 Romina E. Avanzo1
Romina E. Avanzo1 Guadalupe García Liñares
Guadalupe García Liñares Angel H. Romero
Angel H. Romero- 1Laboratorio de Biocatálisis, Departamento de Química Orgánica y UMYMFOR, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Ciudad Universitaria, Buenos Aires, Argentina
- 2Laboratorio de Ingeniería Genética, Instituto de Biomedicina “Dr. Jacinto Convit”, Facultad de Medicina, Universidad Central de Venezuela, Caracas, Venezuela
- 3Grupo de Química Orgánica Medicinal, Facultad de Ciencias, Universidad de la República, Montevideo, Uruguay
Antimalarial drugs based on quinolines have been widely used as leishmanicidal agents for either cutaneous or visceral leishmaniasis models. Herein, we showed the leishmanicidal response against in vitro models of different Leishmania spp. and against in vivo models of eleven key antimalarials, including chloroquine, sitamaquine, amodiaquine, mefloquine, quinine, primaquine, hydroxychloroquine, tafenoquine, quinacrine and moxipraquine. Mechanistic studies and advances in clinical treatment are also discussed. This mini-review aims to show the state of the art in using antimalarial drugs to discover alternative therapies for leishmaniasis treatment.
1 Introduction
Leishmaniasis is one of the most important Neglected Tropical Diseases (NTDs) due to its prevalence in tropical and subtropical regions, being present in 98 countries. That disease is caused by more than 20 species of intracellular parasites of Leishmania (Murray et al., 2005). The disease presents three clinical manifestations: cutaneous leishmaniasis (CL), visceral leishmaniasis (VL) and mucocutaneous leishmaniasis (MCL), registering between 0.7 and 1.3 million new cases and between 26,000 and 65,000 deaths annually (Kumar, 2021; World Health Organization, 2023), being the majority of cases and deaths associated with CL and VL, respectively.
Another challenge within the leishmaniasis field is the absence of vaccines or therapeutic alternatives. Current treatments for leishmaniasis are predominantly chemotherapeutic based on pentavalent antimonials (e.g., Glucantime® and Pentostam®) and pentamidine which are not approved by FDA and other FDA-approved drugs such as amphotericin B and miltefosine (Aronson et al., 2016; Kumari et al., 2022). In general, these commercial drugs present strong side effects (affecting the heart, liver, and kidneys), discomfort during treatment, high cost, low therapeutic efficacy, prolonged treatment duration (30–60 days) and emergence or resistance cases (NIH, 2022). Combination therapies using diverse types of drugs (Mota et al., 2024; Sundar et al., 2024), liposomes and nanoparticles for controlled drug release (Mendes et al., 2020), and repositioning drugs have been used as emerging therapies to improve the efficiency (Chartlon et al., 2017). Alternatively, Drug for Neglected Disease Innovative (DNDi), European and Asian agencies have made great investments, which have allowed them to identify new promising chemotherapeutic entities; however, the failure rate has been too high (only 20 out of 4,200,000 tested) (Drugs for Neglected Diseases initiative, 2023). That situation obligates us to develop new alternatives beyond the classic concept of medicinal chemistry for drug discovery, focusing on key aspects of parasite survival within macrophages. In this sense, quinoline, particularly 4-aminoquinoline, emerges as a privileged scaffold for the development of selective and potent leishmanicidal agents targeting phagolysosome and activating the immune system of the immune-suppressed macrophage (Romero and Delgado, 2025; Del Carpio et al., 2025; Romero, 2019). That type of aminoquinoline is highly attractive from the synthetic point of view because a variety of synthetic strategies is available to functionalize any of the quinoline positions (Delgado et al., 2025; Chanquia et al., 2019). Natural products based on quinolines have also generated active compounds (Yaluf et al., 2025). The relevance of the quinolines is even more notable for the existence of multiple reports concerning the use of antimalarials against Leishmania parasites for in vitro or in vivo models. Antimalarial drugs represent one of the first choices for the repurposing program to discover new chemotherapeutic alternatives against leishmaniasis. Then, this minireview aims to provide a general recopilation of reported examples of eleven antimalarial drugs based on quinolines including chloroquine (CQ), sitamaquine (SQ), amodiaquine (AQ), mefloquine (MQ), quinine (QN), primaquine (PQ), hydroxychloroquine (HCQ), tafenoquine (TFQ), quinacrine (QNA), ferroquine (FQ) and moxipraquine (MXQ) (Figure 1). In particular, the present work pretends to provide general information on the state of the art on the use of antimalarial drugs based on quinoline as leishmanicidal, beginning a condensed analysis of in vitro results against promastigote and amastigote strains of diverse Leishmania spp., followed, if it is available, by the description of in vivo results, use of the combination, mechanistic studies and advance in clinical treatment. Most of the examples are derived from investigations made in the last 25 years, except for a few cases.
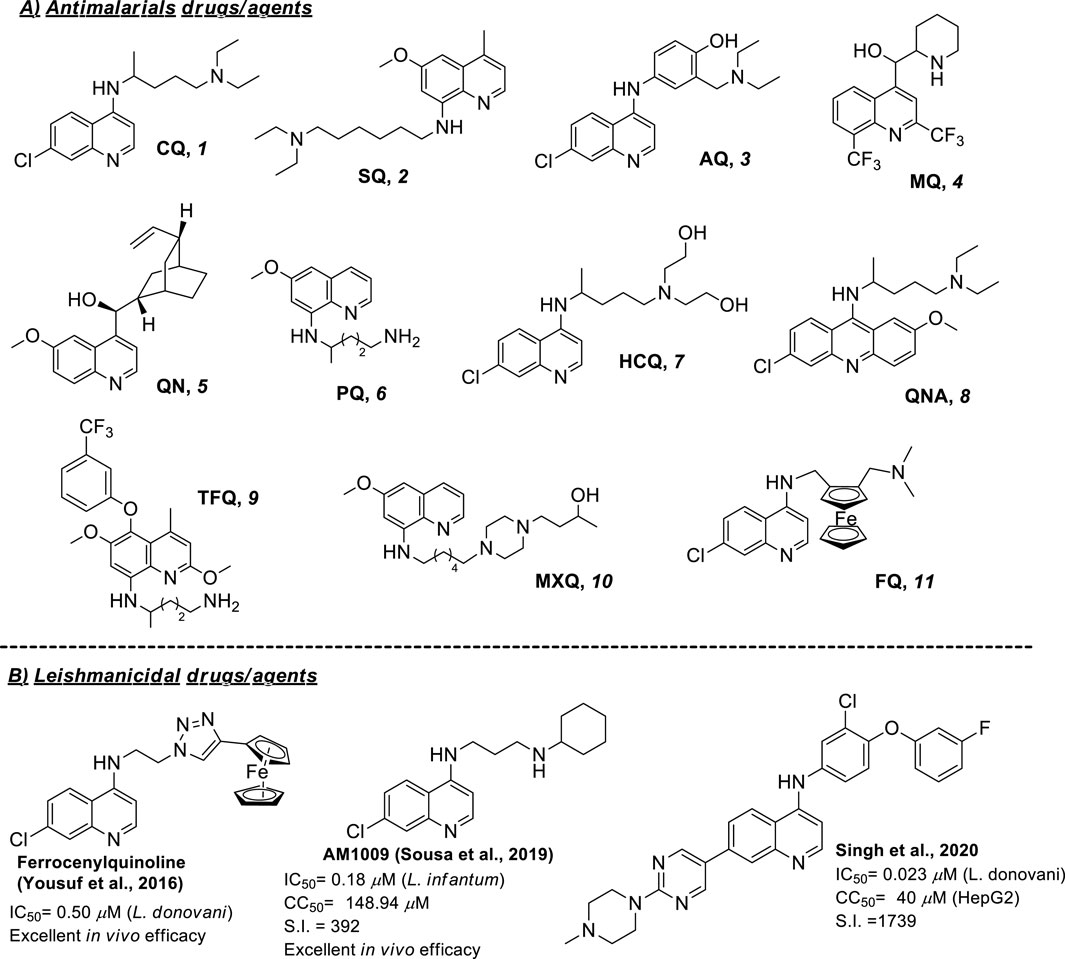
Figure 1. (A) Structure of common antimalarial drugs and, (B) leishmanicidal activity of some key antimalarial quinoline drugs.
2 Antimalarial drugs based on quinoline as leishmanicidal agents
2.1 Chloroquine
Chloroquine represents the most used antimalarial drug as a leishmanicidal agent, with a broad number of studies from in vitro and in vivo models against different types of Leishmania spp. From in vitro studies, against L. amazonensis, CQ displayed EC50 values of more than 50 µM against promastigotes and 0.78 µM against intracellular amastigotes (Rocha et al., 2013). A more recent study reported EC50 values of 4 and 3.77 µM against promastigotes and amastigotes, respectively, of L. amazonensis (Pejara-Rossi et al., 2024). Against L. infantum, EC50 values of 1.3 and 23 µM against promastigote and intracellular amastigote, respectively, were reported (Vale-Costa et al., 2013), whereas EC50 values of 11.3 and 0.5 µM were reported against L. donovani promastigote (Mwololo et al., 2015) and intracellular amastigote (Pomel et al., 2012), respectively. Also, CQ has been assayed against L. major and L. mexicana parasites, but no appreciable response is found under 10 µM treatment (Wijnant et al., 2017). From the cytotoxicity, CQ has exhibited CC50 values of 108 and 157 µM on peritoneal macrophages (Rocha et al., 2013) and THP-1 cells (Pejara-Rossi et al., 2024), respectively, which were significantly lower than those found by using amphotericin and miltefosine.
From in vivo studies using a murine model of CL, infected mice treated with oral chloroquine showed a reduction in lesion size and parasite burden in the draining lymph nodes with an ED50 of 27.29 mg/kg (Rocha et al., 2013; Pejara-Rossi et al., 2024). Further studies based on amastigotes’ ultrastructural analysis showed an accumulation of multivesicular bodies in the cytoplasm of the parasite that suggested an endocytic pathway impairment. Additionally, myelin-like figures were formed, and the Golgi complex was altered.
On the other hand, combination therapy has been employed to enhance the potential of CQ using reference drugs. By 2024, three examples can be found in the literature. The first of them consisted of the combination of CQ with diminazene against in vitro and in vivo models of L. donovani (Mwololo et al., 2015). In vitro evaluation indicated that the combination of diminazene and chloroquine was safer than amphotericin B (higher LC50) and at least nine times more effective (lower IC50 value) than individual treatments in killing promastigotes in culture. Meanwhile, in vivo assays in the murine VL model showed that the combination treatment reduced splenic parasites compared to monotherapies. Later, combination paromomycin-chloroquine therapy was explored against CL models of L. major and L. mexicana. From in vitro assays, the CQ addition (10 µM) to paromomycin reduced the paromomycin-EC50 values against both L. major and L. mexicana. Meanwhile, the in vivo murine CL models showed that the combination therapy only promoted a reduction in lesion progression in a comparable range to paromomycin, but no reduction in parasite burden was found (Wijnant et al., 2017).
The third example showed the use of CQ in combination with amphotericin B against models of CL (L. amazonensis). The combination of chloroquine and amphotericin B showed an additive effect against L. amazonensis. The synergistic effect was tested in murine models, where chloroquine reduced parasitemia by 45% alone and 86% in combination with amphotericin B and modulated Th1 cytokines like IFN-γ, indicating immunomodulatory benefits (Pejara-Rossi et al., 2024).
From clinical trials, significant advances have been achieved by using CQ. Early clinical studies were initiated with CL patients in Pakistan through an intralesional administration. The results indicated that all patients were pathologically and clinically cured after 7 weeks of treatment without adverse effects (4 weeks after completing the therapy). Intralesional CQ was a safe and cost-effective treatment for single lesions of CL, delivering high drug concentrations locally and minimizing systemic exposure (Noor et al., 2005). Another clinical investigation showed that CQ via intralesional provided cure of CL patients with a comparable response to the Glucantime®, although fewer injections of CQ were required than Glucantime®. Patients (60) were treated once weekly for 8 weeks (with additional injections in patients partially responding to treatment) (Yasmin et al., 2011).
The oral CQ treatment was also proved for clinical trials of CL. From 30 patients and based on the healing of the lesions, CQ (under 250 mg three times daily for 20 days) achieved a cure rate of 100% after 3 months, whereas Glucantime® (20 mg/kg for 28 days) promoted a cure rate of 93%. Importantly, no side effects or signs of recurrence were noted in oral CQ treatment, making it an attractive alternative due to its cost, availability, and safety (Khan et al., 2007).
A clinical comparison between intralesional and oral chloroquine administration (250 mg daily) for CL was performed in 86 randomly divided patients with single or multiple lesions. Both administration routes were equally effective (100% cure rate), but intralesional administration required significantly shorter treatment duration and lower total drug dose than oral chloroquine (Hanif et al., 2016). A comparison with oral tetracycline (200 mg daily) in patients showed no significant difference with the CQ treatment (Malik et al., 2019).
However, not all results were in favor of chloroquine as a major candidate for the treatment of CL. A comparison study of oral chloroquine (250 mg twice daily) with intramuscular meglumine antimoniate (810 mg daily) on adult male military patients showed that Glucantime® (84% cure) showed better performance (cure based on lesion healing) than oral CQ (56% cure) (Farooq et al., 2021). Recently, from a group of 64 military CL patients after 8 weeks, a higher efficacy (53.1%) was found for intralesional Glucantime® (53%) than for intralesional chloroquine treatment (18.8%) (Ullah et al., 2024).
2.2 Sitamaquine
Recent in vitro parasite evaluation confirmed the antileishmanial properties of SQ dihydrochloride against a range of Leishmania spp. (Garnier et al., 2006; Mesquita et al., 2014). Against L. aethiopica, SQ displayed EC50 values of 53.6 and 15.4 µM against promastigotes and intracellular amastigotes, respectively. Against L. major, EC50 values of 28.3 and 5.3 µM against promastigotes and intracellular amastigotes, respectively, were reported. Meanwhile, against L. mexicana LV4, SQ displayed EC50 values of 30.9 and 18.9 µM against promastigote and intracellular amastigotes, respectively, whereas against another L. mexicana strain (BEL21), an EC50 of 6.1 µM was reported for the promastigote form. Against L. panamensis promastigotes and amastigotes, EC50 of 36.6 and 5.5 µM were determined, respectively, while against L. amazonensis, an EC50 of 25.8 µM for promastigotes and no activity against intracellular amastigotes. Against L. donovani, EC50 values of 39.9 and 8.8 µM were found against promastigotes and intracellular amastigotes, respectively. Against other L. donovani strains (HU3, BHU3 and BHU11), SQ displayed EC50 of 6.3, 11.4 and 16 μM, respectively (Seifert et al., 2011). Finally, against L. infantum, an EC50 of 2.92 µM has been reported against intracellular amastigotes (Mesquita et al., 2014). Importantly, SQ displayed in vitro activity against L. donovani isolates resistant to sodium stibogluconate (Seifert et al., 2011). Regarding cytotoxicity, SQ has exhibited moderate to low toxicities, finding CC50 values of 67.2, 506 and higher than 60 µM on peritoneal, bone marrow macrophages (Vale-Costa et al., 2012) and kB cells (Yardley et al., 2010), respectively.
In in vivo experiments, SQ was shown to be 708 times more active than Glucantime® against L. donovani in hamsters (Kinnamon et al., 1978). Experiments in CL models (BALB/c mice) of L. major showed that SQ did not provide a significant reduction in the lesion progression and parasite burden (Garnier et al., 2006), which has evidenced the higher potential of SQ for the treatment of VL than for CL.
On the other hand, SQ has been widely studied for combination therapy for either in vitro or in vivo models, more particularly for VL. Against intracellular amastigote of L. donovani HU3 strain, a synergism was found for SQ in combination with pentamidine, whereas an indifferent effect of interaction was identified by using amphotericin B, Glucantime®, miltefosine and paromomycin (Seifert et al., 2011). Against L. infantum intracellular amastigote, SQ has also shown a synergism by using nitazoxanide (Mesquita et al., 2014).
From the mechanism of action, SQ can promote alterations in promastigote morphology (Langreth et al., 1983). It is well documented that SQ internalized/accumulated in membranous organelles such as lysosome (phagolysosome in infected macrophages), acidocalcisomes (López-Martín et al., 2008) and parasite mitochondria (Vercesi and Docampo, 1992; Vercesi et al., 2000). It is suggested that SQ can internalize in membranous organelles by the presence of a long lipophilic chain that could be able to insert into the parasite plasma membrane by interaction with lipid monolayer, whereas the presence of a weak basic group favors the accumulation into parasite through its protonation that facilitates interaction with anionic polar head (e.g., mitochondria) (Dueñas-Romero et al., 2007; Imberta et al., 2014; Loiseau et al., 2011). In summary, it is believed that SQ, once within the mitochondria, dysfunction promotes apoptosis and alterations in morphology (Romero and Delgado, 2025).
Concerning bioavailability, SQ presents a short elimination half-life (about 26 h) compared with miltefosine’s half-life (150–200 h) (Theoharides et al., 1987). From pharmacokinetics, SQ can form metabolites NADPH-dependent (Yeates, 2002), which seem to be derived from the action of different cytochrome P450 isozymes.
Finally, SQ reached phase II studies. The first phase II assay was performed in Kenya, which was positive in 16 patients of VL (Sherwood et al., 1994). Other phase II studies in India with 120 VL patients (Jha et al., 2005) and in Kenya with 95 VL patients (Wasunna et al., 2005) demonstrated that SQ was well tolerated with doses ranging from 1.5 to 3 mg/kg/day. However, some side effects such as vomiting and abdominal pains (about 10%), headache (also about 10%), as well as cyanosis (3%) as a consequence of methemoglobinemia were recognized by SQ treatment. Also, renal adverse effects (nephritic syndrome 3% and glomerulonephritis 2% in India) were observed. Another phase II clinical trial for L. chagasi-infected patients in Brazil showed a lack of efficacy in combination with the emergence of nephrotoxicity (Dietze et al., 2001). All these side effects stopped the progression of SQ as a therapeutic drug.
2.3 Amodiaquine
Amodiaquine is a well-known antimalarial drug that has gained great interest for its potential repurposing as an antileishmanial agent. AQ has been proven against a variety of Leishmania parasites for in vitro models of promastigotes and amastigotes. Against L. infantum, AQ displayed EC50 values of 30.1 and 6.7 µM against promastigotes and intracellular amastigotes (Ribeiro Antinarelli et al., 2023), respectively whereas a significant antiamastigote response (EC50 = 1.4 µM) has been reported against L. donovani (Guglielmo et al., 2009). Meanwhile, against L. amazonensis, L. braziliensis, L. chagasi and L. major parasites, AQ displayed discrete responses against promastigotes giving EC50 values of 40.8, 43, 21.1 and 67.2 µM (Coimbra et al., 2011), respectively. Against amastigotes of L. amazonensis, AQ exhibited an EC50 value of 0.95 µM (De Mello et al., 2004). From the cytotoxicity, AQ has exhibited CC50 values of 90 and 67 µM on kB cells (Guglielmo et al., 2009) and peritoneal macrophages (Ribeiro-Antinarelli et al., 2023), respectively.
On the other hand, AQ has demonstrated good in vivo efficacy response for a model of VL infected with L. donovani, achieving a significant reduction in parasitemia burden under oral administration of AQ and microparticles of hydroxypropylmethylcellulose system loaded with AQ, having no significant differences between them (Nettey et al., 2022).
Further studies showed that AQ promotes a drastic alteration of promastigote shape evidenced by an increase in cell volume with rounding and ribbing as well as a shortened flagellum. Additionally, AQ induced depolarization of the ΔΨm, an increase in ROS and neutral lipids levels, and changes in the cell cycle in promastigotes, without alterations to the permeability of the parasite plasma membrane. For L. infantum-infected macrophages, AQ induced an increase in ROS and NO levels (Ribeiro-Antinarelli et al., 2023).
2.4 Mefloquine
From in vitro studies, MQ has been tested against L. amazonensis and L. donovani. Against L. amazonensis, MQ displayed an effective response with EC50 values of 8.4 and 1.6 μM against promastigotes and intracellular amastigotes, respectively (Rocha et al., 2013). Against L. donovani promastigotes, MQ has shown a discrete activity (EC50 = 48.4 µM) (Yousef et al., 2020). From the cytotoxicity assay, a relative toxicity with a CC50 value of 11.95 µM on peritoneal macrophage has been reported (Rocha et al., 2013).
From in vivo experiments, orally or topically administered, MQ significantly reduced lesion size in infected (L. amazonensis) mice, but it did not reduce the parasite load, indicating that its primary effect may be more related to controlling lesion progression (Rocha et al., 2013). Another in vivo experiment for the CL model of L. amazonensis has demonstrated that MQ presented a limited therapeutic impact under an intramuscular administration (16 mg/kg), promoting only a partial reduction in lesion size (Galvão et al., 2000).
From clinical trials, the potential of MQ for the treatment of CL by L. braziliensis was proven for patients of an endemic region of Brazil. In general, from a group of 10 patients treated with MQ administered via oral (250 mg per day in a single dose for 6 days), only one patient showed an improvement compared with untreated control and comparable with patient treated with Glucantime®, which revealed the limiting impact of the MQ for clinical trials (Laguna-Torres et al., 1999). Previously, MQ promoted an appreciable reduction in lesions for human CL infected with L. panamensis (Landires et al., 1995).
2.5 Quinine
From in vitro studies, QN was more active against promastigotes than the amastigote form. In the case of L. amazonensis, QN exhibited EC50 values of 12.8 and 24.5 µM against promastigotes and intracellular amastigotes, respectively (Pejara-Rossi et al., 2024), whereas it displayed EC50 values of 0.23 and 40.2 µM against promastigotes and intracellular amastigotes, respectively, of L. donovani (Nettey et al., 2016). Regarding cytotoxicity, QN presented a relative toxicity on THP-1 cells, with a CC50 value of 22 µM (Pejara-Rossi et al., 2024). Interestingly, QN in combination with standard drugs such as amphotericin and pentamidine showed synergism against promastigotes of L. donovani, (∼89–90%) (Nettey et al., 2016).
From in vivo experiments, either orally administered QN or QN encapsulated with chitosan microparticles reduced the parasitemia load in the blood and organs (spleen and liver) of mice compared with untreated controls. Results under oral administration were similar to those derived from intraperitoneal administration, demonstrating that QN represents a good choice for the treatment of VL (L. donovani) in mice (Allotey-Babington et al., 2024).
2.6 Primaquine
PQ has been proven against a variety of Leishmania spp. including L. amazonensis, L. infantum, L. major and L. mexicana for in vitro studies. Against L. infantum, PQ displayed a modest response with EC50 values of 32.2 and 40.0 µM against promastigotes and intracellular amastigotes (Vale-Costa et al., 2012), respectively. Against L. amazonensis, no appreciable response against the promastigote form was found under 50 µM treatment (Rocha et al., 2013). Against L. major and L. mexicana, a weak parasite proliferation inhibition (˂ 10%) was found under 10 µM treatment (Rocha et al., 2013). Regarding cytotoxicity, CC50 values of 68.6 and higher than 60 µM were reported on peritoneal (Rocha et al., 2013) and bone marrow macrophages (Vale-Costa et al., 2012), respectively.
From an in vivo CL model of L. major, PQ reduced the lesion size from 3.4 mm for untreated controls to 1.4 and 1.2 mm, under subcutaneous and oral administration, respectively (Beveridge et al., 1980). Results were comparable to those derived from paromomycin and Glucantime®, which promoted a barely higher reduction in lesions to 0.8 mm. Additionally, for an in vivo VL-model of hamsters infected with L. donovani, PQ reduced parasitemia load in a comparable range to Glucantime® (Kinnamon et al., 1978).
2.7 Hydroxychloroquine
Hydroxychloroquine (HCQ), a derivative of chloroquine, has emerged as a safer alternative to CQ for malaria treatment due to its higher efficacy and lower toxicity. In recent decades, due to the knowledge that HCQ has immunomodulatory effects, it is also used for autoimmune diseases (Schrezenmeier and Dörner, 2020). HCQ has also been explored as a potential leishmanicidal against L. amazonensis, showing significant efficacy against intracellular amastigotes, with an IC50 value of 0.67 μM. Against promastigotes of L. amazonensis, no appreciable leishmanicidal response was found under 50 µM treatment. Regarding cytotoxicity, a CC50 value of 140.6 µM on peritoneal macrophages was determined (Rocha et al., 2013), which implied an S.I. of 210. In a murine model, HCQ was less effective than chloroquine; however, its established safety profile, oral bioavailability, and low cost make it a potential agent for the treatment of CL, especially in regions where resistance to traditional treatments was observed (Rocha et al., 2013).
2.8 Quinacrine, tafenoquine, ferroquine and moxipraquine
QNC was evaluated against 2 L. enriettii (wild type) and LePentR50 (resistant pentamidine-strain) and two strains of L. donovani, LdAG83 and LdAG83PentR50 (a resistant pentamidine-strain), under an intracellular amastigote infected macrophage model. QNC displayed EC50 values of 18, 29, 12 and 12 µM against L. enrietti, LePentR50, LdAG83 and LdAG83PentR50, respectively. Also, a synergetic effect was found using pentamidine as a reference drug. Against L. enriettii strain, QNC decreased the EC50 of pentamidine from 26.6 µM to lower values of 16.2, 15.4, 14.3, 9.1 and 7.1 µM under 0.375, 0.75, 1.5, 3.0 and 6.0 µM QNC doses, respectively. Meanwhile, a decrease from 16.2 µM to lower EC50 values of 10.4, 7.1, 4.7, 2.7 and 4.6 µM under 0.375, 0.75, 1.5, 3.0 and 6.0 µM QNC doses, respectively. Against resistant LePentR50 and LdAG83PentR50 strains, a significant reduction in EC50 of pentamidine from 228.6 to 74.7 µM to lower values of 67.8 and 11.8 µM under 6 µM QNC treatment, respectively (Wong et al., 2009).
TFQ has been proven only against in vitro models of L. donovani. For infected models of intracellular amastigote using HU3, DD8, DHU3 and DHU11 host cells, TFQ was able to inhibit the parasite proliferation, giving low EC50 of 1.8, 1.5, 2.3 and 3.7 µM, respectively. The antimalarial drug displayed a high cytotoxicity with a CC50 value of 6.6 on kB cells (Yardley et al., 2010). Meanwhile, MXP was only proven against four in vivo models of CL for infection with L. major, L. panamensis, L. braziliensis and L. mexicana. Against L. major, a significant reduction in lesion size from 3.4 mm (untreated mice) to values of 1.4 and 1.6 mm was found under MTX doses of 25 mg/kg and 50 mg/kg via subcutaneous administration, respectively. A good leishmanicidal response was found under oral administration, giving a reduction in lesion size from 3.4 to 1.75 mm under 100 mg/kg doses (Beveridge et al., 1980). Results were comparable to those derived from paromomycin and Glucantime®, which promoted a barely higher reduction in lesions to 0.8 mm. Against L. mexicana, a reduction in lesion size from 3.57 mm to 0.3 mm was found under MXP treatment, which is comparable with Glucantime® response (0.0 mm). Meanwhile, against L. panamensis, MXP promoted a reduction in lesions from 1.63 mm to 0.44 mm, whereas Glucantime® reduced the lesion to 0.0 mm. Finally, against L. braziliensis, no reduction in lesion size was found. Importantly, MPX presented an acute toxicity, LD50 between 266 and 353 mg/kg. Finally, FQ, which is a chloroquine analogue porting a ferrocenyl group along the dialkyldiamino chain, was inactive at 20 μM against intracellular amastigotes of L. donovani (Pomel et al., 2015).
In this mini-review, we presented an overview of the progress made in the use of antimalarial drugs as a repurposing strategy for treating leishmaniasis. The current treatments have many limitations, so there is an urgent need to search for new and more effective chemotherapeutic agents. CQ is one of the antimalarials most studied as a leishmanicidal agent, showing good in vitro and in vivo results as well as clinical advances using reference drugs within combination therapy, particularly for the case of CL. Meanwhile, SQ also represents a good alternative, mainly against VL models. SQ has successfully reached phase II studies and it represents the second orally active leishmanicidal treatment, although its progression was stopped by methemoglobinemia and nephrotoxicity side effects in treated patients. Despite these effects, SQ chemical structure can be an inspiration for the synthesis design of new compounds because it has a well-defined mechanism, which is associated with the immunological activation of host cells, and mitochondria dysfunction by accumulation in membranous organelles of the parasite. MQ has shown good in vivo results with a limited application in clinical trials. Other antimalarials such as AQ and QN have shown a good profile against VL in vivo models, whereas MXP showed a good response against in vivo CL model and PQ exhibited excellent response for in vivo CL and VL models. TFQ and QNC have been scarcely investigated with good in vitro results, whereas FQ did not show a leishmanicidal response (Table 1). Then, quinoline antimalarials represent a good choice for combination therapy, and they can contribute to a therapeutic effect through an immunostimulant action of the host cell. In addition, the use of quinoline-antimalarial drugs is facilitated by oral treatment due to its use in the protonated form. Future strategies must include the 4-quinoline framework for the development of new compounds as more potent, safer and selective antileishmanial agents.
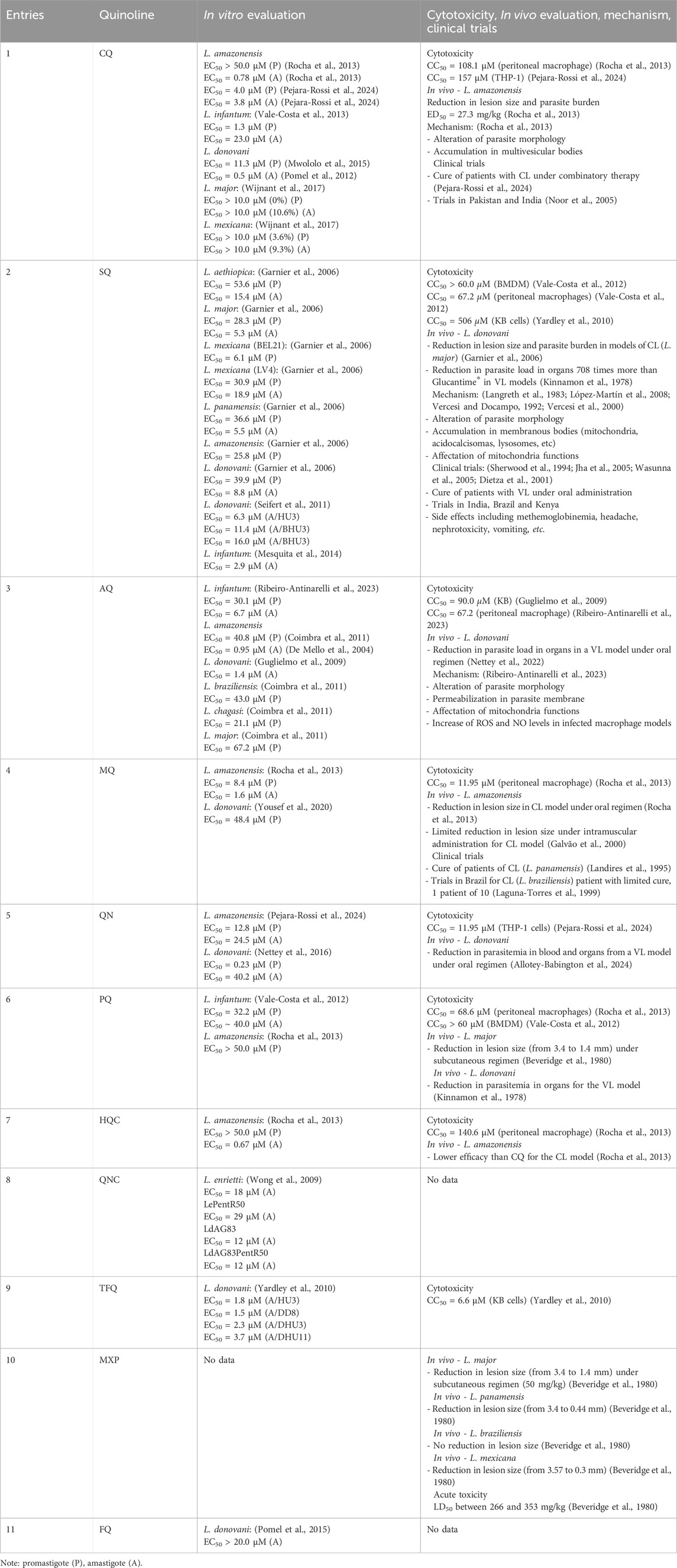
Table 1. Leishmanicidal data for a series of antimarial drugs based on quinolines.
Author contributions
RA: Writing – original draft, Writing – review and editing. GG: Conceptualization, Formal Analysis, Funding acquisition, Investigation, Project administration, Supervision, Visualization, Writing – original draft, Writing – review and editing. NR: Writing – review and editing. AR: Conceptualization, Formal Analysis, Funding acquisition, Investigation, Project administration, Supervision, Visualization, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by PEDECIBA (Programa de Desarrollo de las Ciencias Básicas) under Despegue-Cientifico 2023 funds. AHR thanks to Sistema Nacional de Investigadores (SNI) for grant SNI_2023_1_1013178. GGL and REA thank UBA (UBACYT 20020190100242BA) and CONICET (PIP 11220210100072) for partial financial support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Allotey-Babington, G. L., Nettey, H., Ofori-Gyamera, N. K., Aboadwe-Goode, N., Mensah, A. A., Asiedu-Gyekye, I. J., et al. (2024). Development and evaluation of an oral quinine sulphate sustained release formulation for the management of visceral leishmaniasis. Int. J. Res. Pharm. Sci. 15, 48–55. doi:10.26452/ijrps.v15i4.4720
Aronson, N., Herwaldt, B. L., Libman, M., Pearson, R., Lopez-Velez, R., Weina, P., et al. (2016). Diagnosis and treatment of leishmaniasis: clinical practice guidelines by the infectious diseases society of America (IDSA) and the American society of tropical medicine and hygiene (ASTMH). Clin. Inf. Dis. 63, e202–e264. doi:10.1093/cid/ciw670
Beveridge, E., Caldwell, I. A., Latter, V. S., Neal, R. A., Udall, V., and Waldron, M. M. (1980). The activity against Trypanosoma cruzi and cutaneous leishmaniasis, and toxicity, of moxipraquine (349C59). Trans. Roy. Soc. Trop. Med. Hyg. 74, 43–51. doi:10.1016/0035-9203(80)90010-3
Chanquia, S. N., Larregui, F., Puente, V., Labriola, C., Lombardo, E., and García Liñares, G. (2019). Synthesis and biological evaluation of new quinoline derivatives as antileishmanial and antitrypanosomal agents. Bioorg. Chem. 83, 526–534. doi:10.1016/j.bioorg.2018.10.053
Chartlon, R. L., Rossi-Bergmann, B., Denny, P. W., and Steel, P. G. (2017). Repurposing as a strategy for the discovery of new anti-leishmanials: the-state-of-the-art. Parasitology 145, 219–236. doi:10.1017/s0031182017000993
Coimbra, E. S., da Silva, A. D., Dias, R. M. P., Corrales, R. C. N. R., Bispo, M. L. F., Kaiser, C. R., et al. (2011). Amodiaquine analogs. Synthesis and anti-leishmanial activity. Mediterr. J. Chem. 1, 106–113. doi:10.13171/mjc.1.3.2011.26.09.22
Del Carpio, E., Hernández, L., Lubes, V., Jourdan, F., Cerecetto, H., Scalese, G., et al. (2025). Metal complexes based on quinoline for development of leishmanicidal agents: structure and mechanism of action. Front. Chem. 13, 1586044. doi:10.3389/fchem.2025.1586044
Delgado, F., Benítez, A., Gotopo, L., and Romero, A. (2025). 4-Aminoquinoline: a comprehensive review of synthetic strategies. Front. Chem. 13, 1553975. doi:10.3389/fchem.2025.1553975
De Mello, H., Echevarria, A., Bernardino, A. M., Canto-Cavalheiro, M., and Leon, L. L. (2004). Antileishmanial pyrazolopyridine derivatives: synthesis and structure−activity relationship analysis. J. Med. Chem. 47, 5427–5432. doi:10.1021/jm0401006
Dietze, R., Carvalho, S. F. G., Valli, L. C., Berman, J., Brewer, T., Milhous, W., et al. (2001). Phase II trial of WR6026, an orally administrated 8-aminoquinoline, in the treatment of visceral leishmaniasis caused by Leishmania chagasi. Am. J. Trop. Med. Hyg. 65, 685–689. doi:10.4269/ajtmh.2001.65.685
Drugs for Neglected Diseases initiative. (2023). Available online at: https://www.dndi.org/research-development/drug-discovery/ (Accessed 28 April 2023).
Dueñas-Romero, A. M., Loiseau, P. M., and Saint-Pierre-Chazalet, M. (2007). Interaction of sitamaquine with membrane lipids of Leishmania donovani promastigotes. Biochim. Biophys. Acta 1768, 246–252. doi:10.1016/j.bbamem.2006.07.003
Farooq, M., Farooq, M. S., Bari, A. U., and Malik, T. M. (2021). Comparison of oral chloroquine with systemic meglumine antimoniate in treatment of cutaneous leishmaniasis. Pak. Armed. Forces Med. J. 71, 139–144. doi:10.51253/pafmj.v71i1.3187
Galvão, L. O., Moreira, S., Medeiros, P., Piantino-Lemos, G. J., Cunha, N. F., Parreiras, R. M., et al. (2000). Therapeutic trial in experimental tegumentary leishmaniasis caused by Leishmania amazonensis: a comparative study between mefloquine and aminosidine. Rev. Soc. Bras. Med. Trop. 33, 377–382. doi:10.1590/s0037-86822000000400008
Garnier, T., Brown, M. B., Lawrence, M. J., and Croft, S. L. (2006). In vitro and in vivo studies on a topical formulation of sitamaquine dihydrochloride for cutaneous leishmaniasis. J. Pharm. Pharmacol. 58, 1043–1054. doi:10.1211/jpp.58.8.0004
Guglielmo, S., Bertinaria, M., Rolando, B., Crosetti, M., Fruttero, R., Yardley, V., et al. (2009). A new series of amodiaquine analogues modified in the basic side chain with in vitro antileishmanial and antiplasmodial activity. Eur. J. Med. Chem. 44, 5071–5079. doi:10.1016/j.ejmech.2009.09.012
Hanif, M. M., Akram, K., and Mustafa, G. (2016). Intralesional versus oral chloroquine in cutaneous leishmaniasis: comparison of outcome, duration of treatment and total dose of drug. J. Coll. Physicians Surg. Pak. 26, 260–262.
Imberta, L., Cojeana, S., Libongb, D., Chaminade, P., and Loiseau, P. M. (2014). Sitamaquine-resistance in Leishmania donovani affects drug accumulation and lipid metabolism. Biomed. Pharmacother. 68, 893–897. doi:10.1016/j.biopha.2014.08.009
Jha, T. K., Sundar, S., Thakur, C. P., Felton, J. M., Sabin, A. J., and Horton, J. (2005). A phase II dose-ranging study of sitamaquine for treatment of visceral leishmaniasis in India. Am. J. Trop. Med. Hyg. 73, 1005–1011. doi:10.4269/ajtmh.2005.73.1005
Khan, I., Yasmin, R., and Sidiqui, I. (2007). Chloroquine in cutaneous leishmaniasis. J. Pak. Assoc. Dermatol. 17, 95–100.
Kinnamon, K. E., Steck, E. A., Loizeaux, P. S., Hanson, W. L., Chapman, W. L., and Waits, V. B. (1978). The antileishmanial activity of lepidines. Am. J. Trop. Med. Hyg. 27, 751–757. doi:10.4269/ajtmh.1978.27.751
Kumar, A. (2021). “Leishmaniasis: an overview,” in Developments in immunology, visceral leishmaniasis. Editor A. Kumar (Academic Press), 1–15. doi:10.1016/b978-0-323-91124-5.00012-6
Kumari, S., Kumar, V., Kumar-Tiwari, R., Ravidas, V., Pandey, K., and Kumar, A. (2022). Amphotericin B: a drug of choice for visceral leishmaniasis. Act. Trop. 235, 106661. doi:10.1016/j.actatropica.2022.106661
Laguna-Torres, L. A., Silva, C. A. C., Correia, D., Carvalho, E. M., Magalhães, A. V., and de Oliveira Macêdo, V. (1999). Mefloquina no tratamento da leishmaniose cutânea em uma área endêmica de Leishmania (Viannia) braziliensis. Rev. Soc. Bras. Med. Trop. 32, 529–532. doi:10.1590/S0037-86821999000500010
Landires, E. A. G., Andrial, M., Hosokawa, A., Nonaka, S., and Hashiguchi, Y. (1995). Oral treatment of new world cutaneous leishmaniasis with anti-malarial drugs in Ecuador: a preliminary clinical trial. Jap. J. Trop. Med. Hyg. 23, 151–157. doi:10.2149/tmh1973.23.151
Langreth, S. G., Berman, J. D., Riordan, G. P., and Lee, L. S. (1983). Fine structure alterations in Leishmania tropica within macro phages exposed to antileishmanial drugs in vitro. J. Protozool. 30, 555–561. doi:10.1111/j.1550-7408.1983.tb01421.x
Loiseau, P. M., Cojean, S., and Schrevel, J. (2011). Sitamaquine as a putative antileishmanial drug candidate: from the mechanism of action to the risk of drug resistance. Parasite 18, 115–119. doi:10.1051/parasite/2011182115
López-Martín, C., Pérez-Victoria, J. M., Carvalho, L., Castanys, S., and Gamarro, F. (2008). Sitamaquine sensitivity in Leishmania species is not mediated by drug accumulation in acidocal cisomes. Antimicr. Agent. Chemother. 52, 4030–4036. doi:10.1128/aac.00964-08
Malik, F., Hanif, M. M., and Mustafa, G. (2019). Comparing the efficacy of oral chloroquine versus oral tetracycline in the treatment of cutaneous leishmaniasis. J. Coll. Physicians Surg. Pak. 29, 403–405. doi:10.29271/jcpsp.2019.05.403
Mendes, B., de Oliveira Cardoso, J. M., Fortes De Brito, R. C., Coura-Vital, W., de Oliveira Aguiar-Soares, R. D., and Barbosa Reis, A. (2020). Recent advances and new strategies on leishmaniasis treatment. Appl. Microbiol. Biotech. 104, 8965–8977. doi:10.1007/s00253-020-10856-w
Mesquita, J. T., Tempone, A. G., and Reimão, J. Q. (2014). Combination therapy with nitazoxanide and amphotericin B, Glucantime®, miltefosine and sitamaquine against Leishmania (Leishmania) infantum intracellular amastigotes. Acta Trop. 130, 112–116. doi:10.1016/j.actatropica.2013.11.003
Mota, S., Guedes, B. N., Jain, S., Cardoso, J. C., Severino, P., and Souto, E. B. (2024). Classical and innovative drugs for the treatment of Leishmania infections. Discov. Public Health 21, 122. doi:10.1186/s12982-024-00247-1
Murray, H. W., Berman, J. D., Davies, C. R., and Saravia, N. G. (2005). Advances in leishmaniasis. Lancet 366, 1561–1577. doi:10.1016/s0140-6736(05)67629-5
Mwololo, S. W., Mutiso, J. M., Macharia, J. C., Bourdichon, A. J., and Gicheru, M. M. (2015). In vitro activity and in vivo efficacy of a combination therapy of diminazene and chloroquine against murine visceral leishmaniasis. J. Biomed. Res. 29, 214–223. doi:10.7555/JBR.29.20140072
Nettey, H., Allotey-Babington, G. L., Nguessan, B. B., Afrane, B., Tagoe, M., Ababio, A., et al. (2016). Screening of anti-infectives against Leishmania donovani. Adv. Microbiol. 6, 13–22. doi:10.4236/aim.2016.61002
Nettey, H., Erskine, I. J., Mensah, A. A., Gyamera, N. K. O., Obuobi, C. K., Kumadoh, D., et al. (2022). Oral amodiaquine microparticles repurposed for the treatment of visceral leishmaniasis. Sci. Afr. 17, e01285. doi:10.1016/j.sciaf.2022.e01285
NIH (2022). Adverse effects of antileishmanial medicines. Available online at: https://www.ncbi.nlm.nih.gov/books/NBK581524/(Accessed November 19, 2024).
Noor, S. M., Khan, M. M., and Hussain, D. (2005). Intralesional chloroquine in cutaneous leishmaniasis. J. Pak. Assoc. Dermatol. 15, 18–21.
Pejara-Rossi, N. D., Fialho, S. N., de Jesus Gouveia, A., Santos-Ferreira, A., Azevedo da Silva, M., Do Nascimento-Martinez, L., et al. (2024). Quinine and chloroquine: potential preclinical candidates for the treatment of tegumentary Leishmaniasis. Acta Trop. 252, 107143. doi:10.1016/j.actatropica.2024.107143
Pomel, S., Biot, C., Bories, C., and Loiseau, P. M. (2012). Antiprotozoal activity of ferroquine. Parasitol. Res. 112, 665–669. doi:10.1007/s00436-012-3183-4
Pomel, S., Dubar, F., Forge, D., Loiseau, P. M., and Biot, C. (2015). New heterocyclic compounds: synthesis and antitrypanosomal properties. Bioorg. Med. Chem. 23, 5168–5174. doi:10.1016/j.bmc.2015.03.029
Ribeiro-Antinarelli, L. M., Midlej, V., Silva, E. D. S., Ferraz Coelho, E. A., da Silva, A. D., and Coimbra, E. S. (2023). Exploring the repositioning of the amodiaquine as potential drug against visceral leishmaniasis: the in vitro effect against Leishmania infantum is associated with multiple mechanisms, involving mitochondria dysfunction, oxidative stress and loss of cell cycle control. Chem. Biol. Int. 371, 110333. doi:10.1016/j.cbi.2022.110333
Rocha, V. P. C., Nonato, F. R., Guimarães, E. T., de Freitas, L. A. R., and Soares, M. B. P. (2013). Activity of antimalarial drugs in vitro and in a murine model of cutaneous leishmaniasis. J. Med. Microbiol. 62, 1001–1010. doi:10.1099/jmm.0.058115-0
Romero, A., and Delgado, F. (2025). 4-Aminoquinoline as a privileged scaffold for the design of leishmanicidal agents: structure-property relationships and key biological targets. Front. Chem. 12, 1527946. doi:10.3389/fchem.2024.1527946
Romero, A., Rodriguez, N., Lopez, S. E., and Oviedo, H. (2019). Identification of dehydroxy isoquine and isotebuquine as promising antileishmanial agents. Arch. Pharm. 352 (5), 1800281. doi:10.1002/ardp.201800281
Schrezenmeier, E., and Dörner, T. (2020). Mechanisms of action of hydroxychloroquine and chloroquine: implications for rheumatology. Nat. Rev. Rheumatol. 16, 155–166. doi:10.1038/s41584-020-0372-x
Seifert, K., Munday, J., Syeda, T., and Croft, S. L. (2011). In vitro interactions between sitamaquine and amphotericin B, sodium stibogluconate, miltefosine, paromomycin and pentamidine against Leishmania donovani. J. Antimicrob. Chemother. 66, 850–854. doi:10.1093/jac/dkq542
Sherwood, J. A., Gachihi, G. S., Muigai, R. K., Skillman, D. R., Mugo, M., Rashid, J. R., et al. (1994). Phase 2 efficacy trial of an oral 8-aminoquinoline (WR6026) for treatment of visceral leishmaniasis. Clin. Inf. Dis. 19, 1034–1039. doi:10.1093/clinids/19.6.1034
Sundar, S., Singh, J., Kumar-Singh, K., Agrawal, N., and Kumar, R. (2024). Current and emerging therapies for the treatment of leishmaniasis. Exp. Opin. Orphan Drugs 12, 19–32. doi:10.1080/21678707.2024.2335248
Theoharides, A. D., Kim, M. M., Ashmore, R. W., and Shipley, L. A. (1987). “Identification and quantitation of human urinary metabolites of a candidate 8-aminoquinoline antileishmanial drug (WR 6026),” in Federation proceedings (Bethesda, MD: Federation of American Societies for Experimental Biology), 865.
Ullah, O., Rizwan, M., Raza, N., Zulfiqar, S., Akbar, N., and Ullah, H. (2024). Comparative efficacy of intralesional chloroquine with intralesional meglumine antimoniate in the treatment of cutaneous leishmaniasis. Cureus 16, e56785. doi:10.7759/cureus.56785
Vale-Costa, S., Costa-Gouveia, J., Pérez, B., Silva, T., Teixeira, C., Gomes, P., et al. (2013). N-cinnamoylated aminoquinolines as promising antileishmanial agents. Antimicr. Agent. Chemother. 57, 5112–5115. doi:10.1128/AAC.00557-13
Vale-Costa, S., Vale, N., Matos, J., Tomás, A., Moreira, R., Gomes, P., et al. (2012). Peptidomimetic and organometallic derivatives of primaquine active against Leishmania infantum. Antimicr. Agent. Chemother. 56, 5774–5781. doi:10.1128/AAC.00873-12
Vercesi, A., and Docampo, R. (1992). Ca2+ transport by digitonin-permeabilized Leishmania donovani. Effects of Ca2+, pentamidine and WR-6026 on mitochondrial membrane potential in situ. Biochem. J. 284, 463–467. doi:10.1042/bj2840463
Vercesi, A., Rodrigues, C., Castisti, R., and Docampo, R. (2000). Presence of a Na+/H+ exchanger in acidocalcisomes of Leishmania donovani and their alkalization by anti-leishmanial drugs. FEBS Lett. 473, 203–206. doi:10.1016/S0014-5793(00)01531-3
Wasunna, M. K., Rhashid, J. R., Mbui, J., Kirigi, G., Kinoti, D., Lodenyo, H., et al. (2005). A phase II dose-increasing study of sitamaquine for treatment of visceral leishmaniasis in Kenya. Am. J. Trop. Med. Hyg. 73, 871–876.
Wijnant, G. J., Van Bocxlaer, K., Yardley, V., Murdan, S., and Croft, S. L. (2017). Efficacy of paromomycin-chloroquine combination therapy in experimental cutaneous leishmaniasis. Antimicrob. Agent. Chemother. 61, e00358. doi:10.1128/AAC.00358-17
Wong, L. K., Chan, K. F., Zhao, Y., Chan, T. H., and Chow, L. M. C. (2009). Quinacrine and a novel apigenin dimer can synergistically increase the pentamidine susceptibility of the protozoan parasite Leishmania. J. Antimicr. Chemother. 63, 1179–1190. doi:10.1093/jac/dkp130
World Health Organization (2023). Leishmaniasis. World Health Organization:. Available online at: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis. (Accessed January 12, 2023)
Yaluff, G., Herrera, L. M., Rolon, M., Vega, C., and Cerecetto, H. (2025). The quinoline framework and related scaffolds in natural products with anti-Leishmania properties. Front. Chem. 13, 1571067. doi:10.3389/fchem.2025.1571067
Yardley, V., Gamarro, F., and Croft, S. L. (2010). Antileishmanial and antitrypanosomal activities of the 8-aminoquinoline tafenoquine. Antimicr. Agent. Chemother. 54, 5356–5358. doi:10.1128/AAC.00985-10
Yasmin, R., Khan, I., and Ahmad, S. A. (2011). Response to treatment of cutaneous leishmaniasis with intralesional chloroquine vs intralesional meglumine antimoniate. J. Pak. Assoc. Dermatol. 21, 270–275.
Yeates, C. (2002). Sitamaquine (GlaxoSmithKline/Walter reed army institute). Curr. Opin. Investig. Drugs 3, 1446–1452.
Keywords: quinoline derivatives, antimalarials, leishmaniasis, drugs repurposing, Antiprotozoal activity
Citation: Avanzo RE, García Liñares G, Rodríguez N and Romero AH (2025) A comprehensive revision on the use of quinoline antimalarial drugs as leishmanicidal agents. Front. Chem. 13:1608340. doi: 10.3389/fchem.2025.1608340
Received: 08 April 2025; Accepted: 14 May 2025;
Published: 30 May 2025.
Edited by:
Carmen Gil, Spanish National Research Council (CSIC), SpainReviewed by:
Esther Del Olmo, University of Salamanca, SpainCopyright © 2025 Avanzo, García Liñares, Rodríguez and Romero. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guadalupe García Liñares, bGluYXJlc0Bxby5mY2VuLnViYS5hcg==; Angel H. Romero, YW5nZWwudWN2LnVzYkBnbWFpbC5jb20=
†ORCID: Guadalupe García Liñares, orcid.org/0000-0002-2946-4795; Angel H. Romero, orcid.org/0000-0001-8747-5153