 Orlando G. Elso*†
Orlando G. Elso*† Guadalupe García Liñares*†
Guadalupe García Liñares*†- Laboratorio de Biocatálisis. Departamento de Química Orgánica y UMYMFOR, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires, Ciudad Universitaria, Buenos Aires, Argentina
Leishmaniasis is one of the most widespread parasitic diseases in the world, primarily affecting the poorest and most vulnerable populations. The development of new therapeutic agents that are more efficient, safe, and selective remains a challenge. The quinoline framework emerges as a privileged scaffold for this purpose. This mini-review comprehensively analyses advancements from the last two decades on 2-, 3-, 6-, and 8-substituted quinolines, as well as polysubstituted analogues, as potential antileishmanial agents, focusing on how the position and nature of substituents influence their activity. Although the assays were conducted in different Leishmania species, 2- and 6-substituted quinolones generally show greater activity, often enhanced by the presence of halogen or hydroxyl groups.
1 Introduction
Leishmaniasis is the second most widespread protozoan disease globally. It is a parasitic infection caused by protozoa of the Leishmania genus; more than 20 species and subspecies can infect humans, leading to various clinical manifestations, from localized cutaneous leishmaniasis to visceral leishmaniasis, the more severe form, depending on the parasite species and the host’s immune system (García Liñares et al., 2006; Kumari et al., 2021). This disease is transmitted through the bite of infected female sandflies, and it is estimated that between 700,000 and one million new cases and between 26,000 and 65,000 deaths occur annually (World Health Organization, 2025).
The main drugs used to treat leishmaniasis are pentavalent antimonials, amphotericin B, and pentamidine (Pradhan et al., 2022; Silva Santos et al., 2020), but they present toxicity, primarily affecting the kidneys and heart (Brindha et al., 2021). Furthermore, pentamidine requires hospitalization for administration, which often leads to treatment discontinuation. Considering that: i) the complete eradication of the vector insects for leishmaniasis is infeasible; ii) the effective vaccines are not yet available; iii) the current chemotherapy is still deficient; and iv) there is a high level of resistance to these drugs, the search for new, more potent, selective, and safer compounds is essential.
In line with this, N-based heterocycles have received special attention due to a wide range of biological properties and pharmacological applications (Aatif et al., 2022; Heravi and Zadsirjan, 2020; Marshall et al., 2024). Since the discovery of the natural alkaloid quinine as an antimalarial drug, there has been great interest in the search for substituted quinolines as potential pharmacological agents (Yaluff et al., 2025). The relevance of quinolines is demonstrated in several reports regarding their use as antimalarial drugs (Ajani et al., 2022). Very recently, we have reported a revision on the use of quinoline antimalarial drugs as antileishmanial agents (Avanzo et al., 2025).
Particularly, quinoline-based compounds have arisen as a privileged scaffold for the development of more selective and potent antileishmanial agents. Based on currently used antimalarial drugs, the most significant development to date concerns 4-substituted quinolines (Delgado et al., 2025; Romero and Delgado, 2025). In addition to 4-substituted quinolines such as chloroquine, hydroxychloroquine, mefloquine, or amodiaquine, among others—well-known antimalarial drugs— (Avanzo et al., 2025; Ravindar et al., 2023), new compounds with various activities, such as antifungal (Vandekerckhove et al., 2013), antibacterial (Teng et al., 2018), or antitumor (Afzal et al., 2015), have also been developed. However, it has also been observed that quinolines substituted at other positions have also demonstrated antiparasitic activity (Nefertiti et al., 2018; Rajesh, 2018). Herein, we describe advancements from the last decades on the development of 2-, 3-, 6 and 8-substituted quinolines that display antileishmanial activity. Our focus is on these substitutions because they have yielded compounds with the most promising activity. Substitutions at other positions have been less frequently explored in the context of antileishmanial drug design and thus fall outside the scope of this analysis.
2 Substituted quinolines
2.1 2-Substitution
Given the antileishmanial activity of natural 2-substituted quinoline alkaloids (Fournet et al., 1993; 1996), a library of 2-substituted quinolines was synthesized (Fakhfakh et al., 2003; Franck et al., 2004). Initial in vitro screening against L. amazonensis and L. infantum identified quinolines substituted in the 2-position by a three-carbon alkenyl side chain containing an aldehyde (1), hydroxy (2) or bromine substituent (3) as the most active compounds, with IC50 values between 2 and 4 μM. By contrast, three carbon alkenyl side chains containing carboxylic acid, ester, amide, nitro or phenyl functionalities or larger alkenyl side chains displayed lower activity. To determine if this potent in vitro activity translated to a therapeutic effect, some derivatives were selected for in vivo studies. Compounds were administered by the oral route to a group of mice infected with L. amazonensis, L. infantum, and L. donovani. Interestingly, a disconnect was observed: compound 3, which was the most potent in vitro, showed no activity in vivo, whereas other derivatives with lower in vitro potency demonstrated a significant reduction (>80%) in parasite burden. Compounds 1 and 2 showed satisfactory activity in the visceral leishmaniasis models, with more than an 80% reduction of the parasite burden in the liver and close to 50% reduction in the spleen of mice infected with L. infantum and close to 50% reduction of the parasite burden in the liver of mice infected with L. donovani (Nakayama et al., 2005). Based on these results, the same group synthesized an α,β-unsaturated nitrile (4) with promising in vitro activity against L. donovani amastigotes and significant in vivo efficacy with a significant reduction in parasite burden in the liver (Nakayama et al., 2007).
The synthesis of a group of quinoline-2-carbohydrazides yielded compounds 5a and 5b as the most active against Leishmania (Viannia) panamensis (Coa et al., 2015). These compounds bear a 2-hydroxyphenyl moiety with a second hydroxy group at the 3- or 4- position of the aromatic ring, the presence of which is associated with the improvement of antileishmanial activity. Similar derivatives, with hydroxy groups replaced by methoxy functionalities, showed lower activity (Alodeani et al., 2015).
GDP-mannose pyrophosphorylase, which is involved in the biosynthetic pathway of glycoconjugates, has been recognized as an interesting target for the development of chemotherapeutic agents. Among several selected inhibitors, compound 6 emerged as the most promising antileishmanial agent, with an IC50 of 1.06 μM on axenic amastigotes and 0.63 μM on the RAW264.7 model of L. donovani. However, its selectivity index (SI) of 2.4 was low, limiting its therapeutic potential (Mao et al., 2017).
The primary strength of C-2 substitution lies in the accessibility for introducing different groups, which allows for an easy structural optimization. Despite this, a disconnection between in vitro potency and in vivo efficacy is generally observed.
2.2 3-Substitution
Based on previous studies describing the antileishmanial activity of 3-arylquinolines, this structure was selected as the lead to perform different substitution modifications at various positions (Katsuno et al., 2015; Ortiz et al., 2017). Some 3-substituted quinolines bearing alkenyl, alkynyl, and phenyl groups were synthesized and evaluated against L. amazonensis amastigotes. These analogues exerted low to negligible activity, suggesting this position is less favorable (Fakhfakh et al., 2003). Among a series of synthesized 3-arylamino quinolines, 3- and 4-fluorophenyl derivatives (7a-b) were effective against L. mexicana promastigotes (IC50 = 41.9 μM) with low cytotoxicity (>100 μM) (Chanquia et al., 2019).
Synthesis of hybrid molecules constitutes an attractive strategy for obtaining pharmaceutical compounds, which involves the covalent combination of two biologically active pharmacophores (Viegas-Junior et al., 2007). Several quinoline-based hybrids have been shown to exhibit various activities, including antimalarial, antibacterial, antiviral, antitumoral, and anti-inflammatory (Ammar et al., 2021; Hu et al., 2017; Singh et al., 2022). From this perspective, a library of quinoline-thiazolidinone hybrids was synthesized to target methionine aminopeptidase, an important enzyme for the development of antiprotozoal agents. Compound 8 arose as a promising antileishmanial agent with a twenty-fold higher inhibitory activity against L. donovani aminopeptidase (IC50 = 3.0 μM) compared to the human enzyme (IC50 = 58.0 μM), a good drug-likeness profile, and low cytotoxicity (CC50 > 150 μM) (Bhat et al., 2021).
Simple substitutions at C-3 have generally failed to produce potent compounds but the design of hybrid molecules with complex pharmacophores could yield more selective compounds. Additionally, a general lack of in vivo validation is observed.
2.3 6-Substitution
Substitution on the benzene ring also presents alternatives for the development of new drugs. Based on the concept of molecular hybridization, two series of 6-substituted quinolines linked to either oxadiazole-thiosemicarbazides or 1,3,4-thiadiazoles were synthesized. The in vitro activity against L. major promastigotes was evaluated, showing excellent antileishmanial activity with an IC50 in the submicromolar range. In the first series, fluorinated derivatives 9a and 9b were the most potent with IC50 values of 0.10 μM and 0.15 μM, respectively (Taha et al., 2017). In the second series, dihydroxyphenyl derivatives (10a-d) were the most potent compounds, achieving IC50 values as low as 0.04 µM (Almandil et al., 2019). Docking studies suggested a possible mechanism of action as these compounds are tightly fitted into the active site of pteridine reductase 1, a validated drug target in trypanosomatids. The presence and position of hydroxyl groups influenced antileishmanial activity, indicating that these groups afford polar interactions with residues from the active site of pteridine reductase 1. In the case of fluorinated derivatives, the one with the fluorine atom in the ortho position was the one that exhibited the most significant inhibition, demonstrating that the substitution in this position is essential for its activity.
The primary strength of the 6-substitution is the extremely high in vitro potency achieved through hybridization. In addition, the SAR is relatively clear, with electron-withdrawing and hydrogen-bond-donating groups significantly improving activity. The major limitation is the absence of amastigotes and cytotoxicity assays.
2.4 8-Substitution
The 8-position of the quinoline ring is historically significant due to the antimalarial drug primaquine. A group of N-quinolin-8-yl-arylsulphonamides showed promising activity (Da Silva et al., 2007). Particularly, the dihalogenated compounds 11a-c were very effective against both promastigotes of L. amazonensis and L. chagasi, and significant selectivity indices (SI). More lipophilic derivatives without halogenated substituents have shown somewhat lower activity. Promising results were obtained from assays on the amastigote form of L. amazonensis: significant antileishmanial activity was observed with compounds 11a-c, with IC50 < 1 µM. In contrast, substitution with a hydroxyl or phenyl group resulted in weaker antileishmanial activity against L. amazonensis or L. (V) panamensis amastigotes (Coa et al., 2020; Silva et al., 2018).
Chalcone, furochalcone, and chromone–quinoline hybrids via an alkyl linker have also been tested against L. (Viannia) panamensis (Coa et al., 2017; García et al., 2018). The antileishmanial activity was influenced by the length of the alkyl linker connecting the quinoline ring to its substituent. However, no clear correlation between antiprotozoal activity and alkyl chain length was established, as no consistent trend in activity relative to chain length was observed. Within the furanochalcone series, the derivative which features a three-carbon linker exhibited the highest activity against L. (V) panamensis amastigotes, but it showed high cytotoxicity. In the case of chromone-based series, compounds 12a and 12b, bearing two- and seven-carbon linkers respectively, showed the highest antileishmanial activity (IC50 = 16.9 and 17.0 μM). However, these compounds also displayed considerable cytotoxicity, with SI values below 1. By contrast, compounds 12c and 12d were less active but exhibited low cytotoxicity, with SI values of 4.89 and 5.84, respectively. All chalcone-quinoline hybrids showed moderate activity and low SI.
The low cytotoxicity constitutes the main strength of the 8-substitution pattern. Additionally, some compounds showed very potent activity against the clinically relevant amastigote form.
2.5 Polisubstitution
Considering the groups and substitution positions that have led to compounds with higher activity, new quinolines have been developed, combining different substituents at various positions. For example, a series of 2-styrylquinolines with additional groups at the C-7 position was synthesized and evaluated in vitro against the amastigote form of L. donovani (Loiseau et al., 2011). Compound 13 emerged as the most promising candidate, with IC50 = 1.2 µM and low toxicity (SI = 121.5). Additionally, it was observed that highly hydrophilic groups, such as COOH, dramatically decreased antileishmanial activity, whereas the presence of NO2 groups improved the SI. Compound 2 was taken as the lead structure for the synthesis of a series of quinolines with a hydroxypropenyl group at the two-position. In vitro and in vivo activity against L. donovani was measured (Gopinath et al., 2013). It was observed that the presence of halogens improved metabolic stability compared to the reference compound, and with a morpholine group at position 4, compound 14 emerged as the best candidate with significantly enhanced antileishmanial activity (IC50 = 0.22 μM, SI = 187). Furthermore, in vivo assays using a L. donovani/hamster model, its hydrochloride salt exhibited an 84% inhibition of parasite growth.
Considering the hybridization approach and the known antiparasitic activity of metronidazoles, a series of quinoline-metronidazole hybrids was synthesized and evaluated against L. donovani promastigotes and amastigotes (Upadhyay et al., 2019). Among these hybrids, compound 15 showed more potent activity against L. donovani both in vitro and in vivo and presented a high selectivity index and metabolic stability. Furthermore, the induction of apoptosis via mitochondrial membrane depolarization and an increase in the generation of reactive oxygen species was established as a possible dual mechanism of action. Compound 15 is a promising candidate both as a lead structure for the development of new antileishmanial agents and for clinical trials, taking into account its potency in vitro (IC50 = 3.75 µM in amastigotes) and in vivo assays (>80% reduction of parasite burden in liver and spleen), low toxicity (SI = 57.9) and excellent pharmacokinetic properties.
Consequently, a series of 3-aryl and 3-heteroaryl-N7,N7-dimethylquinoline-2,7-diamine derivatives was synthesized. These compounds were evaluated against L. mexicana amastigotes, showing promising activity (IC50 < 1 μM) (Hammill et al., 2021). According to SAR analysis, no significant variation in antileishmanial activity was observed upon substitution of the aromatic ring, and replacing the aryl group with six membered or bicyclic heterocycles also maintained the activity. However, substitution with five-membered heterocycles reduced activity, suggesting a minimum steric bulk requirement. Among the series, compounds 16 and 17 were the most active and were selected for in vivo experiments, but 16 exhibited toxicity, and 17 failed to suppress lesion progression in a murine model of cutaneous leishmaniasis.
Considering the antileishmanial activity of quinoline and 1,2,3-triazole scaffolds, a series of triazolyl 2-methyl-4-phenylquinoline-3-carboxylate derivatives was synthesized via click chemistry-based molecular hybridization approach and evaluated against L. donovani (Upadhyay et al., 2018). Despite good antileishmanial activity against promastigotes in most cases, only a few compounds showed significant inhibitory activity against intracellular amastigotes. Among them, compounds 18a-b and 19a-b were selected for their evaluation in a L. donovani/golden hamster model. Compounds 18a and 19a showed moderate activity, with 40% and 46% parasite inhibition, respectively, whereas 18b and 19b exhibited poor inhibitory activity.
Clioquinol (20), a dihalogenated 8-hydroxyquinoline, showed promising in vitro activity against L. infantum and L. amazonensis promastigotes and amastigotes, exhibiting low cytotoxicity against murine macrophages (CC50 = 834.4 µM) and human erythrocytes (CC50 = 1.5 × 103 μM), with a higher selectivity index than amphotericin B. As no toxicity was observed when clioquinol was administered to BALB/c mice, it constitutes a promising candidate for subsequent in vivo studies (Tavares et al., 2018). Compound 21, a racemic 8-aminoquinoline derivative, has a strong effect on the mobility and morphology of L. mexicana promastigotes, exhibiting an IC50 value of 1.03 µM, markedly lower compared to glucantime (Isaac-Márquez et al., 2010). Also, 21 exhibited negligible cytotoxicity toward HeLa cells over 120 h.
The obvious strength of polysubstitution is the possibility of designing more active and selective molecules, optimizing pharmacokinetic and pharmacodynamic properties, although at the cost of increased synthetic complexity.
The structures of the most representative compounds are shown in the Figure 1 at the end of the manuscript. The Table 1 summarizes the antileishmania activity data of the most promising quinolines, which exhibit low IC50 values in vitro assays (<10 μM), or significant in vivo inhibition, or high selectivity indexes (SI > 10).
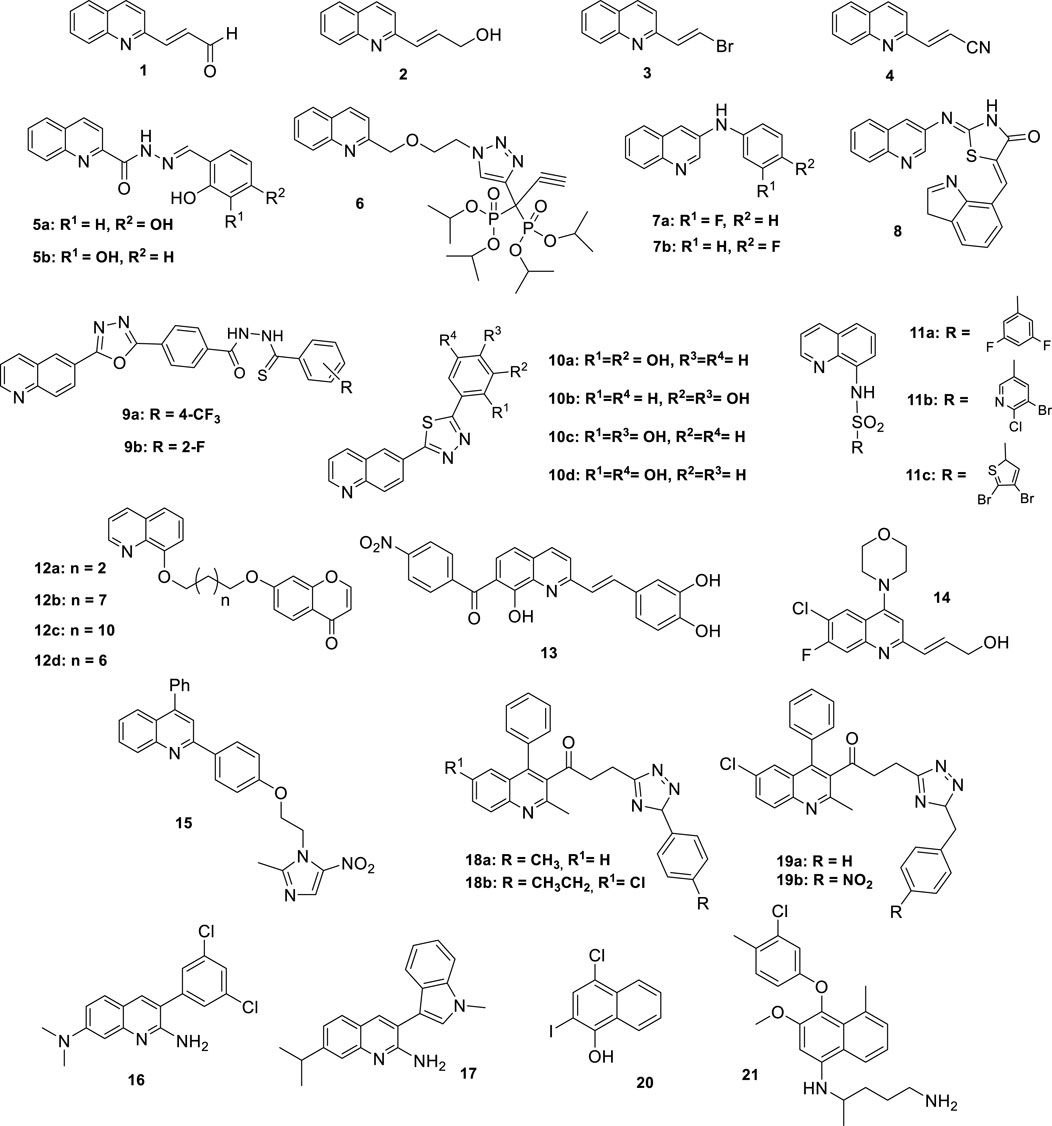
Figure 1. Structures of the more representative substituted quinolines with antileishmanial activity.
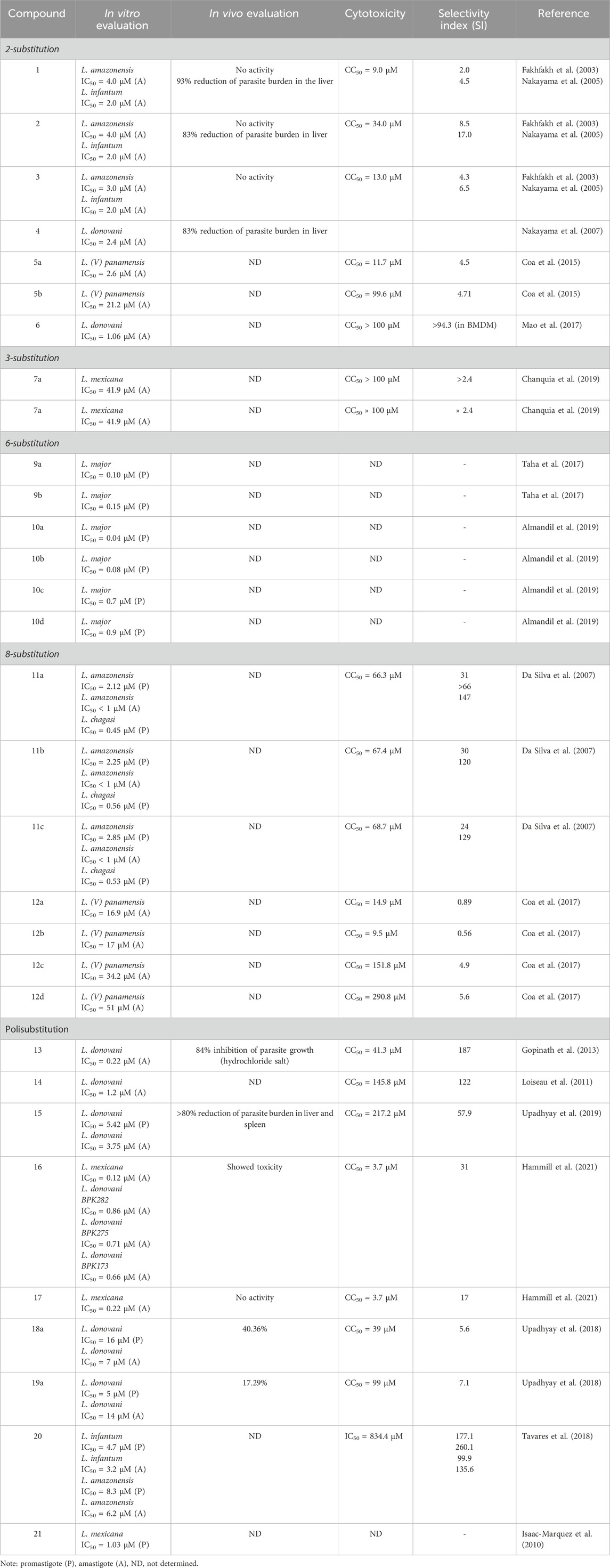
Table 1. Antileishmanial data for selected substituted quinolines.
In summary, in the last decades, the development of new quinoline-derived compounds as potential chemotherapeutic agents for the treatment of leishmaniasis has increased. This remains a significant challenge due to multiple causes, such as the numerous species that cause the disease, diverse manifestations, resistance, and more. The quinoline framework interacts with different receptors, ion channels, and enzymes, making it a privileged scaffold in organic chemistry for the search for bioactive compounds; many natural and synthetic derivatives have been shown to exhibit a wide range of biological properties. In this mini-review, we presented an overview of the impact of substitution position on the antileishmanial properties of substituted quinolines.
Considering all examples mentioned, it can be observed that, among monosubstituted quinolines, 2-substitution results in compounds with greater in vitro (amastigotes) and in vivo activity; 3-substituted quinolines are the least active compounds; and 8-substitution led to compounds with good activity in vitro on amastigotes and high SI, but dependent on Leishmania species. In many cases, for the same type of derivatives, the inclusion of hydroxyl or halogen groups leads to improved antileishmanial activity. In the case of polysubstituted quinolines, substituents in position 2 and/or a hydroxyl group in position 8 seem to be key in the antileishmanial activity and excellent selectivity index values. Among the compounds described, 2, 4, 6, 11a-c, 13, 14, 15, and 20 are the most promising either due to their low IC50 (<10 μM) values against amastigotes and/or high selectivity indexes (SI > 10) SI or good in vivo activity. However, a strict comparison is hindered due to several limitations, like: different Leishmania species with differences in drug sensitivity; in studies reporting very high potency on promastigotes, lack of data about assays against amastigotes, the clinically relevant form of the parasite; heterogeneity in experimental protocols; scarcity of information about molecular targets or action mechanisms. Future strategies should focus not only on a combination of substituents at diverse positions or synthesis of hydrid molecules, but also on systematic studies on structure-activity relationships and mechanisms of action.
Author contributions
OE: Formal Analysis, Writing – original draft, Visualization, Conceptualization, Investigation, Writing – review and editing. GG: Investigation, Formal Analysis, Writing – original draft, Writing – review and editing, Funding acquisition, Visualization, Project administration, Conceptualization.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. UBA (UBACYT 20020190100242BA) and CONICET (PIP 11220210100072).
Acknowledgments
The authors thank UBA (UBACYT 20020190100242BA) and CONICET (PIP 11220210100072) for partial financial support.
Conflict of interest
Authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aatif, M., Raza, M. A., Javed, K., Nashre-ul-Islam, S. M., Farhan, M., and Alam, M. W. (2022). Potential nitrogen-based heterocyclic compounds for treating infectious diseases: a literature review. Antibiotics 11, 1750. doi:10.3390/antibiotics11121750
Afzal, O., Kumar, S., Haider, M. R., Ali, M. R., Kumar, R., Jaggi, M., et al. (2015). A review on anticancer potential of bioactive heterocycle quinoline. Eur. J. Med. Chem. 97, 871–910. doi:10.1016/j.ejmech.2014.07.044
Ajani, O. O., Iyaye, K. T., and Ademosun, O. T. (2022). Recent advances in chemistry and therapeutic potential of functionalized quinoline motifs–a review. RSC Adv. 12, 18594–18614. doi:10.1039/d2ra02896d
Almandil, N. B., Taha, M., Rahim, F., Wadood, A., Imran, S., Alqahtani, M. A., et al. (2019). Synthesis of novel quinoline-based thiadiazole, evaluation of their antileishmanial potential and molecular docking studies. Bioorg. Chem. 85, 109–116. doi:10.1016/j.bioorg.2018.12.025
Alodeani, E. A., Arshad, M., and Izhari, M. A. (2015). Antileishmanial activity and computational studies of some hydrazone derivatives possessing quinoline nucleus. Eur. J. Pharm. Med. Res. 2, 324–328.
Ammar, Y. A., Abd El-Hafez, S. M., Hessein, S. A., Ali, A. M., Askar, A. A., and Ragab, A. (2021). One-pot strategy for thiazole tethered 7-ethoxy quinoline hybrids: synthesis and potential antimicrobial agents as dihydrofolate reductase (DHFR) inhibitors with molecular docking study. J. Mol. Struct. 1242, 130748. doi:10.1016/j.molstruc.2021.130748
Avanzo, R. E., García Liñares, G., Rodríguez, N., and Romero, A. H. (2025). A comprehensive revision on the use of quinoline antimalarial drugs as leishmanicidal agents. Front. Chem. 13, 1608340. doi:10.3389/fchem.2025.1608340
Bhat, S. Y., Bhandari, S., Thacker, P. S., Arifuddin, M., and Qureshi, I. A. (2021). Development of quinoline-based hybrid as inhibitor of methionine aminopeptidase 1 from Leishmania donovani. Chem. Biol. Drug Des. 97, 315–324. doi:10.1111/cbdd.13783
Brindha, J., Balamurali, M. M., and Chanda, K. (2021). An overview on the therapeutics of neglected infectious diseases—leishmaniasis and Chagas diseases. Front. Chem. 9, 622286. doi:10.3389/fchem.2021.622286
Chanquia, S. N., Larregui, F., Puente, V., Labriola, C., Lombardo, E., and García Liñares, G. (2019). Synthesis and biological evaluation of new quinoline derivatives as antileishmanial and antitrypanosomal agents. Bioorg. Chem. 83, 526–534. doi:10.1016/j.bioorg.2018.10.053
Coa, J. C., Castrillón, W., Cardona, W., Carda, M., Ospina, V., Muñoz, J. A., et al. (2015). Synthesis, leishmanicidal, trypanocidal and cytotoxic activity of quinoline-hydrazone hybrids. Eur. J. Med. Chem. 101, 746–753. doi:10.1016/j.ejmech.2015.07.018
Coa, J. C., García, E., Carda, M., Agut, R., Vélez, I. D., Muñoz, J. A., et al. (2017). Synthesis, antileishmanial, trypanocidal and cytotoxic activities of quinoline-chalcone and quinoline-chromone hybrids. Med. Chem. Res. 26, 1405–1414. doi:10.1007/s00044-017-1846-5
Coa, J. C., Yepes, A., Carda, M., Conesa-Milián, L., Upegui, Y., Robledo, S. M., et al. (2020). Synthesis, in silico studies, antiprotozoal and cytotoxic activities of quinoline-biphenyl hybrids. ChemSelect 5, 2918–2924. doi:10.1002/slct.201903835
da Silva, L. E., Joussef, A. C., Pacheco, L. K., da Silva, D. G., Steindel, M., and Rebelo, R. A. (2007). Synthesis and in vitro evaluation of antileishmanial and trypanocidal activities of N-quinolin-8-yl-arylsulfonamides. Bioorg. Med. Chem. 15, 7553–7560. doi:10.1016/j.bmc.2007.09.007
Delgado, F., Benítez, A., Gotopo, L., and Romero, A. (2025). 4-Aminoquinoline: a comprehensive review of synthetic strategies. Front. Chem. 13, 1553975. doi:10.3389/fchem.2025.1553975
Fakhfakh, M. A., Fournet, A., Prina, E., Mouscadet, J. F., Franck, X., Hocquemiller, R., et al. (2003). Synthesis and biological evaluation of substituted quinolines: potential treatment of protozoal and retroviral co-infections. Bioorg. Med. Chem. 11, 5013–5023. doi:10.1016/j.bmc.2003.09.007
Fournet, A., Barrios, A. A., Muñoz, V., Hocquemiller, R., Cavé, A., and Bruneton, J. (1993). 2-substituted quinoline alkaloids as potential antileishmanial drugs. Antimicrob. Agents Chemother. 37, 859–863. doi:10.1128/aac.37.4.859
Fournet, A., Ferreira, M. E., Rojas De Arias, A., Torres De Ortiz, S., Fuentes, S., Nakayama, H., et al. (1996). In vivo efficacy of oral and intralesional administration of 2-substituted quinolines in experimental treatment of new world cutaneous leishmaniasis caused by Leishmania amazonensis. Antimicrob. Agents Chemother. 40, 2447–2451. doi:10.1128/aac.40.11.2447
Franck, X., Fournet, A., Prina, E., Mahieux, R., Hocquemiller, R., and Figadère, B. (2004). Biological evaluation of substituted quinolines. Bioorg. Med. Chem. Lett. 14, 3635–3638. doi:10.1016/j.bmcl.2004.05.026
García, E., Coa, J. C., Otero, E., Carda, M., Vélez, I. D., Robledo, S. M., et al. (2018). Synthesis and antiprotozoal activity of furanchalcone–quinoline, furanchalcone–chromone and furanchalcone–imidazole hybrids. Med. Chem. Res. 27, 497–511. doi:10.1007/s00044-017-2076-6
García Liñares, G., Ravaschino, E. L., and Rodriguez, J. B. (2006). Progresses in the field of drug design to combat tropical Protozoan parasitic diseases. Curr. Med. Chem. 13, 335–360. doi:10.2174/092986706775476043
Gopinath, V. S., Pinjari, J., Dere, R. T., Verma, A., Vishwakarma, P., Shivahare, R., et al. (2013). Design, synthesis and biological evaluation of 2-substituted quinolines as potential antileishmanial agents. Eur. J. Med. Chem. 69, 527–536. doi:10.1016/j.ejmech.2013.08.028
Hammill, J. T., Sviripa, V. M., Kril, L. M., Ortiz, D., Fargo, C. M., Kim, H. S., et al. (2021). Amino-substituted 3-aryl-and 3-heteroarylquinolines as potential antileishmanial agents. J. Med. Chem. 64, 12152–12162. doi:10.1021/acs.jmedchem.1c00813
Heravi, M. M., and Zadsirjan, V. (2020). Prescribed drugs containing nitrogen heterocycles: an overview. RSC Adv. 10, 44247–44311. doi:10.1039/D0RA09198G
Hu, Y. Q., Gao, C., Zhang, S., Xu, L., Xu, Z., Feng, L. S., et al. (2017). Quinoline hybrids and their antiplasmodial and antimalarial activities. Eur. J. Med. Chem. 139, 22–47. doi:10.1016/j.ejmech.2017.07.061
Isaac-Márquez, A. P., McChesney, J. D., Nanayakara, N. D., Satoskar, A. R., and Lezama-Dávila, C. M. (2010). Antileishmanial activity of racemic±8-[(4-Amino-1-methylbutyl) amino]-6-methoxy-4-methyl-5-[3,4-dichlorophenoxy]quinoline. Nat. Prod. Comm. 5, 387–390. doi:10.1177/1934578X1000500309
Katsuno, K., Burrows, J. N., Duncan, K., van Huijsduijnen, R. H., Kaneko, T., Kita, K., et al. (2015). Hit and lead criteria in drug discovery for infectious diseases of the developing world. Nat. Rev. Drug Discov. 14, 751–758. doi:10.1038/nrd4683
Kumari, D., Perveen, S., Sharma, R., and Singh, K. (2021). Advancement in leishmaniasis diagnosis and therapeutics: an update. Eur. J. Pharmacol. 910, 174436. doi:10.1016/j.ejphar.2021.174436
Loiseau, P. M., Gupta, S., Verma, A., Srivastava, S., Puri, S. K., Sliman, F., et al. (2011). In vitro activities of new 2-substituted quinolines against Leishmania donovani. Antimicrob. Agents Chemother. 55, 1777–1780. doi:10.1128/AAC.01299-10
Mao, W., Daligaux, P., Lazar, N., Ha-Duong, T., Cavé, C., van Tilbeurgh, H., et al. (2017). Biochemical analysis of leishmanial and human GDP-Mannose Pyrophosphorylases and selection of inhibitors as new leads. Sci. Rep. 7, 751. doi:10.1038/s41598-017-00848-8
Marshall, C. M., Federice, J. G., Bell, C. N., Cox, P. B., and Njardarson, J. T. (2024). An update on the nitrogen heterocycle compositions and properties of US FDA-approved pharmaceuticals (2013–2023). J. Med. Chem. 67, 11622–11655. doi:10.1021/acs.jmedchem.4c01122
Nakayama, H., Loiseau, P. M., Bories, C., Torres de Ortiz, S., Schinini, A., Rojas de Arias, A., et al. (2005). Efficacy of orally administered 2-substituted quinolines in experimental murine cutaneous and visceral leishmaniases. Antimicrob. Agents Chemother. 49, 4950–4956. doi:10.1128/aac.49.12.4950-4956.2005
Nakayama, H., Desrivot, J., Bories, C., Franck, X., Figadère, B., Hocquemiller, R., et al. (2007). In vitro and in vivo antileishmanial efficacy of a new nitrilquinoline against Leishmania donovani. Biomed. Pharmacother. 61, 186–188. doi:10.1016/j.biopha.2007.02.001
Nefertiti, A. S. G., Batista, M. M., Da Silva, P. B., Batista, D. G. J., Da Silva, C. F., Peres, R. B., et al. (2018). In vitro and in vivo studies of the trypanocidal effect of novel quinolines. Antimicrob. Agents Chemother. 62, e01936-17. doi:10.1128/aac.01936-17
Ortiz, D., Guiguemde, W. A., Hammill, J. T., Carrillo, A. K., Chen, Y., Connelly, M., et al. (2017). Discovery of novel, orally bioavailable, antileishmanial compounds using phenotypic screening. PLoS Neglected Trop. Dis. 11, e0006157. doi:10.1371/journal.pntd.0006157
Pradhan, S., Schwartz, R. A., Patil, A., Grabbe, S., and Goldust, M. (2022). Treatment options for leishmaniasis. Clin. Experim. Dermat. 47, 516–521. doi:10.1111/ced.14919
Rajesh, Y. B. (2018). “Quinoline heterocycles: synthesis and bioactivity,” in Heterocycles-synthesis and biological activities. Editors. B. P. Nandeshwarappa, and S. O. Sadashiv, (IntechOpen). doi:10.5772/intechopen.81239
Ravindar, L., Hasbullah, S. A., Rakesh, K. P., and Hassan, N. I. (2023). Recent developments in antimalarial activities of 4-aminoquinoline derivatives. Eur. J. Med. Chem. 256, 115458. doi:10.1016/j.ejmech.2023.115458
Romero, A., and Delgado, F. (2025). 4-Aminoquinoline as a privileged scaffold for the design of leishmanicidal agents: structure–property relationships and key biological targets. Front. Chem. 12, 1527946. doi:10.3389/fchem.2024.1527946
Silva, E. J., Bezerra-Souza, A., Passero, L. F., Laurenti, M. D., Ferreira, G. M., Fujii, D. G., et al. (2018). Synthesis, leishmanicidal activity, structural descriptors and structure–activity relationship of quinoline derivatives. Future Med. Chem. 10, 2069–2085. doi:10.4155/fmc-2018-0124
Silva Santos, S., de Araujo, R. V., Giarolla, J., El Seoud, O., and Ferreira, E. I. (2020). Searching for drugs for Chagas disease, leishmaniasis and schistosomiasis: a review. Int. J. Antimicrob. Agents 55, 105906. doi:10.1016/j.ijantimicag.2020.105906
Singh, A., Kaur, H., Arora, S., and Bedi, P. M. S. (2022). Design, synthesis, and biological evaluation of novel morpholinated isatin–quinoline hybrids as potent anti-breast cancer agents. Arch. Pharm. 355, 2100368. doi:10.1002/ardp.202100368
Taha, M., Ismail, N. H., Ali, M., Rashid, U., Imran, S., Uddin, N., et al. (2017). Molecular hybridization conceded exceptionally potent quinolinyl-oxadiazole hybrids through phenyl linked thiosemicarbazide antileishmanial scaffolds: in silico validation and SAR studies. Bioorg. Chem. 71, 192–200. doi:10.1016/j.bioorg.2017.02.005
Tavares, G. D. S. V., Mendonça, D. V. C., Lage, D. P., Granato, J. D. T., Ottoni, F. M., Ludolf, F., et al. (2018). Antileishmanial activity, cytotoxicity and mechanism of action of clioquinol against Leishmania infantum and Leishmania amazonensis species. Basic Clin. Pharmacol. Toxic. 123, 236–246. doi:10.1111/bcpt.12990
Teng, P., Li, C., Peng, Z., Marie, V. A., Nimmagadda, A., Su, M., et al. (2018). Facilely accessible quinoline derivatives as potent antibacterial agents. Bioorg. Med. Chem. 26, 3573–3579. doi:10.1016/j.bmc.2018.05.031
Upadhyay, A., Kushwaha, P., Gupta, S., Dodda, R. P., Ramalingam, K., Kant, R., et al. (2018). Synthesis and evaluation of novel triazolyl quinoline derivatives as potential antileishmanial agents. Eur. J. Med. Chem. 154, 172–181. doi:10.1016/j.ejmech.2018.05.014
Upadhyay, A., Chandrakar, P., Gupta, S., Parmar, N., Singh, S. K., Rashid, M., et al. (2019). Synthesis, biological evaluation, structure–activity relationship, and mechanism of action studies of quinoline–metronidazole derivatives against experimental visceral leishmaniasis. J. Med. Chem. 62, 5655–5671. doi:10.1021/acs.jmedchem.9b00628
Vandekerckhove, S., Tran, H. G., Desmet, T., and D’hooghe, M. (2013). Evaluation of (4-aminobutyloxy) quinolines as a novel class of antifungal agents. Bioorg. Med. Chem. Lett. 23, 4641–4643. doi:10.1016/j.bmcl.2013.06.014
Viegas-Junior, C., Danuello, A., da Silva Bolzani, V., Barreiro, E. J., and Fraga, C. A. M. (2007). Molecular hybridization: a useful tool in the design of new drug prototypes. Curr. Med. Chem. 14, 1829–1852. doi:10.2174/092986707781058805
World Health Organization (2025). Leishmaniasis. Available online at: https://www.who.int/es/news-room/fact-sheets/detail/leishmaniasis (Accessed May 18, 2025).
Keywords: quinolines, antiprotozoal activity, substitution, antileishmanial agents, leishmaniasis
Citation: Elso OG and García Liñares G (2025) Quinolines: the role of substitution site in antileishmanial activity. Front. Chem. 13:1645334. doi: 10.3389/fchem.2025.1645334
Received: 11 June 2025; Accepted: 27 August 2025;
Published: 15 September 2025.
Edited by:
Gustavo Benaim, Fundación Instituto de Estudios Avanzados (IDEA), VenezuelaReviewed by:
Jaime Charris, Central University of Venezuela, VenezuelaElena Aguilera, Universidad de la República, Uruguay
Copyright © 2025 Elso and García Liñares. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guadalupe García Liñares, bGluYXJlc0Bxby5mY2VuLnViYS5hcg==; Orlando G. Elso, b2Vsc29AcW8uZmNlbi51YmEuYXI=
†ORCID: Orlando G. Elso, orcid.org/0000-0002-1970-9904; Guadalupe García Liñares, orcid.org/0000-0002-2946-4795