 Fang-Fang Tan1,2*
Fang-Fang Tan1,2* Zhan-Chao Li3,4
Zhan-Chao Li3,4- 1School of Chemistry and Materials, Weinan Normal University, Weinan, China
- 2Sustainable Materials and Chemistry, Department of Wood Technology and Wood-based Composites, University of Göttingen, Göttingen, Germany
- 3School of Chemistry and Environmental Engineering, Sichuan University of Science and Technology, Zigong, China
- 4Key Laboratories of Fine Chemicals and Surfactants in Sichuan Provincial Universities, Zigong, China
Trifluoromethylation stands as a pivotal technology in modern synthetic chemistry, playing an indispensable role in drug design, functional material development, and agrochemical innovation. With the growing emphasis on green chemistry principles, the pursuit of environmentally benign trifluoromethylation strategies has emerged as a critical research frontier. Trifluoroacetic acid (TFA), characterized by its cost-effectiveness, stability, and low toxicity, has become a promising alternative to conventional trifluoromethylation reagents. This review systematically summarizes advancements in photocatalytic decarboxylative trifluoromethylation using TFA and its derivatives over the past decade, focusing on three key activation mechanisms: single-electron transfer (SET), electron donor-acceptor (EDA) complex-mediated pathways, and ligand-to-metal charge transfer (LMCT). This paradigm shift is driven by the intrinsic limitations of conventional thermal decarboxylation, particularly its reliance on harsh conditions and significant environmental burdens. In contrast, photocatalytic strategies enable efficient C–CF3 bond construction under mild conditions, offering a modular platform for synthesizing fluorinated functional molecules. Strategic research priorities should focus on overcoming fundamental challenges, including but not limited to optimizing photosensitizer catalytic efficiency, establishing regioselective manipulation strategies, and engineering multicomponent tandem reaction systems to achieve trifluoromethylation methodologies under mild conditions. Furthermore, the integration of mechanistic investigations with artificial intelligence-driven reaction prediction will accelerate the advancement of precision trifluoromethylation technologies. This progress is anticipated to provide sustainable synthetic solutions for next-generation fluorinated pharmaceuticals and advanced functional materials, effectively bridging the innovation gap between academic research and industrial implementation.
1 Introduction
Trifluoromethylation plays a significant role in contemporary chemistry (Dolbier, 1996; Ni et al., 2015). Compounds containing trifluoromethyl groups (–CF3) exhibit unique properties and broad application potential in pharmaceuticals, pesticides, and materials science (Muller et al., 2007). For instance, the introduction of–CF3 groups can markedly enhance drug bioactivity, metabolic stability, and membrane permeability, offering a robust strategy for developing highly efficient and low-toxicity therapeutics (Dolbier, 1996; Ni et al., 2015; Purser et al., 2008; Muller et al., 2007). Numerous trifluoromethylation reagents have been developed and widely used for the trifluoromethylation of organic molecules, laying a solid theoretical foundation for the development of this field. Examples include trifluorohalomethanes (CF3I, CF3Br) (Guo et al., 2018; Natte et al., 2016; Mckee and Shevlin, 1985), trifluoromethanesulfonic acid derivatives (CF3SO2Cl, (CF3SO2)2O, CF3SO2CH(Me)COPh) (Muralirajan et al., 2021; Ouyang et al., 2018; Liu et al., 2017), Togni reagents (Mejía and Togni, 2012; Charpentier et al., 2015), Umemoto reagents (Cheng et al., 2015; Meucci et al., 2019), TMSCF3 (Krishnamurti et al., 2018; Sun et al., 2019), Langlois reagents (NaSO2CF3) (Wang et al., 2016; Le et al., 2017; Jiang et al., 2021), and Zn(SO2CF3)2 (O’brien et al., 2014; Fujiwara et al., 2012). However, the low atom utilization and the production of hazardous byproducts of conventional trifluoromethylation reagents restrict their use on a large scale.
TFA, a readily available and stable trifluoromethyl source, can generate CF3• radicals via decarboxylation with easily separable byproducts, demonstrating significant potential (Andreev et al., 2013; Arai et al., 2014; Depecker et al., 1999; Ji et al., 2019). However, the high electronegativity of fluorine atoms elevates TFA’s oxidation potential, complicating decarboxylation under mild conditions—a critical bottleneck for its widespread adoption (Andreev et al., 2013; Arai et al., 2014). Photocatalysis, particularly visible-light-driven systems, addresses this challenge by enabling reactive intermediate generation under mild conditions, thus promoting efficient and selective synthetic routes (Prier et al., 2013; Romero and Nicewicz, 2016).
Early studies on photocatalytic TFA decarboxylation relied on ultraviolet or high-energy tungsten lamps, or precious metal catalysts. Alternatively, researchers employed pre-activated or in situ activated derivatives to facilitate CF3• radicals generation under visible light. As illustrated in Scheme 1, three primary pathways exist: A) The SET process is used to generate the CF3• radicals. TFA or trifluoroacetate (CF3CO2[M], where M stands for Na, K or Ag) are oxidized either through holes or at the anode. This results in the formation of CF3CO2•. CO2 is then eliminated, resulting in CF3• radicals (Scheme 1A, path I). Alternatively, TFA derivatives (such as Trifluoroaceticanhydride (TFAA), pentafluoroiodobenzene bis (trifluoroacetate) (FPIFA), trifluoroacetoxybenzimidazole chloride ester (NHBC)) receive an electron from the excited photocatalyst. This results in the formation of their anionic radical intermediates, from which the anionic group (R−/RO−) departs, yielding CF3CO2• or CF3CO•. The subsequent loss of CO2 or CO then generates the CF3• radicals (Scheme 1A, path II). Additionally, trifluoroacetyl oxime (TFAOx) can produce CF3• radicals via an energy transfer (ET) process (Scheme 1A, path III); B) The EDA procedure is used to generate the CF3• radicals. Pyridine N-oxide reacts with TFAA to form an intermediate trifluoroacetoxypyridinium ion. This intermediate acts as an electron acceptor and forms an EDA complex with pyridine N-oxide, which serves as the electron donor. Upon exposure to visible light, electrons are transferred from the donor to the acceptor, leading to the formation of a dearomatized tertiary carbon radical intermediate. This intermediate induces homolytic cleavage of the N–O bond, yielding the CF3CO2•. Subsequent elimination of CO2 results in the formation of the CF3• radicals (Scheme 1B); C) The LMCT process is used to generate the CF3• radicals. The CF3CO2−, obtained from TFA, CF3CO2[M], or TFAA, coordinates with the metal center of a complex. Upon light excitation, the newly formed complex undergoes the LMCT process, producing the CF3CO2•. Subsequently, the elimination of CO2 results in the formation of the CF3• radicals (Scheme 1C).
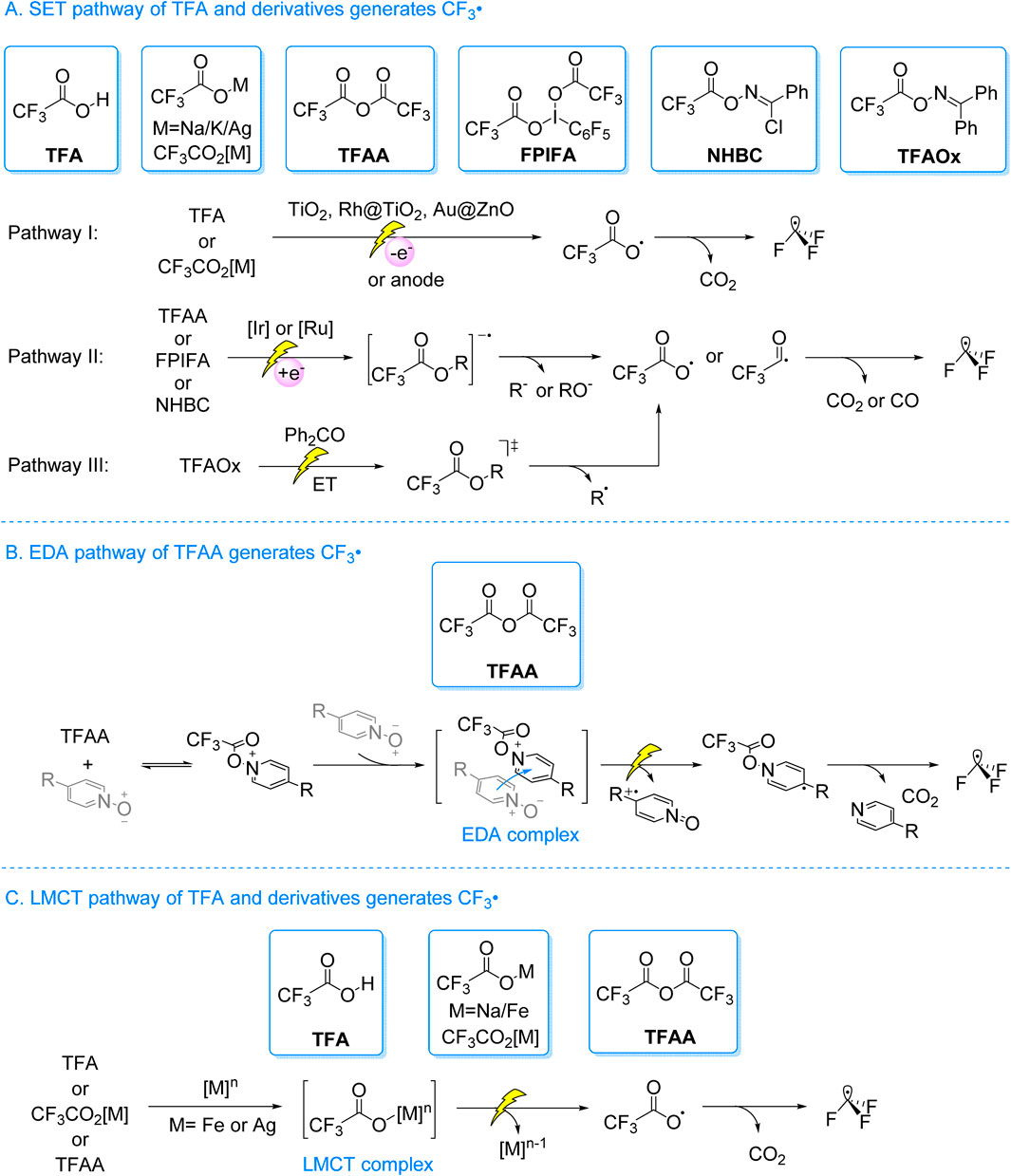
Scheme 1. Three main routes for photocatalytic production of trifluoromethyl from trifluoroacetic acid and its derivatives.
The strategic incorporation of CF3 groups into aromatic systems has become a hallmark of modern medicinal chemistry, as evidenced by the prevalence of this motif in 7.5% of FDA-approved small-molecule drugs (2018 data), with 80% exhibiting direct aromatic-CF3 connectivity—a structural signature shared by therapeutic agents such as Enzalutamide (prostate cancer), Fluoxetine (depression), Lomitapide (hypercholesterolemia), and Regorafenib (leukemia). To address the growing demand for sustainable construction of these pharmacologically privileged scaffolds, this review interrogates cutting-edge advancements in visible-light-mediated trifluoromethylation of (hetero)arenes and alkenes using TFA as a bench-stable CF3• precursor. By deconvoluting three dominant photocatalytic mechanisms—SET, EDA complex activation, and LMCT—we establish comparative mechanistic frameworks that unify radical initiation dynamics across disparate systems. These analyses reveal design principles for optimizing radical generation efficiency and intermediate stabilization, providing actionable strategies to advance trifluoromethylation technologies toward atom-economic and operationally simple synthesis.
2 Photocatalytic decarboxylating trifluoromethylation of TFA and its derivatives
2.1 Catalyst system for SET
The most frequent SET process in photoredox reactions usually entails the photocatalyst’s gain or loss of an electron. The production of active radical intermediates is facilitated by this mechanism (Prier et al., 2013; Romero and Nicewicz, 2016). Studies reveal that radicals like CF3CO2• or CF3CO• undergo a quick decarbonylation process. The CF3• radicals is produced as a result of the removal of CO2 or CO during this process (Sawada et al., 1990; Zhong et al., 2015). The mechanistic understanding of CF3• radical generation via decarboxylation or decarbonylation laid the foundation for developing photocatalytic trifluoromethylation strategies. A key milestone was achieved in 1993 when Mallouk’s group pioneered the photocatalytic trifluoromethylation of aromatic C-H bonds using CF3CO2Ag as the CF3 source and TiO2 as the catalyst under 500 W Hg lamp irradiation (Scheme 2A; Lai and Mallouk, 1993). This work marked the first demonstration of light-induced trifluoroacetate decarboxylative trifluoromethylation. However, the system suffered from metallic Ag deposition-induced catalyst deactivation, requiring excess TiO2 to sustain reactivity, and exhibited limited regioselectivity for substituted arenes, resulting in mixed isomer products. Mechanistic investigations, including hexafluoroethane detection and radical trapping experiments, conclusively identified CF3• radicals as the pivotal intermediates.
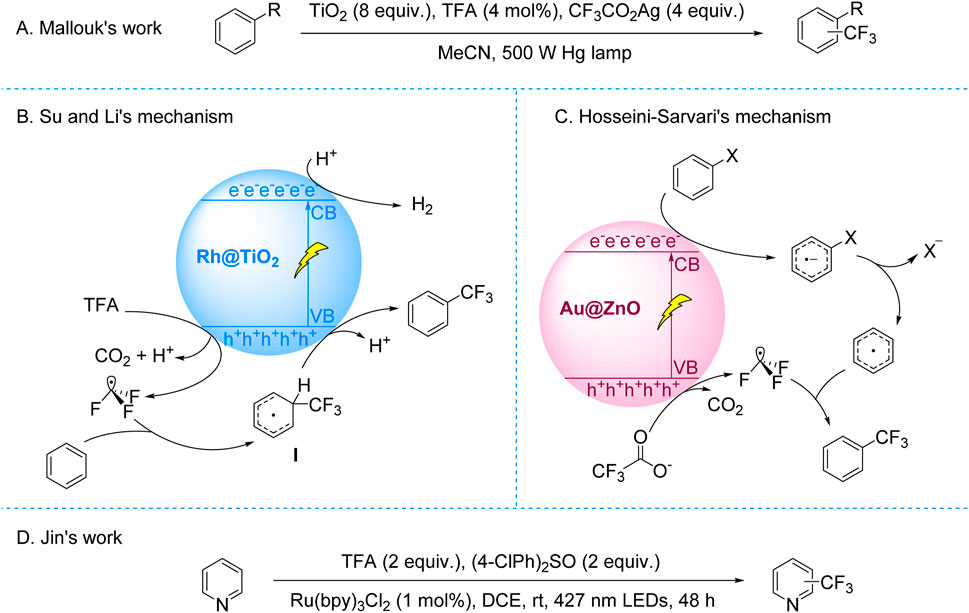
Scheme 2. Photocatalytic decarboxylative trifluoromethylation of TFA and trifluoroacetates. (A) TiO2-catalyzed C–H trifluoromethylation of (hetero)arenes via CF3CO2Ag decarboxylation; (B) Rh@TiO2-catalyzed C–H trifluoromethylation of (hetero)arenes via TFA decarboxylation; (C) Au@ZnO-catalyzed trifluoromethylation of (hetero)arenes via CF3CO2Na decarboxylation; (D) Ru(bpy)3Cl2-catalyzed trifluoromethylation of (hetero)arenes via TFA decarboxylation.
Subsequent advances addressing these challenges expanded both mechanistic understanding and substrate applicability. In 2017, Su and Li developed a Rh@TiO2-mediated photocatalytic system for C–H trifluoromethylation of (hetero)arenes using TFA as a CF3 source under 365 nm UV light. Their mechanistic studies revealed that hole-induced TFA oxidation generated CF3• radicals, which coupled with arenes to form intermediates subsequently oxidized to final products, concurrent with H2 evolution at the conduction band (Scheme 2B; Lin et al., 2017). Complementing this approach, Hosseini-Sarvari’s group employed CF3CO2Na and blue LED irradiation to achieve Au@ZnO-catalyzed trifluoromethylation of diverse C–X (X = I, Br, B) and C–H bonds (Bazyar and Hosseini-Sarvari, 2019). They proposed a dual radical pathway involving conduction band reduction-triggered aryl radical formation and valence band oxidation of CF3CO2− to CF3•, followed by selective radical coupling (Scheme 2C).
The field’s progression toward visible-light compatibility culminated in Jin’s 2020 work employing Ru(bpy)3Cl2 and stoichiometric (4-ClPh)2SO to drive TFA decarboxylative trifluoromethylation under visible light (Scheme 2D). Mechanistic evidence suggested (4-ClPh)2SO facilitates light-induced CF3• generation via TFA decarboxylation, with subsequent dearomative addition to (hetero)arenes forming trifluoromethylated products. Nevertheless, the precise redox synergy between TFA and (4-ClPh)2SO remained unresolved. While these innovations validated TFA-based CF3• generation, persistent limitations—catalyst deactivation, erratic regioselectivity, and UV dependency—underscored the necessity for visible-light systems with enhanced control. This imperative catalyzed the emergence of photoelectrocatalytic paradigms that integrate optical and electrical fields to manipulate radical dynamics, offering new precision in trifluoromethylation strategies.
For instance, the Wu group reported a photoelectrocatalytic platform for direct (hetero)arene C-H trifluoromethylation using TFA, employing a graphite-Pt dual-electrode system under 5 mA current and 390 nm LED irradiation (Qi et al., 2023). This system addressed the scalability challenge by enabling continuous operation without catalyst degradation. Although effective for electron-deficient aromatics, regioselectivity limitations arose from radical-mediated pathways. Mechanistic studies revealed that photoinduced electron-hole pairs at the graphite anode selectively oxidize CF3CO2− through electrostatic interactions, preserving neutral arenes. The generated CF3• radicals undergo dearomative addition to form cyclohexadienyl intermediates, which are subsequently oxidized and deprotonated to regenerate aromaticity, with concurrent H2 evolution at the Pt cathode ensuring charge balance (Scheme 3A). This dual-functional design not only resolved catalyst deactivation but also facilitated late-stage fluorofunctionalization of pharmaceuticals and natural products, demonstrating versatile synthetic applicability.
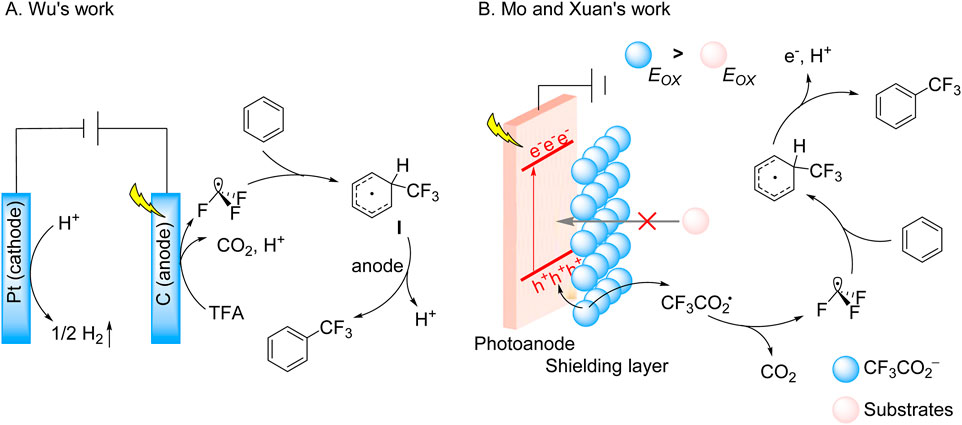
Scheme 3. Photoelectrocatalytic decarboxylative trifluoromethylation of TFA and CF3CO2K. (A) Graphite anode-mediated oxidative TFA decarboxylation for (hetero)arene trifluoromethylation; (B) Mo-doped WO3 anode-mediated oxidative CF3CO2K decarboxylation for (hetero)arene trifluoromethylation.
Building on this, Mo and Xuan introduced an ion-shielded photoelectrocatalytic strategy to address thermodynamic bottlenecks in C–H trifluoromethylation (Chen et al., 2024). Utilizing a Mo-doped WO3 photoanode under 390 nm irradiation with a 2 V bias, CF3CO2− anions formed an electrostatic shielding layer on the anode surface. This layer selectively blocked substrate access to photogenerated holes while promoting preferential oxidation of CF3CO2− to CF3• radicals. These radicals engaged (hetero)arenes via sequential oxidation-dearomatization-deprotonation cascades (Scheme 3B). This study elucidates the mechanistic paradigm for enabling selective electron transfer under extreme anodic polarization while establishing an operationally scalable platform for trifluoromethylation processes.
While photoelectrocatalysis has opened new mechanistic avenues for trifluoromethylation, conventional photocatalytic approaches have simultaneously advanced through systematic engineering of reaction systems. The foundations for these developments were established by early photoredox studies, particularly the seminal 1986 report by Barton and coworkers, who achieved the trifluoromethylation of 1-hydroxypyridine-2-thione using TFAA as the CF3 source (Scheme 4A; Barton et al., 1986). This transformation, which proceeds via light-induced decarboxylation and rearrangement, represented a pioneering example of radical-mediated C–CF3 bond formation. Building upon these fundamental insights, contemporary researchers have developed more sophisticated photocatalytic systems. The Pan group recently demonstrated an efficient, additive-free photoredox protocol utilizing TFAA for the trifluoromethylation of (hetero)aromatics and polarized alkenes (Song et al., 2023). This method exhibits remarkable substrate scope, accommodating natural product derivatives and pharmaceutical scaffolds, while mechanistic studies confirmed the generation of CF3• radicals through visible-light-driven activation of TFAA.
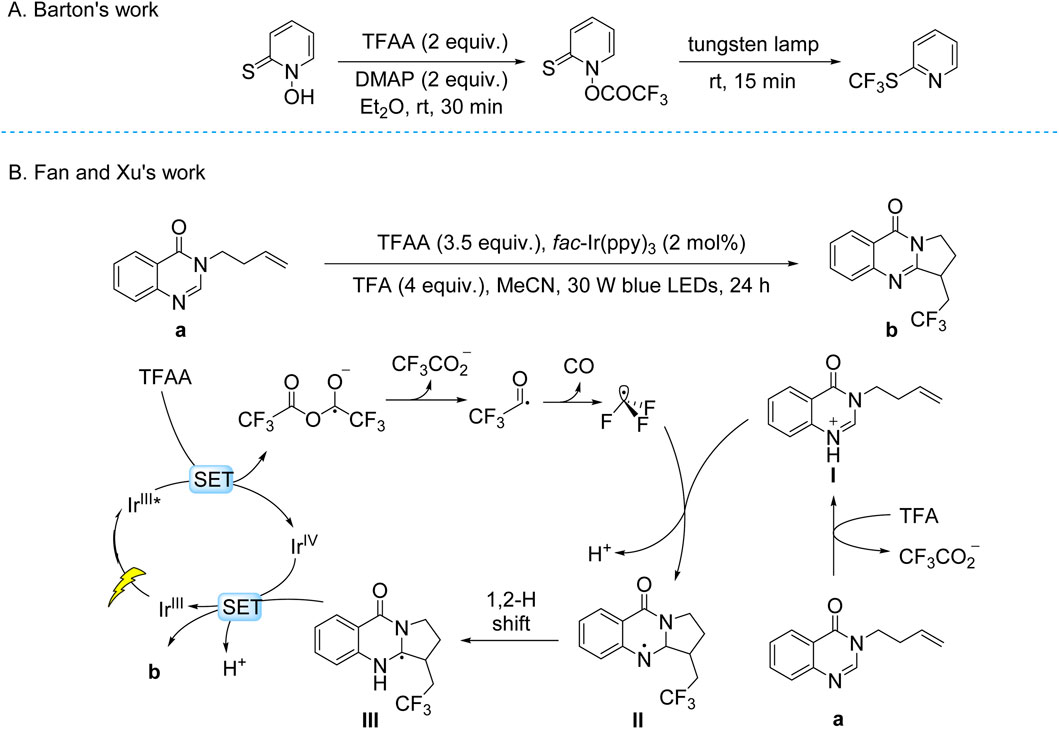
Scheme 4. Photocatalytic decarboxylative trifluoromethylation of TFAA. (A) Trifluoromethylation of 1-hydroxypyridine-2-thione in the early phase using TFAA as a trifluoromethyl source; (B) fac-Ir(ppy)3-catalyzed trifluoromethylation using TFAA as the trifluoromethyl source.
Expanding the synthetic utility of this approach, Fan and Xu group developed a visible-light-mediated tandem trifluoromethylation/cyclization of quinazolinone-tethered alkenes (Zou et al., 2024). Employing fac-Ir(ppy)3 irradiation at 457 nm, this transformation proceeds without strong oxidants to construct tri- and tetracyclic quinazolinones with excellent functional group tolerance. Detailed mechanistic investigations revealed a sequence involving: (i) photoinduced single-electron transfer to TFAA, generating a radical anion that fragments to form CF3CO2•; (ii) decarbonylation to CF3• radicals; and (iii) radical addition followed by cyclization through deprotonation, hydrogen migration, and aromatization steps (Scheme 4B).
These seminal works on TFAA activation exemplify the remarkable progress in photocatalytic trifluoromethylation, yet the field continues to evolve through innovative system design and mechanistic understanding. A prime example of such advancement emerged from the Qing group’s 2018 work, which demonstrated visible-light-driven C–H trifluoromethylation of electron-deficient arenes using Ru(bpy)3(PF6)2 and FPIFA (Yang et al., 2018). The protocol operated under mild conditions, delivering moderate-to-good yields albeit with limited regioselectivity. Mechanistic studies revealed that oxidative quenching of the photoexcited Ru(II)* species by FPIFA generated Ru(III) and a hypervalent iodine intermediate, which subsequently decomposed to release CF3• radicals. These radicals underwent dearomatization with the arene substrate, forming a cyclohexadienyl radical intermediate. This intermediate followed dual oxidation pathways (mediated by Ru(III) or FPIFA) and subsequent deprotonation to yield the trifluoromethylated product (Scheme 5A). Further expanding the reaction scope, Wang, Liang and Wu subsequently developed a hydrotrifluoromethylation protocol for unactivated alkenes using NHBC as a radical precursor (Scheme 5B; Zhang et al., 2019). The proposed mechanism involves oxidative quenching of the photoexcited Ir(III) photocatalyst by NHBC, generating a CF3CO2• that undergoes decarboxylation to produce CF3• radicals. These radicals add to alkenes, followed by HAT-mediated hydrogen abstraction to afford the final hydrotrifluoromethylated product.
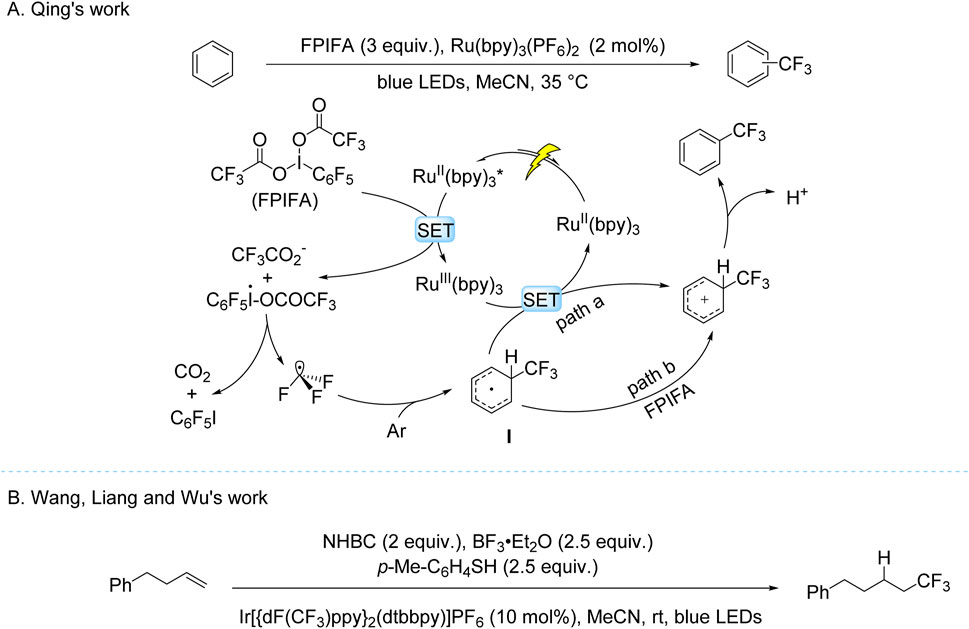
Scheme 5. Photocatalytic single-electron oxidation of TFA derivatives. (A) Ru(bpy)3(PF6)2-catalyzed trifluoromethylation using FPIFA as the trifluoromethyl source; (B) based-Ir-catalyzed trifluoromethylation using NHBC as the trifluoromethyl source.
While SET processes have dominated photoredox trifluoromethylation strategies, recent advances have unveiled the complementary potential of ET mechanisms in radical generation. This paradigm shift is exemplified by the Molander group’s innovative work employing benzophenone-mediated triplet-triplet energy transfer to activate trifluoroacetyl oxime esters (TFAOx) (Majhi et al., 2022). In contrast to conventional SET pathways that rely on redox potentials, this ET approach operates through direct photoexcitation of the oxime ester to its triplet state, triggering N–O bond homolysis and subsequent decarboxylation to generate both CF3• and persistent iminyl radicals. The CF3• radicals undergo kinetically favorable, irreversible and regioselective addition to terminal alkene positions, while the iminyl radicals propagate a chain cycle through interaction with additional oxime ester molecules, ultimately enabling synergistic C–C/C–N bond formation (Scheme 6).
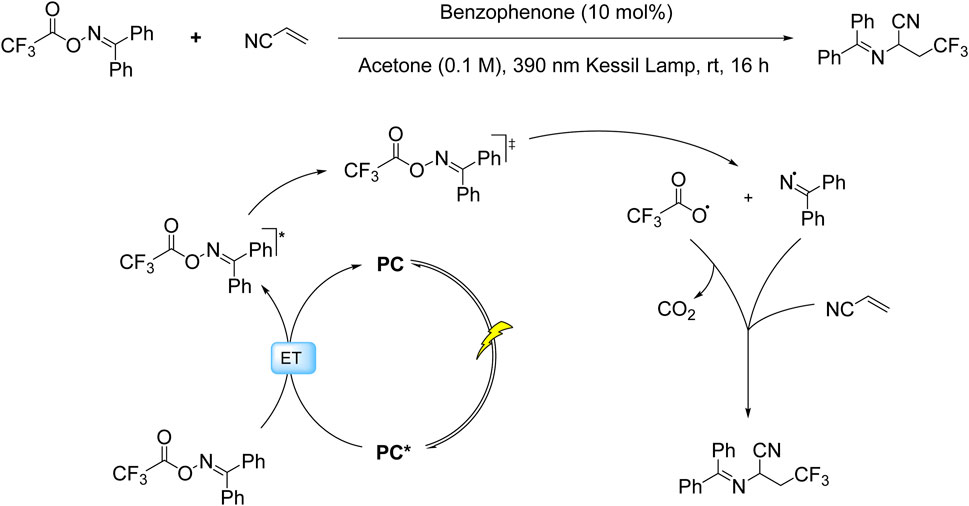
Scheme 6. Photocatalytic ET-mediated decarboxylative trifluoromethylation of alkenes via TFAOx activation.
2.2 Catalyst system for EDA
EDA complexes, formed through non-covalent interactions between electron-rich donors (D) and electron-deficient acceptors (A), enable directional electron transfer under photoexcitation to generate solvent-caged radical ion pairs, which drive diverse reaction pathways such as radical recombination and polarity inversion (Lee and Kochi, 1992; Lima et al., 2016). This mechanism underpins versatile applications in organic synthesis, including catalytic alkylation of carbonyl compounds and stereoselective construction of complex architectures. The EDA paradigm exemplifies a sustainable strategy for achieving spatiotemporal control over reactivity, offering transformative potential for green synthesis of high-value molecules.
The Stephenson group pioneered this approach in 2015, reporting the trifluoromethylation of (hetero)arene C–H bonds using TFAA as the CF3 source, Ru(bpy)3Cl2·6H2O as the photocatalyst, and pyridine N-oxide as the oxidant, with pyridine generated in situ as the base (Beatty et al., 2015). This catalytic system exhibited operational simplicity and scalability to gram-scale synthesis, albeit with moderate regioselectivity and yields. In 2016, the same group expanded on this work, elucidating the mechanism and broadening the substrate scope to include complex (hetero)aromatics, highlighting its potential for pharmaceutical synthesis. Mechanistic studies revealed the formation of a 4-phenyl-1-(trifluoroacetoxy)pyridinium intermediate (I) from TFAA and 4-phenylpyridine N-oxide. Intermediate I generates CF3• radicals through three distinct pathways: (i) EDA complex formation with 4-phenylpyridine N-oxide or aromatic substrates, followed by photoinduced electron transfer to yield radical intermediate II and subsequent release of 4-phenylpyridine and CO2 (Scheme 7, Paths I and II); (ii) Intermediate I oxidizes Ru(II)* via a SET process, forming intermediate II and Ru(III) while releasing 4-phenylpyridine and CO2 simultaneously (Scheme 7, Path III). The resulting CF3• radicals undergo dearomatization via addition to arenes, forming cyclohexadienyl intermediates that re-aromatize upon oxidation to afford the final products (Beatty et al., 2016).
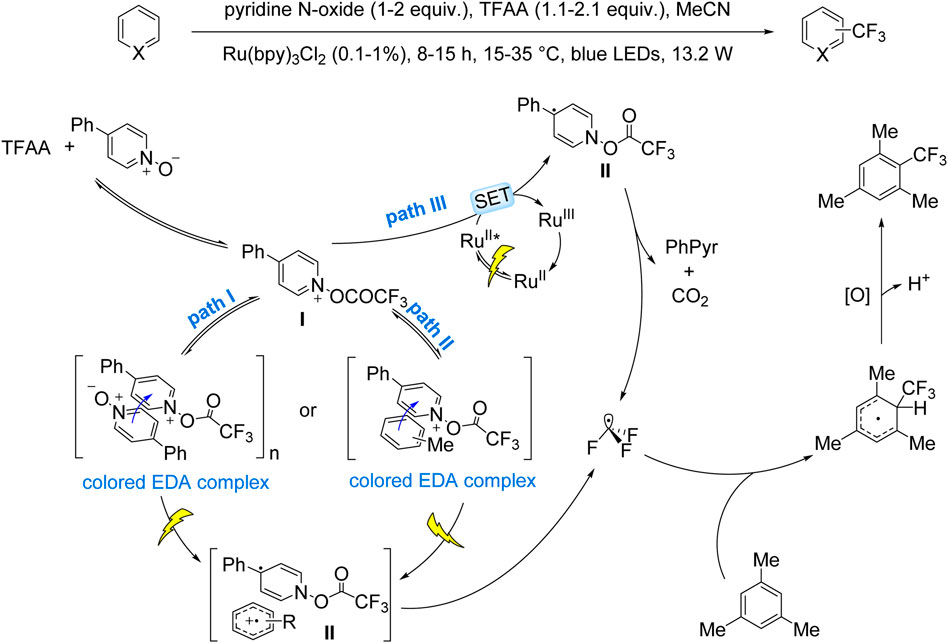
Scheme 7. Stephenson’s pioneering work on EDA-mediated trifluoromethylation.
Building on these foundations, the Leibfarth group adapted the system in 2019 for the trifluoromethylation of industrial plastic waste and commercial plastics (Scheme 8A; Lewis et al., 2019). This work extended the synthetic utility of photocatalytic C–H trifluoromethylation to polymer functionalization, offering a potential strategy for upcycling plastic materials. Concurrently, Zhong and Tong developed a photocatalytic system for the tandem trifluoromethylation and cyclization of secondary benzylamines using TFAA/pyridine N-oxide/Ru(bpy)3Cl2 (Scheme 8B). Mechanistic studies proposed that photoinduced generation of CF3• radicals—via EDA complex-mediated or SET pathways—initiated the transformation. This method demonstrated precise control over regioselectivity in complex heterocycle synthesis (Qi et al., 2019).
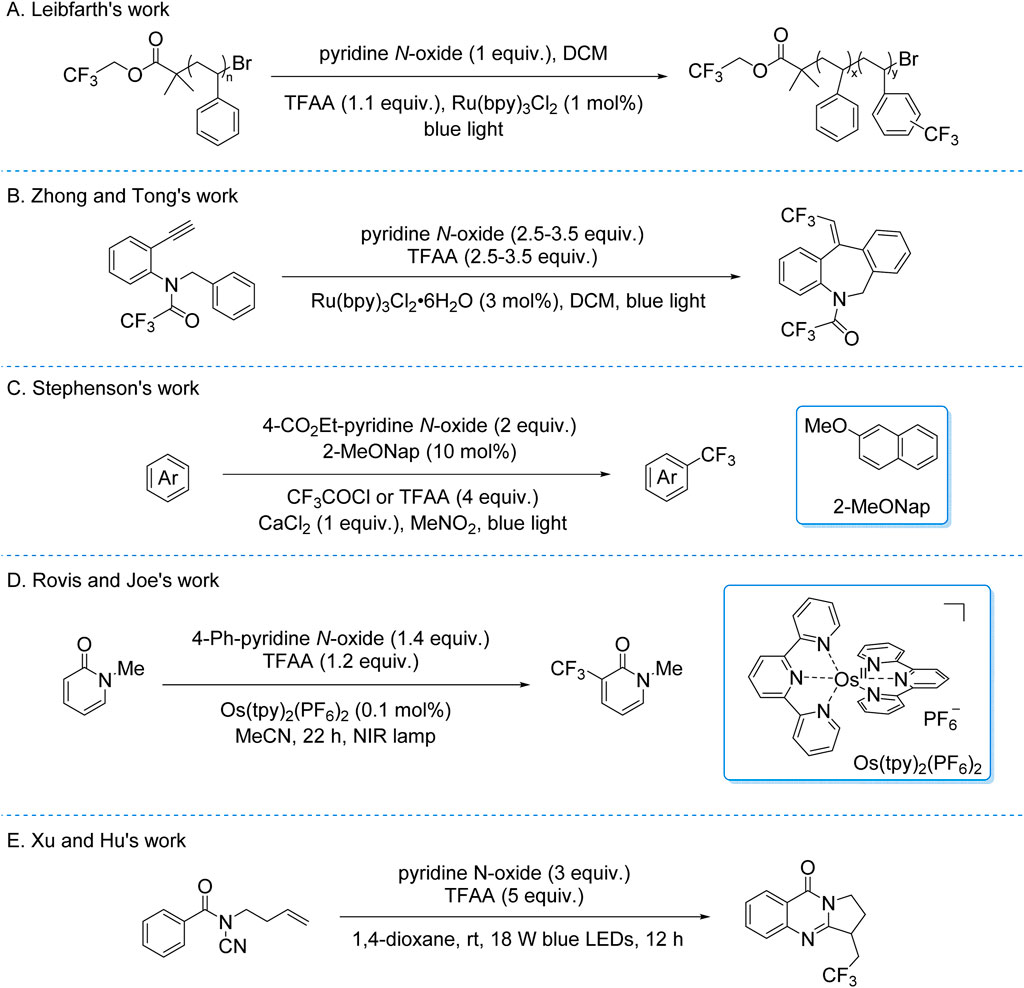
Scheme 8. Photocatalytic generation of CF3• radicals from TFA and derivatives via the EDA process. (A) Leibfarth’s expansion to industrial applications; (B) Advancements in tandem trifluoromethylation and cyclization; (C) Stephenson’s redesign of the EDA platform; (D) Application of Os(tpy)2(PF6)2 as a near-infrared photocatalyst in EDA processes; (E) Tandem trifluoromethylation-cyclization reaction.
In 2020, the Stephenson group redesigned their EDA platform to mitigate back-electron transfer in homogeneous photocatalysis (Scheme 8C; Mcclain et al., 2020). By replacing Ru(bpy)3Cl2 with a dual-component EDA system (2-methoxynaphthalene donor + in situ-generated acylated 4-ethoxycarbonylpyridine N-oxide acceptor from CF3COCl/TFAA), they achieved photosensitizer-free CF3• radicals generation. EDA complex photoexcitation triggered charge transfer, yielding a 2-methoxynaphthalene radical cation and releasing CF3• radicals via acceptor decarboxylation. The radicals engaged in Minisci-type C–H trifluoromethylation of (hetero)arenes, forming a dearomatized intermediate rearomatized by the radical cation to close the catalytic cycle. This strategy achieved moderate yields while eliminating traditional photosensitizers, though catalytic electron donor quantities remained essential. The work demonstrates charge-transfer system engineering to suppress parasitic electron loss, advancing practical late-stage aromatic C–H trifluoromethylation. Furthermore, the Rovis and Joe’s group developed a catalytic system for the trifluoromethylation of 1-methylpyridine-2(1H)-one to synthesize 1-methyl-3-(trifluoromethyl)pyridine-2(1H)-one, employing TFAA/pyridine N-oxide and Os(tpy)2(PF6)2 as the photocatalyst (Scheme 8D). The Os(tpy)2(PF6)2 complex uniquely harnesses near-infrared (NIR) and deep-red (DR) light with minimal energy loss, addressing the ∼25% energy dissipation observed in conventional Ru/Ir-based systems during excited-state quenching (Ravetz et al., 2020).
In 2022, Xu, Hu, et al. developed a photoinduced radical tandem trifluoromethylation/cyclization strategy using TFAA activated by pyridine N-oxide (Scheme 8E; Hu et al., 2022). This methodology leverages an EDA process to generate CF3• radicals, which are captured by N-cyanamide olefins, yielding diverse trifluoromethylated quinazolinones with high efficiency and broad functional group tolerance. While photocatalytic trifluoromethylation strategies exploiting EDA complex mechanisms enable precise C–H activation and heterocycle construction under mild conditions—advancing sustainable fluorofunctionalization for pharmaceutical late-stage modification and complex molecule synthesis—their applicability remains constrained by inherent substrate scope limitations of EDA-mediated processes.
2.3 Catalyst system for LMCT
The LMCT process involves electron transfer from ligand orbitals to empty metal center orbitals, facilitating electron migration from the ligand to the metal center (Juliá, 2022). This process typically occurs in complexes with electrophilic, high-valent metal centers (e.g., Fe(III), Ag(II)) due to the reduced energy barrier for electron transfer. Electron-rich σ- or σ/π-hybridized ligands are more prone to LMCT transitions as they act as intrinsic electron donors (Juliá, 2022). This unique electron transfer mode has opened new pathways for chemical transformations under mild conditions and has been utilized in the trifluoromethylation of organic molecules via TFA decarboxylation (Liu et al., 2024). Mechanistically, a metal catalyst forms a complex with CF3CO2−, which undergoes the LMCT process under visible light to generate CF3CO2•. These radicals drive β-bond cleavage and subsequent transformations, underscoring the potential of LMCT in catalyzing decarboxylative trifluoromethylation of TFA and its salts.
The Juliá-Hernández group developed a visible-light-driven trifluoromethylation of (hetero)arenes using CF3CO2Na as the CF3 source and an Fe(OTf)2/4,4′-dimethoxy-2,2′-bipyridine/K2S2O8 system under 405 nm irradiation (Scheme 9A; Fernandez-Garcia et al., 2024). Mechanistic studies revealed a LMCT mechanism: Fe-ligand complexes underwent photoinduced O–Fe bond homolysis, generating CF3CO2• radicals that decarboxylated to CF3• radicals. These radicals participated in Minisci-type addition to electron-rich (hetero)aromatics, forming intermediates oxidized to final products. The protocol demonstrated broad substrate tolerance, including pyrroles, indoles, and bioactive molecules, with moderate to good yields. However, regioselectivity remained limited for polyfunctional substrates, reflecting inherent challenges in controlling radical pathways in complex systems.
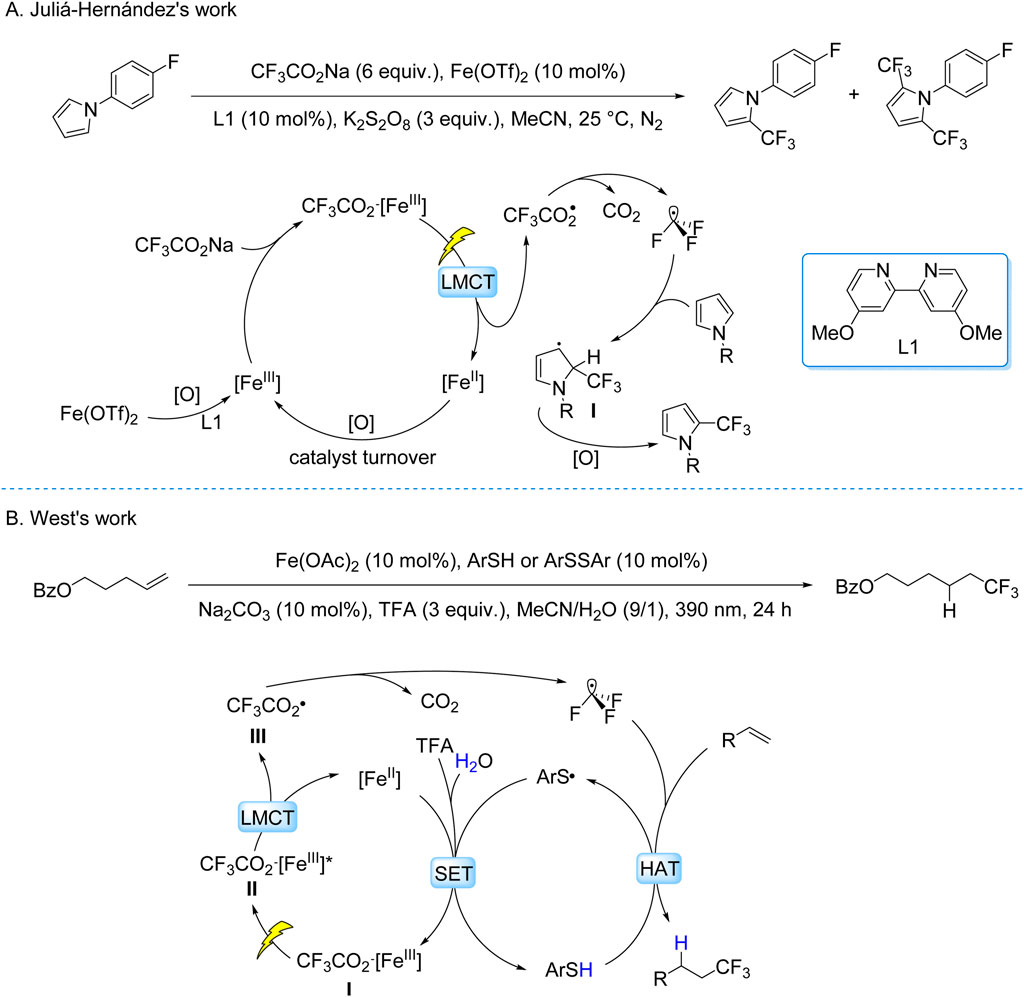
Scheme 9. Photocatalytic generation of •CF3 from TFA and derivatives via the LMCT process. (A) Fe(AcO)2-catalyzed generation of •CF3 from CF3CO2Na via LMCT process; (B) Ag(bpy)2(OTf)2 -catalyzed generation of •CF3 from CF3CO2Na via LMCT process.
The West group developed a redox-neutral hydrofluoroalkylation of alkenes under mild conditions, employing TFA as the CF3 source, Fe(OAc)2 as the catalyst, aryl thiols (ArSH) or disulfides (ArSSAr) as HAT mediators, Na2CO3 as the base, and 390 nm light irradiation (Bian et al., 2023). This dual catalytic system synergizes LMCT and HAT pathways through four key stages: (i) photolytic cleavage of the O–Fe bond in the Fe(II)–TFA coordination complex generates CF3CO2• concurrent with reduction of Fe(III) to Fe(II); (ii) decarboxylation of CF3CO2• produces CF3• radicals; (iii) radical addition to alkenes forms transient carbon-centered radical intermediates; (iv) HAT from ArSH to the intermediate delivers hydrofluoroalkylated products (Scheme 9B). This mechanistically integrated platform enables late-stage installation of C–CF3 bonds in structurally complex bioactive molecules, demonstrating compatibility with over twenty functional groups including electrophilic and protic moieties.
The Nocera group developed a dual-mode catalytic strategy for arene trifluoromethylation using Ag(bpy)2(OTf)2 and CF3CO2Na under visible light irradiation. The reaction proceeds via a LMCT mechanism: photoexcitation of the electrophilic Ag(II) center in the Ag(II)–CF3CO2− coordination complex induces O–Ag bond homolysis, generating CF3• radicals. Two distinct operational modes were engineered—(i) a stoichiometric photocatalytic system (2.0 equiv Ag) to offset incomplete Ag(I)-to-Ag(II) reoxidation, and (ii) a catalytic photoelectrochemical system (2.5 mol% Ag) leveraging anodic oxidation to regenerate Ag(II) from Ag(I), enabling sustained turnover. The CF3• radicals engage in dearomatizing addition to arenes, followed by Ag(II)-mediated rearomatization to furnish trifluoromethylated aromatic products (Scheme 10A; Campbell et al., 2024). This LMCT-driven platform exhibits broad functional group tolerance under mild conditions, enabling scalable synthesis of pharmaceuticals and advanced materials. Notably, the photoelectrochemical variant circumvents high catalyst loadings typical of conventional photoredox systems, showcasing a synergistic integration of light- and electricity-driven processes for energy-efficient bond activation.
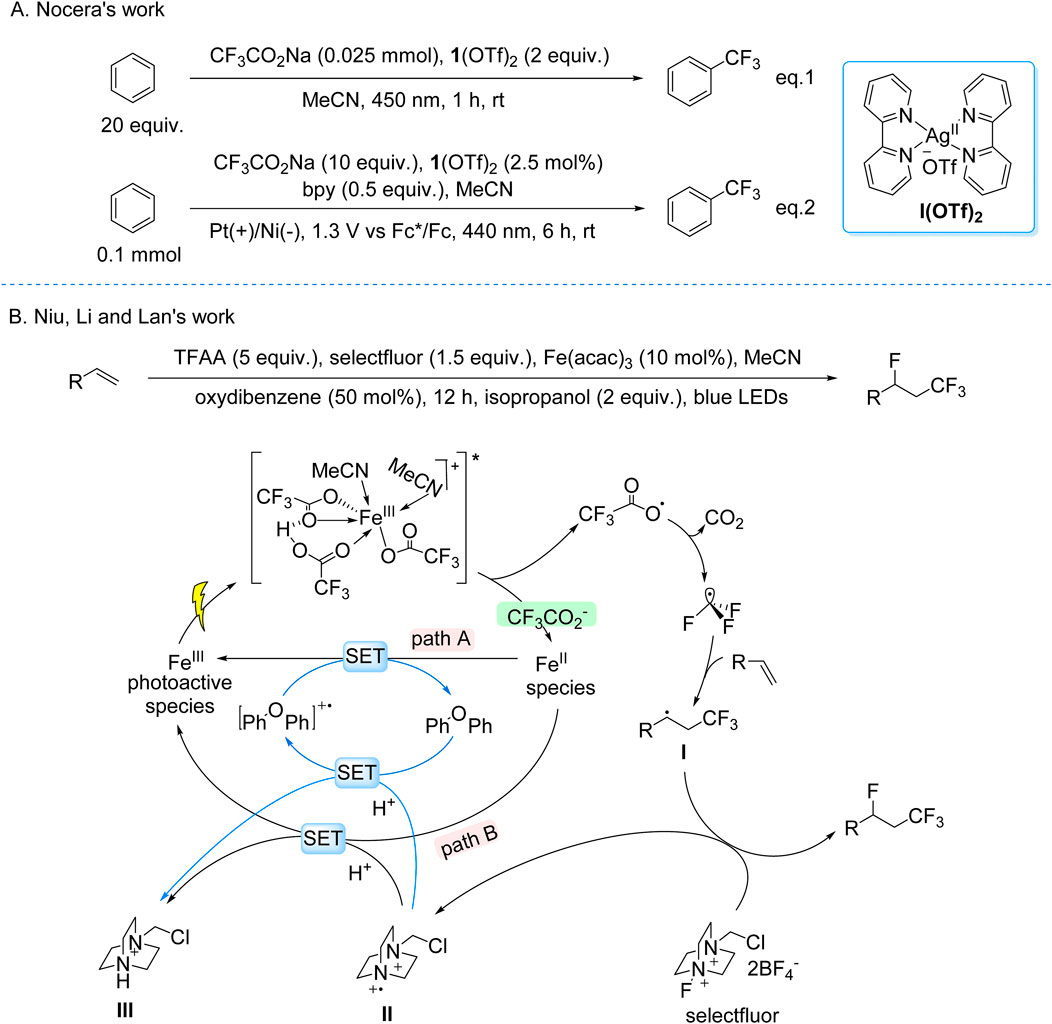
Scheme 10. Photocatalytic generation of •CF3 from TFA and derivatives via the LMCT process. (A) Ag(OTf)2-catalyzed generation of •CF3 from TFA via LMCT process; (B) Fe(acac)3-catalyzed generation of •CF3 from TFAA via LMCT process.
Niu, Li, and Lan recently reported a visible-light-driven photocatalytic fluorotrifluoromethylation of unactivated olefins using Fe(acac)3 as the catalyst, TFAA as the CF3 source, and Selectfluor as the fluorine donor (Scheme 10B). Mechanistic studies revealed that the active species is an in situ-formed Fe(III) complex with CF3CO2−, H+, and acetonitrile, where Brønsted acids enhance LMCT efficiency via hydrogen-bonding interactions. Upon visible-light irradiation, LMCT excitation triggers decarboxylation of the Fe(III) complex, generating CF3• radicals and reducing Fe(III) to Fe(II). The CF3• radicals add to unactivated olefins, forming a carbon-centered radical intermediate, which abstracts a fluorine atom from Selectfluor to yield fluorotrifluoromethylated products. The catalytic cycle is sustained through two pathways: direct Fe(II) reoxidation by intermediate II (Path B) or via a diaryl ether-derived cation radical generated by intermediate II-mediated oxidation (Path A) (Jiang et al., 2024).
LMCT catalysis has emerged as a powerful strategy for trifluoromethylation under mild conditions, leveraging high-valent metal complexes (e.g., Fe(III), Ag(II)) to generate reactive CF3CO2• radicals via photoinduced O–M bond cleavage. These systems exhibit broad substrate scope and operational simplicity, bridging the gap between photoredox and transition-metal catalysis. While challenges in regioselectivity control and catalytic turnover efficiency persist, their ability to enable sustainable transformations underscores LMCT’s potential for mechanistic innovation. This progress highlights LMCT’s transformative role in radical chemistry, prompting further exploration of scalability and selectivity in next-generation synthetic methodologies.
3 Conclusion
This review provides an overview of the recent advancements in photocatalytic decarboxylative trifluoromethylation using TFA and its derivatives at room temperature, with a detailed exploration of three major catalytic systems: SET, EDA, and LMCT. While the SET process has enabled the oxidation and decarboxylation of TFA to some extent, the use of high-energy light sources (such as tungsten, mercury, and UV lamps) has led to lower reaction selectivity. Although noble metal catalysts can activate TFA under visible light, their high cost hinders industrial applications. The EDA process, which induces electron transfer through the formation of electron donor-acceptor complexes under light irradiation, has mitigated some of the stringent conditions required for intermolecular electron transfer but still needs optimization to enhance efficiency and selectivity. The LMCT process, particularly the photoinduced homolysis reactions based on 3d transition metals (e.g., iron), offers a sustainable alternative for visible-light-driven TFA activation, demonstrating the potential for activating high-redox-potential molecules under mild conditions. However, research on this system remains limited, and further expansion to a broader range of transition metal catalysts and applications is needed.
Despite significant progress in the field of TFA and its derivatives for decarboxylative trifluoromethylation, numerous challenges and opportunities remain. On one hand, the development of more efficient, green, and selective catalytic systems remains a key research focus. Future efforts could explore novel ligands or catalyst structures to optimize the LMCT process, enhance the generation efficiency of trifluoromethyl radicals, and thereby improve the activity and selectivity of the reactions. On the other hand, the photoinduced multi-photon redox process, although showing promise, is still in its developmental stage. A deeper understanding of its reaction mechanisms and exploration of how to more precisely control the photon absorption process are needed to achieve the transformation of high-redox-potential and high-bond-energy molecules under milder conditions. Additionally, expanding the substrate scope beyond olefins and arenes to other types of compounds for trifluoromethylation reactions is an important direction to further enrich the scope of organic synthesis. Simultaneously, integrating theoretical calculations with experimental studies will aid in a deeper understanding of the microscopic mechanisms of electron transfer and energy transfer in the reaction processes, providing theoretical guidance for designing more rational catalytic systems. As research continues to advance, it is anticipated that trifluoromethylation reactions will play an increasingly important role in the field of organic synthesis, offering more compounds with unique properties for applications in drug development, materials science, and beyond.
Author contributions
F-FT: Writing – original draft, Writing – review and editing. Z-CL: Writing – review and editing, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by the Natural Science Research Fund of Shaanxi Provincial Department of Education (grant number 22JK0384), the Doctoral Research Start-up Fund of Weinan Normal University (grant number 2022RC18), and the Open Fund of Key Laboratory of Fine Chemicals and Surfactants of Sichuan Province (grant number 2022JXY05). The APC was funded by the Doctoral Research Start-up Fund of Weinan Normal University (grant number 2022RC18).
Acknowledgments
The authors sincerely thank the China Scholarship Council (CSC) for the support provided. We thank the University of Göttingen for the academic collaboration and resource sharing. We deeply appreciate the research team of Professor Kai Zhang for generously providing advanced research facilities.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Andreev, V. N., Grinberg, V. A., Dedov, A. G., Loktev, A. S., Mayorova, N. A., Moiseev, I. I., et al. (2013). Anodic trifluoromethylation of 10-undecylenic acid. Russ. J. Electrochem. 49, 996–1000. doi:10.1134/s1023193513100030
Arai, K., Watts, K., and Wirth, T. (2014). Difluoro-and trifluoromethylation of electron-deficient alkenes in an electrochemical microreactor. ChemistryOpen 3, 23–28. doi:10.1002/open.201300039
Barton, D. H. R., Lacher, B., and Zard, S. Z. (1986). The invention of new radical chain-reactions. Part 13: generation and reactivity of perfluoroalkyl radicals from thiohydroxamic esters. Tetrahedron 42, 2325–2328. doi:10.1016/s0040-4020(01)90613-1
Bazyar, Z., and Hosseini-Sarvari, M. (2019). Au@ZnO core-shell: scalable photocatalytic trifluoromethylation using CF3CO2Na as an inexpensive reagent under visible light irradiation. Org. Process Res. Dev. 23, 2345–2353. doi:10.1021/acs.oprd.9b00225
Beatty, J. W., Douglas, J. J., Cole, K. P., and Stephenson, C. R. J. (2015). A scalable and operationally simple radical trifluoromethylation. Nat. Commun. 6, 7919. doi:10.1038/ncomms8919
Beatty, J. W., Douglas, J. J., Miller, R., Mcatee, R. C., Cole, K. P., and Stephenson, C. R. J. (2016). Photochemical perfluoroalkylation with pyridine N-oxides: mechanistic insights and performance on a kilogram scale. Chem 1, 456–472. doi:10.1016/j.chempr.2016.08.002
Bian, K.-J., Lu, Y.-C., Nemoto, D., Kao, S.-C., Chen, X., and West, J. G. (2023). Photocatalytic hydrofluoroalkylation of alkenes with carboxylic acids. Nat. Chem. 15, 1683–1692. doi:10.1038/s41557-023-01365-0
Campbell, B. M., Gordon, J. B., Raguram, E. R., Gonzalez, M. I., Reynolds, K. G., Nava, M., et al. (2024). Electrophotocatalytic perfluoroalkylation by LMCT excitation of Ag(II) perfluoroalkyl carboxylates. Science 383, 279–284. doi:10.1126/science.adk4919
Charpentier, J., Früh, N., and Togni, A. (2015). Electrophilic trifluoromethylation by use of hypervalent iodine reagents. Chem. Rev. 115, 650–682. doi:10.1021/cr500223h
Chen, Y., He, Y., Gao, Y., Xue, J., Qu, W., Xuan, J., et al. (2024). Scalable decarboxylative trifluoromethylation by ion-shielding heterogeneous photoelectrocatalysis. Science 384, 670–676. doi:10.1126/science.adm8902
Cheng, Y. Z., Yuan, X. G., Ma, J., and Yu, S. Y. (2015). Direct aromatic C-H trifluoromethylation via an electron-donor-acceptor complex. Chem. - Eur. J. 21, 8355–8359. doi:10.1002/chem.201500896
Depecker, C., Marzouk, H., Trevin, S. P., and Devynck, J. (1999). Trifluoromethylation of aromatic compounds via Kolbe electrolysis in pure organic solvent. Study on laboratory and pilot scale. New J. Chem. 23, 739–742. doi:10.1039/A901305I
Dolbier, W. R. (1996). Structure, reactivity, and chemistry of fluoroalkyl radicals. Chem. Rev. 96, 1557–1584. doi:10.1021/cr941142c
Fernandez-Garcia, S., Chantzakou, V. O., and Julia-Hernandez, F. (2024). Direct decarboxylation of trifluoroacetates enabled by iron photocatalysis. Angew. Chem. Int. Ed. 63, e202311984. doi:10.1002/anie.202311984
Fujiwara, Y., Dixon, J. A., O'hara, F., Funder, E. D., Dixon, D. D., Rodriguez, R. A., et al. (2012). Practical and innate carbon-hydrogen functionalization of heterocycles. Nature 492, 95–99. doi:10.1038/nature11680
Guo, Q., Wang, M., Liu, H., Wang, R., and Xu, Z. (2018). Visible-light-promoted dearomative fluoroalkylation of beta-naphthols through intermolecular charge transfer. Angew. Chem. Int. Ed. 57, 4747–4751. doi:10.1002/anie.201800767
Hu, Q., Yu, W.-L., Luo, Y.-C., Hu, X.-Q., and Xu, P.-F. (2022). A photosensitizer–free radical cascade for synthesizing CF3-containing polycyclic quinazolinones with visible light. J. Org. Chem. 87, 1493–1501. doi:10.1021/acs.joc.1c02889
Ji, X., Shi, G., and Zhang, Y. (2019). Progress of trifluoromethylation using trifluoroacetic acid and its derivatives as CF3-sources. Chin. J. Org. Chem. 39, 929–939. doi:10.6023/cjoc201810033
Jiang, X., Lan, Y., Hao, Y., Jiang, K., He, J., Zhu, J., et al. (2024). Iron photocatalysis via Bronsted acid-unlocked ligand-to-metal charge transfer. Nat. Commun. 15, 6115. doi:10.1038/s41467-024-50507-6
Jiang, Y., Li, B., Ma, N., Shu, S., Chen, Y., Yang, S., et al. (2021). Photoredox-catalyst-enabled para-selective trifluoromethylation of tert-butyl arylcarbamates. Angew. Chem. Int. Ed. 60, 19030–19034. doi:10.1002/anie.202105631
Juliá, F. (2022). Ligand-to-Metal charge transfer (LMCT) photochemistry at 3d-metal complexes: an emerging tool for sustainable organic synthesis. ChemCatChem 14, e202200916. doi:10.1002/cctc.202200916
Krishnamurti, V., Munoz, S. B., Ispizua-Rodriguez, X., Vickerman, J., Mathew, T., and Surya Prakash, G. K. (2018). C(sp(2))-H Trifluoromethylation of enamides using TMSCF3: access to trifluoromethylated isoindolinones, isoquinolinones, 2-pyridinones and other heterocycles. Chem. Commun. 54, 10574–10577. doi:10.1039/c8cc04907f
Lai, C. W., and Mallouk, T. E. (1993). A new approach to the photochemical trifluoromethylation of aromatic-compounds. J. Chem. Soc. Chem. Commun. 47, 1359–1361. doi:10.1039/C39930001359
Le, C. C., Wisiner, M. K., Shi, Z.-C., Zhang, R., Conway, D. V., Li, G., et al. (2017). A general small-scale reactor to enable standardization and acceleration of photocatalytic reactions. ACS Cent. Sci. 3, 647–653. doi:10.1021/acscentsci.7b00159
Lee, K. Y., and Kochi, J. K. (1992). Charge-transfer structures of aromatic EDA complexes with N-heteroatom-substituted pyridinium cations. J. Chem. Soc. Perkin Trans. 2, 1011. doi:10.1039/p29920001011
Lewis, S. E., Wilhelmy, B. E., and Leibfarth, F. A. (2019). Upcycling aromatic polymers through C-H fluoroalkylation. Chem. Sci. 10, 6270–6277. doi:10.1039/c9sc01425j
Lima, C. G. S., De, M., Lima, T., Duarte, M., Jurberg, I. D., and Paixão, M. W. (2016). Organic synthesis enabled by light-irradiation of EDA complexes: theoretical background and synthetic applications. ACS Catal. 6, 1389–1407. doi:10.1021/acscatal.5b02386
Lin, J., Li, Z., Kan, J., Huang, S., Su, W., and Li, Y. (2017). Photo-driven redox-neutral decarboxylative carbon-hydrogen trifluoromethylation of (hetero)arenes with trifluoroacetic acid. Nat. Commun. 8, 14353. doi:10.1038/ncomms14353
Liu, J. Y., Cui, Z. W., Bi, J. J., He, X., Ding, Q. J., Zhu, H., et al. (2024). Photocatalytic fluoroalkylation by ligand-to-metal charge transfer. Front. Chem. 12, 1481342. doi:10.3389/fchem.2024.1481342
Liu, P., Liu, W., and Li, C.-J. (2017). Catalyst-free and redox-neutral innate trifluoromethylation and alkylation of aromatics enabled by light. J. Am. Chem. Soc. 139, 14315–14321. doi:10.1021/jacs.7b08685
Majhi, J., Dhungana, R. K., Rentería-Gómez, Á., Sharique, M., Li, L., Dong, W., et al. (2022). Metal-free photochemical imino-alkylation of alkenes with bifunctional oxime esters. J. Am. Chem. Soc. 144, 15871–15878. doi:10.1021/jacs.2c07170
Mcclain, E. J., Monos, T. M., Mori, M., Beatty, J. W., and Stephenson, C. R. J. (2020). Design and implementation of a catalytic electron donor-acceptor complex platform for radical trifluoromethylation and alkylation. ACS Catal. 10, 12636–12641. doi:10.1021/acscatal.0c03837
Mckee, M. L., and Shevlin, P. B. (1985). A theoretical evaluation of the mechanism of acetylene formation in the reactions of atomic carbon with hydrocarbons. J. Am. Chem. Soc. 107, 5191–5198. doi:10.1021/ja00304a027
Mejía, E., and Togni, A. (2012). Rhenium-catalyzed trifluoromethylation of arenes and heteroarenes by hypervalent iodine reagents. ACS Catal. 2, 521–527. doi:10.1021/cs300089y
Meucci, E. A., Nguyen, S. N., Camasso, N. M., Chong, E., Ariafard, A., Canty, A. J., et al. (2019). Nickel(IV)-Catalyzed C-H trifluoromethylation of (Hetero)arenes. J. Am. Chem. Soc. 141, 12872–12879. doi:10.1021/jacs.9b06383
Muller, K., Faeh, C., and Diederich, F. (2007). Fluorine in pharmaceuticals: looking beyond intuition. Science 317, 1881–1886. doi:10.1126/science.1131943
Muralirajan, K., Kancherla, R., Bau, J. A., Taksande, M. R., Qureshi, M., Takanabe, K., et al. (2021). Exploring the structure and performance of Cd–chalcogenide photocatalysts in selective trifluoromethylation. ACS Catal. 11, 14772–14780. doi:10.1021/acscatal.1c04053
Natte, K., Jagadeesh, R. V., He, L., Rabeah, J., Chen, J., Taeschler, C., et al. (2016). Palladium-catalyzed trifluoromethylation of (hetero) arenes with CF3Br. Angew. Chem. Int. Ed. 55, 2782–2786. doi:10.1002/anie.201511131
Ni, C., Hu, M., and Hu, J. (2015). Good partnership between sulfur and fluorine: sulfur-based fluorination and fluoroalkylation reagents for organic synthesis. Chem. Rev. 115, 765–825. doi:10.1021/cr5002386
O'brien, A. G., Maruyama, A., Inokuma, Y., Fujita, M., Baran, P. S., and Blackmond, D. G. (2014). Radical C-H functionalization of heteroarenes under electrochemical control. Angew. Chem. Int. Ed. 53, 11868–11871. doi:10.1002/anie.201407948
Ouyang, Y., Xu, X.-H., and Qing, F.-L. (2018). Trifluoromethanesulfonic anhydride as a low-cost and versatile trifluoromethylation reagent. Angew. Chem. Int. Ed. 57, 6926–6929. doi:10.1002/anie.201803566
Prier, C. K., Rankic, D. A., and Macmillan, D. W. C. (2013). Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev. 113, 5322–5363. doi:10.1021/cr300503r
Purser, S., Moore, P. R., Swallow, S., and Gouverneur, V. (2008). Fluorine in medicinal chemistry. Chem. Soc. Rev. 37, 320–330. doi:10.1039/b610213c
Qi, J., Xu, J., Ang, H. T., Wang, B., Gupta, N. K., Dubbaka, S. R., et al. (2023). Electrophotochemical synthesis facilitated trifluoromethylation of arenes using trifluoroacetic acid. J. Am. Chem. Soc. 145, 24965–24971. doi:10.1021/jacs.3c10148
Qi, X.-K., Zhang, H., Pan, Z.-T., Liang, R.-B., Zhu, C.-M., Li, J.-H., et al. (2019). Photoinduced synthesis of fluorinated dibenz b,e azepines via radical triggered cyclization. Chem. Commun. 55, 10848–10851. doi:10.1039/c9cc04977k
Ravetz, B. D., Tay, N. E. S., Joe, C. L., Sezen-Edmonds, M., Schmidt, M. A., Tan, Y., et al. (2020). Development of a platform for near-infrared photoredox catalysis. ACS Cent. Sci. 6, 2053–2059. doi:10.1021/acscentsci.0c00948
Romero, N. A., and Nicewicz, D. A. (2016). Organic photoredox catalysis. Chem. Rev. 116, 10075–10166. doi:10.1021/acs.chemrev.6b00057
Sawada, H., Nakayama, M., Yoshida, M., Yoshida, T., and Kamigata, N. (1990). Trifluoromethylation of aromatic-compounds with bis(trifluoroacetyl) peroxide. J. Fluor. Chem. 46, 423–431. doi:10.1016/s0022-1139(00)82927-9
Song, Y., Zheng, B., Yang, S., Li, Y., Liu, Q., and Pan, L. (2023). Trifluoromethylations of (Hetero)arenes and polarized alkenes using trifluoroacetic anhydride under photoredox catalysis. Org. Lett. 25, 2372–2376. doi:10.1021/acs.orglett.3c00890
Sun, S.-Z., Xu, H., and Dai, H.-X. (2019). Copper-catalyzed alpha-selective C-H trifluoromethylation of acrylamides with TMSCF3. Chin. Chem. Lett. 30, 969–972. doi:10.1016/j.cclet.2019.02.011
Wang, D., Deng, G.-J., Chen, S., and Gong, H. (2016). Catalyst-free direct C-H trifluoromethylation of arenes in water-acetonitrile. Green Chem. 18, 5967–5970. doi:10.1039/c6gc02000c
Yang, B., Yu, D., Xu, X.-H., and Qing, F.-L. (2018). Visible-light photoredox decarboxylation of perfluoroarene iodine(III) trifluoroacetates for C–H trifluoromethylation of (Hetero)arenes. ACS Catal. 8, 2839–2843. doi:10.1021/acscatal.7b03990
Zhang, W., Zou, Z., Wang, Y., Wang, Y., Liang, Y., Wu, Z., et al. (2019). Leaving group assisted strategy for photoinduced fluoroalkylations using N-hydroxybenzimidoyl chloride esters. Angew. Chem. Int. Ed. 58, 624–627. doi:10.1002/anie.201812192
Zhong, S., Hafner, A., Hussal, C., Nieger, M., and Bräse, S. (2015). Metal-free radical perfluoroalkylation of (hetero) arenes. RSC Adv. 5, 6255–6258. doi:10.1039/c4ra13430c
Keywords: photocatalysis, trifluoromethylation, trifluoroacetic acid (TFA), decarboxylation, radical
Citation: Tan F-F and Li Z-C (2025) Advances in photocatalytic research on decarboxylative trifluoromethylation of trifluoroacetic acid and derivatives. Front. Chem. 13:1602003. doi: 10.3389/fchem.2025.1602003
Received: 28 March 2025; Accepted: 02 May 2025;
Published: 14 May 2025.
Edited by:
Fateh V. Singh, VIT University, IndiaReviewed by:
Wengang Xu, China University of Petroleum (East China), ChinaFrancisco Juliá-Hernández, University of Murcia, Spain
Petros Gkizis, National and Kapodistrian University of Athens, Greece
Jadab Majhi, University of Pennsylvania, United States
Copyright © 2025 Tan and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fang-Fang Tan, ZmFuZ2Zhbmd0YW5AdW5pLWdvZXR0aW5nZW4uZGU=