 Jhe-Sian Lin
Jhe-Sian Lin Zheng-Hao Tzeng
Zheng-Hao Tzeng Jasper S. Dumalaog
Jasper S. Dumalaog Shang-Cheng Hung
Shang-Cheng Hung- 1Institute of Biochemistry and Molecular Biology, National Yang Ming Chiao Tung University, Taipei, Taiwan
- 2Genomics Research Center, Academia Sinica, Taipei, Taiwan
- 3Department of Chemistry, National Tsing Hua University, Hsinchu, Taiwan
- 4Department of Applied Science, National Taitung University, Taitung, Taiwan
- 5Department of Chemistry, National Cheng Kung University, Tainan, Taiwan
QS-21, a potent immunostimulatory saponin obtained from Quillaja saponaria Molina, a soapbark tree native to Chile, has undergone extensive study for its broad application as a vaccine adjuvant against various infectious diseases and cancers. The structure of QS-21, which features a linear oligosaccharide moiety, provides a critical attachment site for both the labile acyl side chain and the distinctive sugar unit that defines each major saponin variant. In this study, we present an efficient synthetic approach to the truncated linear trisaccharide fragment of QS-21, circumventing the challenges associated with the synthesis of the rare sugar D-fucose. The synthesis of this linear trisaccharide enables streamlined access to a homogeneous QS-21.
1 Introduction
Adjuvants play a crucial role in enhancing vaccine effectiveness by stimulating the immune system to produce a robust response (Reed et al., 2013). Among vaccine adjuvants, QS-21 stands out for its potent immunostimulatory properties. It is a natural saponin derived from the bark of the Chilean soapbark tree, Quillaja saponaria (QS) Molina, which contains over 100 structurally related QS saponins due to its diverse composition (Reed et al., 2023). QS-21 is identified as the 21st fraction of 22 obtained from the reverse-phase high-performance liquid chromatography (HPLC) of the semi-purified QS extract with potent adjuvant activity, hence its name (Kensil et al., 1991; Martin et al., 2024). Currently, QS-21 has been the most extensively studied saponin adjuvant for over 28 years (Ragupathi et al., 2011). It has been shown to stimulate both cellular (Th1) and humoral (Th2) immune responses, making it highly effective in enhancing immunogenicity (Fernández-Tejada et al., 2014; Pink and Kieny, 2004). In 2017, QS-21 was first licensed for human use as a vaccine adjuvant, specifically for the herpes zoster vaccine Shingrix® (Lacaille-Dubois, 2019). Extensive clinical studies have demonstrated its strong immunostimulatory effects, significantly improving vaccine efficacy against a range of infectious diseases and cancers (Garçon and Van Mechelen, 2011; Gin and Slovin, 2011).
The structure of QS-21, as illustrated in Figure 1, consists of a quillaic acid triterpene attached to branched trisaccharide and linear oligosaccharide moieties. QS-21 is a mixture of two isomers, apiose- (65%) and xylose-containing (35%) oligosaccharides, attached to the D-xylose ring of the linear trisaccharide group. These two isomers were found to have similar adjuvanticity and toxicity (Ragupathi et al., 2010). A labile acyl side chain containing an arabinofuranose ring is connected to the D-fucose ring of the linear oligosaccharide. Despite its immunostimulatory properties, the instability of the acyl side chain and the challenges associated with its low-yielding purification limit the broader application of QS-21. Moreover, the loss of the lipophilic side chain due to the hydrolysis of the ester linkage results in the loss of its adjuvanticity (Marciani et al., 2001).

Figure 1. Structure of QS-21 and its saponin conjugate 1.
Numerous efforts have been undertaken to synthesize analogs of QS-21 with comparable or enhanced potency to mitigate these challenges. Wang et al. (2005) achieved the pioneering total synthesis of QS-21 and its definitive structural characterization. Additionally, they conducted an efficient semi-synthesis of various QS-21 variants to develop immunoadjuvants with improved chemical stability (Chea et al., 2012; Fernández-Tejada et al., 2016). Liang et al. (2020) patented the synthesis of saponin conjugates, including the truncated QS-21 moiety 1 (Figure 1), which enhances efficacy in both humoral and cell-mediated immunity. The structure of 1 comprises a truncated linear trisaccharide featuring a β-D-Xylp-(1→4)-α-L-Rhap-(1→2)-β-D-Fucp moiety linked to the quillaic acid triterpene.
In this study, we present an efficient and streamlined synthesis of the linear trisaccharide moiety derived from saponin conjugate 1, offering an alternative approach to the synthesis of these QS-21 oligosaccharide units. Two complementary strategies, pre- and post-glycosylation deoxygenation, were explored to address key synthetic challenges. These included the efficient incorporation of the rare sugar D-fucose and the stereoselective construction of oligosaccharides featuring 1,2-trans-glycosidic bonds, both of which are pivotal for constructing the biologically relevant glycan structure.
2 Materials and methods
2.1 General procedures
All moisture-sensitive reactions were carried out under an N2 atmosphere in flame-dried glassware. Solvents such as dichloromethane (CH2Cl2), acetonitrile (CH3CN), and tetrahydrofuran (THF) were distilled using a purification system with activated Al2O3. All commercially obtained reagents were used without additional purification, unless specified otherwise. All distilled water used was purified using a Milli-Q system. Prior to all glycosylations, the starting materials were thoroughly dried under high vacuum in a desiccator. Thin-layer chromatography (TLC) analysis was conducted on Silica Gel 60G F254 glass plates (0.25 mm, E. Merck from Germany). TLC analysis was performed with visualization under ultraviolet light (UV-254 nm), and staining was carried out by spraying with a solution of Hanessian’s reagent containing Ce(NH4)2(NO3)6, (NH4)6Mo7O24, and H2SO4 in water, followed by heating on a hot plate. Flash column chromatography was conducted on Silica Gel 60 (230–400 mesh, E. Merck).
Specific rotations were measured at ambient temperature using a HORIBA SEPA-300 High-Sensitive Polarimeter from Kyoto, Japan at 589 nm (sodium D line) and reported in 10−1⋅deg⋅cm2⋅g–1, with sample concentrations given in g⋅dL–1. IR spectra were recorded on KBr plates using a PerkinElmer Spectrum 100 FT-IR Spectrometer from Waltham, Massachusetts, USA. 1D and 2D NMR spectra were acquired using a Bruker Avance III 600 MHz spectrometer from Billerica, Massachusetts, USA at ambient temperature. Data were recorded as follows: chemical shift in ppm from the solvent resonance used as the internal standard (CDCl3 at 7.26 ppm), multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet), coupling constant in Hz, and integration. 13C NMR spectra were obtained using a 150 MHz spectrometer, and chemical shifts were recorded in ppm relative to the solvent resonance used as the internal standard (CDCl3 at 77.0 ppm). Mass spectra were acquired using an ESI Finnigan LCQ Mass Spectrometer (Thermo Finnigan from Waltham, Massachusetts, USA), performed at the Genomics Research Center.
2.2 Synthetic procedures and characterization data
2.2.1 Benzyl 2,3,4,6-tetra-O-acetyl-β-D-galactopyranoside (9)
To a stirred suspension of 14 (2.0 g, 5.12 mmol) in BnOH (1.1 mL), BF3•Et2O (2.6 mL, 20.5 mmol) was added at 0 °C under an N2 atmosphere. Upon completion of the reaction after 16 h, the mixture was diluted with CH2Cl2, washed with H2O and brine, dried over MgSO4, and then concentrated in vacuo. The residue was purified by column chromatography (silica gel; ethyl acetate/hexane = 1/4) to afford 9 (2.24 g, 75%). The IR spectrum (thin film) showed absorption bands at ν 2,924, 1,749, 1,369, and 1,221 cm–1. The 1H NMR spectrum (600 MHz, CDCl3) exhibited signals at δ 7.35–7.22 (m, 5H, Ar-H), 5.36 (dd, J = 3.5, 1.2 Hz, 1H, H-4), 5.26 (dd, J = 10.5, 7.9 Hz, 1H, H-2), 4.96 (dd, J = 10.5, 3.5 Hz, 1H, H-3), 4.89 (d, J = 12.4 Hz, 1H, Ar-CH2), 4.61 (d, J = 12.3 Hz, 1H, Ar-CH2), 4.49 (d, J = 8.0 Hz, 1H, H-1), 4.22–4.09 (m, 2H, H-6), 3.86 (td, J = 6.7, 1.2 Hz, 1H, H-5), 2.14 (s, 3H, CH3), 2.04 (s, 3H, CH3), 1.99 (s, 3H, CH3), and 1.95 (s, 3H CH3). The 13C NMR spectrum (150 MHz, CDCl3) displayed resonances at δ 170.4 (C), 170.3 (C), 170.1 (C), 169.4 (C), 136.7 (C), 128.4 (CH), 127.9 (CH),127.7 (CH), 99.8 (CH), 70.9 (CH), 70.8 (CH2), 70.7 (CH), 68.8 (CH), 67.0 (CH), 61.2 (CH2), 20.8 (CH3), 20.7 (CH3), 20.7 (CH3), and 20.6 (CH3). High-resolution mass spectrometry (HRMS) (ESI) analysis showed a peak at m/z 456.1869, which is consistent with the calculated value of m/z 456.1864 for C21H26O10NH4 ([M + NH4]+).
2.2.2 Benzyl 3,4-O-isopropylidene-β-D-galactopyranoside (8)
Compound 9 (493.3 mg, 1.83 mmol) and MeOH (6 mL) were added to the flask, and the mixture was stirred at 0 °C for 30 min. NaOMe (19.4 mg, 0.36 mmol) was slowly added to the reaction mixture in 10 mg portions at 10 min intervals, and the mixture was then allowed to warm to room temperature (RT) and react overnight. The reaction progress was monitored by TLC (ethyl acetate/hexane = 1/2). The reaction mixture was neutralized with Dowex® 50W × 8 resin to pH 5–6, filtered directly, and concentrated (45 °C, below 25 mbar) to obtain an off-white solid. The solid was vacuum-dried for over 16 h and used directly in the following steps without additional purification.
To a stirred suspension of the crude intermediate in anhydrous CH3CN (6 mL), 2,2-dimethoxypropane (2,2-DMP, 1.35 mL, 3.65 mmol) and 10-camphorsulfonic acid (CSA, 0.13 g, 0.55 mmol) were added at RT under an N2 atmosphere. Upon completion of the reaction after 30 min, the mixture was diluted with CH2Cl2, washed with H2O and brine, dried over MgSO4, and then concentrated under reduced pressure. The residue was purified by column chromatography (silica gel; ethyl acetate/hexane = 3/2) to yield 8 (301 mg, 53%). The specific rotation was [α]29D +6.16 (c 0.6, CHCl3). The IR spectrum (thin film) showed absorption bands at ν 3,400, 2,919, 1,454, and 1,040 cm–1. The 1H NMR spectrum (600 MHz, CDCl3) exhibited signals at δ 7.34 (d, J = 4.4 Hz, 4H, Ar-H), 7.30 (ddt, J = 8.4, 6.9, 3.6 Hz, 1H, Ar-H), 4.90 (d, J = 11.7 Hz, 1H, Ar-CH2), 4.64 (d, J = 11.7 Hz, 1H, Ar-CH2), 4.27 (d, J = 8.2 Hz, 1H, H-1), 4.13 (dd, J = 5.5, 2.1 Hz, 1H, H-5), 4.07 (dd, J = 7.4, 5.5 Hz, 1H, H-3), 3.97 (dd, J = 12.8, 8.2 Hz, 1H, H-6a), 3.82 (qd, J = 5.4, 4.2, 2.2 Hz, 2H, 4-H, H-6b), 3.60 (t, J = 7.8 Hz, 1H), 2.43 (s, 1H, 2-OH), 2.07–2.02 (m, 1H, 6-OH), 1.59 (s, 3H, CH3), and 1.32 (s, 3H, CH3). The 13C NMR spectrum (150 MHz, CDCl3) displayed resonances at δ 136.6 (C), 128.4 (CH), 128.1 (CH), 127.9 (CH), 110.3 (C), 101.1 (CH), 78.6 (CH), 73.7 (CH), 73.5 (CH), 73.3 (CH), 71.2 (CH2), 62.3 (CH2), 27.9 (CH3), and 26.1 (CH3). HRMS (ESI) analysis showed a peak at m/z 328.1757, which is consistent with the calculated value of m/z 328.1755 for C16H22O6NH4 ([M + NH4]+).
2.2.3 Benzyl 6-O-acetyl-3,4-O-isopropylidene-β-D-galactopyranoside (5)
To a stirred suspension of 8 (76 mg, 0.25 mmol) in anhydrous CH2Cl2 (0.7 mL), Et3N (0.23 g, 1.0 mmol) and Ac2O (24 μL, 0.26 mmol) were added at 0 °C under an N2 atmosphere. Upon completion of the reaction after 2 h, the mixture was diluted with CH2Cl2, washed with H2O and brine, dried over MgSO4, and then concentrated under reduced pressure. The residue was purified by column chromatography (silica gel; ethyl acetate/hexane = 1/2) to yield 5 (66.9 mg, 76%). The specific rotation was [α]28D +12.1 (c 1.0, CHCl3). The IR spectrum (thin film) showed absorption bands at ν 3,454, 2,987, 1,741, 1,242, and 1,076 cm–1. The 1H NMR spectrum (600 MHz, CDCl3) exhibited signals at δ 7.34 (d, J = 4.3 Hz, 4H, Ar-H), 7.30 (q, J = 4.4 Hz, 1H, Ar-H), 4.91 (d, J = 11.5 Hz, 1H, Ar-CH2), 4.60 (d, J = 11.6 Hz, 1H, Ar-CH2), 4.41–4.35 (m, 2H, H-6), 4.21 (d, J = 8.4 Hz, 1H, H-1), 4.13–4.09 (m, 1H, H-4), 4.04 (t, J = 6.5 Hz, 1H, H-3), 3.94 (t, J = 6.0 Hz, 1H, H-5), 3.60 (t, J = 7.9 Hz, 1H, H-2), 2.45 (s, 1H, 2-OH), 2.10 (d, J = 1.5 Hz, 3H, CH3), 1.50 (s, 3H, CH3), and 1.32 (s, 3H, CH3). The 13C NMR spectrum (150 MHz, CDCl3) displayed resonances at δ 170.8 (C), 136.7 (C), 128.5 (CH), 128.3 (CH), 128.1 (CH), 110.6 (C), 101.8 (CH), 78.6 (CH), 73.5 (CH), 73.4 (CH), 71.2 (CH), 70.9 (CH2), 63.5 (CH2), 28.1 (CH3), 26.3 (CH3), and 20.9 (CH3). HRMS (ESI) analysis showed a peak at m/z 370.1861, which is consistent with the calculated value of m/z 370.1860 for C18H24O7NH4 ([M + NH4]+).
2.2.4 Benzyl 6-O-methanesulfonyl-3,4-O-isopropylidene-β-D-galactopyranoside (20)
To a stirred suspension of 8 (60 mg, 0.194 mmol) in anhydrous CH2Cl2 (2.4 mL), Et3N (27.1 μL, 0.194 mmol) and MsCl (15.1 μL, 0.388 mmol) were added at 0 °C under an N2 atmosphere. Upon completion of the reaction after 1 h, the mixture was diluted with CH2Cl2, washed with H2O and brine, dried over MgSO4, and then concentrated under reduced pressure. The residue was purified by column chromatography (silica gel; ethyl acetate/hexane = 1/3) to afford 20 (47.5 mg, 63%). The specific rotation was [α]28D +7.80 (c 0.5, CHCl3). The IR spectrum (thin film) showed absorption bands at ν 3,492, 2,922, 1,712, 1,355, and 1,073 cm–1. The 1H NMR spectrum (600 MHz, CDCl3) exhibited signals at δ 7.34 (s, 3H, Ar-H), 7.38–7.27 (m, 2H, Ar-H), 4.90 (d, J = 11.6 Hz, 1H, Ar-CH2), 4.62 (d, J = 11.6 Hz, 1H, Ar-CH2), 4.51–4.42 (m, 2H, H-6), 4.25 (d, J = 8.3 Hz, 1H, H-1), 4.13 (dd, J = 5.6, 2.3 Hz, 1H, H-4), 4.11–4.04 (m, 2H, 3-H, H-5), 3.60 (td, J = 7.9, 1.9 Hz, 1H, H-2), 3.04 (s, 3H, SO2CH3), 2.40 (d, J = 2.4 Hz, 1H, OH), 1.50 (s, 3H, CH3), and 1.32 (s, 3H, CH3). The 13C NMR spectrum (150 MHz, CDCl3) displayed resonances at δ 136.5 (C), 128.61 (CH), 128.28 (CH), 128.24 (CH), 110.7 (C), 100.83 (CH), 78.6 (CH), 73.4 (CH), 73.0 (CH), 71.2 (CH2), 68.7 (CH2), 37.38 (CH3), 28.00 (CH3), and 26.31 (CH3). HRMS (ESI) analysis showed a peak at m/z 406.1535, which is consistent with the calculated value of m/z 406.1530 for C17H24O8SNH4 ([M + NH4]+).
2.2.5 Benzyl 6-deoxy-6-iodo-3,4-O-isopropylidene-β-D-galactopyranoside (21)
To a solution of 20 (20.2 mg, 0.059 mmol) in DMF (2.0 mL), tetrabutylammonium iodide (TBAI) (19.2 mg, 0.059 mmol) and KI (29.7 mg, 0.179 mmol) were added. The solution was stirred at 120 °C for 26 h, then cooled to RT, diluted with water, and extracted using ethyl acetate. The combined organic layer was washed with saturated aqueous Na2S2O3 and brine and then dried over anhydrous Na2SO4. The organic layer was evaporated, and the residue was purified by silica gel chromatography (silica gel; ethyl acetate/hexane = 1/3) to afford 21 (24.8 mg, 72%). The specific rotation was [α]28D +12.1 (c 0.5, CHCl3). The IR spectrum (thin film) showed absorption bands at ν 3,432, 2,920, and 1,065 cm–1. The 1H NMR spectrum (600 MHz, CDCl3) exhibited signals at δ 7.40–7.32 (m, 4H, Ar-H), 7.34–7.27 (m, 1H, Ar-H), 4.95 (d, J = 11.6 Hz, 1H, Ar-CH2), 4.65 (d, J = 11.7 Hz, 1H, Ar-CH2), 4.27 (dd, J = 5.5, 2.3 Hz, 1H, H-4), 4.22 (d, J = 8.3 Hz, 1H, H-1), 4.05 (dd, J = 7.4, 5.4 Hz, 1H, H-3), 3.88 (ddd, J = 7.3, 6.7, 2.3 Hz, 1H, H-5), 3.59 (ddd, J = 8.3, 7.4, 2.3 Hz, 1H, H-2), 3.43 (dd, J = 7.0, 0.9 Hz, 2H, H-6a, H-6b), 2.34 (d, J = 2.3 Hz, 1H, OH), 1.50 (s, 3H, CH3), and 1.34 (d, J = 0.8 Hz, 3H, CH3). The 13C NMR spectrum (150 MHz, CDCl3) displayed resonances at δ 136.6 (C), 128.6 (CH), 128.5 (CH), 128.2 (CH), 110.29 (C), 100.62 (CH), 78.7 (CH), 74.0 (CH), 73.8 (CH), 73.5 (CH), 70.79 (CH2), 28.1 (CH3), 26.2 (CH3), and 1.86 (CH2). HRMS (ESI) analysis showed a peak at m/z 438.0777, which is consistent with the calculated value of m/z 438.0772 for C16H21IO5NH4 ([M + NH4]+).
2.2.6 Benzyl 3,4-O-isopropylidene-β-D-fucopyranoside (10)
To a solution of 21 (10.1 mg, 0.024 mmol) in THF/MeOH = 10/1 (2 mL), Pd(OH)2/C (20% wt, 10.1 mg) was added. The resulting suspension was stirred under H2(g) at room temperature and atmospheric pressure for 6 h. The reaction mixture was filtered through Celite and concentrated under reduced pressure. The residue was purified by silica gel chromatography (silica gel; ethyl acetate/hexane = 1/3) to afford 10 (6.72 mg, 95%). The specific rotation was [α]29D −28.5 (c 0.1, CHCl3). The IR spectrum (thin film) showed absorption bands at ν 3,445, 2,926, and 1,071 cm–1. The 1H NMR spectrum (600 MHz, CDCl3) displayed signals at δ 7.37–7.31 (m, 4H, Ar-H), 7.29 (ddt, J = 8.8, 6.6, 3.0 Hz, 1H, Ar-H), 4.92 (dd, J = 11.6, 1.6 Hz, 1H, Ar-CH2), 4.57 (dd, J = 11.6, 1.8 Hz, 1H Ar-CH2), 4.21 (dd, J = 8.3, 1.6 Hz, 1H, H-1), 4.03–3.98 (m, 1H, H-3), 4.00–3.96 (m, 1H, H-4), 3.84 (qd, J = 6.6, 3.5 Hz, 1H, H-5), 3.61–3.55 (m, 1H, H-2), 2.35 (s, 1H, OH), 1.52 (s, 3H, CH3), 1.43 (dd, J = 6.6, 1.7 Hz, 3H, H-6), and 1.34 (s, 3H, CH3). The 13C NMR spectrum (150 MHz, CDCl3) exhibited resonances at δ 137.0 (C), 128.5 (CH), 128.3 (CH), 128.0 (CH), 109.9 (C), 100.9 (CH), 78.7(CH), 76.3 (CH), 73.6 (CH), 70.8 (CH2), 69.2 (CH), 28.2 (CH3), 26.3 (CH3), and 16.6 (CH3). HRMS (ESI) analysis showed a peak at m/z 312.1810, which is consistent with the calculated value of m/z 312.1805 for C16H22O5NH4 ([M + NH4]+).
2.2.7 2,3,4-Tri-O-acetyl-α,β-D-xylopyranoside (12)
A 12-L reaction flask and a 1-L addition funnel were dried and allowed to cool to room temperature. D-Xylose (500 g, 3.33 mol), Et3N (2.8 L, 19.9 mol), and DMAP (40.7 g, 0.33 mol) were added to the flask, and the mixture was cooled to 0 °C and stirred for 30 min. Ac2O (1.57 L, 16.6 mol) was placed in an addition funnel and added dropwise over approximately 1 h. The mixture changed from pale yellow and turbid to dark brown while remaining turbid. The reaction was stirred for approximately 3 h. The reaction progress was monitored by TLC (ethyl acetate/hexane = 1/1). The reaction mixture was quenched by pouring into 2 L of ice water, extracted with ethyl acetate, and washed with saturated aqueous NaHCO3 solution. The organic layer was dried over MgSO4, filtered, concentrated (45 °C, below 20 mbar), and used directly in the subsequent steps. Mechanical stirring was set up for a 20-L reaction flask. Compound 12 and THF were added to the flask, and the mixture was cooled to 0 °C and stirred for 30 min. BnNH2 (610.2 g, 5.59 mol) was added dropwise using a 1-L addition funnel. The mixture was allowed to warm to room temperature and react overnight. The reaction progress was monitored by TLC (ethyl acetate/hexane = 1/1). The reaction mixture was quenched with 1 N HCl(aq) and neutralized to a pH of 0–2. The mixture was extracted twice with ethyl acetate/brine, dried over MgSO4, filtered, and concentrated (45 °C, below 30 mbar). The crude product was then subjected to 151G3 flash column chromatography in three batches. The 151G3 flash column (diameter: 18 cm; height: 20 cm; volume: approximately 5 L) was loaded with crude product (approximately 300 g) that had been premixed with silica gel (approximately 300 g) in an ethyl acetate/hexane = 1/2 mixture. The column was packed with 2 L of silica gel, followed by the addition of the crude solution. The column was eluted with an ethyl acetate/hexane = 1/2 mixture (approximately 3 L per 8 fractions). The eluent was concentrated (45 °C, below 15 mbar) and allowed to stand overnight, yielding a precipitate. The solid was washed with hexane, dried under vacuum, and afforded a white solid (561 g) with a yield of 62% (over two steps). The specific rotation was [α]28D +58.4 (c 1.0, CHCl3). The IR spectrum (thin film) showed absorption bands at ν 3,393, 2,947, 1,757, 1,230, and 1,052 cm–1.
2.2.8 p-Methylphenyl 2,3-O-isopropylidene-1-thio-α-L-rhamnopyranoside (7)
A 5-L reaction flask was dried and allowed to cool to room temperature. Compound 13 (300 g, 0.757 mol) and MeOH (3 L) were added to the flask, and the mixture was stirred at 0 °C for 30 min. NaOMe (40.9 g, 0.757 mol) was slowly added to the reaction mixture in 10 g portions at 10 min intervals, and the mixture was allowed to warm to room temperature and react overnight. The reaction progress was monitored by TLC (ethyl acetate/hexane = 2/1). The reaction mixture was neutralized with Dowex® 50W × 8 resin (prewashed twice with MeOH, approximately 330 g) to a pH of 5–6, filtered directly, and concentrated (45 °C, below 25 mbar) to obtain an off-white solid. The solid was dried under vacuum for over 16 h and then used directly in the subsequent steps without further purification.
Crude intermediates, CH3CN, 2,2-DMP (182.4 mL, 1.48 mol), and CSA (51.6 g, 0.222 mol), were added to the dried 5-L reaction flask, and the mixture was stirred at room temperature for 60 min. The reaction progress was monitored by TLC (ethyl acetate/hexane = 2/1 and ethyl acetate/hexane = 1/3). If the reaction was incomplete, the mixture was directly concentrated to dryness and step 2 was repeated with the addition of CH3CN and 2,2-DMP. Upon completion of the reaction, the mixture was quenched with Et3N, concentrated to dryness, and subjected to thin-layer chromatography using ethyl acetate/hexane = 1/2 as the eluent. The concentrated product was then subjected to azeotropic distillation with toluene (45 °C, below 25 mbar). The resulting solid was allowed to precipitate and washed thrice with hexane. The product was dried under vacuum to obtain 7 (169.2 g, 72% over two steps) as a yellow solid. The specific rotation was [α]28D −196.2 (c 1.0, CHCl3). The IR spectrum (thin film) showed absorption bands at ν 3,447, 2,984, and 1,065 cm–1.
2.2.9 p-Methylphenyl 2,3,4-tri-O-acetyl-β-D-xylopyranosyl-(1→4)-2,3-O-isopropylidene-1-thio-α-L-rhamnopyranoside (15)
Compound 12 (50 g, 0.181 mol), CCl3CN (500 mL), and Cs2CO3 (176.9 g, 0.543 mol) were sequentially added to the reaction flask and stirred at RT for approximately 30 min. The reaction progress was monitored by TLC (ethyl acetate/hexane = 1/2). The reaction mixture was filtered through Celite and concentrated to obtain crude product 6. This crude product was then subjected to vacuum drying for 16 h together with compound 7 (56.2 g, 0.181 mol). Then, 5 Å molecular sieve powder was activated by heating and allowed to cool to room temperature. It was then added to the reaction flask along with CH2Cl2, and the mixture was stirred for 1 h. The reaction mixture was cooled to −78 °C, followed by the addition of 6.5 mL of TMSOTf. The mixture was stirred for 2 h. The temperature of the reaction mixture was gradually increased to 0 °C, and an additional 16.4 mL of TMSOTf was added. The mixture was stirred for 3 h. The reaction progress was monitored by TLC (ethyl acetate/hexane = 1/2). The reaction mixture was filtered through Celite, neutralized with Na2CO3(aq), and extracted with CH2Cl2. The organic layer was dried over MgSO4, filtered, and concentrated. The residue was purified by column chromatography (silica gel; ethyl acetate/hexane = 2/3) to yield 15 (42.73 g, 53%). The specific rotation was [α]28D −193.6 (c 1.0, CHCl3). The IR spectrum (thin film) showed absorption peaks at ν 2,984, 1,757, 1,230, and 1,054 cm–1. The 1H NMR spectrum (600 MHz, CDCl3) displayed signals at δ 7.36–7.30 (m, 2H, Ar-H), 7.10 (d, J = 7.9 Hz, 2H, Ar-H), 5.62 (s, 1H, H-1), 5.18 (t, J = 8.5 Hz, 1H, H-2′), 4.99 (d, J = 6.8 Hz, 1H, H-1′), 4.96–4.86 (m, 2H, H-4′, H-3′), 4.27 (d, J = 5.5 Hz, 1H, H-2), 4.13–4.02 (m, 3H, H-3, H-5, H-5a′), 3.57 (dd, J = 10.0, 7.5 Hz, 1H, H-4), 3.33 (dd, J = 11.8, 8.6 Hz, 1H, H-5b′), 2.31 (s, 3H, CH3), 2.08 (s, 3H, CH3), 2.02 (s, 6H, CH3), 1.49 (s, 3H, CH3), 1.33 (s, 3H, CH3), and 1.18 (d, J = 6.2 Hz, 3H, H-6). The 13C NMR spectrum (150 MHz, CDCl3) exhibited resonances at δ 170.1 (C), 169.9 (C), 169.7 (C), 138.0 (C), 132.7 (CH), 129.9 (CH), 129.3 (C), 109.6 (C), 99.4 (CH), 84.05 (CH), 79.0 (CH), 77.9 (CH), 77.0 (CH), 71.4 (CH), 71.0 (CH), 69.1 (CH), 65.56 (CH), 62.0 (CH2), 27.9 (CH3), 26.5 (CH3), 21.2 (CH3), 20.8 (CH3), 20.7 (CH3), 17.3 (CH3), and 14.2 (CH3). HRMS (ESI) analysis showed a peak at m/z 586.2319, which is consistent with the calculated value of m/z 586.2317 for C27H36O11SNH4 ([M + NH4]+).
2.2.10 p-Methylphenyl 2,3,4-tri-O-acetyl-β-D-xylopyranosyl-(1→4)-2,3-di-O-acetyl-1-thio-α-L-rhamnopyranoside (4)
A solution of 15 (4.6 g, 8.1 mmol) in 2% HCl/MeOH (100 mL) was prepared and stirred for 16 h. The resulting mixture was evaporated and then azeotropically distilled with toluene (50 mL) twice under reduced pressure. After drying under high vacuum, the crude syrup was treated with Ac2O (2.2 mL, 23.1 mmol), Et3N (5.2 mL, 38.1 mmol), and DMAP (9 mg, 0.074 mmol) in CH2Cl2 under an N2 atmosphere at RT. Upon completion of the reaction after 2 h, the mixture was diluted with CH2Cl2, washed with H2O and brine, dried over MgSO4, and then concentrated under reduced pressure. The residue was purified by column chromatography (silica gel; ethyl acetate/hexane = 2/3) to yield 4 (3.8 g, 81%). The specific rotation was [α]28D −132.9 (c 1.0, CHCl3). The IR spectrum (thin film) showed absorption bands at ν 2,940, 1,751, 1,223, and 1,054 cm–1. The 1H NMR spectrum (600 MHz, CDCl3) exhibited signals at δ 7.34–7.30 (m, 2H, Ar-H), 7.09 (d, J = 7.9 Hz, 2H, Ar-H), 5.37 (dd, J = 3.4, 1.6 Hz, 1H, H-2), 5.24 (d, J = 1.6 Hz, H-1), 5.20 (dd, J = 9.7, 3.4 Hz, 1H, 1H, H-3), 5.12 (t, J = 9.2 Hz, 1H, H-3′), 4.94 (td, J = 9.3, 5.4 Hz, 1H, H-4′), 4.88 (dd, J = 9.5, 7.6 Hz, 1H, H-2′), 4.64 (d, J = 7.6 Hz, 1H, H-1′), 4.22 (dq, J = 9.5, 6.2 Hz, 1H, H-5), 4.10 (dd, J = 11.7, 5.4 Hz, 1H, H-5′a), 3.70 (t, J = 9.6 Hz, 1H, H-4), 3.32 (dd, J = 11.8, 9.6 Hz, 1H, H-5′b), 2.29 (s, 3H, CH3), 2.08 (d, J = 13.0 Hz, 6H, CH3), 2.03–1.98 (m, 9H, CH3), and 1.29 (d, J = 6.2 Hz, 3H, CH3). The 13C NMR spectrum (150 MHz, CDCl3) displayed resonances at δ 170.3 (C), 169.9 (C), 169.9 (C), 169.6 (C), 169.6 (C), 138.2 (C), 132.7 (CH), 129.9 (CH), 129.3 (C), 101.07 (CH), 85.7 (CH), 76.5 (CH), 72.2 (CH), 71.7 (CH), 71.6 (CH), 71.1 (CH), 69.2 (CH), 68.1(CH), 62.5 (CH2), 21.1 (CH3), 21.0 (CH3), 20.9 (CH3), 20.8 (CH3), 20.7 (CH3), 20.5 (CH3), and 17.4 (CH3). HRMS (ESI) analysis showed a peak at m/z 630.2215, which is consistent with the calculated value of m/z 630.2215 for C28H36O13SNH4 ([M + NH4]+).
2.2.11 Benzyl 2,3,4-tri-O-acetyl-β-D-xylopyranosyl-(1→4)-2,3-di-O-acetyl-α-L-rhamnopyranosyl-(1→2)-6-O-acetyl-3,4-O-isopropylidene-β-D-galactopyranoside (16)
To a stirred suspension of 4 (53.0 mg, 0.086 mmol), 5 (25.4 mg, 0.072 mmol), and activated 5 Å molecular sieve powder in anhydrous CH2Cl2 (1.6 mL), NIS (19.5 mg, 0.086 mmol) and TfOH (1.5 μL, 0.017 mmol) were added at 0 °C under an N2 atmosphere. Upon completion of the reaction after 30 min, the reaction was quenched by the addition of Et3N, saturated NaHCO3, and 10% Na2S2O3 aqueous solution. After warming and stirring at RT for 1 h, the reaction mixture was filtered, diluted with CH2Cl2, washed with 10% Na2S2O3 aqueous solution, saturated NaHCO3, and brine, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by column chromatography (silica gel; ethyl acetate/hexane = 1/2) to yield 16 (46.1 mg, 76%) as a white solid. The specific rotation was [α]26D −70.2 (c 1.5, CHCl3). The IR spectrum (thin film) showed absorption peaks at ν 2,935, 1,748, 1,223, and 1,051 cm–1. The 1H NMR spectrum (600 MHz, CDCl3) exhibited signals at δ 7.37–7.32 (m, 2H, Ar-H), 7.32–7.27 (m, 3H, Ar-H), 5.23–5.16 (m, 2H, H-2′, H-3′), 5.13–5.05 (m, 2H, H-1′, H-3′), 4.93 (td, J = 9.0, 5.3 Hz, 1H, H-4′), 4.89–4.80 (m, 2H, H-2″, Ar-CH2), 4.64–4.57 (m, 2H, H-1″, Ar-CH2), 4.39–4.26 (m, 3H, H-1, H-6a, H-6b), 4.15–4.10 (m, 1H, H-3), 4.07 (ddd, J = 11.5, 6.0, 3.5 Hz, 3H, H-4, H-5′, H-5a’’), 3.87 (ddd, J = 7.3, 4.7, 2.2 Hz, 1H, H-5), 3.74–3.68 (m, 1H, H-2), 3.58 (t, J = 9.6 Hz, 1H, H-4′), 3.31 (dd, J = 11.9, 9.2 Hz, 1H, H-5b’’), 2.10 (d, J = 7.8 Hz, 6H), 2.05 (s, 3H, CH3), 2.01 (s, 3H, CH3), 1.97 (d, J = 9.4 Hz, 6H, CH3), 1.47 (s, 3H, CH3), 1.27 (s, 3H, CH3), and 1.07 (d, J = 6.1 Hz, 3H, H-6′). The 13C NMR spectrum (150 MHz, CDCl3) displayed resonances at δ 170.79 (C), 170.25 (C), 170.03 (C), 169.88 (C), 169.86 (C), 169.28 (C), 136.45 (C), 128.57 (CH), 128.53 (CH), 128.05 (CH), 110.62 (C), 101.02 (CH), 98.72 (CH), 96.24 (CH), 79.72 (CH), 76.49 (CH), 75.80 (CH), 73.65 (CH), 72.15 (CH), 71.70 (CH), 71.14 (CH), 70.80 (CH), 70.48 (CH2), 70.01 (CH), 69.30 (CH), 66.82 (CH), 63.45 (CH2), 62.41 (CH2), 27.87 (CH3), 26.32 (CH3), 21.03 (CH3), 20.88 (CH3), 20.74 (CH3), 20.69 (CH3), 20.42 (CH3), and 17.20 (CH3). HRMS (ESI) analysis showed a peak at m/z 858.3399, which is consistent with the calculated value of m/z 858.3390 for C39H52O20NH4 ([M + NH4]+).
2.2.12 Benzyl 2,3,4-tri-O-acetyl-β-D-xylopyranosyl-(1→4)-2,3-di-O-acetyl-α-L-rhamnopyranosyl-(1→2)-6-O-methanesulfonyl-3,4-O-isopropylidene-β-D-galactopyranoside (17)
To a stirred suspension of 16 (50.3 mg, 0.059 mmol) in anhydrous CH2Cl2 (5.0 mL), 7%–8% Mg(OMe)2 (0.5 mL) was added at 0 °C under an N2 atmosphere. Upon completion of the reaction after 1 h, the mixture was diluted with CH2Cl2, washed with H2O and brine, dried over MgSO4, and then concentrated under reduced pressure. The resulting mixture was evaporated and then azeotropically distilled with toluene (50 mL) twice under reduced pressure to generate crude intermediate 3. After drying under high vacuum, the crude syrup containing 3 was treated with Et3N (16.6 μL, 0.119 mmol) and MsCl (9.2 μL, 0.119 mmol) at 0 °C in anhydrous CH2Cl2 (5 mL) under an N2 atmosphere. Upon completion of the reaction after 1 h, the mixture was diluted with CH2Cl2, washed with H2O and brine, dried over MgSO4, and then concentrated under reduced pressure. The residue was purified by column chromatography (silica gel; ethyl acetate/hexane = 2/3) to yield 17 (36.2 mg, 70% over two steps). The specific rotation was [α]28D −38.2 (c 1.0, CHCl3). The IR spectrum (thin film) showed absorption bands at ν 2,935, 1,693, 1,295, and 1,193 cm–1. The 1H NMR spectrum (600 MHz, CDCl3) exhibited signals at δ 7.39–7.33 (m, 2H, Ar-H), 7.30 (td, J = 6.4, 1.7 Hz, 3H, Ar-H), 5.23–5.16 (m, 2H, H-2′, H-3′), 5.14–5.08 (m, 2H, H-1′, H-3″), 4.94 (td, J = 9.1, 5.4 Hz, 1H, H-4″), 4.89–4.82 (m, 2H, 2-H″, ArCH2), 4.64–4.60 (m, 2H, H-1′, ArCH2), 4.49–4.39 (m, 2H, H-6a, H-6b), 4.34 (d, J = 8.1 Hz, 1H, H-1), 4.17 (dd, J = 6.9, 5.6 Hz, 1H, H-3), 4.10–4.03 (m, 3H, H-4, H-5′, H-5a), 4.00 (ddd, J = 7.7, 4.4, 2.2 Hz, 1H, H-5), 3.71 (dd, J = 8.2, 6.9 Hz, 1H, H-2), 3.59 (t, J = 9.6 Hz, 1H, H-4′), 3.31 (dd, J = 11.8, 9.3 Hz, 1H, H-5b’’), 3.04 (s, 3H, SO2CH3), 2.11 (s, 3H, CH3), 2.06 (s, 3H, CH3), 2.02 (s, 3H, CH3), 1.98 (s, 3H, CH3), 1.97 (s, 3H, CH3), 1.47 (s, 3H, CH3), 1.27 (s, 3H, CH3), and 1.09 (d, J = 6.2 Hz, 3H, H-6′). The 13C NMR spectrum (150 MHz, CDCl3) displayed resonances at δ 170.2 (C), 169.9 (C), 169.8 (C), 169.7 (C), 169.3 (C), 136.2 (C), 128.6 (CH), 128.5 (CH), 128.1 (CH), 110.8 (C), 101.0 (CH), 98.9 (CH), 96.3 (CH), 79.8 (CH), 76.4 (CH), 75.7 (CH), 73.1 (CH), 72.1 (CH), 71.6 (CH), 71.1 (CH), 70.8 (CH2), 70.7 (CH), 69.9 (CH), 69.3 (CH), 68.7 (CH2), 66.9 (CH), 62.4 (CH2), 37.4 (CH3), 27.8 (CH3), 26.3 (CH3), 20.9 (CH3),20.7 (CH3), 20.6 (CH3), 20.4 (CH3), and 17.2 (CH3). HRMS (ESI) analysis showed a peak at m/z 894.3065¸ which is consistent with the calculated value of m/z 894.3060 for C38H52O21SNH4 ([M + NH4]+).
2.2.13 Benzyl 2,3,4-tri-O-acetyl-β-D-xylopyranosyl-(1→4)-2,3-di-O-acetyl-α-L-rhamnopyranosyl-(1→2)-6-deoxy-6-iodo-3,4-O-isopropylidene-β-D-galactopyranoside (18)
To a solution of 17 (10.2 mg, 0.012 mmol) in DMF (2 mL), tetrabutylammonium iodide (TBAI) (4.3 mg, 0.012 mmol) and KI (5.8 mg, 0.034 mmol) were added. The solution was stirred at 120 °C for 26 h, then cooled to room temperature, diluted with water, and extracted using ethyl acetate. The combined organic layer was washed with saturated aqueous Na2S2O3 and brine and then dried over anhydrous Na2SO4. The organic layer was concentrated in vacuo, and the residue was purified by silica gel chromatography (silica gel; ethyl acetate/hexane = 2/3) to yield 18 (6.3 mg, 58%). The specific rotation was [α]27D 29.4 (c 0.25, CHCl3). The IR spectrum (thin film) showed absorptions bands at ν 3,401, 2,920, 1,749, 1,221, and 1,052 cm–1. The 1H NMR spectrum (600 MHz, CDCl3) exhibited signals at δ 7.33 (td, J = 13.9, 7.0 Hz, 5H, Ar-H), 5.20 (dd, J = 9.0, 2.6 Hz, 2H, H-2′, H-3), 5.14–5.06 (m, 2H, H-1′, H-3″), 4.97–4.85 (m, 3H, H-2″, H-4″, Ar-CH2), 4.68 (d, J = 11.7 Hz, 1H, Ar-CH2), 4.63 (d, J = 7.5 Hz, 1H, H-1″), 4.28 (d, J = 8.2 Hz, 1H, H-1), 4.21 (dd, J = 5.6, 2.3 Hz, 1H, H-3), 4.15–4.05 (m, 3H, H-4, H-5″, H-5a’’), 3.81 (dt, J = 10.1, 3.9 Hz, 1H, H-5), 3.71 (t, J = 7.5 Hz, 1H, H-2), 3.59 (t, J = 9.4 Hz, 1H, H-4′), 3.41 (td, J = 9.7, 9.2, 6.9 Hz, 2H, H-6a, H-6b), 3.32 (dd, J = 11.8, 9.2 Hz, 1H, H-5b’’), 2.11 (d, J = 1.8 Hz, 3H, CH3), 2.02 (d, J = 1.7 Hz, 3H, CH3), 1.98 (s, 3H, CH3), 1.97 (s, 3H, CH3), 1.47 (s, 3H, CH3), 1.29 (s, 3H, CH3), and 1.10 (d, J = 6.0 Hz, 3H, H-6′). The 13C NMR spectrum (150 MHz, CDCl3) displayed resonances at δ 170.2 (C), 170.0 (C), 169.7 (C), 169.2 (C), 136.4 (C), 128.6 (C), 128.5 (CH), 128.1 (CH), 110.4 (C), 101.0 (CH), 98.6 (CH), 96.1 (CH), 79.8, 75.5(CH), 74.2 (CH), 73.4 (CH), 72.1 (CH), 71.6 (CH), 71.1 (CH), 70.4 (CH2), 70.0 (CH), 69.3 (CH), 66.8 (CH), 62.4 (CH2), 27.8 (CH3), 26.3 (CH2), 21.0 (CH2), 20.7 (CH3), 20.7 (CH3), 20.4 (CH3), and 17.2 (CH3). HRMS (ESI) analysis showed a peak at m/z 926.2298, which is consistent with the calculated value of m/z 926.2302 for C37H49IO18NH4 ([M + NH4]+).
2.2.14 Benzyl 2,3,4-tri-O-acetyl-β-D-xylopyranosyl-(1→4)-2,3-di-O-acetyl-α-L-rhamnopyranosyl-(1→2)-3,4-O-isopropylidene-β-D-fucopyranoside (19)
From 4 and 10: A mixture of acceptor 10 (5.0 mg, 0.017 mmol, 1 equiv), donor 4 (17.7 mg, 1.7 equiv, 0.029 mmol), and freshly activated AW-500 MS (25 mg) in dry CH2Cl2 (1.5 mL) was stirred at room temperature for 1 h under an N2 atmosphere and then cooled to 0 °C. NIS (6.5 mg, 1.7 equiv, 0.029 mmol) and TfOH (0.3 μL, 0.2 equiv, 0.003 mmol) were added to the reaction mixture. After constant stirring at 0 °C for 2 more hours, the mixture was filtered through a pad of Celite, the solids were washed with CH2Cl2, and the filtrate was washed with a mixture of saturated NaHCO3(aq) and 20% Na2S2O3 solution and then with water. The organic layer was dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The residue was purified by flash column chromatography (silica gel; ethyl acetate/hexane = 2/3) to afford trisaccharide 19 (12.7 mg, 95%).
From 18: To a solution of 18 (10.1 mg, 0.011 mmol) in a co-solvent of THF/MeOH (10/1, 2 mL), Pd(OH)2/C (20% wt, 20.2 mg) was added. The resulting suspension was stirred under H2(g) at room temperature and atmospheric pressure for 6 h. The reaction mixture was filtered through Celite and concentrated under reduced pressure. The residue was purified by silica gel chromatography (silica gel; ethyl acetate/hexane = 2/3) to yield 19 (8.2 mg, 95%).
The specific rotation was [α]28D +38.0 (c 0.1, CHCl3). The IR spectrum (thin film) showed absorption bands at ν 2,919, 1,750, 1,222, and 1,073 cm–1. The 1H NMR spectrum (600 MHz, CDCl3) exhibited signals at δ 7.34 (t, J = 7.0 Hz, 2H, Ar-H), 7.31–7.26 (m, 3H, Ar-H), 5.24–5.17 (m, 2H, H-2′, H-3′), 5.13–5.07 (m, 2H, H-1′, H-3″), 4.97–4.90 (m, 1H, H-4″), 4.90–4.83 (m, 2H, H-2″, Ar-CH2), 4.62 (dd, J = 7.4, 2.1 Hz, H-1″), 4.58 (dd, J = 11.7, 2.0 Hz, 1H, Ar-CH2), 4.29 (dd, J = 8.2, 1.8 Hz, 1H, H-1), 4.12–4.04 (m, 3H, H-3, H-5′, H-5a’’), 3.93 (dt, J = 4.6, 2.0 Hz, 1H, H-4), 3.77 (qd, J = 6.6, 3.5 Hz, 1H, H-5), 3.73–3.67 (m, 1H, H-2), 3.57 (td, J = 9.6, 2.1 Hz, 1H, H-4′), 3.31 (ddd, J = 11.5, 9.1, 2.0 Hz, 1H, H-5b’’), 2.11 (s, 3H, CH3), 2.05 (s, 3H, CH3), 2.02 (s, 3H, CH3), 1.97 (s, 3H, CH3), 1.96 (s, 3H, CH3), 1.49 (s, 3H, CH3), 1.40 (dd, J = 6.5, 1.8 Hz, 3H, H-6), 1.29 (s, 3H, CH3), and 1.05 (dd, J = 6.5, 2.2 Hz, 3H, H-6′). The 13C NMR spectrum (150 MHz, CDCl3) displayed resonances at δ 170.2 (C), 170.0 (C), 169.8 (C), 169.8 (C), 169.2(C), 136.8 (C), 128.6 (CH), 128.4 (CH), 127.9 (CH), 110.0 (C), 100.9 (CH), 98.8 (CH), 96.2 (CH), 79.9 (CH), 75.8 (CH), 72.1 (CH), 71.8 (CH), 71.1 (CH), 70.3 (CH2), 70.1 (CH), 69.3 (CH), 68.8 (CH), 66.7 (CH), 62.4 (CH2), 28.0 (CH3), 26.4 (CH3), 21.0 (CH3), 20.7 (CH3), 20.7 (CH3), 20.4 (CH3), 17.2 (CH3), and 16.6 (CH3). HRMS (ESI) analysis showed a peak at m/z 800.3337, which is consistent with the calculated value of m/z 800.3335 for C37H50O18NH4 ([M + NH4]+).
2.2.15 2,3,4-tri-O-acetyl-β-D-xylopyranosyl-(1→4)-2,3-di-O-acetyl-α-L-rhamnopyranosyl-(1→2)-3,4-O-isopropylidene-β-D-fucopyranoside (2)
From 18: To a solution of 18 (7.1 mg, 0.008 mmol) in THF/MeOH (1/1, 1 mL), Pd(OH)2/C (20% wt, 35.5 mg) was added. The resulting suspension was stirred under H2(g) at room temperature and atmospheric pressure for 7 h. The reaction mixture was filtered through Celite and concentrated under reduced pressure. The residue was purified by silica gel chromatography (silica gel; ethyl acetate/hexane = 2/3) to yield 2 (4.8 mg, 89%, α:β = 1.1:1.0).
From 19: To a solution of 19 (10.4 mg, 0.012 mmol) in THF/MeOH (1/1, 1 mL), Pd(OH)2/C (20% wt, 52.0 mg) was added. The resulting suspension was stirred under H2(g) at room temperature and atmospheric pressure for 4 h. The reaction mixture was filtered through Celite and concentrated under reduced pressure. The residue was purified by silica gel chromatography (silica gel; ethyl acetate/hexane = 2/3) to afford 2 (7.4 mg, 89%, α:β = 1.5:1.0).
The IR spectrum (thin film) showed absorption bands at ν 2,919, 1,750, 1,222, and 1,073 cm–1. The 1H NMR spectrum (600 MHz, CDCl3) exhibited signals at δ 5.24 (ddd, J = 22.0, 3.8, 1.8 Hz, 2H), 5.21–5.15 (m, 3H), 5.11 (td, J = 9.1, 2.8 Hz, 2H), 5.06 (d, J = 2.1 Hz, 2H), 4.96–4.90 (m, 2H), 4.85 (dd, J = 9.5, 7.7 Hz, 2H), 4.61 (td, J = 7.1, 6.5, 2.7 Hz, 3H), 4.37 (d, J = 7.1 Hz, 1H), 4.31 (dd, J = 7.2, 5.7 Hz, 1H), 4.15 (t, J = 6.2 Hz, 1H), 4.10 (dd, J = 11.8, 5.4 Hz, 2H), 4.07–4.02 (m, 2H), 3.98 (dd, J = 5.8, 2.1 Hz, 1H), 3.82–3.75 (m, 2H), 3.62 (t, J = 9.6 Hz, 2H), 3.57 (t, J = 7.1 Hz, 1H), 3.36–3.29 (m, 2H), 3.26 (dd, J = 6.3, 1.2 Hz, 1H), 2.85 (d, J = 4.1 Hz, 1H), 2.13 (d, J = 1.6 Hz, 6H), 2.05 (d, J = 1.6 Hz, 6H), 2.01 (d, J = 1.5 Hz, 6H), 1.99 (d, J = 1.5 Hz, 6H), 1.98 (d, J = 4.6 Hz, 6H), 1.49 (d, J = 7.7 Hz, 6H), 1.38 (d, J = 6.5 Hz, 3H), 1.33 (d, J = 6.6 Hz, 3H), 1.31 (d, J = 2.9 Hz, 6H), and 1.28–1.26 (m, 6H). The 13C NMR spectrum (150 MHz, CDCl3) displayed resonances at δ 170.2 (C), 170 (C), 169.9 (C), 169.8 (C), 169.6 (C), 169.5 (C), 110.0 (C), 109.1 (C), 101.1 (CH),101.0 (CH), 97.5 (CH), 97.1 (CH), 96.1 (CH), 94.8 (CH), 91.6 (CH), 78.6 (CH), 76.3 (CH), 76.3 (CH), 75.9 (CH), 75.4 (CH), 75.3 (CH), 75.1 (CH), 72.2 (CH), 72.1 (CH), 71.4 (CH), 71.2 (CH), 71.1 (CH), 70.1 (CH), 70.0 (CH), 69.2 (CH), 68.7 (CH), 67.3 (CH), 67.1 (CH), 63.4 (CH), 62.5 (CH2), 62.4 (CH2), 28.0 (CH3), 27.9 (CH3), 26.2 (CH3), 26.1 (CH3), 21.0 (CH3), 20.9 (CH3), 20.7 (CH3), 20.6 (CH3), 20.5 (CH3), 17.6 (CH3), 17.5 (CH3), 16.6 (CH3), and 16.5 (CH3). HRMS (ESI) analysis showed a peak at m/z 710.2867, which was consistent with the calculated value of m/z 710.2866 for C30H44O18NH4 ([M + NH4]+).
3 Results and discussion
3.1 Retrosynthesis
A linear trisaccharide of interest consists of D-xylose, L-rhamnose, and the rare D-fucose. Key challenges, such as the synthesis of the rare D-fucose unit and the stereocontrolled installation of 1,2-trans glycosidic bonds between each sugar ring, must be addressed to effectively generate the target linear trisaccharide. The target linear trisaccharide 2 can be synthesized using two [2 + 1] glycosylation strategies between a common disaccharide donor 4 and acceptors 5 and 10, as shown in Scheme 1.

Scheme 1. Structure of linear trisaccharide moiety 2 from QS-21 and its retrosynthesis. Ac, acetyl; Bn, benzyl; Tol, p-methylphenyl.
In the first retrosynthetic strategy, the target trisaccharide was envisioned to be obtained through a glycosylation reaction between glycosyl donor 4 and glycosyl acceptor 5. The latter can be derived via regioselective acetylation of compound 8, which, in turn, can be obtained through deacylation, followed by acetonide protection of compound 9. After glycosylation, the D-galactose reducing end in 3 would be converted into a D-fucose moiety through deoxygenation at the C6 position. On the other hand, the second retrosynthetic strategy was to synthesize the target trisaccharide from donor 4 and acceptor 10. Prior to glycosylation, D-galactose in 8 would be converted into a D-fucose acceptor 10. Disaccharide donor 4, used in both approaches, is synthesized via [1 + 1] glycosylation of D-xylose-derived donor 6 with L-rhamnose-derived acceptor 7, ensuring efficient assembly. The glycosylation reactions are anticipated to yield 1,2-trans-glycosidic linkages facilitated by the adjacent acetyl (Ac) group at the C2 position.
3.2 Synthesis of the monosaccharide precursors
The truncated linear trisaccharide domain was synthesized beginning with the preparation of monosaccharide building blocks. In Scheme 2A, D-xylose building block 12 was synthesized starting from DMAP-catalyzed per-O-acetylation of D-xylose to generate 11 in 93% yield. Regioselective anomeric deacetylation with benzylamine (BnNH2) then afforded 12 in 68% yield. To prepare L-rhamnose-derived acceptor 7 (Scheme 2B), an acetonide protecting group was introduced via a sequential two-step process involving Zemplén deacylation, followed by isopropylidenation of the 2,3-cis-diol group using 2,2-DMP and CSA. This acetonide protection sequence resulted in the formation of 7 in 72% yield over two steps.

Scheme 2. Synthesis of the monosaccharide building blocks 12 (A), 7 (B), and 5 (C). Reagents and conditions: (a) Ac2O, DMAP, Et3N, 0 °C, 3 h, 93%; (b) BnNH2, THF, 0 °C, 16 h, 68%; (c) NaOMe, MeOH, 0 °C, 1 h; (d) 2,2-DMP, CSA, MeCN, RT, 1 h, 72% over 2 steps; (e) BnOH, BF3•Et2O, 0 °C, 16 h, 75%; (f) NaOMe, MeOH, 0 °C to RT, 5 h; (g) 2,2-DMP, CSA, MeCN, RT, 30 min, 48% over two steps; (h) Ac2O, Et3N, CH2Cl2, 0 °C to RT, 3 h, 76%. DMAP, 4-dimethylaminopyridine; BnNH2, benzylamine; THF, tetrahydrofuran; Me, methyl; 2,2-DMP, 2,2-dimethoxypropane; CSA, 10-camphorsulfonic acid.
In Scheme 2C, per-O-acetylated D-galactose underwent BF3•Et2O-catalyzed benzyl group substitution at the anomeric position using benzyl alcohol (BnOH) to afford 9 in 75% yield. Subsequently, acetonide protection of 9 was performed analogously to the synthesis of 7, resulting in the formation of 8 in 48% yield over two steps. The acetonide group was installed to protect the target trisaccharide prior to its final functionalization, when it was attached to the acyl side chain of QS-21. Finally, regioselective acetylation of the primary alcohol at the C6 position produced 2-alcohol 5 in 76% yield.
3.3 Synthesis of the common disaccharide donor
The synthesis of common disaccharide donor 4 depicted in Scheme 3 involves the efficient assembly of compounds 6 and 7. Conversion of 12 to a glycosyl trichloroacetimidate 6 using Cl3CCN was carried out, and then it was directly coupled with 7 through a TMSOTf-promoted glycosylation to afford disaccharide 15 in 53% yield. Neighboring group participation of the 2-O-Ac group from D-xylose resulted in a β-linked disaccharide (1JC–H = 162.9 Hz). Analysis of the nondecoupled HSQC spectra distinguished the α- and β-anomers, with the α-anomers displaying a JC1–H1 coupling of approximately 170 Hz for the anomeric carbon and proton, while the β-anomers exhibited around 160 Hz (Bock and Pedersen, 1974). Hydrolysis of the isopropylidene ketal in 15 using 2% HCl/MeOH, followed by acetylation, afforded common disaccharide donor 4 in 81% yield over two steps.

Scheme 3. Synthesis of the common disaccharide donor 4. Reagents and conditions: (a) Cl3CCN, Cs2CO3, RT, 0.5 h; (b) TMSOTf, AW-500, CH2Cl2, –78 °C to 0 °C, 5 h, 53%; (c) 2% HCl/MeOH, RT, 16 h; (d) Ac2O, DMAP, Et3N, EtOAc, 0 °C, 16 h, 81% over two steps. TMSOTf, trimethylsilyl trifluoromethanesulfonate; EtOAc, ethyl acetate.
3.4 Synthesis of the truncated linear trisaccharide
The synthesis of linear trisaccharide 2 was conducted using two distinct approaches. The first strategy, illustrated in Scheme 4, involved an NIS/TfOH-promoted [2 + 1] glycosylation between thioglycoside donor 4 and acceptor 5. This reaction resulted in the successful formation of α-1→2-linked L-Rha-D-Gal 16 in 76% yield (1JC–H = 172.5 Hz), through the neighboring group participation of the 2-O-Ac moiety from the donor.
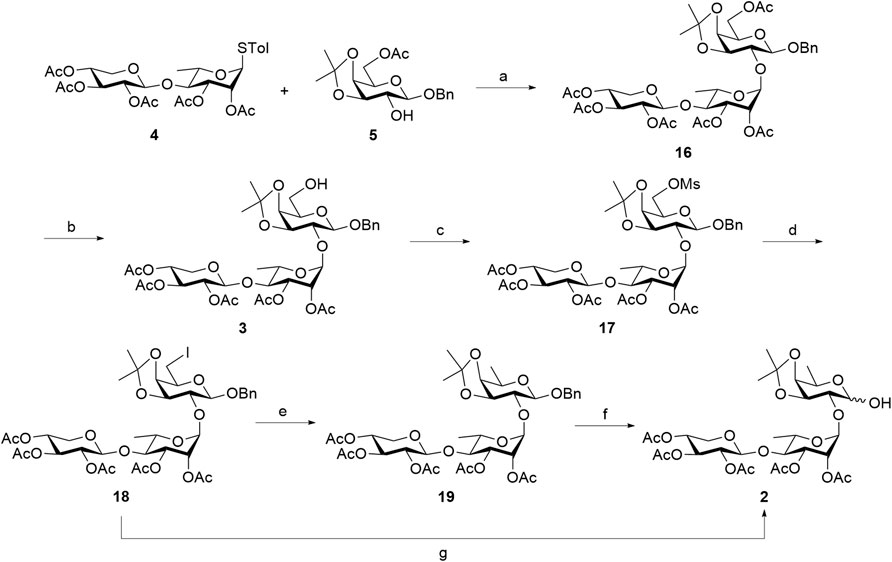
Scheme 4. First approach in the synthesis of truncated linear trisaccharide domain 2. Reagents and conditions: (a) NIS, TfOH, AW-500, CH2Cl2, 0 °C, 2 h, 76%; (b) 7%–8% Mg(OMe)2 in MeOH, CH2Cl2, 0 °C, 1 h; (c) Et3N, MsCl, CH2Cl2, 0 °C, 1 h, 70% (over two steps); (d) TBAI, KI, DMF, 120 °C, 22 h, 58%; (e) H2(g), Pd(OH)2/C, THF/MeOH (10/1), RT, 6 h, 95%; (f) H2(g), Pd(OH)2/C, THF/MeOH (1/1), RT, 4 h, 89%; (g) H2(g), Pd(OH)2/C, THF/MeOH (10/1), RT, 7 h, 89%. NIS, N-iodosuccinimide; TfOH, trifluoromethanesulfonic acid; MsCl, methanesulfonyl chloride; TBAI, tetra-n-butylammonium iodide; DMF, N,N-dimethylformamide.
Upon obtaining D-galactose-containing trisaccharide 16, post-glycosylation deoxygenation at the C6 position was performed to transform the reducing end into a rare D-fucose moiety. First, the primary 6-O-Ac group of D-galactose was selectively removed using 7%–8% Mg(OMe)2 in MeOH, generating 6-alcohol 3, which was used directly in the next step without further purification. From this intermediate, deoxygenation was initially planned by introducing an iodide group at the C6 position, which could then be removed via hydrogenolysis (Lemieux and Levine, 1962). However, attempts to directly substitute the 6-hydroxy group in compound 3 with iodine on a 50 mg scale using the Mitsunobu reaction [PPh3, diisopropyl azodicarboxylate (DIAD), and methyl iodide] were unsuccessful. No product formation was observed, even after varying the solvent or increasing the reaction temperature. As shown in Table 1, the reaction in CH2Cl2 at 0 °C–22 °C (Entry 1) and 22 °C (Entry 2) failed to produce desired product 18, and the starting material was largely recovered. Changing the solvent to THF and conducting the reaction at 0 °C–22 °C (Entry 3) or 60 °C (Entry 4) also did not lead to successful installation of a 6-iodo group at the reducing-end sugar. This may be attributed to the low reactivity of the hydroxy group toward direct iodine substitution.
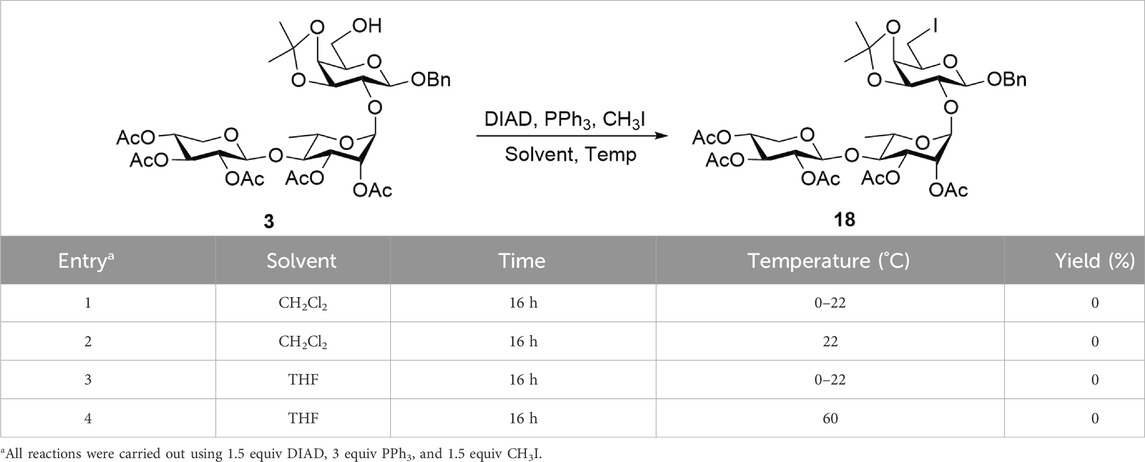
Table 1. Direct substitution of the iodine atom through the Mitsunobu reaction.
To overcome this key synthetic challenge, a methanesulfonyl (or mesyl) group was introduced at the O6 position prior to substitution with iodine to convert the hydroxy group to a more reactive leaving group. Following the deprotection of the 6-O-Ac group in trisaccharide 16 to form compound 3, mesylation with mesyl chloride (MsCl) furnished 17 in 70% yield over two steps. Various reaction conditions were then explored to convert 17 into the C6-iodinated trisaccharide 18, as summarized in Table 2. At a 10 mg scale, treatment of 17 with TBAI (1 equiv) and NaI (3 equiv) in DMF at 90 °C (Entry 1) did not yield 18, even after 48 h, and the starting material remained unreacted. Increasing the temperature to 120 °C (Entry 2) also failed to produce the desired product. A trace amount of 18 was observed when KI (3 equiv) was used in place of NaI at 90 °C for 22 h (Entry 3). Gratifyingly, increasing the temperature to 120 °C under these conditions (Entry 4) successfully afforded the C6-iodinated trisaccharide 18 in 58% yield. This likely results from the higher solubility and enhanced nucleophilicity of iodide ions from KI in DMF at elevated temperature, facilitating more efficient substitution. The conversion from mesyl to iodide caused an upfield shift of the two H6 protons to 3.41 ppm and the disappearance of the mesyl CH3 proton signal at 3.04 ppm, suggesting the efficient formation of 18. The final functionalization of trisaccharide 18 involves hydrogenolysis to remove the benzyl group and transform the galactosyl iodide moiety into a rare D-fucose residue. In our initial attempt, hydrogenolysis of compound 18 was carried out using Pd(OH)2/C (Degussa type, 20 wt%) at a loading rate of 1 g catalyst per gram of 18 under a hydrogen atmosphere. Under the initial conditions, using THF as the solvent, the reaction proceeded very slowly. Switching to an increased catalyst loading of 2 g Pd(OH)2 per gram of 18 and THF/MeOH (10/1) as co-solvent significantly improved the reaction efficiency, enabling selective removal of the iodine atom while preserving the benzyl group and affording compound 19 with a D-fucose-reducing end in 95% yield. The formation of D-fucose in 19 from the D-galactose-reducing end in 18 was evidenced by the presence of methyl protons at the C6 position (1H NMR: d, δ 1.44 ppm, J = 6.5 Hz). Furthermore, hydrogenolysis of 19 in the THF/MeOH (1/1) co-solvent using Pd(OH)2/C at a loading rate of 5 g catalyst per gram of starting material then furnished target linear trisaccharide 2 in 89% yield (α:β = 1:1).
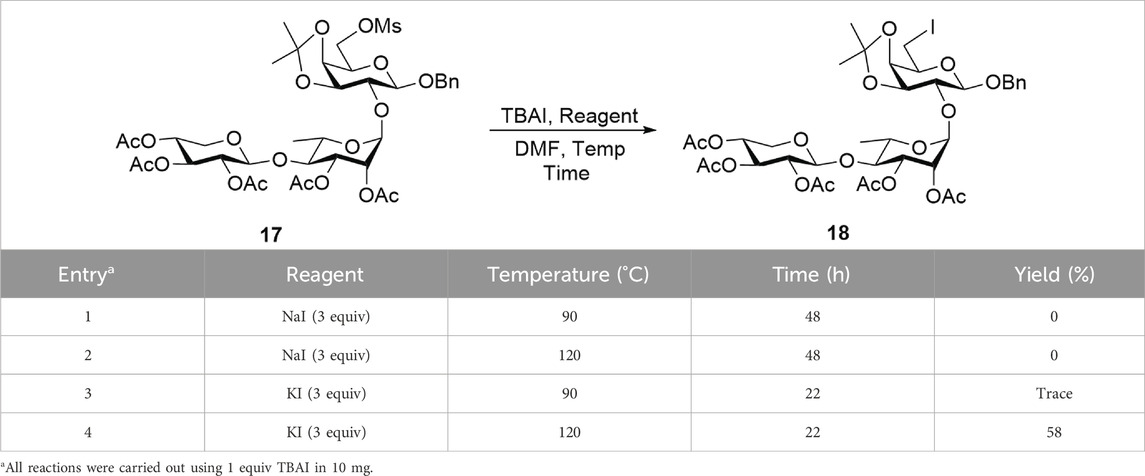
Table 2. Substitution of the iodine atom with mesylated compound 17.
In our second attempt to carry out the final functionalization, we tried to synthesize trisaccharide 2 directly from 18 by investigating the effect of Pd(OH)2/C loading on the hydrogenolysis reaction. Hydrogenolysis of C–O bonds, such as those in benzyl protecting groups, typically requires harsher conditions than the cleavage of C–I bonds (Ahluwalia, 2023). Hydrogenolysis was performed using Pd(OH)2/C at a loading rate of 5 g catalyst per gram of 18 under an H2 atmosphere, with THF/MeOH (1/1) as the co-solvent. This condition effectively removed both the iodine atom and the benzyl protecting group from 18, directly producing trisaccharide 2 in 89% yield. The successful synthesis of final compound 2 from 18 was confirmed by the disappearance of benzyl proton-NMR peaks and the detection of methyl protons at the C6 position (1H NMR: d, δ 1.38 ppm, J = 6.5 Hz).
Our second strategy in the synthesis of the truncated linear trisaccharide domain of QS-21 utilizes a pre-glycosylation deoxygenation approach from Scheme 1. Conversion of the D-galactose unit into rare D-fucose was conducted prior to [2 + 1] glycosylation with D-xylose and L-rhamnose-derived donor 4. This strategy was anticipated to be more efficient than the initial approach due to its reduced number of steps since the 6-deoxygenation reaction is only conducted on a monosaccharide.
The pre-glycosylation deoxygenation strategy commenced with the transformation of isopropylidenated D-galactose 8 into D-fucose through a series of mesylation, iodine substitution, and selective hydrogenolysis reactions (Scheme 5). Mesylation of 2,6-diol 8 at the C6 position furnished 20 in 63% yield, as indicated by the presence of mesyl methyl protons at 3.04 ppm. Following the optimized conditions from Table 2, the substitution of the OMs group with an iodine atom using TBAI and KI in DMF afforded 21 in 72% yield. Finally, D-fucose acceptor 10 was obtained following selective hydrogenolysis at the C6 position in THF/MeOH (10/1), preserving the integrity of the benzyl group prior to the [2 + 1] glycosylation step. The reaction was carried out using 1 g of Pd(OH)2/C per gram of 21 to achieve a high yield of 95% for compound 10, mirroring the synthesis of trisaccharide 19. The detection of an upfield chemical shift for the methyl C6-protons at 1.43 ppm of 10 indicates the successful conversion of D-galactose into rare D-fucose.

Scheme 5. Synthesis of acceptor 10 through pre-glycosylation deoxygenation. Reagents and conditions: (a) Et3N, MsCl, CH2Cl2, 0 °C, 2 h, 63%; (b) TBAI, KI, DMF, 120 °C, 22 h, 72%; (c) H2(g), Pd(OH)2/C, THF/MeOH (10/1), RT, 7 h, 95%.
With synthesized D-fucose acceptor 10 and disaccharide donor 4 in hand, we proceeded with [2 + 1] glycosylation to generate the trisaccharide unit 19 (Scheme 6). Using NIS/TfOH in CH2Cl2 at 0 °C, glycosylation proceeded with exclusive α-stereoselectivity (1JC–H = 173.2 Hz) to furnish trisaccharide 19 in 95% yield. Neighboring group participation of the 2-O-Ac group led to α-selectivity. Afterward, benzyl group deprotection through hydrogenolysis with H2 and Pd(OH)2/C in THF/MeOH (1/1) produced target linear trisaccharide 2 in 89% yield (α:β = 1.5:1.0). The structures of the final compound were confirmed through NMR spectroscopic and mass spectrometric analyses (see Supplementary Material).

Scheme 6. [2 + 1] glycosylation approach for the synthesis of truncated linear trisaccharide 2 using the pre-glycosylation deoxygenation strategy. Reagents and conditions: (a) NIS, TfOH, AW-500, CH2Cl2, 0 °C, 2 h, 95%; (b) H2(g), Pd(OH)2/C, THF/MeOH (1/1), RT, 7 h, 89%.
Using trisaccharide 19 as a representative example, all glycan backbone 1H protons were examined through the 1H, 13C, COSY, HSQC, HMBC, and 1D total correlation spectroscopy (1D TOCSY) experiments in detail. Following the identification of anomeric carbons and protons using HSQC, the complex 1H NMR spectrum was then deconvoluted into three distinct spectroscopic patterns by selectively exciting anomeric 1H nuclei at a given frequency using 1D TOCSY NMR. Figure 2 depicts the identification of protons from D-xylose (blue spectrum), L-rhamnose (red spectrum), and D-fucose (green spectrum) through a combined 1D TOCSY NMR and 2D COSY analysis. This combined analysis, along with HSQC and HMBC, confirms the connectivity of each sugar ring from trisaccharide 19, as shown in Figure 2.
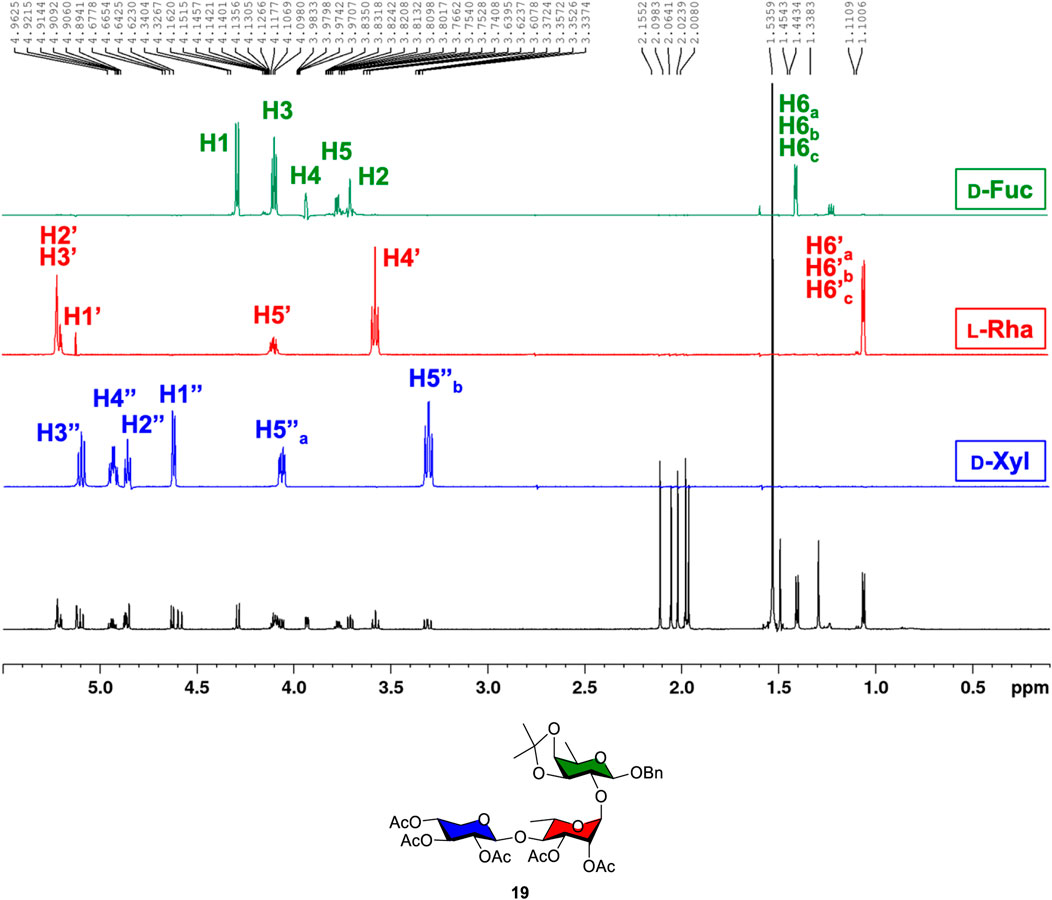
Figure 2. 1D TOCSY analysis of trisaccharide 19 resolving the complex 1H NMR spectrum into three resolve spectra corresponding to the ring protons of D-xylose (blue), L-rhamnose (red), and D-fucose (green).
Comparing the post-glycosylation and pre-glycosylation deoxygenation strategies, target trisaccharide 2 was obtained in overall yields of 7.8% and 15.6%, respectively. The pre-glycosylation deoxygenation route required only a single-step transformation after the [2 + 1] glycosylation reaction to reach the final product, whereas the post-glycosylation pathway involved three additional steps. Although the pre-glycosylation approach includes a three-step synthesis of the deoxygenated D-fucose acceptor, this added effort is offset by the reduced number of steps following glycosylation. Overall, the higher yield and greater step economy highlight the pre-glycosylation deoxygenation strategy as the more efficient route to the truncated linear trisaccharide fragment of QS-21.
4 Conclusion
We have successfully accomplished an efficient synthesis of the truncated linear trisaccharide domain 2 of QS-21. Two synthetic strategies were used: post-glycosylation and pre-glycosylation deoxygenation. The latter proved to be more efficient, yielding a higher overall yield with fewer reaction steps. En route, we established an efficient synthesis of the rare D-fucose moiety from D-galactose. We believe that this efficient synthesis of the linear trisaccharide domain will be applicable to the synthesis of pure and homogeneous QS-21 structures and their analogs for use as vaccine adjuvants.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author contributions
J-SL: Data curation, Formal analysis, Writing – original draft, Methodology. Z-HT: Data curation, Formal analysis, Investigation, Writing – review and editing, Methodology, Conceptualization. JD: Data curation, Formal analysis, Writing – original draft, Writing – review and editing. S-CH: Writing – original draft, Visualization, Conceptualization, Project administration, Writing – review and editing, Funding acquisition, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the National Science and Technology Council (NSTC) of Taiwan (NSTC 111-2639-M-001-001-ASP and NSTC 112-2639-M-001-001-ASP). S-CH also acknowledges being awarded the Taiwan Bio-Development Foundation (TBF) Chair in Biotechnology.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2025.1650302/full#supplementary-material
References
Ahluwalia, V. K. (2023). “Hydrogenolysis,” in Reduction in organic synthesis, 115–121. doi:10.1007/978-3-031-37686-3_4
Bock, K., and Pedersen, C. A. (1974). Study of 13C–H Coupling Constants in Hexopyranoses. J. Chem. Soc. Perkin Trans. 2 (293), 293–297. doi:10.1039/P29740000293
Chea, E. K., Fernández-Tejada, A., Damani, P., Adams, M. M., Gardner, J. R., Livingston, P. O., et al. (2012). Synthesis and preclinical evaluation of QS-21 variants leading to simplified vaccine adjuvants and mechanistic probes. J. Am. Chem. Soc. 134, 13448–13457. doi:10.1021/ja305121q
Fernández-Tejada, A., Chea, E. K., George, C., Pillarsetty, N., Gardner, J. R., Livingston, P. O., et al. (2014). Development of a minimal saponin vaccine adjuvant based on QS-21. Nat. Chem. 6, 635–643. doi:10.1038/nchem.1963
Fernández-Tejada, A., Tan, D. S., and Gin, D. Y. (2016). Development of improved vaccine adjuvants based on the saponin natural product QS-21 through chemical synthesis. Acc. Chem. Res. 49, 1741–1756. doi:10.1021/acs.accounts.6b00242
Garçon, N., and Van Mechelen, M. (2011). Recent clinical experience with vaccines using MPL- and QS-21-containing adjuvant systems. Expert Rev. Vaccines 10, 471–486. doi:10.1586/erv.11.29
Gin, Y., and Slovin, F. (2011). Enhancing immunogenicity of cancer vaccines: QS-21 as an immune adjuvant. Curr. Drug. Ther. 6, 207–212. doi:10.2174/157488511796391988
Kensil, C. R., Patel, U., Lennick, M., and Marciani, D. (1991). Separation and characterization of saponins with adjuvant activity from Quillaja saponaria Molina cortex. J. Immunol. 146, 431–437. doi:10.4049/jimmunol.146.2.431
Lacaille-Dubois, M.-A. (2019). Updated insights into the mechanism of action and clinical profile of the immunoadjuvant QS-21: a review. Phytomedicine 60, 152905. doi:10.1016/j.phymed.2019.152905
Lemieux, R. U., and Levine, S. (1962). The products of the prevost reaction on D-glucal triacetate. Can. J. Chem. 40, 1926–1932. doi:10.1139/v62-296
Liang, P.-H., Lai, Y.-H., Chang, C.-K., and Chaw, C.-W. (2020). Saponin conjugate and vaccine or pharmaceutical composition comprising the same.
Marciani, D. J., Pathak, A. K., Reynolds, R. C., Seitz, L., and May, R. D. (2001). Altered immunomodulating and toxicological properties of degraded Quillaja saponaria Molina saponins. Int. Immunopharmacol. 1, 813–818. doi:10.1016/S1567-5769(01)00016-9
Martin, L. B. B., Kikuchi, S., Rejzek, M., Owen, C., Reed, J., Orme, A., et al. (2024). Complete biosynthesis of the potent vaccine adjuvant QS-21. Nat. Chem. Bio. 20, 493–502. doi:10.1038/s41589-023-01538-5
Pink, J. R., and Kieny, M.-P. (2004). 4th Meeting on novel adjuvants currently in/close to human clinical testing. Vaccine 22, 2097–2102. doi:10.1016/j.vaccine.2004.01.021
Ragupathi, G., Damani, P., Deng, K., Adams, M. M., Hang, J., George, C., et al. (2010). Preclinical evaluation of the synthetic adjuvant SQS-21 and its constituent isomeric saponins. Vaccine 28, 4260–4267. doi:10.1016/j.vaccine.2010.04.034
Ragupathi, G., Gardner, J. R., Livingston, P. O., and Gin, D. Y. (2011). Natural and synthetic saponin adjuvant QS-21 for vaccines against cancer. Expert Rev. Vaccines 10, 463–470. doi:10.1586/erv.11.18
Reed, S. G., Orr, M. T., and Fox, C. B. (2013). Key roles of adjuvants in modern vaccines. Nat. Med. 19, 1597–1608. doi:10.1038/nm.3409
Reed, J., Orme, A., El-Demerdash, A., Owen, C., Martin, L. B. B., Misra, R. C., et al. (2023). Elucidation of the pathway for biosynthesis of saponin adjuvants from the soapbark tree. Science 379, 1252–1264. doi:10.1126/science.adf3727
Keywords: QS-21, linear trisaccharide, glycosylation, carbohydrate chemistry, vaccine adjuvant
Citation: Lin J-S, Tzeng Z-H, Dumalaog JS and Hung S-C (2025) A streamlined synthetic approach to the truncated linear trisaccharide fragment of QS-21. Front. Chem. 13:1650302. doi: 10.3389/fchem.2025.1650302
Received: 19 June 2025; Accepted: 13 August 2025;
Published: 23 September 2025.
Edited by:
Naohiko Yoshikai, Tohoku University, JapanReviewed by:
Mattan Hurevich, Hebrew University of Jerusalem, IsraelSagnik Sengupta, University of Texas Southwestern Medical Center, United States
Copyright © 2025 Lin, Tzeng, Dumalaog and Hung. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shang-Cheng Hung, c2NodW5nQGdhdGUuc2luaWNhLmVkdS50dw==