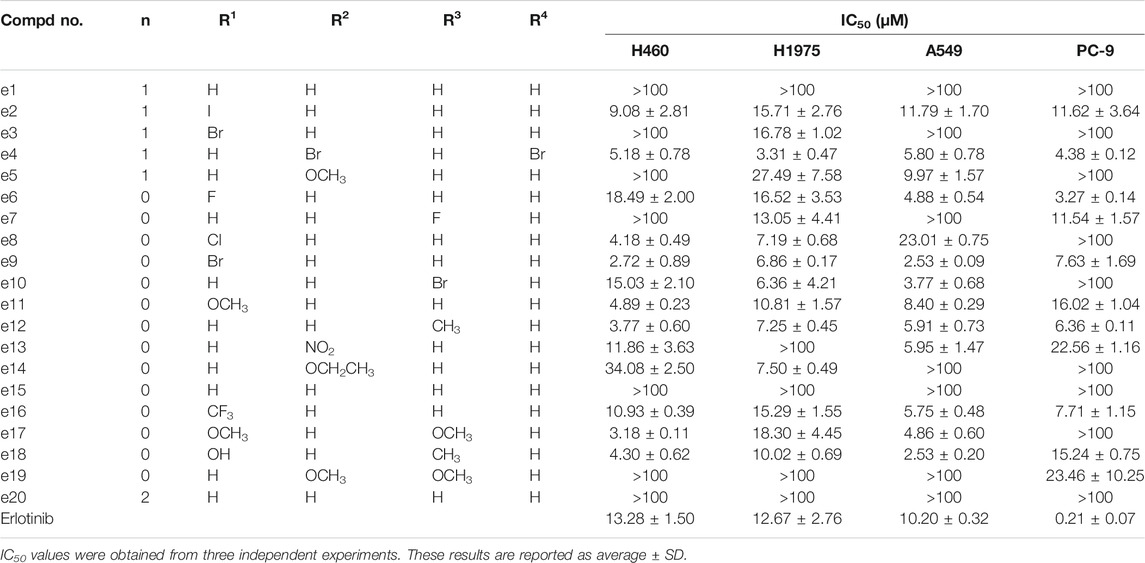
TABLE 1. The antitumor activities of the designed compounds against NSCLC cells in vitro.
Cell Apoptosis Assay
To establish whether the antiproliferative efficacy is associated with apoptosis, PC-9 and H460 cells were treated with compound e4 or e12 of various concentrations (4, 8 and 12 μM) for 48 h, and then assessed with flow cytometry. As shown in Figures 2A,B, we determined significant cell apoptosis in both PC-9 and H460 cells exposed to various concentrations of e4 or e12.
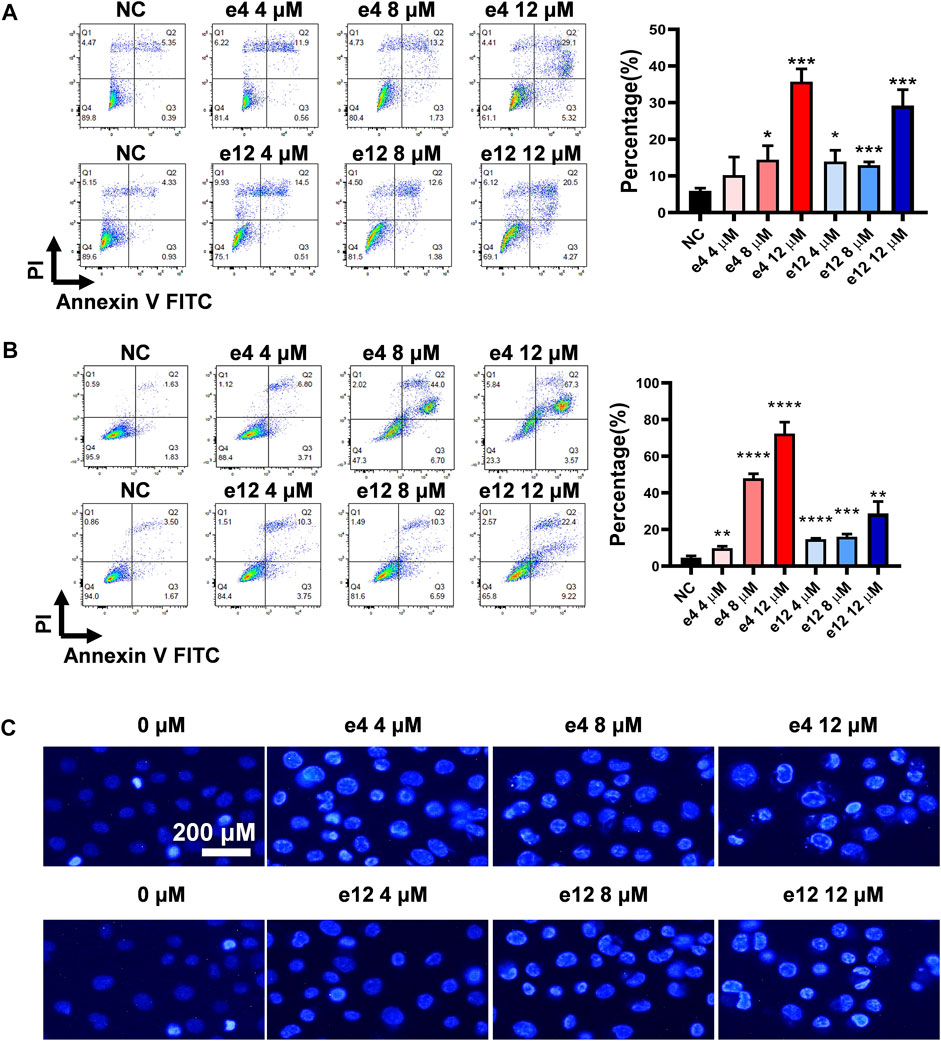
FIGURE 2. Compounds e4 and e12 induced apoptosis of PC-9 and H460 cells. PC-9 (A) and H460 (B) cells were exposed to compound e4 or e12 for 48 h. Flow cytometry analysis data from three independent experiments were summarized and shown. NC, negative control. *p-value < 0.05, **p-value < 0.01 and ***p-value < 0.001 (unpaired t-test). (C) Representative images of Hoechst 33342 staining of PC-9 cells exposed to compound e4 or e12 for 48 h. Images were analyzed with a fluorescence microscope (×200, original magnification).
To further verify the impact of compounds e4 and e12 on apoptosis of NSCLC cells, PC-9 cells were treated with either compound e4 or e12 at various concentrations for 48 h and then stained with dye Hoechst 33342. We compared PC-9 cells to cells treated with 0.1% DMSO, and they exhibited features typical of apoptosis such as condensed cell nuclei, fragmented chromatin and cell shrinkage (Brady, 2004) (Figure 2C). Taken together, these results indicate that compounds e4 and e12 induced cell apoptosis, leading to exceptional antiproliferative activities against NSCLC cells.
Cell Cycle Assay
To investigate the effects of compounds e4 and e12 on various phases of the cell cycle, PC-9 cells were treated with compound e4 or e12 of various concentrations for 24 h, and H460 cells were treated with compound e4 or e12 of various concentrations for 12 h. As shown in Figures 3A,B, the results of flow cytometry indicated that compounds e4 and e12 induced higher percentages of PC-9 (Figure 3A) and H460 (Figure 3B) cells in the G0/G1 phrase during incubation.
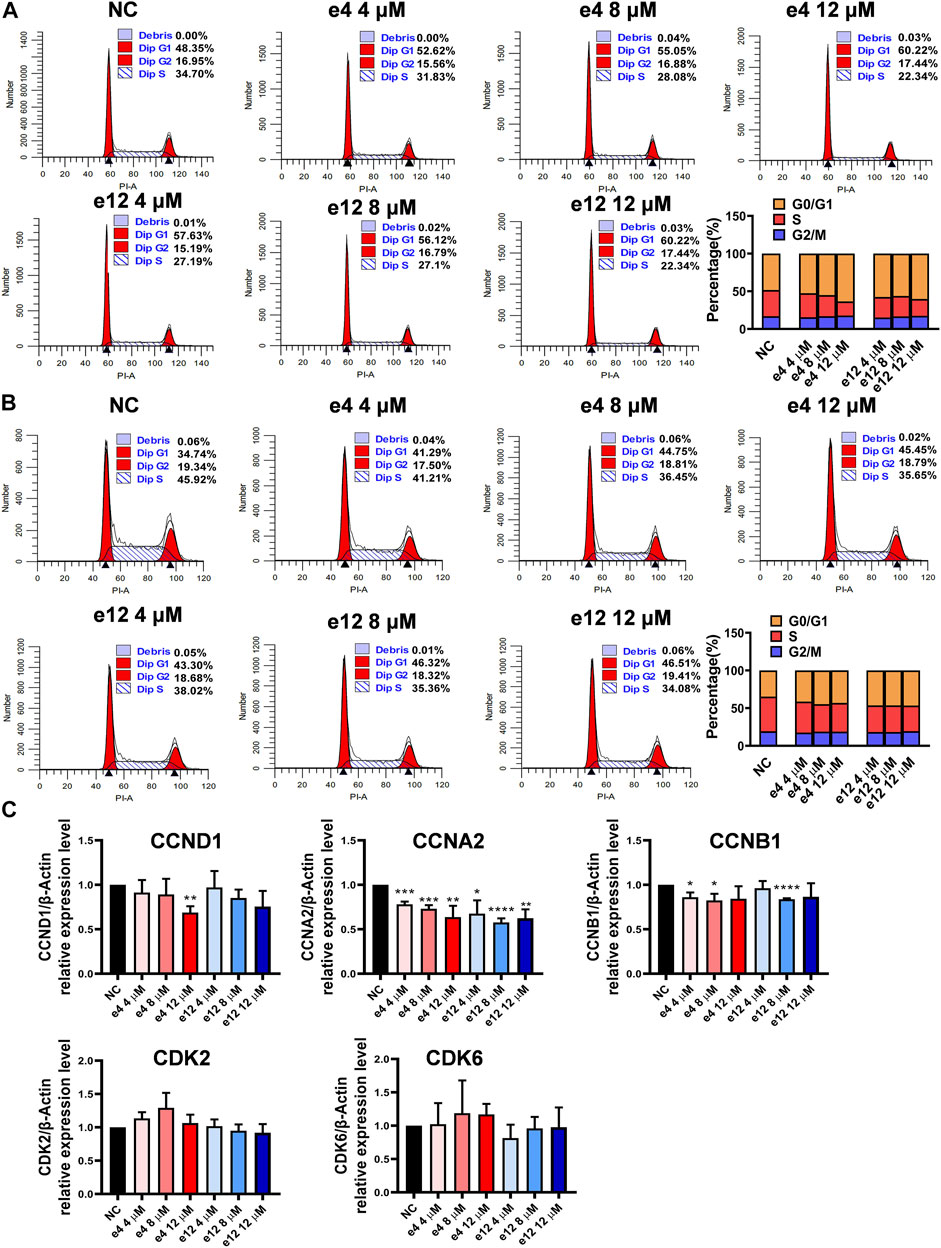
FIGURE 3. Cell cycle arrests induced by compounds e4 and e12 in PC-9 and H460 cells. PC-9 (A) and H460 (B) cells were exposed to either compound e4 or e12 at various concentrations respectively. The cell cycle and the proportions of each phase were detected through flow cytometry. This experiment was repeated three times independently with similar results. (C) Cell cycle-related gene analysis by qPCR in PC-9. PC-9 cells were treated with compound e4 or e12 at various concentrations for 16 h. Genes including CCND1, CCNA2, CCNB1, CDK2 and CDK6 were quantified. The results are reported as a mean ± SD of values from three independent experiments (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 vs. Ctrl). NC, negative control.
To better understand the mechanisms underlying cell cycle arrest induced by e4 and e12 in NSCLC cells, we assessed the expression levels of a few genes involved in cell cycle regulation, including cyclin D1 (CCND1), cyclin A2 (CCNA2), cyclin B1 (CCNB1), cyclin-dependent kinase 2 (CDK2), and cyclin-dependent kinase 6 (CDK6) in PC-9 by qPCR. As shown in Figure 3C, compared with DMSO control, e4 and e12 significantly decreased RNA levels of CCNA2 and CCNB1 in PC-9 cells over 16 h of incubation. In addition, e4 also negatively regulated the RNA expression of CCND1 at the concentration of 12 μM. CCNA2 is known to bind and activate CDK2 and thus promotes the transition through G1/S and G2/M. CCNB1 and CCND1 are essential to mitosis and G1/S transition respectively. While G2/M arrest weas not detected, the changes in these gene expression profiles interpret the G1/S arrests in PC-9 cells treated with e4 and e12. PC-9 cells exposed to the compounds for various periods should be examined further. In conclusion, e4 and e12 impeded cell proliferation by arresting the cell cycle at the G0/G1 phases in PC-9 and H460 cells, which was further confirmed by the decreased expression of genes involved in regulation of the cell cycle in PC-9 cells.
In Vivo Antitumor Activity
Inspired by the pronounced EGFR activities and the antitumor activities of these derivatives in vitro, we next determine the in vivo antitumor efficacy using mouse tumor models. NSG mice harboring PC-9 tumors were dosed with compound e4 (25 mg/kg, qd) and Erlotinib (25 mg/kg, qd) by intraperitoneal injection for 7 consecutive days (Figure 4A). As shown in Figure 4C, treatment with e4 or Erlotinib exhibited similar potency for tumor growth inhibition over treatment with vehicle. There was no significant difference with respect to growth rate between groups treated with e4 and Erlotinib (Figures 4B,D). No significant difference was detected in the changes of body mass between the three groups after treatment (Figure 4E), indicating that compound e4 had no obvious toxicity in vivo.
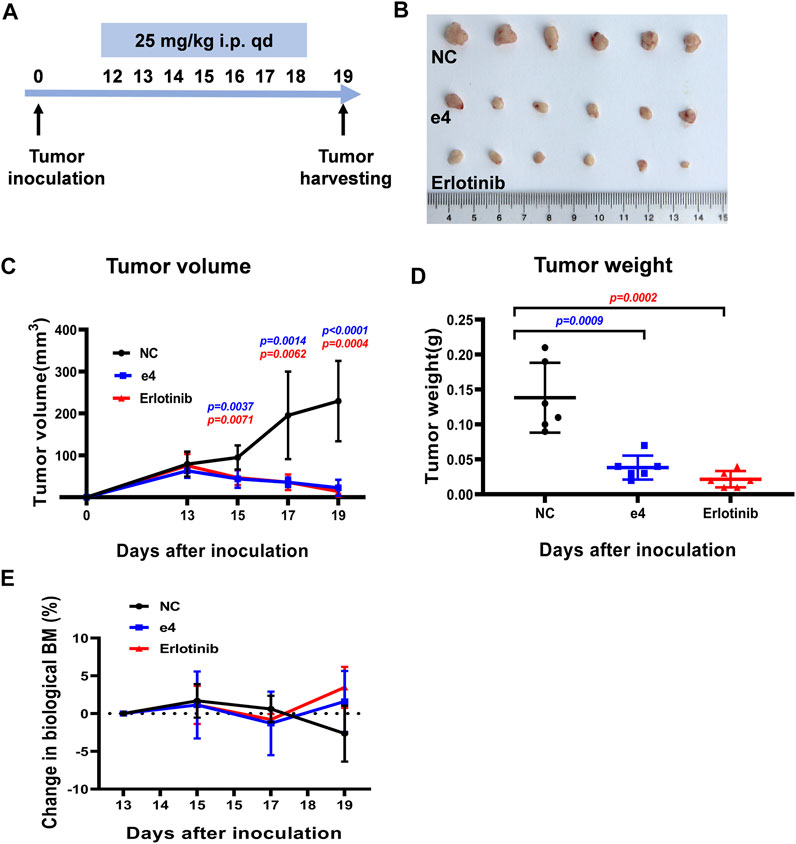
FIGURE 4. Compound e4 inhibits tumor growth of PC-9 xenograft in NSG mice. (A) Schematic diagram of the experimental procedure. (B) The images of the tumors formed by PC-9 cells treated with vehicle, compound e4 or Erlotinib 19 days after inoculation (n = 6). (C) Growth curves for the PC-9 tumors from the NSG mice after treatment with vehicle, compound e4 or Erlotinib (n = 6, 9 and 7 respectively). (D) The tumor weight of PC-9 xenograft and the corresponding statistical results were presented (n = 6). (E) The changes in the body mass of the mice after treatment. NC, negative control. Statistical significance was evaluated by the Student’s t-test.
EGFR Inhibition Study
Aberrant EGFR signaling in NSCLC promotes cell proliferation and survival, and Erlotinib inhibits the EGFR tyrosine kinase and its transduction of downstream signaling in NSCLC cells. To clarify whether the antiproliferative efficacy of the new compounds was associated with EGFR inhibitory activities, all the new compounds were assayed for their activities to inhibit EGFR tyrosine phosphorylation in vitro using ELISA. Erlotinib was served as a positive control. The results were shown as IC50 values in Table 2. Under these conditions, the IC50 of Erlotinib was 4.8 nM, which was similar to previously reported values (Moyer et al., 1997; Akita and Sliwkowski, 2003) (IC50 = 2 nM). As illustrated in Table 1, while all the Erlotinib derivatives showed less ability to inhibit EGFR tyrosine phosphorylation (IC50 values ranged from 0.09 to 63.02 μM), e2, e6, e15 and e20 displayed favorable EGFR inhibitory activities (IC50 values < 1 μM), especially compound e15, whose IC50 value was 0.09 μM.
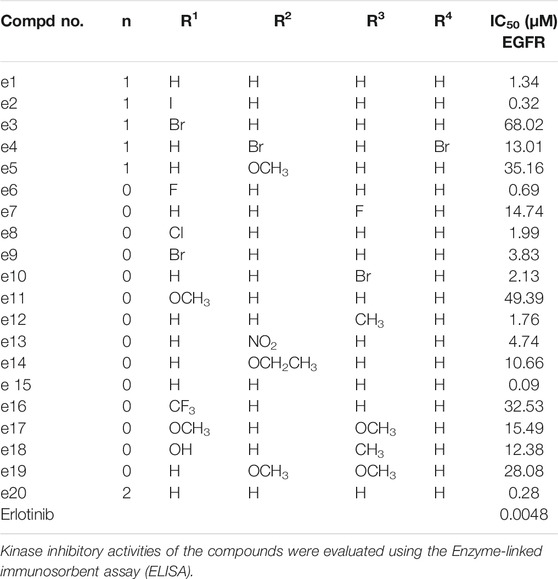
TABLE 2. EGFR inhibitory activities of the derivatives.
We also performed western blot analysis to evaluate the impact of e4 and e12 on EGFR signaling in PC-9 and H460 cells. The results showed that e4 and e12 suppressed the phosphorylation of EGFR and AKT in both PC-9 and H460 cells, especially at a concentration of 10 μM, consistent with their potent antiproliferation activities in NSCLC cells (Figure 5). In addition, we conducted molecular docking studies to explore the potential binding modes of compound e4 in the active site of EGFR. It showed that e4 has a desirable binding affinity with EGFR but a different binding mode from that of Erlotinib (Supplementary Figure S21).
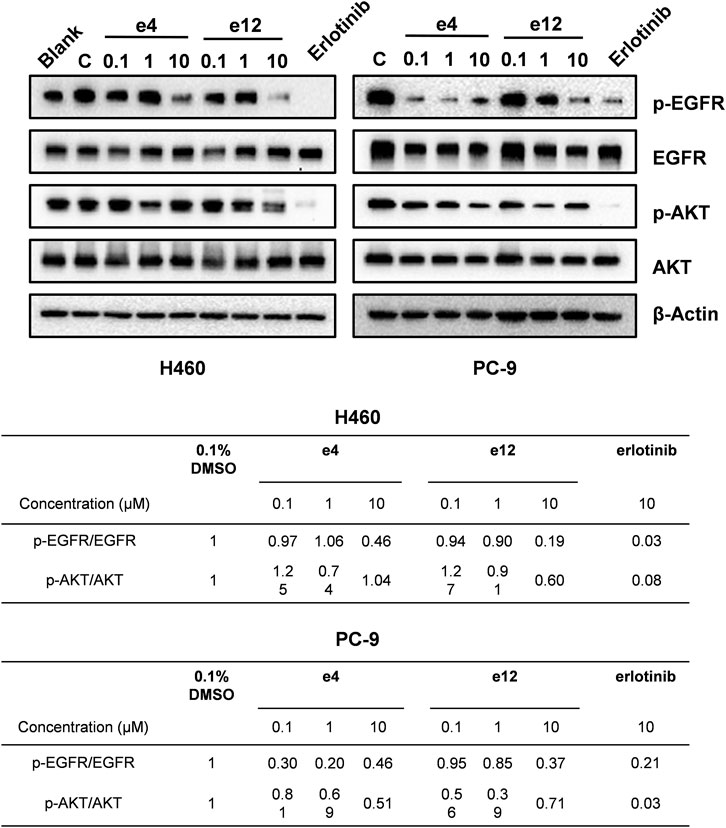
FIGURE 5. Compounds e4 and e12 suppressed EGFR phosphorylation and its transduction of downstream signaling in NSCLC cells. Protein bands (images on the top) and quantification (tables at the bottom) are presented.
Discussion
The survival rate of NSCLC patients has improved considerably since the adoption of EGFR-TKIs. Nevertheless, the constant emergence of novel mutations after treatment finally lead to their ineffectiveness. In this study, a new class of Erlotinib derivatives containing 1,2,3-triazole ring were designed and synthesized. Cell-based assay discovered that PC-9 cells that harbored an EGFR-activating mutation, is still most sensitive to Erlotinib. However, several of the Erlotinib derivatives (e.g., compounds e4, e9 and e12) showed more robust inhibitory activity against NSCLC cells including H460, H1975 and A549, compared with Erlotinib. We confirmed that these Erlotinib derivatives hah slightly lower but still adequate EGFR tyrosine kinase inhibitory activity when compared with that of Erlotinib. We suspect that the antiproliferative activities of the new compounds against NSCLC cells probably incompletely depend on the EGFR inhibitory effects, H460 and A549 cells do not express EGFR-activating mutations, and H1975 cells, which are resistant to Erlotinib, whereas are sensitive to some Erlotinib derivatives. This indicates that they may act via such mechanisms as the cancerous inhibitor of protein phosphatase 2A (CIP2A) pathway, through which Erlotinib induces apoptosis in NSCLC cells that do not have EGFR mutations; however, this should be investigated further (Wang et al., 2014). In addition, we confirmed that compound e4 restrains growth of EGFR-activating-mutant PC-9 xenografts and its antitumor efficacy in vivo is close to that of Erlotinib. As all considered above, we conclude that these Erlotinib-1,2,3-triazole compounds with EGFR tyrosine kinase inhibitory activities may serve as novel antitumor agents against NSCLC and additional mechanisms merit further investigation.
Materials and Methods
Materials
The 4-chloro-6,7-bis (methoxy ethoxy) quinazolinone, 3-aminophenylacetylene and Azido compounds were purchased from Acros Organics (United States). The Dulbecco’s Modified Eagle medium (DMEM), RPMI-1640 medium, phosphate buffered saline (PBS), fetal bovine serum (FBS) and trypsin were purchased from Gibco (United States). Cell Counting Kit-8 (CCK-8) was purchased from Dojindo Molecular Technologies Inc. (Japan). Annexin V/Propidium iodide (PI) staining kit was purchased from BD Biosciences (United States). Hoechst 33342 and RIPA lysis buffer was purchased from Beyotime Biotechnology CO., LTD. (China). Cell-cycle staining kit was purchased from MULTISCIENCES (LIANKE) BIOTECH, CO., LTD. (China). RNA-Quick Purification Kit was purchased from Shanghai Yishan Biotechnology CO., LTD. (China). High Capacity cDNA Reverse Transcription Kit was purchased from Applied Biosystems (United States). TB Green™ Premix Ex Taq™ II (Tli RNaseH Plus) was purchased from Takara Bio Inc. (Japan). The primary antibodies of EGFR, p-EGFR (Tyr1068), Akt, AKT1 (phospho S473) and the secondary antibodies of anti-rabbit and anti-mouse were purchased from Cell Signaling Technology, Inc. (United States). NSCLC cell lines PC-9, H460, A549 and H1975 were purchased from ATCC.
Chemistry
The general procedures of preparation for Erlotinib and compounds e1–e20 were described in the section of results. The structures of the all target compounds including Erlotinib were confirmed by nuclear magnetic resonance (1H NMR and 13C NMR) and high-resolution mass spectrometry (HRMS) as below.
The 4-chloro-6, 7-bis (methoxy ethoxy) quinazolinone (3.1 g, 10 mmol) was suspended in 50 ml of isopropanol and 3-aminophenylacetylene (1.3 g, 11 mmol) was added. The suspension was stirred at room temperature for 5 min and then stirred at 85°C under nitrogen overnight. The orange solution was concentrated to obtain an orange solid residue which was recrystallized from EtOAc to yield Erlotinib as an off white solid (3.2 g). 1H NMR (600 MHz, DMSO-d6): δ 9.48 (s, 1H), 8.51 (s, 1H), 8.00 (s, 1H), 7.91 (d, J = 9.5 Hz, 1H), 7.87 (s, 1H), 7.41 (t, J = 7.9 Hz, 1H), 7.27–7.17 (m, 2H), 4.30 (d, J = 15.1 Hz, 4H), 4.21 (s, 1H), 3.78 (d, J = 31.1 Hz, 4H), 3.38 (s, 3H), 3.36 (s, 3H);13C NMR (150 Hz, DMSO-d6): 156.59, 154.15, 153.27, 148.61, 147.49, 140.28, 129.37, 126.81, 125.21, 123.02, 122.21, 109.39, 108.69, 103.65, 83.97, 81.03, 70.59, 70.52, 68.85, 68.52, 58.87, 58.82; HR MS(ESI)m/z: calcd for C22H23O4N3Na [M+Na]+ 416.1581, found 416.1585.
Aryl-azido (1.2 mmol) and Erlotinib (1.0 mmol) were added to 20 ml mixed solvent (water/tert-butanol/tetrahydrofuran = 1:1:1). The reaction was catalyzed with copper sulfate pentahydrate (0.1 mmol) and sodium ascorbate (0.2 mmol), and the mixture then stirred at 60°Cfor 5 h. When the reaction ended (monitored by TLC), the mixture was extracted with dichloromethane (20 ml × 3). The combined organic phase was washed successively with water and brine, dried over sodium sulfate and concentrated in vacuo. Finally, the residue was purified through column chromatography (CH2Cl2/MeOH) to obtain the desired compounds e1–e20 as crystalline powder.
[3-(1-Benzyl-1H-[1,2,3]triazol-4-yl)-phenyl]-[6,7-bis-(2-methoxy-ethoxy)-quinazolin-4-yl]-amine (e1). Purity 99.1%; m.p.89–92°C; 1H NMR (600 MHz, DMSO-d6): δ 9.56 (s, 1H), 8.67 (s, 1H), 8.49 (s, 1H), 8.27 (s, 1H), 7.95–7.86 (m, 2H), 7.56 (d, J = 7.7 Hz, 1H), 7.51–7.28 (m, 6H), 7.24 (s, 1H), 5.67 (s, 2H), 4.31 (d, J = 21.7 Hz, 4H), 3.78 (d, J = 32.4 Hz, 4H), 3.39 (s, 3H), 3.36 (s, 3H). 13C NMR (150 Hz, DMSO-d6): 156.83, 154.06, 153.40, 148.56, 147.44, 147.12, 140.52, 136.50, 131.39, 129.48, 129.30, 128.66, 128.42, 122.28, 122.12, 120.79, 119.23, 109.43, 108.68, 103.69, 70.60, 70.54, 68.83, 68.51, 58.88, 58.82, 53.53; HR MS (ESI) m/z: calcd for C29H30O4N6Na [M+Na]+ 549.2221, found 549.2231.
[6,7-Bis-(2-methoxy-ethoxy)-quinazolin-4-yl]-{3-[1-(2-iodo-benzyl)-1H-[1,2,3]triazol-4-yl]-phenyl}-amine (e2). Purity 98.4%; m.p.93–96°C; 1H NMR (600 MHz, DMSO-d6): δ 9.63 (s, 1H), 8.64 (s, 1H), 8.54 (s, 1H), 8.32 (s, 1H), 8.06–7.90 (m, 3H), 7.63 (d, J = 7.7 Hz, 1H), 7.50 (dd, J = 16.4, 8.0 Hz, 2H), 7.28 (s, 1H), 7.20 (dd, J = 11.8, 7.6 Hz, 2H), 5.75 (s, 2H), 4.35 (d, J = 20.8 Hz, 4H), 3.83 (d, J = 31.5 Hz, 4H), 3.43 (s, 3H), 3.41 (s, 3H). 13C NMR (150 Hz, DMSO-d6): 156.86, 154.09, 153.34, 148.57, 147.30, 146.93, 140.50, 140.00, 138.36, 131.32, 130.80, 130.17, 129.49, 129.38, 122.52, 122.38, 120.88, 119.30, 109.42, 108.58, 103.72, 99.70, 70.59, 70.54, 68.84, 68.52, 58.88, 58.83, 58.03; HR MS(ESI)m/z: calcd for C29H29O4N6INa [M+Na]+ 675.1187, found 675.1196.
[6,7-Bis-(2-methoxy-ethoxy)-quinazolin-4-yl]-{3-[1-(2-bromo-benzyl)-1H-[1,2,3]triazol-4-yl]-phenyl}-amine (e3). Purity 99.3%; m.p.94–97°C; 1H NMR (600 MHz, DMSO-d6): δ 9.60 (s, 1H), 8.63 (s, 1H), 8.50 (s, 1H), 8.27 (s, 1H), 7.98–7.84 (m, 2H), 7.72 (d, J = 7.9 Hz, 1H), 7.58 (d, J = 7.7 Hz, 1H), 7.46 (dt, J = 11.5, 7.7 Hz, 2H), 7.34 (t, J = 8.3 Hz, 1H), 7.29–7.16 (m, 2H), 5.76 (s, 2H), 4.31 (d, J = 20.5 Hz, 4H), 3.78 (d, J = 31.6 Hz, 4H), 3.38 (s, 3H), 3.36 (s, 3H). 13C NMR (150 Hz, DMSO-d6): 156.88, 154.10, 153.31, 148.58, 147.22, 146.92, 140.47, 135.28, 133.42, 131.32, 131.00, 129.94, 129.50, 128.83, 123.37, 122.53, 122.41, 120.91, 119.33, 109.41, 108.53, 103.72, 87.36, 70.60, 70.53, 68.84, 68.52, 58.88, 58.83, 53.63; HR MS(ESI)m/z: calcd for C29H29O4N6BrNa [M+Na]+ 627.1331, found 627.1336.
[6,7-Bis-(2-methoxy-ethoxy)-quinazolin-4-yl]-{3-[1-(3,5-dibromo-benzyl)-1H-[1,2,3]triazol-4-yl]-phenyl}-amine (e4). Purity 98.9%; m.p.102–105°C; 1H NMR (600 MHz, DMSO-d6): δ 9.58 (s, 1H), 8.72 (s, 1H), 8.49 (s, 1H), 8.28 (s, 1H), 7.99–7.89 (m, 2H), 7.86 (s, 1H), 7.64 (s, 2H), 7.57 (d, J = 7.6 Hz, 1H), 7.47 (t, J = 7.9 Hz, 1H), 7.24 (s, 1H), 5.70 (s, 2H), 4.35–4.26 (m, 4H), 3.83–3.73 (m, 4H), 3.39 (s, 3H), 3.36 (s, 3H); 13C NMR (150 Hz, DMSO-d6): 156.84, 154.06, 153.39, 148.56, 147.45, 147.22, 140.83, 140.56, 133.73, 131.21, 130.67, 129.51, 123.23, 122.41, 120.83, 119.30, 109.44, 108.68, 103.71, 70.61, 70.54, 68.84, 68.51, 58.88, 58.82, 52.01; HR MS(ESI)m/z: calcd for C29H29O4N6Br2Na [M+Na]+683.0612, found 683.0624.
[6,7-Bis-(2-methoxy-ethoxy)-quinazolin-4-yl]-{3-[1-(3-methoxy-phenyl)-1H-[1,2,3]triazol-4-yl]-phenyl}-amine (e5). Purity 99.5%; m.p.85–88°C; 1H NMR (600 MHz, DMSO-d6): δ 9.61 (s, 1H), 8.66 (s, 1H), 8.27 (s, 1H), 8.01 (s, 1H), 7.91 (d, J = 7.9 Hz, 1H), 7.57 (d, J = 7.6 Hz, 1H), 7.46 (t, J = 7.9 Hz, 1H), 7.32 (t, J = 7.9 Hz, 2H), 6.97 (s, 1H), 6.93 (d, J = 7.9 Hz, 2H), 5.63 (s, 2H), 4.32 (d, J = 9.1 Hz, 4H), 3.80 (d, J = 9.0 Hz, 4H), 3.76 (s, 3H), 3.39 (s, 3H), 3.37 (s, 3H).13C NMR (150 Hz, DMSO-d6): 159.96, 156.75, 154.04, 148.63, 147.09, 140.46, 137.91, 131.41, 130.47, 129.50, 122.35, 122.13, 120.87, 120.51, 119.31, 114.27, 113.98, 108.94, 103.82, 87.75, 70.60, 70.54, 68.85, 68.54, 58.88, 58.83, 55.61, 53.46, 22.56; HR MS(ESI)m/z: calcd for C30H33O5N6 [M+H]+ 557.2512, found 557.2508.
[6,7-Bis-(2-methoxy-ethoxy)-quinazolin-4-yl]-{3-[1-(2-fluoro-phenyl)-1H-[1,2,3]triazol-4-yl]-phenyl}-amine (e6). Purity 98.6%; m.p.83–86°C; 1H NMR (600 MHz, DMSO-d6): δ 9.62 (s, 1H), 9.11 (s, 1H), 8.51 (s, 1H), 8.39 (s, 1H), 8.04–7.86 (m, 3H), 7.73–7.59 (m, 3H), 7.56–7.46 (m, 2H), 7.25 (s, 1H), 4.37–4.28 (m, 4H), 3.83–3.74 (m, 4H), 3.39 (s, 3H), 3.37 (s, 3H). 13C NMR (150 Hz, DMSO-d6): 156.85, 155.22, 154.08, 153.41, 148.57, 147.40, 140.64, 131.88, 130.76, 129.61, 126.56, 126.08, 123.40, 122.74, 121.08, 119.48, 117.75, 117.62, 109.46, 108.69, 103.71, 70.61, 70.54, 68.85, 68.51, 58.88, 58.82; HR MS(ESI)m/z: calcd for C28H27O4N6FNa [M+Na]+ 553.1970, found 553.1979.
[6,7-Bis-(2-methoxy-ethoxy)-quinazolin-4-yl]-{3-[1-(4-fluoro-phenyl)-1H-[1,2,3]triazol-4-yl]-phenyl}-amine (e7). Purity 97.9%; m.p.88–91°C; 1H NMR (600 MHz, DMSO-d6): δ 9.73 (s, 1H), 9.32 (s, 1H), 8.57 (s, 1H), 8.38 (s, 1H), 8.14–7.86 (m, 4H), 7.67 (d, J = 7.6 Hz, 1H), 7.52 (dt, J = 12.5, 8.3 Hz, 3H), 7.26 (s, 1H), 4.41–4.25 (m, 4H), 3.85–3.72 (m, 4H), 3.39 (s, 3H), 3.37 (s, 3H); 13C NMR (150 Hz, DMSO-d6): 162.98, 161.35, 156.98, 156.96, 154.21, 153.11, 148.67, 147.80, 140.47, 133.71, 130.95, 129.63, 122.88, 122.82, 120.45, 119.61, 117.36, 117.20, 108.32, 103.79, 70.59, 70.53, 68.87, 68.57, 58.89, 58.83; HR MS(ESI)m/z: calcd for C28H27O4N6FNa [M+Na]+ 553.1970, found 553.1979.
[6,7-Bis-(2-methoxy-ethoxy)-quinazolin-4-yl]-{3-[1-(2-chloro-phenyl)-1H-[1,2,3]triazol-4-yl]-phenyl}-amine (e8). Purity 99.0%; m.p.131–134°C; 1H NMR (600 MHz, DMSO-d6): δ 9.62 (s, 1H), 9.08 (s, 1H), 8.51 (s, 1H), 8.40 (s, 1H), 7.94 (d, J = 10.6 Hz, 2H), 7.85–7.79 (m, 2H), 7.70–7.61 (m, 3H), 7.52 (t, J = 7.9 Hz, 1H), 7.25 (s, 1H), 4.36–4.27 (m, 4H), 3.84–3.74 (m, 4H), 3.39 (s, 3H), 3.37 (s, 3H); 13C NMR (150 Hz, DMSO-d6): 156.86, 154.09, 153.40, 148.58, 147.44, 146.99, 140.64, 135.03, 131.08, 130.89, 129.61, 129.13, 129.01, 128.96, 124.19, 122.67, 121.04, 119.48, 109.44, 108.66, 103.72, 70.61, 70.54, 68.85, 68.52, 58.88, 58.83; HR MS(ESI)m/z: calcd for C28H27O4N6ClNa [M+Na]+ 569.1675, found 569.1678.
[6,7-Bis-(2-methoxy-ethoxy)-quinazolin-4-yl]-{3-[1-(2-bromo-phenyl)-1H-[1,2,3]triazol-4-yl]-phenyl}-amine (e9). Purity 97.7%; m.p.93–97°C; 1H NMR (600 MHz, DMSO-d6): δ 9.63 (s, 1H), 9.05 (s, 1H), 8.51 (s, 1H), 8.40 (s, 1H), 8.00–7.89 (m, 3H), 7.77 (dd, J = 7.8, 1.5 Hz, 1H), 7.67 (t, J = 7.7 Hz, 2H), 7.60 (t, J = 8.6 Hz, 1H), 7.52 (t, J = 7.9 Hz, 1H), 7.25 (s, 1H), 4.35–4.27 (m, 4H), 3.83–3.75 (m, 4H), 3.39 (s, 3H), 3.37 (s, 3H); 13C NMR (150 Hz, DMSO-d6): 156.87, 154.10, 153.39, 148.58, 147.40, 146.92, 140.63, 136.72, 134.14, 132.58, 130.94, 129.61, 129.49, 129.24, 124.20, 122.64, 121.02, 119.47, 109.44, 108.64, 103.72, 100.00, 70.61, 70.54, 68.85, 68.52, 58.88, 58.83; HR MS(ESI)m/z: calcd for C28H27O4N6BrNa [M+Na]+ 613.1169, found 613.1180.
[6,7-Bis-(2-methoxy-ethoxy)-quinazolin-4-yl]-{3-[1-(4-bromo-phenyl)-1H-[1,2,3]triazol-4-yl]-phenyl}-amine (e10). Purity 98.2%; m.p.105–108°C; 1H NMR (600 MHz, DMSO-d6): δ 9.63 (s, 1H), 9.37 (s, 1H), 8.51 (s, 1H), 8.38 (s, 1H), 7.96 (dd, J = 16.5, 7.6 Hz, 4H), 7.86 (d, J = 8.8 Hz, 2H), 7.66 (d, J = 7.6 Hz, 1H), 7.53 (t, J = 7.9 Hz, 1H), 7.25 (s, 1H), 4.32 (d, J = 24.8 Hz, 4H), 3.79 (d, J = 34.8 Hz, 4H), 3.39 (s, 3H), 3.37 (s, 3H); 13C NMR (150 Hz, DMSO-d6): 156.86, 154.09, 153.40, 148.58, 147.97, 147.44, 140.63, 136.32, 133.32, 130.82, 129.62, 122.79, 122.38, 121.82, 121.01, 120.19, 119.48, 109.43, 108.66, 103.68, 70.60, 70.54, 68.84, 68.51, 58.88, 58.82; HR MS(ESI)m/z: calcd for C28H27O4N6BrNa [M+Na]+ 613.1169, found 613.1177.
[6,7-Bis-(2-methoxy-ethoxy)-quinazolin-4-yl]-{3-[1-(2-methoxy-phenyl)-1H-[1,2,3]triazol-4-yl]-phenyl}-amine (e11). Purity 98.7%; m.p.87–90°C; 1H NMR (600 MHz, DMSO-d6): δ 9.62 (s, 1H), 8.94 (s, 1H), 8.50 (s, 1H), 8.36 (s, 1H), 7.98–7.91 (m, 2H), 7.69 (dd, J = 21.0, 7.7 Hz, 2H), 7.58 (t, J = 7.9 Hz, 1H), 7.50 (t, J = 7.9 Hz, 1H), 7.36 (d, J = 8.2 Hz, 1H), 7.25 (s, 1H), 7.19 (t, J = 7.6 Hz, 1H), 4.32 (d, J = 25.5 Hz, 4H), 3.90 (s, 3H), 3.79 (d, J = 34.6 Hz, 4H), 3.39 (s, 3H), 3.37 (s, 3H); 13C NMR (150 Hz, DMSO-d6): 156.88, 154.08, 153.41, 152.32, 148.56, 147.45, 146.67, 140.57, 131.39, 131.21, 129.52, 126.43, 126.21, 123.94, 122.54, 121.36, 121.04, 119.43, 113.49, 109.45, 108.67, 103.73, 68.84, 68.51, 58.88, 58.52, 56.65; HR MS(ESI)m/z: calcd for C29H30O5N6Na [M+Na]+ 565.2170, found 565.2172.
[6,7-Bis-(2-methoxy-ethoxy)-quinazolin-4-yl]-[3-(1-p-tolyl-1H-[1,2,3]triazol-4-yl)-phenyl]-amine (e12). Purity 97.4%; m.p.95–98°C; 1H NMR (600 MHz, DMSO-d6): δ 9.63 (s, 1H), 9.28 (s, 1H), 8.51 (s, 1H), 8.37 (s, 1H), 7.95 (d, J = 9.9 Hz, 2H), 7.87 (d, J = 8.3 Hz, 2H), 7.67 (d, J = 7.6 Hz, 1H), 7.52 (t, J = 7.9 Hz, 1H), 7.45 (d, J = 8.3 Hz, 2H), 7.25 (s, 1H), 4.32 (d, J = 25.1 Hz, 4H), 3.79 (d, J = 35.1 Hz, 4H), 3.39 (s, 3H), 3.37 (s, 3H), 2.51 (s, 3H); 13C NMR (150 Hz, DMSO-d6): 156.88, 154.09, 153.40, 148.58, 147.70, 147.40, 140.59, 138.83, 134.89, 131.06, 130.75, 129.57, 122.69, 121.02, 120.36, 120.02, 119.46, 109.43, 108.64, 103.69, 70.60, 70.54, 68.84, 68.52, 58.88, 58.82, 21.07; HR MS(ESI)m/z: calcd for C29H31O4N6 [M+H]+ 527.2401, found 527.2410.
[6,7-Bis-(2-methoxy-ethoxy)-quinazolin-4-yl]-{3-[1-(3-nitro-phenyl)-1H-[1,2,3]triazol-4-yl]-phenyl}-amine (e13). Purity 99.2%; m.p.98–101°C; 1H NMR (600 MHz, DMSO-d6): δ 9.64 (s, 1H), 9.60 (s, 1H), 8.83 (t, J = 2.1 Hz, 1H), 8.50 (d, J = 12.4 Hz, 2H), 8.41 (s, 1H), 8.37 (d, J = 7.5 Hz, 1H), 7.99–7.94 (m, 3H), 7.69 (d, J = 7.8 Hz, 1H), 7.55 (t, J = 7.9 Hz, 1H), 7.25 (s, 1H), 4.32 (d, J = 25.9 Hz, 4H), 3.79 (d, J = 36.0 Hz, 4H), 3.39 (s, 3H), 3.37 (s, 3H); 13C NMR (150 Hz, DMSO-d6): 156.86, 154.09, 153.40, 149.06, 148.59, 147.45, 140.67, 137.70, 132.09, 130.64, 129.68, 126.40, 123.61, 122.92, 121.03, 120.61, 119.55, 115.08, 109.43, 108.66, 103.67, 70.61, 70.54, 68.84, 68.52, 58.88, 58.83; HR MS(ESI)m/z: calcd for C28H27O6N7Na [M+Na]+ 580.1915, found 580.1923.
[6,7-Bis-(2-methoxy-ethoxy)-quinazolin-4-yl]-{3-[1-(3-ethoxy-phenyl)-1H-[1,2,3]triazol-4-yl]-phenyl}-amine (e14). Purity 98.3%; m.p.110–114°C; 1H NMR (600 MHz, DMSO-d6): δ 9.63 (s, 1H), 9.36 (s, 1H), 8.51 (s, 1H), 8.37 (s, 1H), 7.95 (d, J = 10.1 Hz, 2H), 7.67 (d, J = 7.7 Hz, 1H), 7.55 (d, J = 36.4 Hz, 4H), 7.25 (s, 1H), 7.08 (d, J = 10.2 Hz, 1H), 4.32 (d, J = 24.9 Hz, 4H), 4.17 (q, J = 7.0 Hz, 2H), 3.79 (d, J = 35.2 Hz, 4H), 3.39 (s, 3H), 3.37 (s, 3H), 1.39 (t, J = 7.0 Hz, 3H); 13C NMR (150 Hz, DMSO-d6): 159.98, 156.87, 154.08, 153.42, 148.57, 147.84, 147.49, 140.61, 138.13, 131.37, 130.97, 129.59, 122.77, 121.01, 120.19, 119.48, 115.27, 112.27, 109.46, 108.69, 106.46, 103.68, 70.61, 70.54, 68.84, 68.51, 64.12, 58.88, 58.82, 15.02; HR MS(ESI)m/z: calcd for C30H32O5N6Na [M+Na]+ 579.2326, found 579.2332.
[6,7-Bis-(2-methoxy-ethoxy)-quinazolin-4-yl]-[3-(1-phenyl-1H-[1,2,3]triazol-4-yl)-phenyl]-amine (e15). Purity 96.8%; m.p.137–140°C; 1H NMR (600 MHz, DMSO-d6): δ 9.63 (s, 1H), 9.34 (s, 1H), 8.50 (s, 1H), 8.38 (s, 1H), 7.99 (d, J = 7.6 Hz, 2H), 7.94 (d, J = 9.8 Hz, 2H), 7.66 (dd, J = 16.3, 8.6 Hz, 3H), 7.53 (d, J = 18.2 Hz, 2H), 7.24 (s, 1H), 4.32 (d, J = 25.9 Hz, 4H), 3.78 (d, J = 35.2 Hz, 4H), 3.39 (s, 3H), 3.36 (s, 3H); 13C NMR (150 Hz, DMSO-d6): 156.88, 154.08, 153.42, 148.58, 147.81, 147.45, 140.61, 137.13, 130.99, 130.42, 129.59, 129.21, 122.73, 121.03, 120.49, 120.15, 119.48, 109.44, 108.66, 103.69, 70.61, 70.54, 68.84, 68.51, 58.88, 58.82; HR MS(ESI)m/z: calcd for C28H28O4N6Na [M+Na]+ 535.2064, found 535.2069.
[6,7-Bis-(2-methoxy-ethoxy)-quinazolin-4-yl]-{3-[1-(2-trifluoromethyl-phenyl)-1H-[1,2,3]triazol-4-yl]-phenyl}-amine (e16). Purity 97.7%; m.p.113–116°C; 1H NMR (400 MHz, DMSO-d6): δ 9.62 (s, 1H), 9.05 (s, 1H), 8.51 (s, 1H), 8.39 (s, 1H), 8.08 (d, J = 7.1 Hz, 1H), 7.93 (d, J = 61.3 Hz, 5H), 7.65 (d, J = 7.8 Hz, 1H), 7.52 (t, J = 7.9 Hz, 1H), 7.24 (s, 1H), 4.32 (d, J = 18.3 Hz, 4H), 3.78 (d, J = 25.0 Hz, 4H), 3.39 (s, 3H), 3.37 (s, 3H); 13C NMR (100 Hz, DMSO-d6): 156.83, 154.04, 153.35, 148.54, 147.33, 146.97, 140.62, 134.57, 131.77, 130.75, 129.84, 129.63, 127.97, 125.55, 125.24, 124.74, 122.72, 121.94, 121.01, 119.49, 109.39, 108.53, 103.59, 70.57, 70.50, 68.78, 68.47, 58.85, 58.80; HR MS (ESI)m/z: calcd for C29H27O4N6F3Na [M+Na]+ 603.1938, found 603.1945.
[6,7-Bis-(2-methoxy-ethoxy)-quinazolin-4-yl]-{3-[1-(2,4-dimethoxy-phenyl)-1H-[1,2,3]triazol-4-yl]-phenyl}-amine (e17). Purity 98.5%; m.p.85–88°C; 1H NMR (400 MHz, DMSO-d6): δ 9.66 (s, 1H), 8.88 (s, 1H), 8.55 (s, 1H), 8.39 (s, 1H), 8.02–7.92 (m, 2H), 7.70 (d, J = 7.8 Hz, 1H), 7.63 (d, J = 8.7 Hz, 1H), 7.54 (t, J = 7.9 Hz, 1H), 7.29 (s, 1H), 6.92 (d, J = 2.5 Hz, 1H), 6.78 (dd, J = 8.8, 2.6 Hz, 1H), 4.37 (d, J = 19.0 Hz, 4H), 3.93 (s, 3H), 3.92 (s, 3H), 3.84 (d, J = 25.5 Hz, 4H), 3.44 (s, 3H), 3.42 (s, 3H); 13C NMR (100 Hz, DMSO-d6): 161.72, 156.84, 154.01, 153.70, 153.37, 148.52, 146.49, 140.50, 131.29, 129.49, 127.48, 124.06, 122.46, 120.99, 119.62, 119.34, 108.60, 105.72, 103.61, 99.99, 70.57, 70.51, 68.79, 68.47, 58.86, 58.80, 56.66, 56.18; HR MS (ESI) m/z: calcd for C30H32O6N6Na [M+Na]+ 595.2276, found 595.2285.
2-(4-{3-[6,7-Bis-(2-methoxy-ethoxy)-quinazolin-4-ylamino]-phenyl}-[1,2,3]triazol-1-yl)-5-methyl-phenol (e18)¶. Purity 98.8%; m.p.100–103°C; 1H NMR (400 MHz, DMSO-d6): δ 9.66 (s, 1H), 8.98 (s, 1H), 8.55 (s, 1H), 8.40 (s, 1H), 7.99 (d, J = 12.3 Hz, 2H), 7.73 (dd, J = 14.5, 7.8 Hz, 2H), 7.63 (t, J = 7.9 Hz, 1H), 7.55 (t, J = 7.9 Hz, 1H), 7.41 (d, J = 7.7 Hz, 1H), 7.29 (s, 1H), 7.24 (t, J = 7.6 Hz, 1H), 4.37 (d, J = 19.7 Hz, 4H), 3.95 (s, 3H), 3.83 (d, J = 25.8 Hz, 4H), 3.44 (s, 3H), 3.42 (s, 3H); 13C NMR (100 Hz, DMSO-d6): 156.85, 154.02, 153.39, 152.29, 148.52, 147.37, 146.65, 140.53, 131.40, 131.18, 129.53, 126.45, 126.17, 123.98, 122.55, 121.35, 121.03, 119.39, 113.45, 109.40, 108.58, 103.60, 70.57, 70.51, 68.78, 68.47, 58.87, 58.81, 56.62; HR MS(ESI)m/z: calcd for C29H30O5N6Na [M+Na]+ 565.2170, found 565.2175.
N-(3-(1-(3,4-dimethoxyphenyl)-1H-1,2,3-triazol-4-yl)phenyl)-6,7-bis(2-methoxyethoxy)quinazolin-4-amine (e19). Purity 97.9%; m.p.106–109°C; 1H NMR (400 MHz, DMSO-d6): δ 9.70 (s, 1H), 9.32 (s, 1H), 8.57 (s, 1H), 8.40 (s, 1H), 8.01 (d, J = 10.2 Hz, 2H), 7.71 (d, J = 7.7 Hz, 1H), 7.58 (d, J = 27.3 Hz, 3H), 7.30 (s, 1H), 7.24 (d, J = 8.8 Hz, 1H), 4.37 (d, J = 19.7 Hz, 4H), 3.95 (s, 3H), 3.91 (s, 3H), 3.84 (d, J = 25.9 Hz, 4H), 3.44 (s, 3H), 3.42 (s, 3H).);13C NMR (100 Hz, DMSO-d6): 157.31, 154.49, 153.84, 150.26, 149.82, 149.00, 147.98, 147.89, 141.03, 131.55, 130.97, 129.97, 123.11, 121.40, 120.60, 119.84, 112.96, 109.86, 109.09, 105.43, 104.09, 71.03, 70.96, 69.26, 68.93, 59.30, 59.24, 56.81, 56.73; HR MS(ESI) m/z: calcd for C30H32O6N6Na [M+Na]+ 595.2276, found 595.2281.
[6,7-Bis-(2-methoxy-ethoxy)-quinazolin-4-yl]-[3-(1-phenethyl-1H-[1,2,3]triazol-4-yl)-phenyl]-amine (e20). Purity 98.4%; m.p.109–112°C; 1H NMR (600 MHz, DMSO-d6): δ 9.56 (s, 1H), 8.53 (s, 1H), 8.49 (s, 1H), 8.24 (s, 1H), 7.93 (s, 1H), 7.89 (d, J = 8.9 Hz, 1H), 7.51 (d, J = 7.7 Hz, 1H), 7.45 (t, J = 7.8 Hz, 1H), 7.29 (t, J = 7.4 Hz, 2H), 7.22 (dd, J = 13.1, 6.9 Hz, 4H), 4.68 (t, J = 7.3 Hz, 2H), 4.31 (d, J = 22.8 Hz, 4H), 3.78 (d, J = 32.9 Hz, 4H), 3.38 (s, 3H), 3.36 (s, 3H), 3.24 (t, J = 7.3 Hz, 2H); 13C NMR (150 Hz, DMSO-d6): 156.84, 154.06, 153.39, 148.56, 147.42, 146.59, 140.51, 138.11, 131.54, 129.48, 129.17, 127.08, 122.19, 121.88, 120.69, 119.17, 109.43, 108.66, 103.68, 70.60, 70.53, 68.83, 68.51, 58.87, 58.82, 51.12, 36.04; HR MS(ESI)m/z: calcd for C30H32O4N6Na [M+Na]+563.2377, found 563.2381.
Biological Assay
Cell Culture
The human NSCLC cells PC-9, H460, A549 and H1975 were cultured in RPMI-1640 complete growth medium containing 100 U/ml penicillin–streptomycin and 10% FBS. The cells were incubated in a humidified atmosphere at 37°C containing 5% CO2. The compounds were dissolved in DMSO to make a 20 μM stock solution and diluted to the concentration of working solutions with complete growth medium before administration.
CCK-8 Assay
Cells were seeded in 96-well plates: 3,000–5,000 cells/well in 100 ul. One day after seeding, the test compounds at concentration between 1.0 and 16.0
Flow Cytometry Detection for Cell Apoptosis
Cell-apoptosis analysis was carried out by flow cytometry using the Annexin V/PI apoptosis kit according to the manufacturer’s manual. Briefly, PC-9 or H460 (3 × 104–5 × 104/well) cells were seeded in 12-well plates for 24 h, and then treated with 0.1% DMSO (as control) or various concentrations of compound e4 or e12 for 48 h, respectively. Cells were harvested, washed with PBS, and then incubated with 100ul of 1X Annexin V binding buffer containing 1 μl PI and 1 μl FITC Annexin V for 15 min at RT in the dark. 200 µl of 1X binding buffer were added to each tube for flow cytometry analysis (BD LSRFortessa™ Flow Cytometer). The percentages of apoptotic cells were analyzed using FlowJo soft.
Hoechst Staining for Cell Apoptosis
PC-9 cells were seeded in 96-well plates, then treated with 0.1% DMSO (as control) or various concentrations of compound e4 or e12 for 48 h. Culture medium from the cells was removed and replaced with medium containing dye Hoechst 33342. Cells were incubated at 37°C for 5–15 min, washed with PBS twice, and then observed and imaged under the Nikon Ti-U microscope.
Flow Cytometry Detection for Cell Cycle
PC-9 or H460 (2 × 105–3 × 105/well) cells were seeded in six-well plates for 24 h, and then treated with 0.1% DMSO (as control), various concentrations of either compound e4 or e12 for 24 h (PC-9) or 12 h (H460), respectively. The treated cells were harvested, washed with PBS, and then stained using the cell-cycle staining kit according to the manufacturer’s manual. The distribution of cell-cycle phases with different DNA contents was determined by flow cytometry (BD LSRFortessa™ Flow Cytometer) and analyzed using ModFit LT software.
qPCR Assay for Cell Cycle-Related Gene Analysis
PC-9 cells were incubated in 12-well plates for 24 h, and then treated with 0.1% DMSO, various concentrations of either compound e4 or e12 for 16 h. Total RNA of cells was extracted and cDNA was synthesized. The primers for the target products were designed as in Table 3.

TABLE 3. Primer sequences for qPCR analysis.
In Vivo Antitumor Activity Study
PC-9 xenografts were established by subcutaneous transplantation of cancer cells into the flanks of NSG mice (male, 4–6-week old). Once the tumors in the transplanted mice become palpable, the mice were divided randomly into three groups: one group was injected intraperitoneally with vehicle (5% DMSO, 30% PEG300 and 65% ddH2O; qd), one with compound e4 (25 mg/kg, qd) and one with Erlotinib (25 mg/kg, qd) in vehicle every day for 7 consecutive days. Tumor volumes were measured using calipers and calculated with the formula: V = length × width2/2. The mouse body mass was recorded as well. 19 days after inoculation, the mice were sacrificed and the tumors were harvested and weighed.
EGFR Kinase Assay
Kinase inhibitory activities of compounds were evaluated using the Enzyme-linked immunosorbent assay (ELISA). The kinase enzyme of EGFR was purchased from Carna Bioscience (Kobe, Japan). A total of 10 ng/ml anti phosphotyrosine (PY713) antibody (abcam, Cambridge Science Park, United Kingdom) was precoated in 96-well ELISA plates. Active kinases were incubated with indicated drugs in 1 X reaction buffer (50 mmol/L HEPES pH 7.4, 20 mmol/L MgCl2, 0.1 mmol/L MnCl2, 1 mmol/L DTT) containing 20 μmol/L substrate (NH2-ETVYSEVRK-biotin) at 25°C for 1 h. Then, a total of 3 μmol/L ATP was added and the reaction was continued for 2 h. The products of reaction were transferred into 96-well ELISA plates containing antibody and incubated at 25°C for 30 min. After incubation, the wells were washed with PBS and then incubated with horseradish peroxidase (HRP)-conjugated streptavidin. The wells were visualized using 3,3′,5,5′-tetramethylbenzidine (TMB), and chromogenic reaction was ended with 2 mol/L H2SO4, the absorbance was read with a multimode plate reader (PerkinElmer, United States) at 450 nm.
Western Blot Analysis
H460 cells were seeded in six-well plates with serum-free medium overnight, and then treated complete medium containing 0.1% DMSO, various concentrations (0.1, 1 and 10 μM) of either compound e4 or e12 for 2 h, respectively. Complete medium with or without Erlotinib (10 μM) were used as control. Then the H460 cells were treated with complete medium containing EGF (10 ng/ml) for 5 min. Then the cells were harvested and washed with PBS, centrifuged, resuspended in 100–200 μl of RIPA lysis buffer containing Protease and Phosphatase Inhibitor Cocktail (1X), and then centrifuged for 5 min at 12,000 rpm at 4°C. The supernatant was used for protein analysis. Total protein extracts were separated by 7.5% SDS polyacrylamide gel electrophoresis and transferred onto NC membranes. Membrane was blocked with 5% skimmed milk or BSA in TBS-T (10 mM Tris [pH 7.4], 150 mM NaCl, and 0.1% Tween20) for 1 h at RT, then incubated with a 1:1,000 dilution of anti-EGFR (D38B1), anti-Akt (pan) (C67E7),anti-p-EGFR (Tyr1068) andanti-AKT1 (phospho S473) [EP2109Y] primary antibodyor 1:5,000 dilution of anti-β-Actin (8H10D10) primary antibody overnight at 4°C. Then the membrane was washed 5 min (X3) and incubated at 1:3,000 dilution of anti-rabbit secondary antibodies or 1:1,000 dilution of anti-mouse secondary antibodies for 1 h at RT. After washing with TBST 5 min (X3), protein bands were developed by chemiluminescence. The protein levels were quantified by the gray values of the bands in the resulting images by ImageJ, and the control group was used as the standard. For PC-9, cells were incubated in six-well plates with complete medium overnight, and then treated with 0.1% DMSO, various concentrations (0.1, 1 and 10 μM) of either compound e4 or e12 for 12 h. Erlotinib (10 μM) was used as control. The PC-9 cells were harvested and analyzed as above.
Statistical Analysis
All values are presented as means ± SD. Significant differences were evaluated using GraphPad Prism 8 software. The Student’s t-test was used to determine significant differences between two groups. A two-tailed p-value of <0.05 was considered statistically significant in all experiments.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics Statement
The animal study was reviewed and approved by Care and Use of Laboratory Animals of Southern Medical University.
Author Contributions
GS and LM carried out the experiments and wrote the manuscript with support from WD, SX, JZ, and JY. WL conceived and planned the experiments of chemistry part. MY devised the biological part of the study. WL, MY, and KY helped supervise the project and conceived the original idea.
Funding
This work was supported by the National Natural Science Foundation of China (81972488, 81701836), the Eighth Affiliated Hospital of Sun Yat-sen University Outstanding Youth Reserve Talent Science Fund (FBJQ2019002), the Scientific and Technological Project of Henan Province (No. 192102310142), and the Shenzhen Science and Technology Program (JCYJ20210324115209026).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2021.789030/full#supplementary-material
References
Akita, R. W., and Sliwkowski, M. X. (2003). Preclinical Studies with Erlotinib (Tarceva). Semin. Oncol. 30 (3H), 15–24. doi:10.1016/s0093-7754(03)70011-6
Bhatia, P., Sharma, V., Alam, O., Manaithiya, A., Alam, P., Kahksha, , et al. (2020). Novel Quinazoline-Based EGFR Kinase Inhibitors: A Review Focussing on SAR and Molecular Docking Studies (2015-2019). Eur. J. Med. Chem. 204, 112640. doi:10.1016/j.ejmech.2020.112640
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A., and Jemal, A. (2018). Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA: A Cancer J. Clinicians 68 (6), 394–424. doi:10.3322/caac.21492
Chmielecki, J., Foo, J., Oxnard, G. R., Hutchinson, K., Ohashi, K., Somwar, R., et al. (2011). Optimization of Dosing for EGFR-Mutant Non-small Cell Lung Cancer with Evolutionary Cancer Modeling. Sci. Transl. Med. 3 (90), 90ra59. doi:10.1126/scitranslmed.3002356
Cross, D. A. E., Ashton, S. E., Ghiorghiu, S., Eberlein, C., Nebhan, C. A., Spitzler, P. J., et al. (2014). AZD9291, an Irreversible EGFR TKI, Overcomes T790M-Mediated Resistance to EGFR Inhibitors in Lung Cancer. Cancer Discov. 4 (9), 1046–1061. doi:10.1158/2159-8290.CD-14-0337
Dungo, R. T., and Keating, G. M. (2013). Afatinib: First Global Approval. Drugs 73 (13), 1503–1515. doi:10.1007/s40265-013-0111-6
Elkamhawy, A., Farag, A. K., Viswanath, A. N. I., Bedair, T. M., Leem, D. G., Lee, K.-T., et al. (2015a). Targeting EGFR/HER2 Tyrosine Kinases with a New Potent Series of 6-substituted 4-anilinoquinazoline Hybrids: Design, Synthesis, Kinase Assay, Cell-Based Assay, and Molecular Docking. Bioorg. Med. Chem. Lett. 25 (22), 5147–5154. doi:10.1016/j.bmcl.2015.10.003
Elkamhawy, A., Viswanath, A. N. I., Pae, A. N., Kim, H. Y., Heo, J.-C., Park, W.-K., et al. (2015b). Discovery of Potent and Selective Cytotoxic Activity of New Quinazoline-Ureas against TMZ-Resistant Glioblastoma Multiforme (GBM). Eur. J. Med. Chem. 103, 210–222. doi:10.1016/j.ejmech.2015.08.001
Gridelli, C., Rossi, A., Carbone, D. P., Guarize, J., Karachaliou, N., Mok, T., et al. (2015). Non-small-cell Lung Cancer. Nat. Rev. Dis. Primers 1, 15009. doi:10.1038/nrdp.2015.9
Helfrich, B. A., Raben, D., Varella-Garcia, M., Gustafson, D., Chan, D. C., Bemis, L., et al. (2006). Antitumor Activity of the Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Inhibitor Gefitinib (ZD1839, Iressa) in Non-small Cell Lung Cancer Cell Lines Correlates with Gene Copy Number and EGFR Mutations but Not EGFR Protein Levels. Clin. Cancer Res. 12 (23), 7117–7125. doi:10.1158/1078-0432.CCR-06-0760
Herbst, R. S., and Bunn, P. A. (2003). Targeting the Epidermal Growth Factor Receptor in Non-small Cell Lung Cancer. Clin. Cancer Res. 9 (16), 5813–5824.
Hirsch, F. R., Scagliotti, G. V., Mulshine, J. L., Kwon, R., Curran, W. J., Wu, Y.-L., et al. (2017). Lung Cancer: Current Therapies and New Targeted Treatments. The Lancet 389 (10066), 299–311. doi:10.1016/s0140-6736(16)30958-8
Kan, Z., Jaiswal, B. S., Stinson, J., Janakiraman, V., Bhatt, D., Stern, H. M., et al. (2010). Diverse Somatic Mutation Patterns and Pathway Alterations in Human Cancers. Nature 466 (7308), 869–873. doi:10.1038/nature09208
Kobayashi, K., and Hagiwara, K. (2013). Epidermal Growth Factor Receptor (EGFR) Mutation and Personalized Therapy in Advanced Nonsmall Cell Lung Cancer (NSCLC). Targ Oncol. 8 (1), 27–33. doi:10.1007/s11523-013-0258-9
Lynch, T. J., Bell, D. W., Sordella, R., Gurubhagavatula, S., Okimoto, R. A., Brannigan, B. W., et al. (2004). Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non-small-cell Lung Cancer to Gefitinib. N. Engl. J. Med. 350 (21), 2129–2139. doi:10.1056/NEJMoa040938
Maddili, S. K., Katla, R., Kannekanti, V. K., Bejjanki, N. K., Tuniki, B., Zhou, C.-H., et al. (2018). Molecular Interaction of Novel Benzothiazolyl Triazolium Analogues with Calf Thymus DNA and HSA-Their Biological Investigation as Potent Antimicrobial Agents. Eur. J. Med. Chem. 150, 228–247. doi:10.1016/j.ejmech.2018.02.056
Moyer, J. D., Barbacci, E. G., Iwata, K. K., Arnold, L., Boman, B., Cunningham, A., et al. (1997). Induction of Apoptosis and Cell Cycle Arrest by CP-358,774, an Inhibitor of Epidermal Growth Factor Receptor Tyrosine Kinase. Cancer Res. 57 (21), 4838–4848.
Muhsin, M., Graham, J., and Kirkpatrick, P. (2003). Gefitinib. Nat. Rev. Cancer 3 (8), 556–557. doi:10.1038/nrc1159
Paez, J. G., Jänne, P. A., Lee, J. C., Tracy, S., Greulich, H., Gabriel, S., et al. (2004). EGFR Mutations in Lung Cancer: Correlation with Clinical Response to Gefitinib Therapy. Science 304 (5676), 1497–1500. doi:10.1126/science.1099314
Qi, W.-X., Shen, Z., Lin, F., Sun, Y.-J., Min, D.-L., Tang, L.-N., et al. (2012). Comparison of the Efficacy and Safety of EFGR Tyrosine Kinase Inhibitor Monotherapy with Standard Second-Line Chemotherapy in Previously Treated Advanced Non-small-cell Lung Cancer: a Systematic Review and Meta-Analysis. Asian Pac. J. Cancer Prev. 13 (10), 5177–5182. doi:10.7314/apjcp.2012.13.10.5177
Saeedi, M., Mohammadi-Khanaposhtani, M., Pourrabia, P., Razzaghi, N., Ghadimi, R., Imanparast, S., et al. (2019). Design and Synthesis of Novel Quinazolinone-1,2,3-Triazole Hybrids as New Anti-diabetic Agents: In Vitro α-glucosidase Inhibition, Kinetic, and Docking Study. Bioorg. Chem. 83, 161–169. doi:10.1016/j.bioorg.2018.10.023
Safavi, M., Ashtari, A., Khalili, F., Mirfazli, S. S., Saeedi, M., Ardestani, S. K., et al. (2018). Novel quinazolin-4(3H )-one Linked to 1,2,3-triazoles: Synthesis and Anticancer Activity. Chem. Biol. Drug Des. 92 (1), 1373–1381. doi:10.1111/cbdd.13203
Schettino, C., Bareschino, M. A., Ricci, V., and Ciardiello, F. (2008). Erlotinib: an EGF Receptor Tyrosine Kinase Inhibitor in Non-small-cell Lung Cancer Treatment. Expert Rev. Respir. Med. 2 (2), 167–178. doi:10.1586/17476348.2.2.167
Sequist, L. V., Waltman, B. A., Dias-Santagata, D., Digumarthy, S., Turke, A. B., Fidias, P., et al. (2011). Genotypic and Histological Evolution of Lung Cancers Acquiring Resistance to EGFR Inhibitors. Sci. Transl. Med. 3 (75), 75ra26. doi:10.1126/scitranslmed.3002003
Travis, W. D., Brambilla, E., and Riely, G. J. (2013). New Pathologic Classification of Lung Cancer: Relevance for Clinical Practice and Clinical Trials. Jco 31 (8), 992–1001. doi:10.1200/JCO.2012.46.9270
Tsao, M.-S., Sakurada, A., Cutz, J.-C., Zhu, C.-Q., Kamel-Reid, S., Squire, J., et al. (2005). Erlotinib in Lung Cancer - Molecular and Clinical Predictors of Outcome. N. Engl. J. Med. 353 (21533-4406), 133–144. doi:10.1056/nejmoa050736
Keywords: erlotinib, 1,2,3-triazole, NSCLC, EGFR, anti-tumor
Citation: Sun G, Mao L, Deng W, Xu S, Zhao J, Yang J, Yao K, Yuan M and Li W (2022) Discovery of a Series of 1,2,3-Triazole-Containing Erlotinib Derivatives With Potent Anti-Tumor Activities Against Non-Small Cell Lung Cancer. Front. Chem. 9:789030. doi: 10.3389/fchem.2021.789030
Received: 04 October 2021; Accepted: 10 December 2021;
Published: 07 January 2022.
Edited by:
Zhenjun Yang, Peking University, ChinaCopyright © 2022 Sun, Mao, Deng, Xu, Zhao, Yang, Yao, Yuan and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kaitai Yao, a3R5YW8xOTMxQDE2My5jb20=; Miaomiao Yuan, eXVhbm1tMjAxOUAxNjMuY29t; Wei Li, bGl3ZWlncUAxNjMuY29t