 Gajendra Singh Thakur1
Gajendra Singh Thakur1 Ajay Kumar Gupta1
Ajay Kumar Gupta1 Dipti Pal1
Dipti Pal1 Yogesh Vaishnav1
Yogesh Vaishnav1 Neeraj Kumar2
Neeraj Kumar2 Sivakumar Annadurai3
Sivakumar Annadurai3 Sanmati Kumar Jain1*†
Sanmati Kumar Jain1*†- 1Drug Discovery and Research Laboratory, Department of Pharmacy, Guru Ghasidas Vishwavidyalaya (A Central University), Bilaspur, Chhattisgarh, India
- 2Department of Pharmaceutical Chemistry, Bhupal Nobles’ College of Pharmacy, Udaipur, Rajasthan, India
- 3Department of Pharmacognosy, College of Pharmacy, King Khalid University, Abha, Saudi Arabia
Introduction: One of the foremost contributors to mortality worldwide is cancer. Chemotherapy remains the principal strategy for cancer treatment. A significant factor leading to the failure of cancer chemotherapy is the phenomenon of multidrug resistance (MDR) in cancer cells. The primary instigator of MDR is the over expression of P-glycoprotein (P-gp), a protein that imparts resistance and facilitates the ATP-dependent efflux of various anticancer agents. Numerous efforts have been made to inhibit P-gp function with the aim of restoring the effectiveness of chemotherapy due to its broad specificity. The main objective has been to create compounds that either serve as direct P-gp inhibitors or interact with cancer therapies to modulate transport. Despite substantial in vitro achievements, there are currently no approved drugs available that can effectively “block” P-gp mediated resistance. Cabozantinib (CBZ), a multi-kinase inhibitor, is utilized in the treatment of various carcinomas. CBZ has been shown to inhibit P-gp efflux activity, thereby reversing P-gp mediated MDR. Consequently, P-gp has emerged as a critical target for research in anti-cancer therapies.
Methods: The purpose of this study was to computationally identify new andsafer analogues of CBZ using bioisosteric approach, focusing on improved pharmacokinetic properties andreduced toxicity. The physicochemical, medicinal, and ADMET profiles of generated analogues were computed using the ADMETLab 3.0 server. We also predicted the drug likeness (DL) and drug score (DS) of analogues. The molecular docking studies of screened analogues against the protein (PDB ID: 3G5U) were conducted using AutoDock Vina flowing by BIOVIA Discovery Studio for visualizing interactions.Molecular dynamics (MD) simulation of docked ligands was done using Schrödinger suite.
Results and Discussion: The docking scores for the ligands CBZ01, CBZ06, CBZ11, CBZ13, CBZ25, CBZ34, and CBZ38 ranged from −8.0 to −6.4 kcal/mol against the protein (PDB ID: 3G5U). A molecular dynamics (MD) simulation of CBZ01, CBZ13, and CBZ38 was conducted using the Schrödinger suite, revealing that these complexesmaintained stability throughout the 100 ns simulation.
Conclusion: An integrated computational approach combining bioisosteric approach, molecular docking, drug likeness calculations, and MD simulations highlights the promise of ligands CBZ01 and CBZ13 as candidates for the development of potential anticancer agents for the treatment of various cancers.
1 Introduction
Cancer continues to be a primary cause of worldwide mortality. The International Agency for Research on Cancer (IARC), a division of the World Health Organization (WHO), which is dedicated to cancer studies, has published its recent estimates regarding the global cancer burden. An estimated 20 million new cancer cases and over 9.7 million cancer-related deaths occurred in 2022. Around 53.5 million people survived for at least 5 years after their diagnosis. Lung cancer has emerged as the most prevalent cancer globally, with 2.5 million new cases, representing 12.4% of all new diagnoses. Following lung cancer, female breast cancer ranked second in prevalence at 11.6%, succeeded by colorectal cancer (9.6%), prostate cancer (7.3%), and stomach cancer (4.9%). Forecasts show that the number of new cancer cases could escalate to 35 million by 2050, which marks an increase of 77% compared to the figures recorded in 2022. This significant rise in the global cancer burden is largely attributed to an aging population, overall population growth, and shifts in exposure to various risk factors, many of which are linked to socioeconomic development. Key contributors to the rising incidence of cancer include tobacco use, alcohol consumption, and obesity, while air pollution remains a significant environmental risk factor (Ferlay et al., 2024; Sung et al., 2021).
The treatment of cancer is recognized as a particularly challenging endeavour, encompassing methods such as chemotherapy, radiotherapy, and surgical interventions (Cao et al., 2024; Bray et al., 2024; Liu B. et al., 2024). A significant obstacle in contemporary cancer research is the emergence of drug resistance and the recurrence of cancer, which often undermine the effectiveness of even the most potent anti-cancer therapies (Anand et al., 2023; Khan et al., 2024). In the context of chemotherapy, one of the primary reasons for treatment failure is the phenomenon of multidrug resistance (MDR) observed in cancer cells. The over expression of P-glycoprotein (P-gp), an ATP-binding cassette transporter, is a key factor contributing to MDR, as it enhances the efflux of various anti-cancer agents from the cells. P-gp was the first protein identified to be linked to drug resistance (Bukowski et al., 2020; Lin et al., 2016a; Lin et al., 2016b; Goebel et al., 2021; Zhang et al., 2022; Garrigues et al., 2002). P-gp, identified in 1976, is a membrane glycoprotein with an estimated molecular weight of 170 kDa, found in drug-resistant Chinese hamster ovary cells. It consists of two transmembrane domains (TMDs) and two nucleotide-binding domains (NBDs) (Kim and Chen, 2018; Tian et al., 2023; Sun et al., 2023). The NBDs, located in the cytoplasm, facilitate the movement of substrates by transferring energy across cellular membranes, while the TMDs, composed of six transmembrane helices, provide substrate selectivity (Vasiliou et al., 2009; Johnson and Chen, 2018).
P-gp is recognized as a significant MDR transporter, particularly in relation to its role in conferring resistance to cancer chemotherapy. Its expression has been found to be elevated in various tumor types, such as osteosarcoma, kidney cancer, liver cancer, breast cancer, gastric cancer, lung cancer, and colorectal cancer, which contributes to the development of chemotherapy resistance (Karthika et al., 2022; Heming et al., 2022; Sharom, 2011). Cabozantinib (CBZ), a small-molecule, multitargeted tyrosine kinase inhibitor, is utilized in the treatment of several cancers, including metastatic medullary thyroid cancer, RCC, and HCC. Patients undergoing CBZ therapy may experience a range of toxicities, including hepatotoxicity and renal impairment, which can be severe or potentially life-threatening (Srigadha et al., 2023; Markowitz and Fancher, 2018; Choueiri et al., 2015). According to LiverTox, the hepatotoxicity likelihood score of cabozantinib is E*, unproven but probably rare cause of clinically apparent liver damage (National Center for Biotechnology Information, 2025). According to DrugBank, cabozantinib has extensive plasma protein binding (≥99.7%) (National Center for Biotechnology Information, 2025). The toxicities of cabozantinib may affect the patient’s quality of life. The most common adverse events (AEs) are diarrhea, fatigue, hypertension, hand-foot syndrome, weight loss, nausea, stomatitis, gastrointestinal perforation, hypothyroidism and myelotoxicity (Schmidinger and Danesi, 2018). Adverse reactions were recorded from clinic reports and the most common were hypertension, mucositis/hand-foot skin reaction (HFSR), or gastrointestinal toxicity (Martini et al., 2022). Cabozantinib has a higher risk of hepatotoxicity (Wang K. et al., 2024). Krens et al. (2022) reported that cabozantinib is registered at a fixed dose of 60 mg. However, 46%–62% of patients in pivotal studies required dose reduction due to toxicity. Consequently, it is crucial to modify the structure of the CBZ molecule to develop analogues that are less toxic and safer. Adverse effects associated with CBZ treatment include diarrhea, hypertension, hand-foot syndrome, weight loss, reduced appetite, stomatitis, and nausea (Schwartz et al., 2020; Rimassa et al., 2019; Zuo et al., 2015). Numerous reports have documented hepatotoxicity and a variety of dose-dependent side effects linked to CBZ (Barnhill et al., 2020; Andrade et al., 2019; Chiruvella et al., 2020). Due to these toxicities, it is imperative to modify the structure of the CBZ molecule to create safer and less toxic analogues.
The development of a lead chemical into a pharmaceutical agent presents significant challenges and often incurs high costs. Most candidates fail primarily due to pharmacokinetic and metabolic complications rather than a lack of efficacy. Even when a lead molecule exhibits the desired pharmacological effect, it may still present adverse side effects, characteristics that hinder its bioavailability, or chemical structures that impede its metabolism and elimination from the body. To address these issues, researchers employ the strategy of bioisosterism, which involves the selective modification of lead compounds to create safer and more effective medications. Bioisosterism is often perceived as a qualitative and intuitive concept. The common physicochemical properties of a set of bioisosteres are believed to contribute to their capacity to elicit similar biological responses. By leveraging an understanding of pharmacophores and physicochemical features, researchers are increasingly substituting bioisosteres for functional groups, thereby enhancing the potential for the development of innovative therapeutic agents. The foundational work of Langmuir in 1919 laid the groundwork for the bioisosterism approach to modifying lead compounds. Through the bioisosteric method, chemists can adjust various characteristics of the lead compound, including its size, shape, electrical distribution, polarizability, dipole moment, polarity, lipophilicity, and pKa, while ensuring effective binding to the target. Consequently, the strategic application of the bioisosteric method allows for the modification of lead compounds to yield more favorable therapeutic drugs with enhanced potency, selectivity, improved physical and metabolic properties, and minimized side effects (Giordano et al., 2022; Das et al., 2022; Jayashree et al., 2022).
Structure-based drug design represents a highly effective and powerful approach within the broader context of drug discovery. The drug development process, which encompasses combinatorial chemistry, various screening methodologies, and the assessment of parameters such as absorption, distribution, metabolism, excretion, and toxicity (ADMET), can be significantly accelerated through the use of computational resources (Anderson, 2003; Jiang et al., 2018; Nie et al., 2020; Ejalonibu et al., 2021; Gao et al., 2023; Li et al., 2024). Recently, molecular docking has become an essential element of in silico drug discovery. This technique focuses on predicting the atomic-level interactions between proteins and small molecules. The accessibility of free software for conducting docking simulations of protein-ligand systems has facilitated a growing number of studies utilizing this approach, with tools like AutoDock, ArgusLab, and GOLD providing docking estimates for a variety of receptor-ligand interactions. The docking interactions suggest the most favorable docked conformers based on the overall energy of the system. Additionally, it assists in identifying the specific amino acids of the protein that interact with the test molecules, thereby helping to evaluate the affinity of the tested molecule for the target protein (Bhagat et al., 2021; Du et al., 2016; Ferreira et al., 2015; Duan et al., 2024; Kang et al., 2018). To elucidate the molecular basis of protein function, molecular dynamics (MD) simulation is the predominant computational method employed to investigate the structure and dynamic behavior of proteins. Based on the docking scores and interactions, we selected three complexes of CBZ analogues for MD simulations (Salo-Ahen et al., 2020; Gao et al., 2019; Naresh and Guruprasad, 2020; Ajmal et al., 2016). The primary objective of this study is to modify various groups within the CBZ molecule, specifically phenyl, amide cyclopropyl, and cyclopropyl groups (Figure 1). The goal is to create CBZ analogues that are both safer and more effective. Additionally, we conducted ADMET predictions, molecular docking analyses, and MD simulations on the chosen CBZ analogues. The overall workflow of the present study is shown in Figure 2.
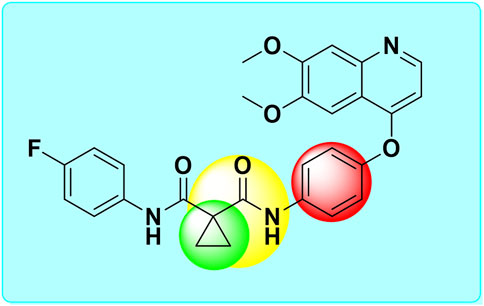
Figure 1. Structure of cabozantinib and its bioisosterically modified groups [Cyclopropyl (green circle), amide cyclopropyl (yellow circle), and phenyl (red circle)].

Figure 2. The overall work flow of the present study.
2 Materials and methods
2.1 Designing of CBZ Bioisosteres
The smile notation for CBZ analogues was acquired from DrugBank, a prominent chemical information platform. The bioisosteres of CBZ were created utilizing the MolOpt software, an online tool that produces bioisosteres through data mining, similarity assessments, and AI generative models (Shan and Ji, 2020; Subbaiah and Meanwell, 2021).
2.2 Pharmacokinetic and toxicological (ADMET) Profile Predictions
The pharmacokinetic and toxicological profile were forecasted utilizing the ADMETLab 3.0 online software. This comprises 119 quantitative and qualitative predictable endpoints, which effectively and thoroughly assess ADMET characteristics for novel ligands that exhibit ADMET properties similar to those observed in mammals (Dulsat et al., 2023; Gupta et al., 2024; Lou et al., 2023; Liu M. et al., 2024).
2.3 Drug likeness (DL) and drug score (DS) prediction
The primary variables leading to the failure of drug candidates in clinical trials are often intolerable toxicity levels or unfavourable pharmacokinetic characteristics. Therefore, it is crucial to conduct evaluations of DL and DS in the early stages of the drug development process. The calculations for drug score and drug likeness were performed utilizing the Osiris property explorer (Sun et al., 2022).
2.4 Molecular docking studies
Molecular docking is essential in drug discovery and structural molecular biology for predicting the main binding mode(s) of a ligand with a protein of known 3D structure. In this context, commonly used docking-related terminology (such as Apo protein, positive control, native ligand, and co-crystal inhibitors) is employed to elucidate the core principles of molecular docking, which encompass binding affinity, binding orientation, and ligand interactions. The docking analysis was conducted using AutoDock Vina (ADV) as the primary tool, and the interactions were evaluated with Discovery studio software (De Ruyck et al., 2016; Trott and Olson, 2010; Wang Z. et al., 2024; Ikwu et al., 2020). P-gp is integral to the process of cellular detoxification, as it facilitates the removal of a wide range of chemically diverse toxins. However, it is also linked to the phenomenon of multidrug resistance (MDR) in cancer treatments. Among the various transporters associated with MDR, P-gp is the most significant member of the ATP-binding cassette (ABC). P-gp demonstrates remarkable poly-specificity, enabling it to recognize a broad spectrum of compounds with molecular weights ranging from 330 Da to 4,000 Da. The X-ray crystallographic analysis of apo-P-gp with a resolution of 3.8 Å shows an internal cavity that is approximately 6,000 Å3, with a separation of 30 Å between the two nucleotide binding domains (NBD). In addition, two additional P-gp structures, which complex with cyclic peptide inhibitors, illustrate different drug binding sites within the inner cavity that have a stereoselectivity influenced by hydrophobic and aromatic interactions. Therefore, the P-gp protein, which has the ability to absorb a variety of substrates, is a defining feature of its function and makes a structural understanding of the poly-specific drug binding necessary for the rational development of anticancer agents and MDR inhibitors (PDB ID: 3G5U), chosen for the present study.
2.4.1 Protein preparation
The crystal structure of the P-gp (PDB ID: 3G5U) was acquired from the Protein Data Bank (Aller et al., 2009). To prepare the protein for docking studies, we initially introduced hydrogen atoms, applied Kollman charges, and eliminated water molecules. Subsequently, we saved the modified structure in PDBQT format after addressing the missing atoms (Sastry et al., 2013).
2.4.2 Ligand preparation
The ligands 2D chemical structures were created utilizing ChemDraw. Subsequently, the 2D representations of CBZ and their corresponding analogues were converted into 3D structures through Chem3D software. The newly developed CBZ bioisosteres underwent energy minimization in Chem3D and were subsequently saved in SDF format. Using OpenBabel, the ligands were transformed into MOL2 format (Zielesny, 2005). These ligands were then incorporated into ADV tool and saved in pdbqt format to facilitate the docking process. Additionally, protein was also dragged into the ADV tool, and a grid box was prepared that defines the boundary for the docking process.
2.4.3 Protein–ligand interactions using ADV
Utilizing the ADV software, we conducted docking study involving the ligands and the protein. The entire active site of the protein was encompassed within a grid box with the size of dimensions at X = 84, Y = 84 and Z = 84. The center of the grid was positioned at X = 28, Y = 86, and Z = 40. The default configurations for other docking parameters, such as ADV settings, crossover rates, and gene mutation rates, were maintained. The interaction between ligand and amino acid residue of protein were studied using Discovery Studio to produce 2D and 3D pose of interactions. The binding site of the target protein includes LEU64, PHE724, GLN942, MET945, PHE974 and VAL978, which are used to determine potential binding sites of the target protein with respect to the designed ligands (Xiang et al., 2015).
2.5 Molecular dynamics (MD) simulation
The MD simulation aims to explore the dynamic behavior and stability of protein-ligand complexes. We selected three complexes based on their interaction profiles and docking scores. The simulations were executed on an Acer workstation operating with Ubuntu 22.04. The Desmond program, part of the Schrödinger suite, was utilized to perform the MD simulations and to assess the docking of molecules, evaluating the efficacy of the predicted ligands (Elekofehinti et al., 2021). The protein-ligand complexes were constructed using the ‘System Builder’ tool. Following a reduction in volume, we opted for the SPC water model configured in an orthorhombic arrangement. The periodic boundary conditions for the X, Y, and Z-axes of the protein-ligand complex were established at 10 × 10 × 10 Å. Additionally, the crystal structure of the P-gp (PDB ID: 3G5U) served as a reference, illustrating its ability to accommodate 50.447 mM sodium and 53.855 mM chloride ions. Ions and salts within a 20 Å radius were omitted from the neutralization simulation. Before commencing the MD simulations, we applied the OPLS 2005 force field to minimize the energy of the complex, facilitating its transition to an equilibrium state. The OPLS 2005 force field, known as Optimized Potentials for Liquid Simulations, is a well-known force field in molecular mechanics designed to effectively simulate molecular interactions, particularly in the context of small organic molecules and biomolecular systems. This version represents an advance over the original OPLS force field and provides greater precision for simulations (González, 2011; Banks et al., 2005). To refine the complexes, we employed a minimization approach based on the steepest descent method. The complexes were subsequently heated to 300 K, achieving equilibrium after 1000 steps, with a time step of 100 ns. The final production run for the complexes was carried out over a period of 100 ns.
2.5.1 Binding free energy calculations
The binding affinity of ligand-protein complexes, represented by binding free energy, was assessed through the binding energies protocol available in the Desmond program. The complexes CBZ01-3G5U and CBZ13-3G5U, which were produced following molecular dynamics (MD) simulations, underwent analysis for binding energy estimation. Both Poisson-Boltzmann and generalized Born models, in conjunction with surface area continuum solvation methods (MM-PBSA and MM-GBSA), were utilized for solvation analysis. Furthermore, binding energy calculations were performed without considering solvation effects. The free energy derived from the MM-PBSA method was computed using the gmx_MMPBSA tool, which necessitates the input of “.top” and “.trr” files. To generate these files, the Desmond Composite Model System files (.cms) were initially converted using the InterMol software (https://github.com/shirtsgroup/InterMol), leading to the creation of “.gro” and “.top” files. Subsequently, the Desmond trajectory was imported into VMD and saved in the “.trr” format. After preparing all required input files, MM-GBSA calculations were carried out using the gmx_MMPBSA tool. The MM-GBSA approach integrates molecular dynamics simulations with thermodynamic principles, enabling the calculation of the total binding free energy between a ligand and a protein, as illustrated in Equation 1 (Wang et al., 2019; Bouricha and Hakmi, 2024; Matore et al., 2023).
3 Results and discussion
3.1 Bioisosteres of cabozantinib
Bioisosterism represents a strategy in medicinal chemistry that employs a lead compound as a primary method for molecular modification, aimed at the rational development of new pharmaceuticals (Karmacharya et al., 2021). We have applied the bioisosterism to improve ADMET profile and reduce undesirable toxic effects. MolOpt produced 592 analogues of CBZ, targeting various groups including phenyl, amide cyclopropyl, and cyclopropyl within the CBZ drug framework. The compounds that were screened are detailed in Supplementary Table S1.
3.2 Physicochemical properties prediction
The aim of molecular property prediction is to ascertain the physicochemical, bioactive, toxicological, and other characteristics of a target compound based on its molecular structure. Supplementary Table S1 presents the predicted molecular properties for CBZ bioisosteres. All analogues complies with Lipinski’s rule of five, suggesting that these drug candidates possess favorable absorption and bioavailability. Furthermore, all analogues demonstrated a commendable topological polar surface area (TPSA) score, highlighting their capability to permeate cell membranes and reach target sites in the body.
3.3 Medicinal properties prediction
In the initial stages of drug development, the selection of molecules based on their drug-likeness is of paramount importance. This concept encompasses eight characteristics that are indicative of drug-related properties. The quantitative estimation of drug-likeness (QED) scores for the designed analogues, including CBZ01-02, CBZ05, and CBZ23-CBZ24, fall below the desirable QED score threshold (>0.67) but exceed the score of a standard drug (0.31). In the realm of drug design, predicting synthetic accessibility is a vital task that entails evaluating the ease of laboratory synthesis for a specific molecule. The synthetic accessibility scores for all designed analogues were determined to be within an acceptable range (<6). A research team from the Medicinal Chemistry Department of in silico Medicine has introduced the original descriptor MCE-18, which outlines the essential features of “next-generation” molecules and examines the evolution of medicinal chemistry over time (Ivanenkov et al., 2019). We utilized MCE-2018 to evaluate the efficacy of newly designed molecules such as CBZ03-04, CBZ05, CBZ12-14, CBZ16, CBZ18, CBZ23-25, CBZ27-29, CBZ31, CBZ33, CBZ37, CBZ39, CBZ48, CBZ49-50, CBZ57, and CBZ63, which yielded scores exceeding 63. Consequently, these analogues require visual inspection to determine their drug-likeness and target profiles. With the exception of CBZ07, CBZ08, CBZ26, CBZ45, CBZ47, CBZ64-66 and CBZ80, all designed analogues met the acceptance criteria of Lipinski’s rule of five (Karami et al., 2022). Additionally, Pfizer’s rule was satisfied by all analogues, indicating favorable physicochemical properties with potential for cellular permeability. The GT rule found accepted for anlogues such as CBZ05-06, CBZ12, CBZ14-16, CBZ25, CBZ27-29, CBZ31, CBZ35-36, CBZ52-54, and CBZ59. The medicinal properties of CBZ analogues are presented in Table 1.
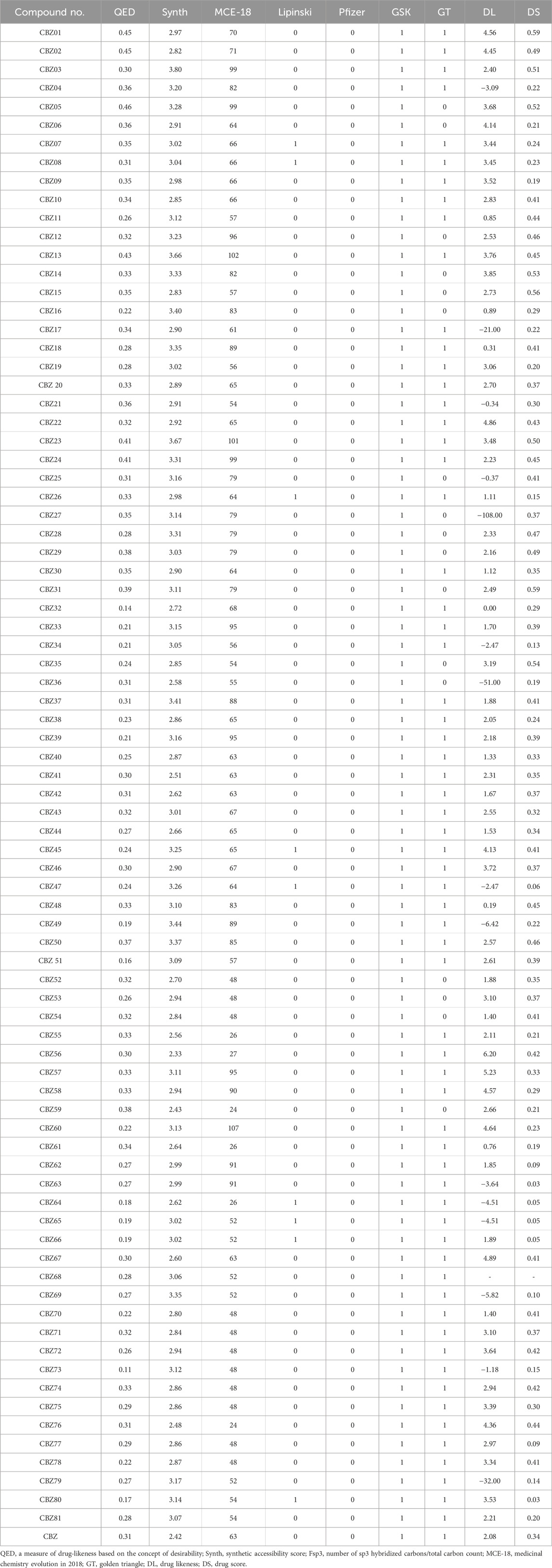
Table 1. Medicinal, drug likeness (DL) and drug score (DS) properties of CBZ analogues.
3.4 Prediction of DS and DL score
DL and DS are qualitative metrics utilized in drug design to assess the “drug-like” characteristics of a molecule, particularly in relation to factors such as bioavailability. These metrics are extensively integrated into the early stages of lead and drug discovery (Bickerton et al., 2012). During the initial phases of drug development, it is essential to screen compounds based on their drug-likeness and DS. Accurately predicting a compound’s drug-likeness is vital, as it offers valuable insights that can enhance the likelihood of transforming lead compounds into viable drugs, necessitating various drug-like attributes (Lagu et al., 2022). The DL score of compounds can indicate their potential effectiveness and safety. The DS serves as a comprehensive metric that consolidates factors such as toxicity concerns, cLogP, logs, molecular weight, and drug-likeness into a singular value. Among the analogues, CBZ56-57 and CBZ69 exhibit higher DL scores, while CBZ01-02, CBZ22, CBZ45, CBZ67, and CBZ76 demonstrate scores ranging from 4 to 4.5, surpassing the standard drug CBZ, which has a score of 2.08. The DL and DS scores of analogues are tabulated in Table 1.
3.5 Pharmacokinetic profile
Pharmacokinetics plays a crucial role in the drug discovery process by guiding the optimization of a compound’s absorption, distribution, metabolism, and excretion (ADME) properties. The primary objective is to ensure that a lead compound achieves a concentration-time profile within the body that supports the desired efficacy and safety outcomes. By incorporating ADMET data into the drug design framework, researchers can improve a compound’s solubility, permeability, and stability, ultimately developing clinical drug candidates capable of maintaining therapeutic concentrations for the required duration while minimizing potential risks. The predictable Caco-2 permeability scores offer valuable insights into the ability of substances to traverse intestinal cell membranes, which is a critical aspect of oral drug absorption. The Caco-2 scores for compounds CBZ01, CBZ04, CBZ06-15, CBZ17, CBZ19-21, CBZ24-31, CBZ33, CBZ34, CBZ37-47, CBZ49-51, CBZ55, CBZ64-66, CBZ77, and CBZ80 ≤ −5.15 log cm/s, indicating effective transport across intestinal membranes and favorable permeability. Conversely, the Caco-2 scores for compounds CBZ02, CBZ03, CBZ05, CBZ16, CBZ18, CBZ22, CBZ23, CBZ32, CBZ35, CBZ36, CBZ48, CBZ52-54, CBZ56-63, CBZ67-76, CBZ78, and CBZ79 fall ≥ −5.15, suggesting potentially poor permeability. Madin-Darby Canine Kidney (MDCK) cells are acknowledged as a reliable in vitro model for evaluating permeability, providing critical insights into the absorption efficiency of chemical substances within the body. The MDCK permeability scores for all evaluated compounds exceed 2 × 10⁻⁶ cm/s, indicating excellent MDCK permeability. This finding suggests that these compounds are likely to exhibit favorable permeability characteristics, making them promising candidates for effective systemic absorption.
The results show that all compounds reach a praising human intestinal absorption score (HIA) between 0 and 0.3, which indicates the significant potential for effective absorption in the human gastrointestinal tract. This favorable absorption profile implies that these compounds are likely to have high oral bioavailability, which is essential for the maintenance of the therapeutic drug level in systemic circulation. In addition, none of the compounds are classified as poorly absorbed (HIA+ with a value of more than 0.7), which reduces the likelihood of bioavailability problems associated with inadequate intestinal absorption. In addition, the results show that all compounds from 0 to 1 have values, which reflects the different probabilities of penetrating the blood-brain barrier (BBB). The findings further indicate that all compounds demonstrate the varying probabilities of traversing the blood-brain barrier (BBB). Plasma protein binding (PPB) is crucial in influencing the absorption, distribution, and pharmacodynamics of pharmaceuticals. The degree to which drugs associate with plasma proteins affects the concentration of the unbound drug that is available for therapeutic action. Compounds such as CBZ02, CBZ03, CBZ05, CBZ08, CBZ14-16, CBZ18, CBZ23, CBZ26, CBZ27, CBZ28, CBZ35, CBZ36, CBZ48, and CBZ51 demonstrate PPB values of 90% or less, indicating strong binding properties that promote an optimal equilibrium between the concentration of free drug and its therapeutic efficacy. In addition, compounds like CBZ34, CBZ41, CBZ52, CBZ53, CBZ55-66, CBZ68, CBZ69, CBZ71-73, CBZ75, and CBZ77-81 exhibit PPB values exceeding 90%, similar to CBZ, which has a PPB of 97.74%. The volume of distribution at steady state (VDss) serves as a pharmacokinetic indicator of how extensively a drug is distributed in the body relative to its plasma concentration. The expected VDss is measured in L/kg, with an optimal range of 0.04–20 L/kg indicated. All compounds show a projected VDSS score that falls in the range from 0.04 to 20 L/kg, which indicates superior distribution properties, with the exception of CBZ04, CBZ26, CBZ40-42, CBZ47, CBZ48 and CBZ51.
The unbound fraction (Fu) in plasma is a significant pharmacokinetic parameter that influences a drug’s efficacy and distribution. It represents the proportion of a drug that remains unbound to serum proteins, enabling it to traverse cellular membranes and exert its pharmacological effects. The Fu values for compounds CBZ01-03, CBZ05-10, CBZ13-18, CBZ20, CBZ23-28, CBZ31-33, CBZ35-37, CBZ39, CBZ43, CBZ44, CBZ46-48, CBZ50, and CBZ51 are greater than 5%. These compounds exhibit favorable unbound drug fractions, suggesting their potential to effectively penetrate cellular membranes and reach their designated targets, thereby indicating their promise for further drug development. In contrast, other compounds, including CBZ, present Fu values below 5%. The results show that all compounds serve as substrates for CYP3A4, a key and prevailing enzyme within the CYP450 family, which is essential for the metabolism of phase I. CYP3A4 is crucial for the oxidative metabolism of numerous medicines and endogenous compounds that occur mainly in the liver and intestine.
Plasma clearance (CL) is an indicator of the body’s ability to eliminate a drug from the plasma. This parameter directly affects the overall drug exposure and is crucial for determining the appropriate dosage required to maintain a stable plasma concentration. The compounds CBZ01-13, CBZ15-23, CBZ26, CBZ30, CBZ32-34, CBZ36-49, CBZ51-53, CBZ55, CBZ57, CBZ59-61, CBZ63, CBZ69, CBZ71-74, and CBZ76-78 exhibit CL scores ranging from 0 to 5 mL/min/kg, indicating excellent clearance profiles. Their elimination rates are effectively regulated, ensuring consistent drug exposure and optimal dosing. The half-life (T1/2) serves as a measure of the interplay between clearance and volume of distribution. A precise evaluation of these two parameters provides comprehensive insight into the pharmacokinetics of drugs within the body. The T1/2 values for the compounds CBZ01-11, CBZ13, CBZ17, CBZ18, CBZ20, CBZ22, CBZ23, CBZ26, CBZ31-33, CBZ37-40, CBZ42, CBZ44-49, and CBZ51 exceed 0.903, indicating that these compounds are suitable for dosing that maintains therapeutic drug levels. In contrast, the remaining compounds exhibit T1/2 values below 0.903, similar to that of CBZ, which implies the need for multiple dosing strategies. The pharmacokinetic (ADME) profile of CBZ analogues is mentioned in Table 2.
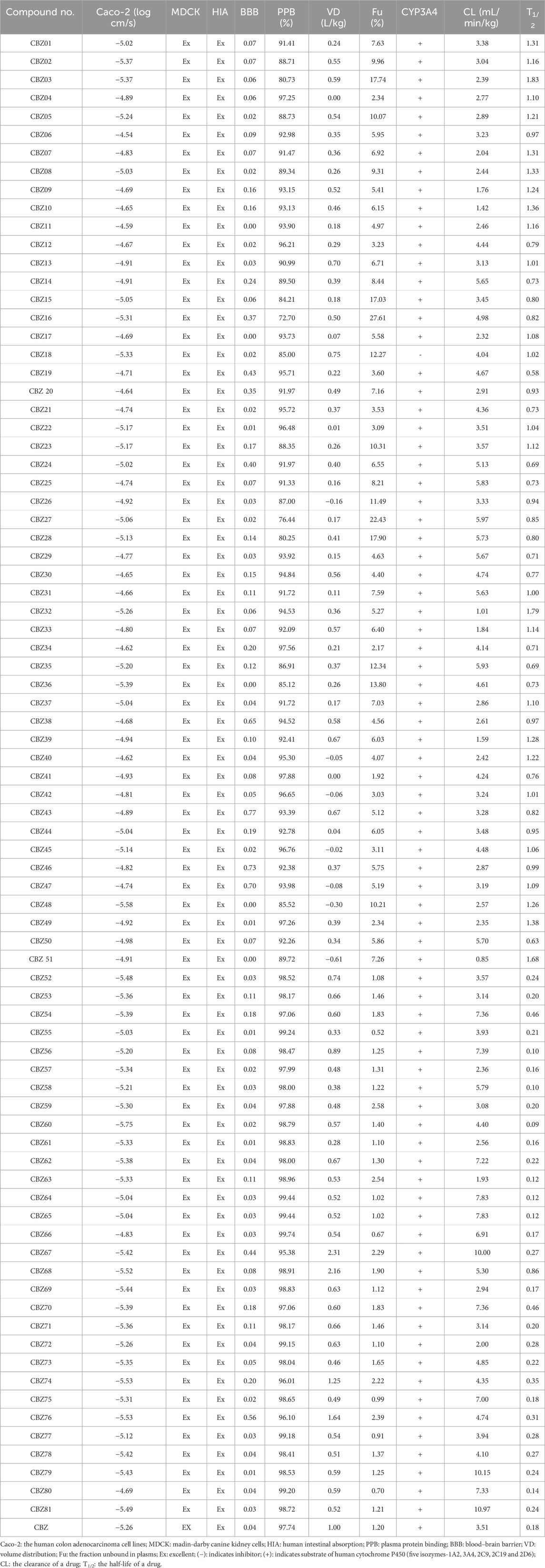
Table 2. ADME properties of CBZ analogues.
3.6 Prediction of toxicity characteristics
In the initial stages of drug development, it is essential to precisely predict the ADMET properties to identify molecules that exhibit optimal pharmacokinetics while minimizing toxicity. The results indicated that all analogues exhibited lower human hepatotoxicity (H-HT) and drug-induced liver injury (DILI), compared to the reference drug (CBZ), suggesting a reduced risk of toxicity. The hERG (cardiotoxicity) score of all compounds, such as CBZ07, CBZ09, CBZ10, CBZ14, CBZ15, CBZ17, CBZ19, CBZ25-27, CBZ29, CBZ34, CBZ36, CBZ40, CBZ43, CBZ45-48, CBZ50, CBZ51, CBZ57, CBZ63-65, CBZ69 and CBZ72 show less than 0.3, indicating a lower risk of cardiotoxicity compared to the standard drug (0.604). Researchers have advocated for the use of mutagenicity data as a relatively quick and cost-effective method to assess long-term human health risks, including cancer in somatic cells and heritable mutations in germ cells. Given the strong correlation between mutagenicity and carcinogenicity, this assay is frequently employed in evaluating the mutagenic potential of compounds. The mutagenicity scores for certain analogues, including CBZ02, CBZ05, CBZ12-14, CBZ18, CBZ21, CBZ23-24, CBZ28, CBZ30, CBZ34, CBZ48, CBZ49, CBZ56-57, CBZ59-61, CBZ63, and CBZ79-CBZ81, fall within a safer range (from 0 to 0.3), indicating their suitability for non-mutagenic effects. Assessing acute toxicity in mammals, such as rats or mice, is a critical component of the safety evaluation for drug candidates, with toxicity testing conducted to identify potential adverse reactions. The ROA scores (ranging from 0 to 0.3) for certain analogues, including CBZ53, CBZ55-58, CBZ60, CBZ72, and CBZ75, also indicate a safer profile. Carcinogenicity represents one of the numerous toxicological endpoints associated with chemicals, which raises considerable concern due to its harmful effects on human health. As the landscape of chemical exposure and cancer epidemiology evolves, it is imperative that the assessment of carcinogenicity adapts accordingly. The carcinogenicity scores (ranging from 0 to 0.3) for certain analogues, including CBZ53, CBZ55, CBZ63, CBZ68, CBZ71, CBZ73, CBZ74, and CBZ78, indicate a safer profile, making them appropriate for minimizing carcinogenic risks. The NR-AR and NR-AR-LBD scores of analogues found within the range of 0–0.3. The toxicity parameter scores are presented in Table 3.
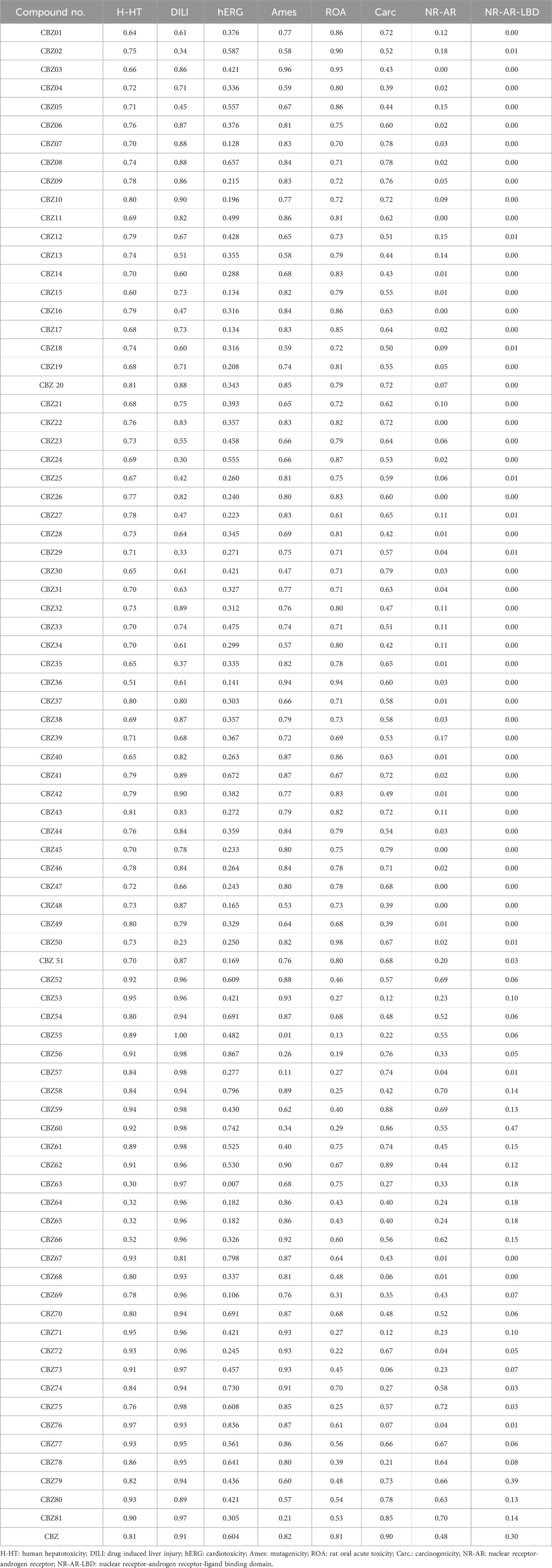
Table 3. Toxicity profile of CBZ analogues.
3.7 Molecular docking analysis
Before the ligand’s docking study begins, we ensured the validity of the docking procedure by downloading a co-crystalized ligand receptor complex from the protein database (PDB ID: 3G5U). The structure of the co-crystallized ligand and the associated protein was prepared and saved in pdbqt format. Then we carried out a redocking experiment with the native ligand, under which Autodock Vina was used to transform the original ligand into the active site of the protein. We then evaluated the docked pose against the crystallographic pose by calculating the Root Mean Square Deviation (RMSD). The results showed that the RMSD value of the optimal pose (0.48 Å) is within the acceptable threshold of 2 Å.
The interaction of designed ligands (Supplementary Table S1) and their docking score is being shown in Table 4, CBZ01 serves as a bioisostere of CBZ, characterized by the substitution of the phenyl group with a piperazine ring. The overall structure of CBZ01 closely resembles that of CBZ. Notably, the ligand CBZ01 exhibited the most favorable binding score of −8.0 kcal/mol. In the docked ADV complex, the residue GLN942 of the target protein forms hydrogen bonds with the oxygen of the methoxy group present in the quinoline ring of the ligand, with a distance of 1.9722 Å. The hydrogen bonds involving GLN942 are similar to those observed in CBZ. Additionally, residues GLN128 (2.5708 Å), PHE938 (2.9146 Å), and THR (2.9649 Å) demonstrated other hydrogen bond interactions with the carbonyl, phenyl, and fluorine atoms of the analogue. Furthermore, PHE934 established a halogenic bond with the fluorine atom of the fluorophenyl group of the ligand at a distance of 3.4667 Å. Figures 3A, 4A depict the 2D and 3D pose of the CBZ01, respectively.
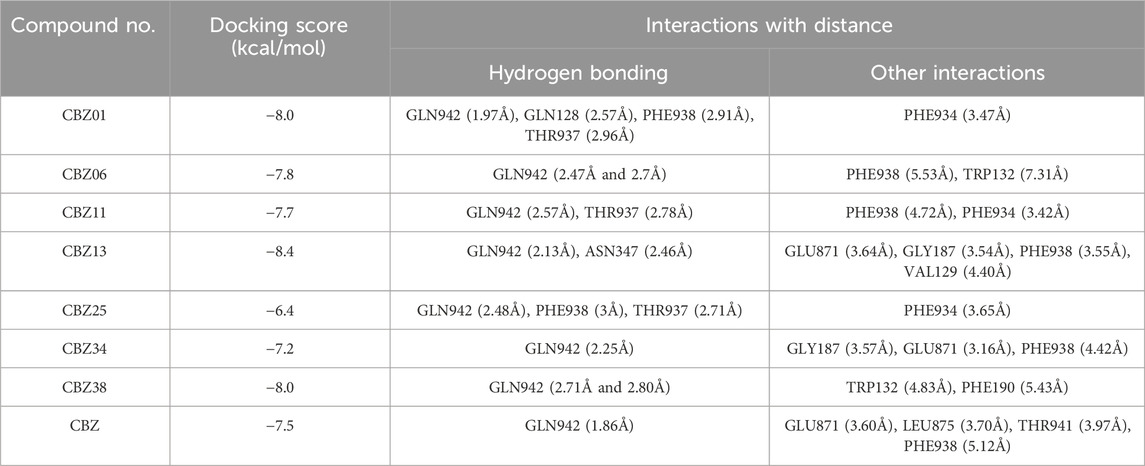
Table 4. Docking score and interaction of the selected CBZ analogues.
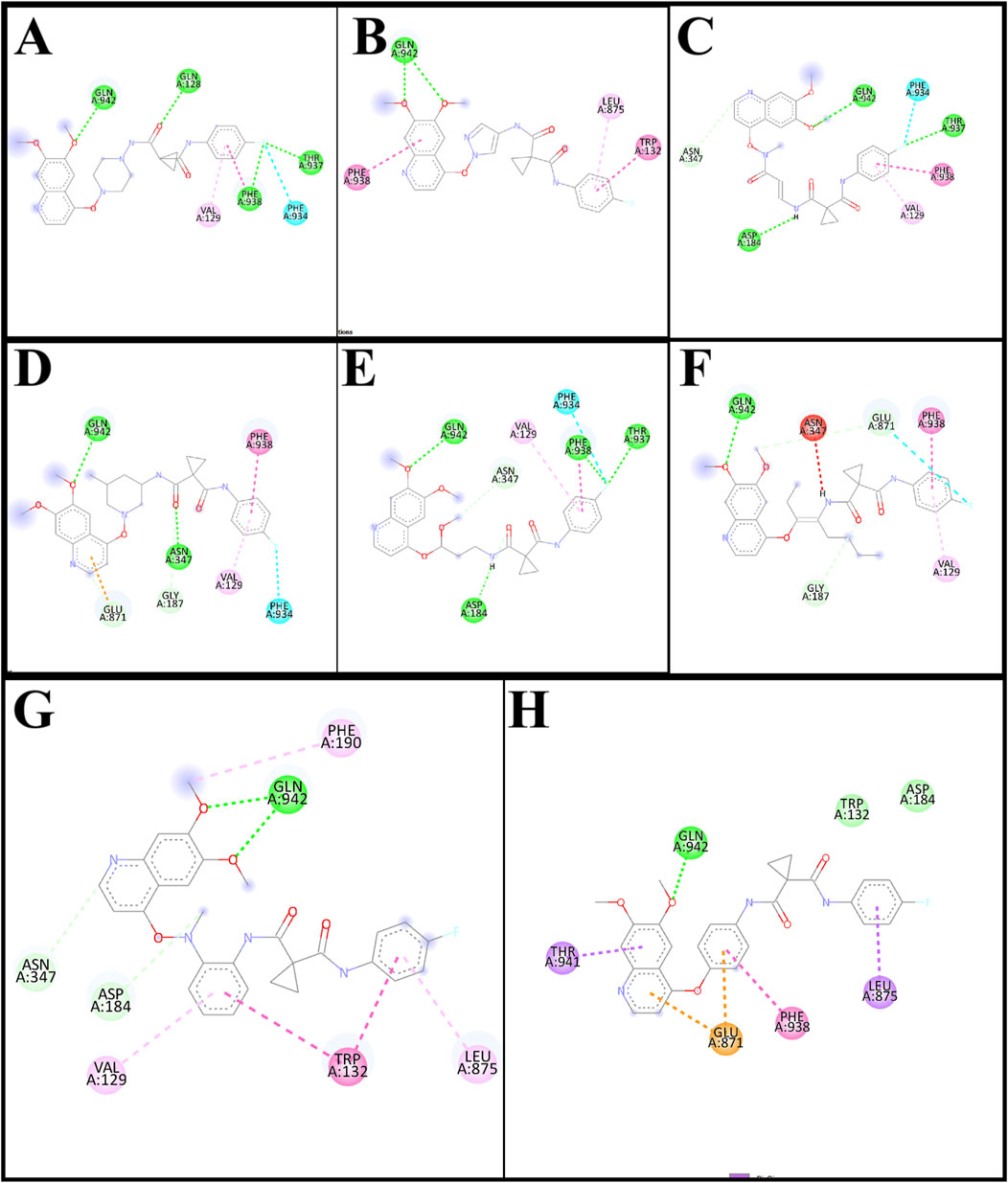
Figure 3. 2D pose of ligands CBZ01 (A), CBZ06 (B), CBZ11 (C), CBZ13 (D), CBZ25 (E), CBZ34 (F), CBZ38 (G) and Cabozantinib (H).
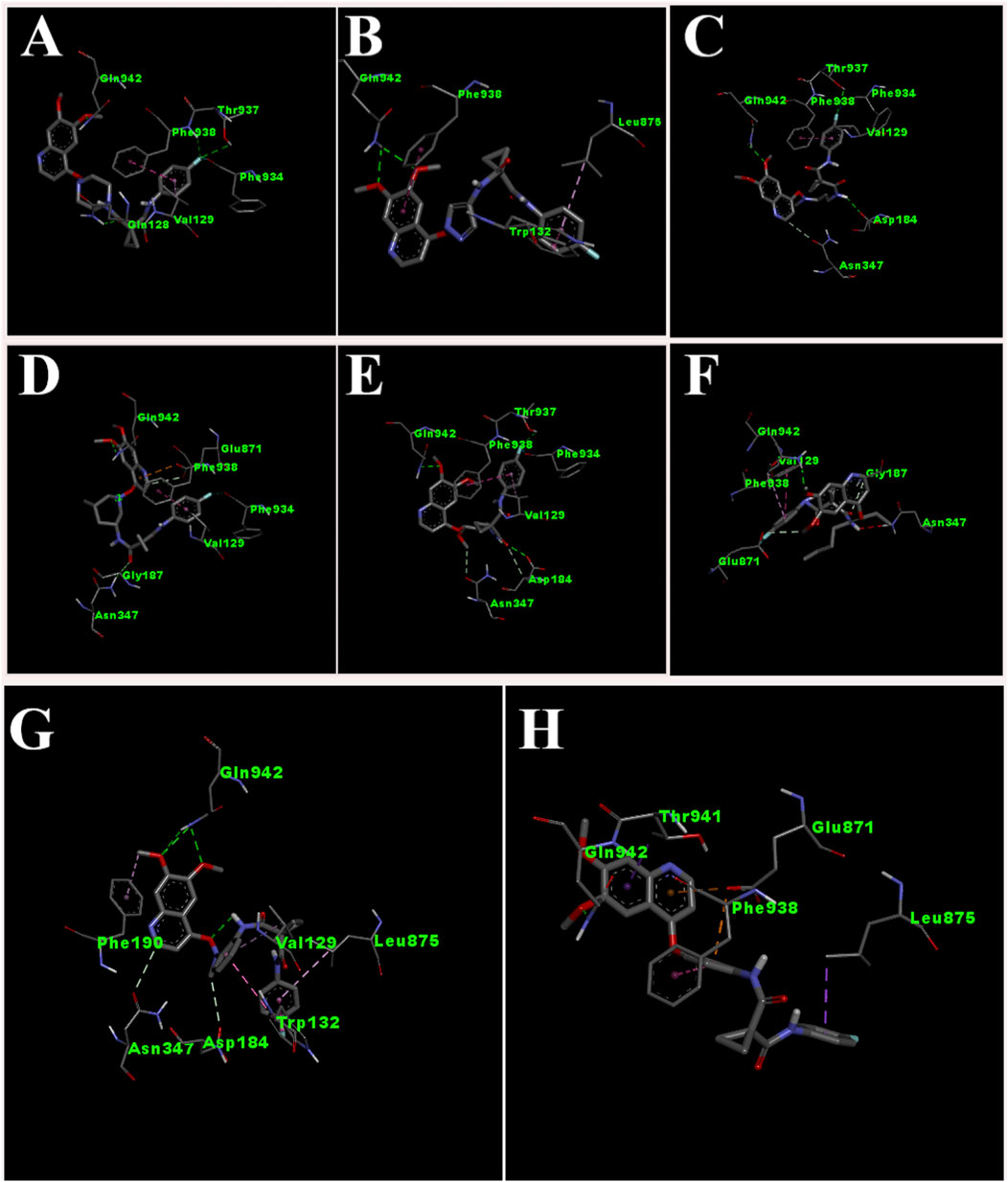
Figure 4. 3D pose of ligands CBZ01 (A), CBZ06 (B), CBZ11 (C), CBZ13 (D), CBZ25 (E), CBZ34 (F), CBZ38 (G) and Cabozantinib (H).
CBZ06 serves as a bioisostere of CBZ, characterized by the substitution of the phenyl group with a pyrazole ring. The overall structure of CBZ06 closely resembles that of CBZ. Notably, the ligand CBZ06 exhibited the most advantageous binding score of −7.8 kcal/mol. In the docked ADV complex, the residue GLN942 of the target protein establishes hydrogen bonds with the oxygen atoms of the methoxy groups present in the quinoline ring, at distances of 2.4658 Å and 2.7057 Å. The hydrogen bonding interactions involving GLN942 are similar to those observed with CBZ. Additionally, residues PHE938 (5.5257 Å) and TRP132 (7.031 Å) demonstrate π-π stacking interactions with the quinoline and phenyl rings of CBZ, respectively. Figures 3B, 4B depict the 2D and 3D interactions of the ligand CBZ06, respectively.
CBZ11 serves as a bioisostere of CBZ, characterized by the substitution of the phenyl ring with a methyl amino-3-oxoprop-1-en-1-yl group. The overall structure of CBZ11 closely resembles that of CBZ. Notably, the ligand CBZ11 exhibited the most favorable binding score of −7.7 kcal/mol. In the docked ADV complex, the residue GLN942 of the target protein forms hydrogen bonds with the oxygen atoms of the methoxy groups present in the quinoline ring of the ligands, maintaining a distance of 2.5741 Å. The hydrogen bonds formed with GLN942 which was similar observed in the standard (CBZ). Additionally, residue THR937 demonstrates another hydrogen bond interaction with the amino and fluorine atoms of the analogue at a distance of 2.7844 Å. Furthermore, PHE934 forms a halogenic bond with the fluorine atom of the fluorophenyl group of the ligand, measured at a distance of 3.4229 Å PHE938 also engages in a π-π stacked interaction with the phenyl ring of the fluorophenyl group of the ligand, at a distance of 4.7230 Å. Figures 3C, 4C depict the 2D and 3D pose of the CBZ11, respectively.
CBZ13 serves as a bioisostere of CBZ, characterized by the substitution of the phenyl ring with a methylpiperidin-3-yl group. The overall structure of CBZ13 closely resembles that of CBZ. Notably, the ligand CBZ13 exhibited the most advantageous binding score of −8.4 kcal/mol. In the docked ADV complex, the residue GLN942 of the target protein engages in hydrogen bonding with the oxygen atoms of the methoxy groups located on the quinoline ring of the ligands, maintaining a distance of 2.1263 Å. Additionally, residue ASN347, at a distance of 2.4628 Å, displays another hydrogen bond interaction with the carbonyl group of the analogues. Residues GLU871 (3.6375 Å) and GLY187 (3.5368 Å) exhibit C-H bonding interactions with the quinoline ring and the carbonyl group, respectively. Furthermore, PHE938 and VAL129 establish π-π stacking and π-alkyl interactions with the fluorophenyl group of the ligand, at distances of 3.5515 Å and 4.3974 Å, respectively. Figures 3D, 4D depict the 2D and 3D pose of the CBZ13, respectively.
CBZ25 serves as a bioisostere of CBZ, characterized by the substitution of the phenyl ring with a methoxy propyl group. The overall structure of CBZ25 closely resembles that of CBZ. Notably, the ligand CBZ25 exhibited the most favorable binding score of −6.4 kcal/mol. In the docked ADV complex, the residue GLN942 of the target protein forms hydrogen bonds with the oxygen atoms of the methoxy groups present in the quinoline ring of the ligand, with a distance of 2.4776 Å. Additionally, residue PHE938 (2.9955 Å) and THR937 (2.7086 Å) demonstrate other hydrogen bond interactions with the fluorine atom of the CBZ analogue. Furthermore, PHE934 establishes a halogen bond with the fluorine atom of the fluorophenyl group of the ligand at a distance of 3.6510 Å. Figures 3E, 4E depict the 2D and 3D pose of the ligand CBZ25, respectively.
CBZ34 serves as a bioisostere of CBZ, characterized by the substitution of the phenyl ring with an oct-3-en-4-yl group. The overall structure of CBZ34 closely resembles that of CBZ. Notably, the ligand CBZ34 exhibited the most favorable binding score of −7.2 kcal/mol. In the docked ADV complex, the residue GLN942 of the target protein forms hydrogen bonds with the oxygen atoms of the methoxy groups located on the quinoline ring of the ligands, with a distance of 2.2471 Å. Additionally, residue GLY187 demonstrated a C-H bond with the carbonyl group of the analogues at a distance of 3.5685 Å. Furthermore, GLU871 established a halogen bond with the fluorine atom of the fluorophenyl group of the ligand, at a distance of 3.1584 Å. Lastly, residue PHE938 exhibited a π-π interaction with the phenyl ring of CBZ34 at a distance of 4.4169 Å. Figures 3F, 4F depict the 2D and 3D pose of the CBZ34, respectively.
CBZ38 serves as a bioisostere of CBZ, characterized by the substitution of the phenyl ring with a methyl amino phenyl group. The overall structure of CBZ38 closely resembles that of CBZ. The ligand CBZ38 exhibited the most favorable binding score of −8.0 kcal/mol. In the docked ADV complex, the residue GLN942 of the target protein forms hydrogen bonds with the oxygen atoms of the methoxy groups present in the quinoline ring of the ligand, at distances of 2.7084 Å and 2.7971 Å. Additionally, residue TRP132 demonstrates a π-π interaction at a distance of 4.8259 Å, while PHE190 forms a π-alkyl bond with the methoxy group of the quinoline in the ligand at a distance of 5.4208 Å. Figures 3G, 4G depict the 2D and 3D poseof the CBZ38, respectively.
The docking interaction of the standard drug (CBZ) exhibited a favorable binding score of −7.5 kcal/mol. The residue GLN942 of the target protein engages in hydrogen bonding with the oxygen atom of the methoxy group present in the quinoline ring, with a distance of 1.861 Å. The hydrogen bonds involving GLN942 are consistent across all ligands. Additionally, the residue GLU871 demonstrates a π-anionic interaction at a distance of 3.60 Å. The residue PHE190 forms a π-π interaction with the phenyl ring of quinoline in CBZ at a distance of 5.12 Å, while LEU875 shows a π-σ interaction. Figures 3H, 4H illustrate the 2D and 3D pose of the standard drug CBZ, respectively.
The comprehensive docking analysis showed that the designed ligands of CBZ, especially CBZ01 and CBZ13, had multiple interactions compared to CBZ itself. In the case of CBZ01, four hydrogen bonds were identified with residues GLN942, GLN128, PHE938, and THR937 of the target protein with distances of 1.97, 2.57, 2.91, and 2.96 Å, respectively. Furthermore, CBZ13 exhibited two hydrogen bonds with the target protein residues GLN942 and ASN347 of the target protein with distances of 2.13 and 2.46 Å, respectively. In contrast, CBZ showed a single interaction with residue GLN942 at a distance of 1.86 Å through hydrogen bonding. This could be the result of a change in the docking score between the parent compound cabozantinib (−7.5) and its analogues CBZ1 (−8.0) and CBZ13 (−8.4). The common amino acid residue GLN942 matches the key residue reported by Xiang et al. (2015). The authors suggested that residue GLN942 may play a crucial role in the reversal of P-gp-mediated MDR by CBZ analogues, which is achieved by inhibiting P-gp transporter function. Further validation of the docking study of these two ligands was carried out using a molecular dynamics (MD) simulation study. Examination of the MD simulation study revealed that the analogues CBZ01 and CBZ13 were found to be stable over the period of 100 ns.
3.8 Molecular dynamics (MD) simulation
MD simulations represent a computational approach that effectively models the physical behaviour of atoms and molecules, thereby facilitating advancements in drug discovery. The dynamic properties of molecular atoms were investigated through MD simulations. This approach encompasses a series of algorithms aimed at assessing and predicting the stability of protein-ligand complexes. It is acknowledged as a powerful independent method for precisely detecting alterations at both molecular and atomic levels. To comprehend the stability of protein-ligand complexes, it is crucial to investigate the interactions between ligand molecules and proteins. The assessment of a ligand’s stability and dynamic behaviour in relation to the protein relies heavily on MD simulations. We examined the MD simulation at 100 ns SPC water model-based simulation utilizing the simulation interaction diagram (SID). This examination yielded valuable insights into the deviations, fluctuations, and intermolecular interactions that transpired during the simulation. MD simulation was performed for the CBZ01, CBZ13, and CBZ38. Among these, the MD simulations for ligands CBZ01 and CBZ13 demonstrated stable complexes with the target protein, whereas the MD simulations for compound CBZ38 did not remain within the chosen parameters.
3.8.1 RMSD and RMSF
The root mean square deviation (RMSD) quantifies the deviation of an atom’s molecule from its original structure or target over time. It is employed to evaluate the fluctuations in the protein’s backbone (C and N) during the 100 nsrun. Throughout the MD simulation, only minor variations in RMSD values related to the protein backbone were detected. For the protein complexed with the CBZ01, the ligand displayed a fluctuation of 2.5 Å, while the backbone RMSD initially ranged from 0 to 4.2 Å at the 0.50 ns? In contrast, when the protein was associated with the ligand CBZ13, the initial RMSD deviations for the ligand and protein were recorded at 2.8 Å and 2.9 Å, respectively. The P-gp complexed with CBZ01 showed an average RMSD of 1.57 Å, whereas the ligand exhibited an RMSD of 2.03 Å at the 100 ns? Conversely, the P-gp in complex with CBZ13 demonstrated an average RMSD of 1.61 Å, with the ligand showing an RMSD of 0.75 Å at the same time point. Initially, a lower deviation was observed from 0 to 25 ns, with slight fluctuations noted for two frames. Stable complexes (CBZ01) were identified from 25 to 50 ns, followed by a consistent deviation value with minimal changes from 50 to 100 ns? In the case of complex CBZ13, a lower deviation was also noted from 0 to 25 ns, with slight fluctuations observed for three frames. Stable complexes (CBZ01) were again identified from 25 to 50 ns and from 50 to 80 ns, followed by a consistent deviation value with minor changes from 80 to 100 ns? Overall, a stable RMSD value with slight deviations was observed from 80 to 100 ns, indicating that both complexes-maintained stability throughout the 100 ns simulation. The RMSD values for ligands CBZ01 and CBZ13 are illustrated in Figures 5A, B, respectively.
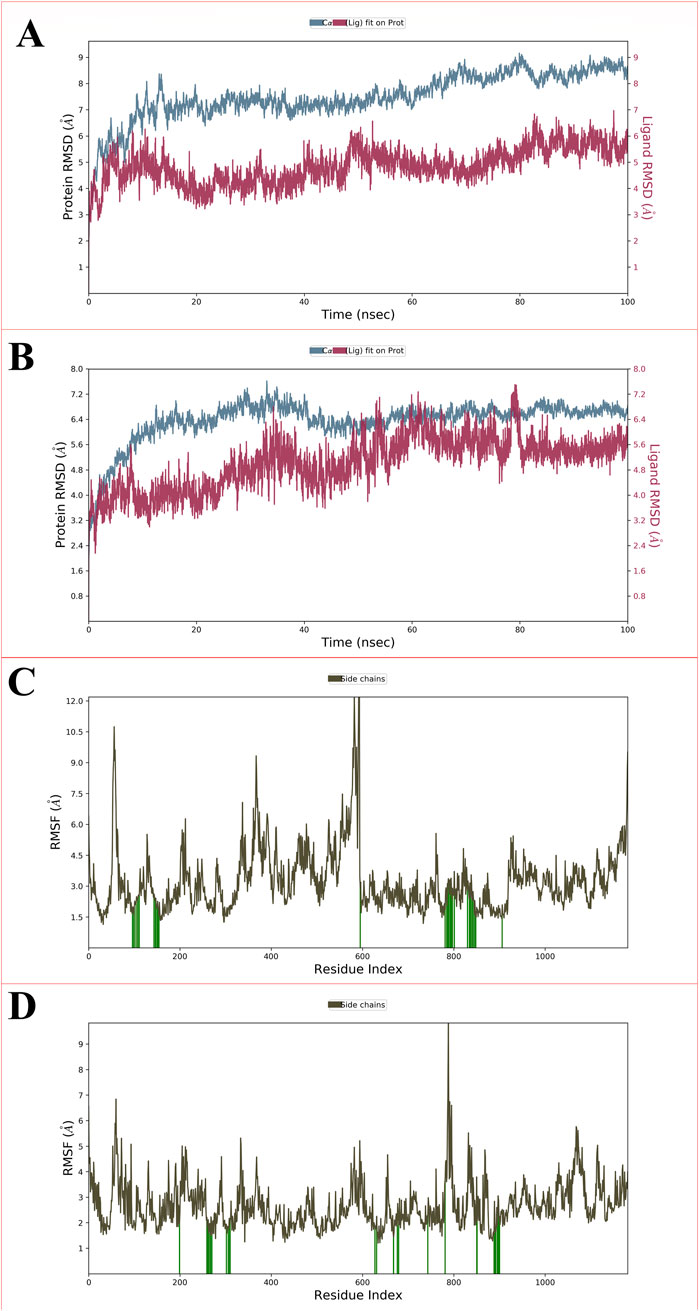
Figure 5. Showing the RMSD for P-glycoprotein with ligand CBZ01 (A) and ligand CBZ13 (B); The protein-RMSF plot of P-glycoprotein concerning the CBZ01 (C) and ligand CBZ13 (D) throughout the 100 ns run.
The root mean square fluctuation (RMSF) serves to measure the fluctuation of individual residues from their average fluctuation over a specified time period. The peaks identified in the protein-RMSF graph indicate the residues that display the most significant fluctuations throughout the simulation. Secondary structural components, such as alpha helices and beta strands, tend to exhibit less fluctuation than loop regions, owing to their enhanced rigidity relative to the unstructured portions of the protein. The green vertical bars on the graph denote protein residues that interact with the ligand. The variability among the other amino acid residues is considerably lower, reflecting a diminished level of fluctuation. During the 100 ns simulation, the ligands CBZ01 and CBZ13 showed minimal alterations in their interactions with each other, likely attributable to their effective engagement with various amino acid residues. The overall fluctuation observed is relatively low, providing valuable insights for future studies involving proteins with both ligands, CBZ01 and CBZ13. Additionally, the rigidity of the protein is reinforced by hydrogen bonds, pi-pi stacking, and the presence of secondary structural elements. In both scenarios illustrated in Figures 5C,D (CBZ01 and CBZ13), the fluctuations remain below 2 Å, indicating promising results.
3.8.2 Intermolecular interactions
MD simulations facilitate the examination of intermolecular interactions, which are defined as the attractive or repulsive forces that exist between molecules. Over the course of a 100 ns simulation, we explored various binding interactions between the ligand and the protein. Numerous intramolecular interactions were identified in both complexes, encompassing hydrophobic, polar, water-mediated, and pi-pi stacking interactions. The presence of N, O, and NH atoms led to the formation of hydrophilic, hydrophobic, and cationic interactions across different percentiles. No direct interactions with carbon molecules were observed. Furthermore, the direction of the arrows indicates both donors and acceptors. The residue TRP132 engaged with the phenyl group through hydrophobic interactions. The carbonyl group of the amide was linked to LEU875 and LYS930 via a water bridge, exhibiting both hydrophobic and cationic characteristics. LYS930 also established a cationic interaction with the carbonyl group of the amide. The amino acid residue ASN179 interacted with the methoxy group of the ligands through polar interactions facilitated by a water bridge. Additionally, the nitrogen atom of the quinoline ring formed an anionic interaction with GLU887 through a water bridge. Figure 6A depicts the intramolecular interaction between the CBZ01 complex and the target protein.
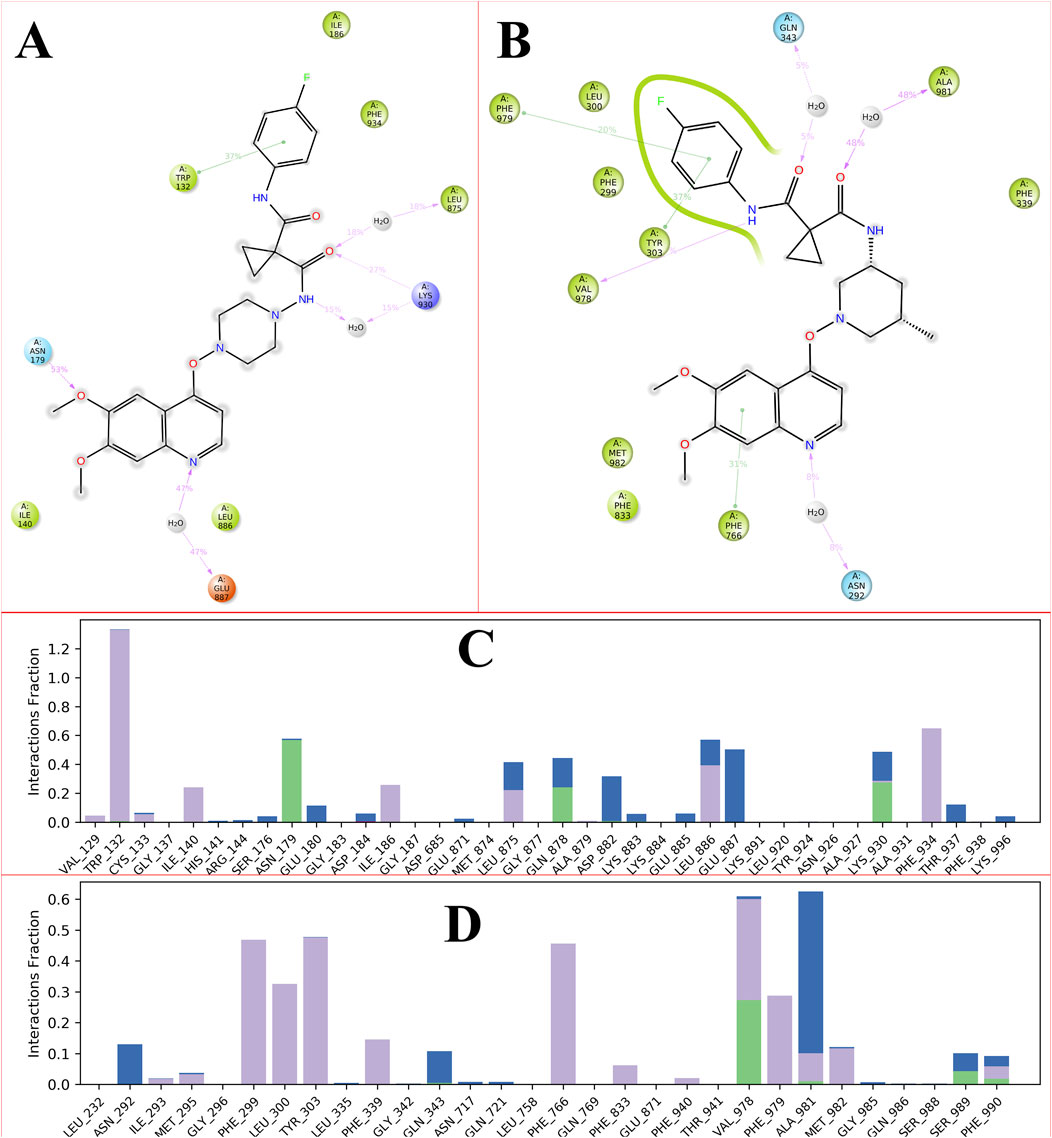
Figure 6. Showing the 2D-Summary of interacting atoms of P-glycoprotein with CBZ01 (A) and ligand CBZ13 (B); The count of interactions in histogram form for P-glycoprotein with CBZ01 (C) and ligand CBZ13 (D) throughout the 100 ns run.
The carbonyl group of the amide interacted with the GLN343 and ALA981 residues through polar and hydrophobic interactions, respectively. Moreover, the PHE979 and TYR303 residues formed hydrophobic interactions with the phenyl group and the carbonyl of the amide group, respectively. The NH of the amide and the phenyl group of the quinoline ring displayed hydrophobic interactions with the VAL292 and PHE766 residues, respectively. Additionally, the nitrogen atom of the quinoline ring established a hydrogen bond with the PHE766 residue, mediated by a water bridge. The intramolecular interactions of the CBZ13 complex with the target protein are depicted in Figure 6B. Figure 6C, D provide the histogram for ligands CBZ01 and CBZ13. The interactions between the protein and ligand are classified into four categories: ionic, hydrophobic, hydrogen bonding, and water bridges. The histogram illustrates that the stacked bar charts are standardized over the trajectory. The findings from the MD simulation indicate that both complexes, CBZ01 and CBZ13, demonstrated stability, characterized by lower RMDS and RMSF values, along with favorable interactions.
3.8.3 Binding free energy calculations and residue decomposition
Numerous methods are available to assess the binding free energy of protein-ligand complexes. MM-PBSA and MM-GBSA are currently the leading techniques due to their effectiveness in predicting the interactions between small molecules and biological entities (Genheden and Ryde, 2015). Energies associated with the protein (PDB ID: 3G5U), along with those of the CBZ01-3G5U and CBZ13-3G5U complexes, as well as the entropy derived from these methodologies, were documented. An MM-GBSA analysis was carried out to determine whether variations in binding mode can be differentiated in the predicted binding-free energy between the two systems.
The study demonstrated that the total energetic component breakdown for the CBZ01-3G5U and CBZ13-3G5U complexes was −40 kcal/mol and −50 kcal/mol, respectively, as illustrated in Figures 7A, B. For the ligand CBZ01, an extensive analysis of the free-energy components revealed that ΔGgas (−65 kcal/mol) was predominantly influenced within the protein environment. A minor decrease in ∆Evdwalls (−55 kcal/mol) and a significant reduction in ∆EEEl (−15 kcal/mol) contributed to the overall decline in ΔGgas (−40 kcal/mol). In the case of ligand CBZ13, a thorough examination of the free-energy components indicated that ΔGgas (−70 kcal/mol) was primarily affected within the protein. There was a slight decrease in ∆Evdwalls (−60 kcal/mol) and a notable reduction in ∆EEEl (−15 kcal/mol), leading to an overall decrease in ΔGgas (−50 kcal/mol).
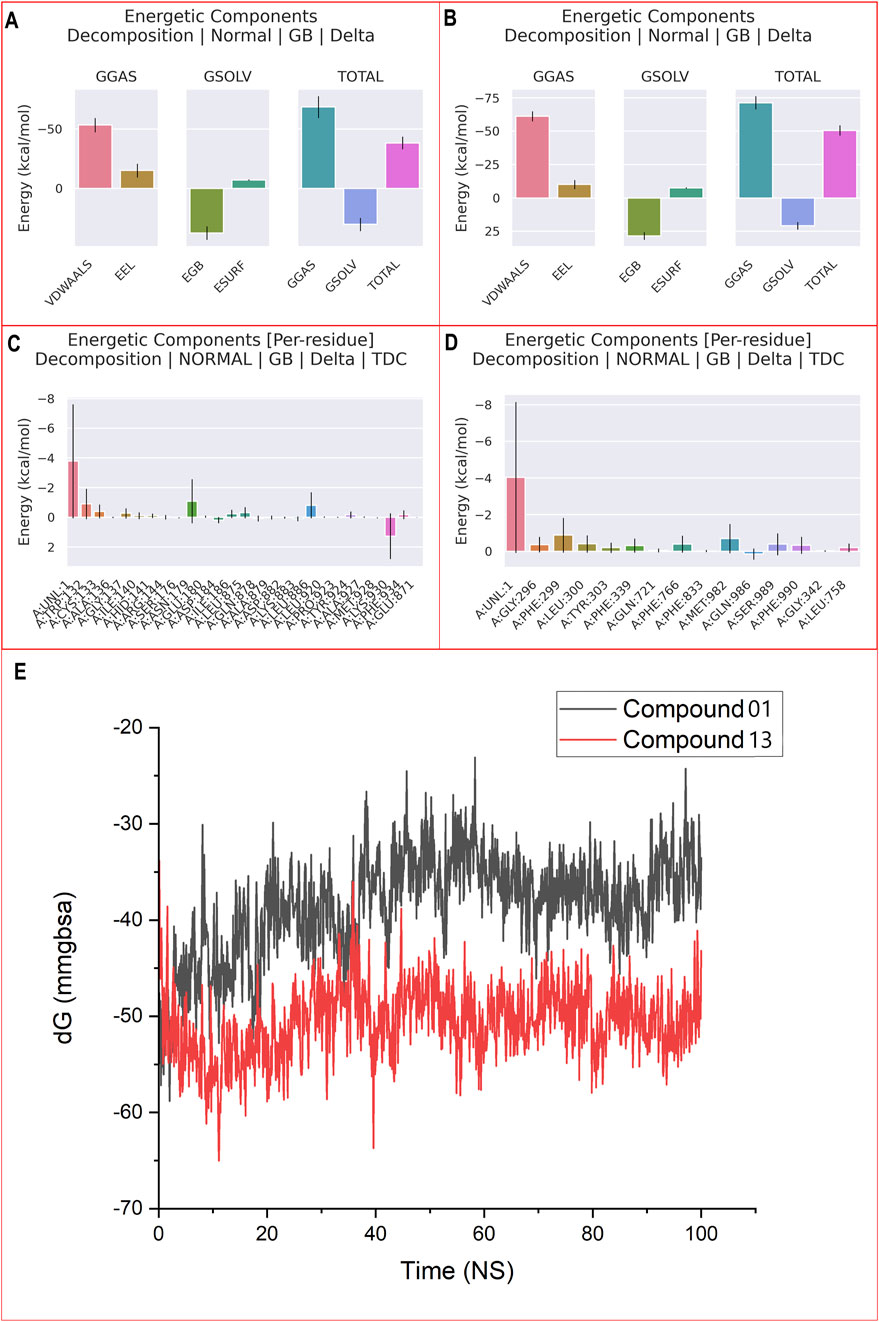
Figure 7. MM-PBSA binding free energy for ligand CBZ01-3G5U (A) and CBZ13-3G5U complexes (B); per residue free energy decomposition for ligand CBZ01-3G5U (C) and CBZ13-3G5U complexes (D); and MM-GBSA plot for CBZ01-3G5U and CBZ13-3G5U complexes (E).
A detailed per-residue free energy decomposition analysis was performed on the CBZ01-3G5U and CBZ13-3G5U complexes to assess the contributions of various amino acid residues surrounding the binding site to the overall binding free energy (Figures 7C,D). The addition of 3G5U in the context of CBZ01 led to a reduction in energy contributions from several critical residues, particularly 136, 140, 141, 144, 176, 184, 186, 878, 879, 882, 883, 920, 923, 924, 927, 928, and 871. Similarly, the incorporation of 3G5U with respect to CBZ13 resulted in decreased energy contributions from key residues, namely, 296, 303, 721, 833, 342, and 758. The free energies of the CBZ01-3G5U and CBZ13-3G5U complexes were determined to be −38.40 and −50.56 kcal/mol, respectively, through the application of the GBSA solvation method (Figure 7E). We have computed the binding free energy for each five-frame interval over the course of the 100 ns simulation, totalling 5,000 frames (Supplementary Table S2). These results suggest that the CBZ13-3G5U complex shows considerable stability and a strong affinity for the P-gp receptor and thus effectively acts as an anti-cancer agent.
4 Conclusion
Certain cancers demonstrate varying degrees of resistance to medications, which significantly undermines the efficacy of chemotherapy in achieving favorable treatment outcomes. The cell membranes are characterized by the presence of P-gp, a crucial protein that expels several foreign substances from cells and may contribute to resistance to chemotherapeutic agents. The CBZ, a tyrosine kinase inhibitor, is utilized in the treatment of various types of cancer. P-gp plays a role in mediating multidrug resistance (MDR), a challenge that can be addressed by CBZ through the direct inhibition of its export mechanism. Consequently, P-gp represents a vital target for the development of anti-cancer therapeutics. Patients undergoing treatment with CBZ may experience a range of side effects, including liver dysfunction, hypertension, hand-foot syndrome, reduced appetite, and general malaise. To address these issues, modifications to the scaffold of the CBZ molecule are necessary to create safer, less toxic, and more effective agents against MDR. In this research, we developed novel CBZ analogues employing a bioisosteric approach. We assessed the pharmacokinetic and toxicological profiles of the newly designed CBZ bioisosteres using ADMETlab 3.0. Following the screening process, the selected ligands were docked against the target protein (PDB ID: 3G5U) utilizing ADV, and their interactions were examined using Discovery studio. The docking scores for the ligands ranged from −6.4 to −8.4 kcal/mol. The ligands CBZ01, CBZ06, CBZ11, CBZ13, CBZ25, CBZ34, and CBZ38 demonstrated favorable interactions with the target protein. The amino acid residue GLN942 was identified as a critical residue in the binding process, potentially contributing to the inhibitory activity of P-gp. We selected the top three ligands, CBZ01, CBZ13, and CBZ38, for a 100 ns MD simulation using the Schrödinger suite, based on their docking scores and interactions. The trajectory analysis indicated that CBZ01 and CBZ13 maintained stability when complexed with the target protein. This combined computational approach suggests that CBZ01 and CBZ13 may be promising candidates for further development of potential anticancer agents.
Further experimental studies are currently underway to substantiate the anti-cancer properties of the designed CBZ analogues.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
GT: Writing–review and editing, Data curation, Investigation, Methodology, Writing–original draft. AG: Data curation, Investigation, Methodology, Writing–original draft, Writing–review and editing. DP: Data curation, Writing–review and editing. YV: Writing–review and editing, Formal Analysis, Validation. NK: Writing–review and editing, Data curation, Investigation, Methodology. SA: Writing–review and editing, Formal Analysis, Funding acquisition, Validation. SJ: Formal Analysis, Conceptualization, Supervision, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The author SA would like to extend his appreciation to the Deanship of Research and Graduate Studies at King Khalid University for funding this work through small group research under grant number RGP1/161/45.
Acknowledgments
The authors would like to thank Head, Department of Pharmacy, Guru Ghasidas Vishwavidyalaya, Koni, Bilaspur (Chhattisgarh), India for proving all necessary facilities.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2025.1543075/full#supplementary-material
References
Ajmal, M. R., Abdelhameed, A. S., Alam, P., and Khan, R. H. (2016). Interaction of new kinase inhibitors cabozantinib and tofacitinib with human serum alpha-1 acid glycoprotein. A comprehensive spectroscopic and molecular docking approach. Spectrochim. Acta A Mol. Biomol. Spectrosc. 159, 199–208. doi:10.1016/j.saa.2016.01.049
Aller, S. G., Yu, J., Ward, A., Weng, Y., Chittaboina, S., Zhuo, R., et al. (2009). Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323, 1718–1722. doi:10.1126/science.1168750
Anand, U., Dey, A., Chandel, A. K. S., Sanyal, R., Mishra, A., Pandey, D. K., et al. (2023). Cancer chemotherapy and beyond: current status, drug candidates, associated risks and progress in targeted therapeutics. Genes Dis. 10, 1367–1401. doi:10.1016/j.gendis.2022.02.007
Anderson, A. C. (2003). The process of structure-based drug design. Chem. Biol. 10, 787–797. doi:10.1016/j.chembiol.2003.09.002
Andrade, R. J., Chalasani, N., Björnsson, E. S., Suzuki, A., Kullak-Ublick, G. A., Watkins, P. B., et al. (2019). Drug-induced liver injury. Nat. Rev. Dis. Prim. 5, 58. doi:10.1038/s41572-019-0105-0
Banks, J. L., Beard, H. S., Cao, Y., Cho, A. E., Damm, W., Farid, R., et al. (2005). Integrated modeling program, applied chemical theory (IMPACT). J. Comp. Chem. 26, 1752–1780. doi:10.1002/jcc.20292
Barnhill, M. S., Steinberg, J. M., Jennings, J. J., and Lewis, J. H. (2020). Hepatotoxicty of agents used in the management of inflammatory bowel disease: a 2020 update. Curr. Gastroenterol. Rep. 22, 47–12. doi:10.1007/s11894-020-00781-3
Bhagat, R. T., Butle, S. R., Khobragade, D. S., Wankhede, S. B., Prasad, C. C., Mahure, D. S., et al. (2021). Molecular docking in drug discovery. J. Pharm. Res. Int. 33, 46–58. doi:10.9734/JPRI/2021/v33i30B31639
Bickerton, G. R., Paolini, G. V., Besnard, J., Muresan, S., and Hopkins, A. L. (2012). Quantifying the chemical beauty of drugs. Nat. Chem. 4, 90–98. doi:10.1038/nchem.1243
Bouricha, E. M., and Hakmi, M. (2024). Investigating lasofoxifene efficacy against the Y537S + F404V double-mutant estrogen receptor alpha using molecular dynamics simulations. Bioinform. Biol. Insights 18, 11779322241288703. doi:10.1177/11779322241288703
Bray, F., Laversanne, M., Sung, H., Ferlay, J., Siegel, R. L., Soerjomataram, I., et al. (2024). Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 74, 229–263. doi:10.3322/caac.21834
Bukowski, K., Kciuk, M., and Kontek, R. (2020). Mechanisms of multidrug resistance in cancer chemotherapy. Int. J. Mol. Sci. 21, 3233. doi:10.3390/ijms21093233
Cao, W., Qin, K., Li, F., and Chen, W. (2024). Comparative study of cancer profiles between 2020 and 2022 using global cancer statistics (GLOBOCAN). J. Natl. Cancer Cent. 4, 128–134. doi:10.1016/j.jncc.2024.05.001
Chiruvella, V., Annamaraju, P., and Guddati, A. K. (2020). Management of nephrotoxicity of chemotherapy and targeted agents: 2020. Am. J. Cancer Res. 10, 4151–4164.
Choueiri, T. K., Escudier, B., Powles, T., Mainwaring, P. N., Rini, B. I., Donskov, F., et al. (2015). Cabozantinib versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 373, 1814–1823. doi:10.1056/NEJMoa1510016
Das, B., Baidya, A. T., Mathew, A. T., Yadav, A. K., and Kumar, R. (2022). Structural modification aimed for improving solubility of lead compounds in early phase drug discovery. Bioorg. Med. Chem. Lett. 56, 116614. doi:10.1016/j.bmc.2022.116614
De Ruyck, J., Brysbaert, G., Blossey, R., and Lensink, M. F. (2016). Molecular docking as a popular tool in drug design, an in-silico travel. Adv. Appl. Bioinform. 9, 1–11. doi:10.2147/AABC.S105289
Du, X., Li, Y., Xia, Y. L., Ai, S. M., Liang, J., Sang, P., et al. (2016). Insights into protein–ligand interactions: mechanisms, models, and methods. Int. J. Mol. Sci. 17, 144. doi:10.3390/ijms17020144
Duan, J. L., Wang, C. C., Yuan, Y., Hui, Z., Zhang, H., Mao, N. D., et al. (2024). Design, synthesis, and structure–activity relationship of novel pyridazinone-based PARP7/HDACs dual inhibitors for elucidating the relationship between antitumor immunity and HDACs inhibition. J. Med. Chem. 67, 4950–4976. doi:10.1021/acs.jmedchem.4c00090
Dulsat, J., López-Nieto, B., Estrada-Tejedor, R., and Borrell, J. I. (2023). Evaluation of free online ADMET tools for academic or small biotech environments. Molecules 28, 776. doi:10.3390/molecules28020776
Ejalonibu, M. A., Ogundare, S. A., Elrashedy, A. A., Ejalonibu, M. A., Lawal, M. M., Mhlongo, N. N., et al. (2021). Drug discovery for Mycobacterium tuberculosis using structure-based computer-aided drug design approach. Int. J. Mol. Sci. 22, 13259. doi:10.3390/ijms222413259
Elekofehinti, O. O., Iwaloye, O., Josiah, S. S., Lawal, A. O., Akinjiyan, M. O., and Ariyo, E. O. (2021). Molecular docking studies, molecular dynamics and ADME/tox reveal therapeutic potentials of STOCK1N-69160 against papain-like protease of SARS-CoV-2. Mol. Divers. 25, 1761–1773. doi:10.1007/s11030-020-10151-w
Ferlay, J., Ervik, M., Lam, F., Laversanne, M., Colombet, M., Mery, L., et al. (2024). Global cancer observatory: cancer today. Lyon, France: International Agency for Research on Cancer. Available at: https://gco.iarc.who.int/today (Accessed January 22, 2025).
Ferreira, L. G., Dos Santos, R. N., Oliva, G., and Andricopulo, A. D. (2015). Molecular docking and structure-based drug design strategies. Molecules 20, 13384–13421. doi:10.3390/molecules200713384
Gao, X., Tang, J., Liu, H., Liu, L., and Liu, Y. (2019). Structure–activity study of fluorine or chlorine-substituted cinnamic acid derivatives with tertiary amine side chain in acetylcholinesterase and butyrylcholinesterase inhibition. Drug Dev. Res. 80, 438–445. doi:10.1002/ddr.21515
Gao, Y., Duan, J., Dang, X., Yuan, Y., Wang, Y., He, X., et al. (2023). Design, synthesis and biological evaluation of novel histone deacetylase (HDAC) inhibitors derived from β -elemene scaffold. J. Enzyme Inhib. Med. Chem. 38, 2195991. doi:10.1080/14756366.2023.2195991
Garrigues, A., Escargueil, A. E., and Orlowski, S. (2002). The multidrug transporter, P-glycoprotein, actively mediates cholesterol redistribution in the cell membrane. Proc. Natl. Acad. Sci. 99, 10347–10352. doi:10.1073/pnas.162366399
Genheden, S., and Ryde, U. (2015). The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 10, 449–461. doi:10.1517/17460441.2015.1032936
Giordano, D., Biancaniello, C., Argenio, M. A., and Facchiano, A. (2022). Drug design by pharmacophore and virtual screening approach. Pharmaceutical 15, 646. doi:10.3390/ph15050646
Goebel, J., Chmielewski, J., and Hrycyna, C. A. (2021). The roles of the human ATP-binding cassette transporters P-glycoprotein and ABCG2 in multidrug resistance in cancer and at endogenous sites: future opportunities for structure-based drug design of inhibitors. Cancer Drug Resist 4, 784–804. doi:10.20517/cdr.2021.19
Gonzalez, M. A. (2011). Force fields and molecular dynamics simulations. Collect. SFN 12, 169–200. doi:10.1051/sfn/201112009
Gupta, A. K., Vaishnav, Y., Jain, S. K., Annadurai, S., and Kumar, N. (2024). Exploring novel Apalutamide analogues as potential therapeutics for prostate cancer: design, molecular docking investigations and molecular dynamics simulation. Fron. Chem. 12, 1418975. doi:10.3389/fchem.2024.1418975
Heming, C. P., Muriithi, W., Macharia, L. W., Niemeyer Filho, P., Moura-Neto, V., and Aran, V. (2022). P-glycoprotein and cancer: what do we currently know? Heliyon 8, e11171. doi:10.1016/j.heliyon.2022.e11171
Ikwu, F. A., Shallangwa, G. A., and Mamza, P. A. (2020). QSAR, QSTR, and molecular docking studies of the anti-proliferative activity of phenylpiperazine derivatives against DU145 prostate cancer cell lines. Beni-Suef. Univ. J. Basic Appl. Sci. 9, 35. doi:10.1186/s43088-020-00054-y
Ivanenkov, Y. A., Zagribelnyy, B. A., and Aladinskiy, V. A. (2019). Are we opening the door to a new era of medicinal chemistry or being collapsed to a chemical singularity? J. Med. Chem. 62, 10026–10043. doi:10.1021/acs.jmedchem.9b00004
Jayashree, B. S., Nikhil, P. S., and Paul, S. (2022). Bioisosterism in drug discovery and development-an overview. Med. Chem. 18, 915–925. doi:10.2174/1573406418666220127124228
Jiang, C., Sun, T., Xiang, D., Wei, S., and Li, W. (2018). Anticancer activity and mechanism of xanthohumol: a prenylated flavonoid from hops (Humulus lupulus L.). Front. Pharmacol. 9, 530. doi:10.3389/fphar.2018.00530
Johnson, Z. L., and Chen, J. (2018). ATP binding enables substrate release from multidrug resistance protein 1. Cell 172, 81–89.e10. doi:10.1016/j.cell.2017.12.005
Kang, L., Gao, X., Liu, H., Men, X., Wu, H., Cui, P., et al. (2018). Structure–activity relationship investigation of coumarin–chalcone hybrids with diverse side-chains as acetylcholinesterase and butyrylcholinesterase inhibitors. Mol. Divers. 22, 893–906. doi:10.1007/s11030-018-9839-y
Karami, T. K., Hailu, S., Feng, S., Graham, R., and Gukasyan, H. J. (2022). Eyes on lipinski’s rule of five: a new “rule of thumb” for physicochemical design space of ophthalmic drugs. J. Ocul. Pharmacol. Ther. 38, 43–55. doi:10.1089/jop.2021.0069
Karmacharya, U., Guragain, D., Chaudhary, P., Jee, J. G., Kim, J. A., and Jeong, B. S. (2021). Novel pyridine bioisostere of cabozantinib as a potent c-met kinase inhibitor: synthesis and anti-tumor activity against hepatocellular carcinoma. Int. J. Mol. Sci. 22, 9685. doi:10.3390/ijms22189685
Karthika, C., Sureshkumar, R., Zehravi, M., Akter, R., Ali, F., Ramproshad, S., et al. (2022). Multidrug resistance of cancer cells and the vital role of P-glycoprotein. Life 12, 897. doi:10.3390/life12060897
Khan, S. U., Fatima, K., Aisha, S., and Malik, F. (2024). Unveiling the mechanisms and challenges of cancer drug resistance. Cell Commun. Signal. 22, 109. doi:10.1186/s12964-023-01302-1
Kim, Y., and Chen, J. (2018). Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science. 359, 915–919. doi:10.1126/science.aar7389
Krens, S. D., van Boxtel, W., Uijen, M. J. M., Jansman, F. G. A., Desar, I. M. E., Mulder, S. F., et al. (2022). Exposure-toxicity relationship of cabozantinib in patients with renal cell cancer and salivary gland cancer. Int. J. Cancer. 150, 308–316. doi:10.1002/ijc.33797
Lagu, S. B., Yejella, R. P., Nissankararao, S., Bhandare, R. R., Golla, V. S., Lokesh, B. V., et al. (2022). Antitubercular activity assessment of fluorinated chalcones, 2-aminopyridine-3-carbonitrile and 2-amino-4H-Pyran-3-Carbonitrile derivatives: in vitro, molecular docking and in-silico drug likeliness studies. PLoS One 17, e0265068. doi:10.1371/journal.pone.0265068
Li, Y., Zhou, L., Qian, F., Fang, Q., Yu, Z., Cui, T., et al. (2024). ScImmOmics: a manually curated resource of single-cell multi-omics immune data. Nucleic Acids Res. 53, D1162–D1172. doi:10.1093/nar/gkae985
Lin, X., Liao, Y., Chen, X., Long, D., Yu, T., and Shen, F. (2016a). Regulation of oncoprotein 18/stathmin signaling by ERK concerns the resistance to taxol in nonsmall cell lung cancer cells. Cancer biother. Radiopharm. 31, 37–43. doi:10.1089/cbr.2015.1921
Lin, X., Yu, T., Zhang, L., Chen, S., Chen, X., Liao, Y., et al. (2016b). Silencing op18/stathmin by RNA interference promotes the sensitivity of nasopharyngeal carcinoma cells to taxol and high-grade differentiation of xenografted tumours in nude mice. Basic Clin. Pharmacol. Toxicol. 119, 611–620. doi:10.1111/bcpt.12633
Liu, B., Zhou, H., Tan, L., Siu, K. T. H., and Guan, X. Y. (2024). Exploring treatment options in cancer: tumor treatment strategies. Signal Transduct. Target Ther. 9, 175. doi:10.1038/s41392-024-01856-7
Liu, M., An, R., Wu, Z., Dai, L., Zeng, Q., and Chen, W. (2024). The trajectory of oral mucositis in Head and neck cancer patients undergoing radiotherapy and its influencing factors. Ear Nose Throat J., 1455613241228211. doi:10.1177/01455613241228211
Lou, Y., Song, F., Cheng, M., Hu, Y., Chai, Y., Hu, Q., et al. (2023). Effects of the CYP3A inhibitors, voriconazole, itraconazole, and fluconazole on the pharmacokinetics of osimertinib in rats. PeerJ 11, e15844. doi:10.7717/peerj.15844
Lounnas, V., Ritschel, T., Kelder, J., McGuire, R., Bywater, R. P., and Foloppe, N. (2013). Current progress in structure-based rational drug design marks a new mindset in drug discovery. Comput. Struct. Biotechnol. J. 5, e201302011. doi:10.5936/csbj.201302011
Markowitz, J. N., and Fancher, K. M. (2018). Cabozantinib: a multitargeted oral tyrosine kinase inhibitor. Pharmacotherapy 38, 357–369. doi:10.1002/phar.2076
Martini, D. J., Evans, S. T., Liu, Y., Shabto, J. M., Uner, O. E., Olsen, T. A., et al. (2022). Analysis of toxicity and clinical outcomes in full versus reduced starting dose cabozantinib in metastatic renal cell carcinoma patients. Clin. Genitourin. Cancer. 20, 53–59. doi:10.1016/j.clgc.2021.11.004
Matore, B. W., Roy, P. P., and Singh, J. (2023). Discovery of novel VEGFR2-TK inhibitors by phthalimide pharmacophore based virtual screening, molecular docking, MD simulation and DFT. J. Biomol. Struct. Dyn. 41, 13056–13077. doi:10.1080/07391102.2023.2178510
Naresh, G. K. R. S., and Guruprasad, L. (2020). Structural dynamics of cabozantinib bound TAM receptor tyrosine kinases. J. Biomol. Struct. Dyn. 39, 1–20. doi:10.1080/07391102.2020.1730968
National Center for Biotechnology Information (2025). PubChem compound summary for CID 25102847. Cabozantinib. Available at: https://pubchem.ncbi.nlm.nih.gov/compound/Cabozantinib (Accessed January 1, 2025).
Nie, Y., Li, D., Peng, Y., Wang, S., Hu, S., Liu, M., et al. (2020). Metal organic framework coated MnO2 nanosheets delivering doxorubicin and self-activated DNAzyme for chemo-gene combinatorial treatment of cancer. Int. J. Pharm. 585, 119513. doi:10.1016/j.ijpharm.2020.119513
Rimassa, L., Danesi, R., Pressiani, T., and Merle, P. (2019). Management of adverse events associated with tyrosine kinase inhibitors: improving outcomes for patients with hepatocellular carcinoma. Cancer Treat. Rev. 77, 20–28. doi:10.1016/j.ctrv.2019.05.004
Salo-Ahen, O. M., Alanko, I., Bhadane, R., Bonvin, A. M., Honorato, R. V., Hossain, S., et al. (2020). Molecular dynamics simulations in drug discovery and pharmaceutical development. Processes 9, 71. doi:10.3390/pr9010071
Sastry, G. M., Adzhigirey, M., Day, T., Annabhimoju, R., and Sherman, W. (2013). Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 27, 221–234. doi:10.1007/s10822-013-9644-8
Schmidinger, M., and Danesi, R. (2018). Management of adverse events associated with cabozantinib therapy in renal cell carcinoma. Oncologist 23, 306–315. doi:10.1634/theoncologist.2017-0335
Schwartz, G., Darling, J. O., Mindo, M., and Damicis, L. (2020). Management of adverse events associated with cabozantinib treatment in patients with advanced hepatocellular carcinoma. Target Onco 15, 549–565. doi:10.1007/s11523-020-00736-8
Shan, J., and Ji, C. (2020). MolOpt: a web server for drug design using bioisosteric transformation. Curr. Comput-Aided Drug Des. 16, 460–466. doi:10.2174/1573409915666190704093400
Sharom, F. J. (2011). The P-glycoprotein multidrug transporter. Essays Biochem. 50, 161–178. doi:10.1042/bse0500161
Shivakumar, D., Williams, J., Wu, Y., Damm, W., Shelley, J., and Sherman, W. (2010). Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J. Chem. Theory Comp. 6, 1509–1519. doi:10.1021/ct900587b
Srigadha, V. K., Prabhash, K., Noronha, V., Joshi, A., Patil, V. M., Menon, N., et al. (2023). Cabozantinib: a narrative drug review. Cancer Res. Stat. Treat. 6, 74–87. doi:10.4103/crst.crst_9_23
Subbaiah, M. A. M., and Meanwell, N. A. (2021). Bioisosteres of the phenyl ring: recent strategic applications in lead optimization and drug design. J. Med. Chem. 64, 14046–14128. doi:10.1021/acs.jmedchem.1c01215
Sun, D., Gao, W., Hu, H., and Zhou, S. (2022). Why 90% of clinical drug development fails and how to improve it? Acta Pharm. Sin. B 12, 3049–3062. doi:10.1016/j.apsb.2022.02.002
Sun, D., Li, X., Nie, S., Liu, J., and Wang, S. (2023). Disorders of cancer metabolism: the therapeutic potential of cannabinoids. Biomed. Pharmacother. 157, 113993. doi:10.1016/j.biopha.2022.113993
Sung, H., Ferlay, J., Siegel, R. L., Laversanne, M., Soerjomataram, I., Jemal, A., et al. (2021). Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249. doi:10.3322/caac.21660
Tian, Y., Lei, Y., Wang, Y., Lai, J., Wang, J., and Xia, F. (2023). Mechanism of multidrug resistance to chemotherapy mediated by P-glycoprotein. Int. J. Oncol. 63, 1–19. doi:10.3892/ijo.2023.5567
Trott, O., and Olson, A. J. (2010). AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461. doi:10.1002/jcc.21334
Vasiliou, V., Vasiliou, K., and Nebert, D. W. (2009). Human ATP-binding cassette (ABC) transporter family. Hum. Genomics. 3, 281–310. doi:10.1186/1479-7364-3-3-281
Wang, E., Sun, H., Wang, J., Wang, Z., Liu, H., Zhang, J. Z. H., et al. (2019). End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem. Rev. 119, 9478–9508. doi:10.1021/acs.chemrev.9b00055
Wang, K., Yin, J., Chen, J., Ma, J., Si, H., and Xia, D. (2024). Inhibition of inflammation by berberine: molecular mechanism and network pharmacology analysis. Phytomedicine 128, 155258. doi:10.1016/j.phymed.2023.155258
Wang, Z., Jiang, L., Lv, X., Yin, H., Wang, Z., Li, W., et al. (2024). Higher risk of hepatotoxicity associated with cabozantinib in cancer patients. Crit. Rev. Oncol.Hematol. 196, 104298. doi:10.1016/j.critrevonc.2024.104298
Xiang, Q. F., Zhang, D. M., Wang, J. N., Zhang, H. W., Zheng, Z. Y., Yu, D. C., et al. (2015). Cabozantinib reverses multidrug resistance of human hepatoma HepG2/adr cells by modulating the function of P-glycoprotein. Liver Int. 35, 1010–1023. doi:10.1111/liv.12524
Xiong, G., Wu, Z., Yi, J., Fu, L., Yang, Z., Hsieh, C., et al. (2021). ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties. Nucleic. Acids Res. 49, W5–W14. doi:10.1093/nar/gkab255
Zhang, L., Shi, H., Tan, X., Jiang, Z., Wang, P., and Qin, J. (2022). Ten-gram-scale mechanochemical synthesis of ternary lanthanum coordination polymers for antibacterial and antitumor activities. Front. Chem. 10, 898324. doi:10.3389/fchem.2022.898324
Zielesny, A. (2005). Chemistry software package Chem office ultra 2005. J. Chem. Inf. Model. 45, 1474–1477. doi:10.1021/ci050273j
Keywords: anti-cancer agent, newer analogues, bioisosteric approach, cabozantinib, molecular docking, MD simulation
Citation: Thakur GS, Gupta AK, Pal D, Vaishnav Y, Kumar N, Annadurai S and Jain SK (2025) Designing novel cabozantinib analogues as p-glycoprotein inhibitors to target cancer cell resistance using molecular docking study, ADMET screening, bioisosteric approach, and molecular dynamics simulations. Front. Chem. 13:1543075. doi: 10.3389/fchem.2025.1543075
Received: 10 December 2024; Accepted: 04 February 2025;
Published: 27 February 2025.
Edited by:
Afzal Basha Shaik, Jawaharlal Nehru Technological University, Kakinada, IndiaReviewed by:
Vidyasrilekha Sanapalli, SVKM'S NMIMS University, IndiaBhaskara Rao Karri, University of Minnesota Twin Cities, United States
Copyright © 2025 Thakur, Gupta, Pal, Vaishnav, Kumar, Annadurai and Jain. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sanmati Kumar Jain, c2FubWF0aWphaW43MkB5YWhvby5jby5pbg==
†ORCID: Sanmati Kumar Jain, orcid.org/0000-0002-4798-7151