 Timur M. Volkov1
Timur M. Volkov1 Yury E. Tsvetkov1
Yury E. Tsvetkov1 Dmitry V. Yashunsky1
Dmitry V. Yashunsky1 Anton N. Kuznetsov1Oleg D. Sclyarov2Olesia V. Babicheva2Dmitry O. Zherdev3Liliya I. Mukhametova3
Anton N. Kuznetsov1Oleg D. Sclyarov2Olesia V. Babicheva2Dmitry O. Zherdev3Liliya I. Mukhametova3 Sergei A. Eremin3
Sergei A. Eremin3 Vadim B. Krylov1
Vadim B. Krylov1 Nikolay E. Nifantiev1*
Nikolay E. Nifantiev1*- 1Laboratory of Glycoconjugate Chemistry, N. D. Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Moscow, Russia
- 2Russian State Centre of Quality and Standardization of Veterinary Drugs and Feeds, Moscow, Russia
- 3Department of Chemistry, Moscow State University, Moscow, Russia
Pathogenic bacteria of the genus Brucella cause a severe threat for public health and agricultural economics. The World Health Organization considers brucellosis to be one of the most serious and also neglected zoonotic diseases. The use of traditional whole-cell brucellosis vaccines complicates the differentiation between infected and vaccinated animals (DIVA). Moreover, diagnostics based on lipopolysaccharide of Brucella are susceptible to false positive results. Structural features of Brucella O-antigens make synthetic oligosaccharides promising agents for the development of diagnostic tools and vaccines against brucellosis. Here we report the synthesis of spacer-armed di-, tri-, tetra- and penta-4,6-dideoxy-4-formamido-α-(1→2)-d-mannopyranosides which are related to the A-epitope of Brucella O-antigen. The key α-(1→2)-linked disaccharide thioglycoside donor was synthesized by employing the strategy of orthogonal glycosylation of thioglycoside acceptor with trichloroacetimidate donor. Sequential block-wise assembly yielded a series of desired compounds, which were subsequently deprotected and converted into target molecules and then into their fluorescein-labeled conjugates. The obtained conjugates were employed as tracers in a fluorescence polarization assay (FPA) to detect anti-Brucella immunoglobulins. Among the studied compounds, the trisaccharide conjugate showed the greatest difference in median FP signals between Brucella-positive and Brucella-negative sera samples making it a promising candidate for developing FP diagnostic assays. The decreased FP signal in the cases of tetra- and pentasaccharide tracers can be associated with the known “propeller-effect” due to the rotational mobility of the part bearing the fluorescent label and of the fluorescein itself and/or the enlarging of the distance between the fluorescein part and the antibody-oligosaccharide complex. This observation demonstrates the advantages of using synthetic relatively small synthetic tracers with well-defined structure in comparison with heterogeneous fluorescein-labelled O-polysaccharides which are in use today in spite of the fact that they contain poorly characterized amounts of label attached along the polysaccharide chains.
Introduction
Brucellosis is a zoonotic infection primarily affecting domestic animals, caused by Gram-negative Brucella spp. (Khurana et al., 2021). Upon contact with infected animals or their products, the infection can be transmitted to humans, leading to serious acute and chronic illnesses with non-specific symptoms similar to those of malaria or influenza (Franco et al., 2007).
Early diagnosis is crucial for the prevention, control of the spread, and treatment of brucellosis. Currently, its detection is based on several principles (El Ayoubi et al., 2024; Kuznetsov et al., 2025; Tian et al., 2020), including the monitoring of specific antibodies to the Brucella O-polysaccharide in animal and human sera. For this purpose the preparations containing Brucella O-polysaccharide are used most often as marker antigens in the serodiagnosis of brucellosis (Godfroid et al., 2010). In particular, the systematic review and meta-analysis of the accuracy of serological tests for bovine brucellosis showed that indirect enzyme-linked immunosorbent assay (ELISA) and FPA are relatively easy to perform and interpret, and the test that showed the best overall accuracy was FPA (Andrade et al., 2024). Fluorescence polarization assay (FPA) is a homogeneous technique that involves only a single mix-and-measure step. Results can be obtained within minutes, making FPA a promising method for rapid detection of biomarkers in disease diagnostics (Jolley and Nasir, 2003). The availability of portable instruments capable of measuring fluorescence polarization signals outside the laboratory has made FPA another option for on-site infection detection. FPA has proven to be effective for the detection of antibodies against bacterial contamination, especially for the detection of antibodies to gram-negative bacteria (Jolley and Nasir, 2003).
The O-polysaccharide is an α-(1→2)-linked polymer of 4,6-dideoxy-4-formamido-d-mannopyranose (perosamine) terminated by one or more tetrasaccharides containing a central α-(1→3)-glycosidic bond (Figure 1, on the left) (Kubler-Kielb and Vinogradov, 2013). The antigenic determinants, represented by the α-(1→2)-linked chain and the terminal tetrasaccharide with the central α-(1→3)-bond, are referred to as the A epitope and the M epitope, respectively (Kubler-Kielb and Vinogradov, 2013). The ratio of A and M epitopic fragments in Brucella O-polysaccharides depends on the type of the serotype, and in some cases the M epitope can be even absent (Kubler-Kielb and Vinogradov, 2013).
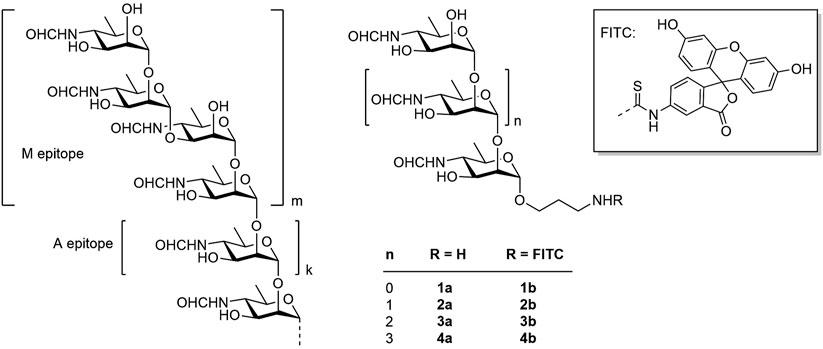
Figure 1. Structure of the Brucella O-polysaccharide (on the left), target spacered oligosaccharides 1a-4a and fluorescein labeled conjugates 1b-4b prepared in this work.
The use of the natural Brucella O-polysaccharide as the diagnostic antigen has some disadvantages. In particular, the cultivation of bacteria and the isolation, purification, and standardization of lipopolysaccharides is a challenging task. Another complication is related to the existence of bacterial species that produce O-polysaccharides made from N-acylated d-perosamine. These include, for example, Yersinia enterocolitica O:9 (Caroff et al., 1984), Escherichia coli 0157 (Perry and Bundle, 1990), Vibrio cholerae O1 (Kenne et al., 1982), and some others. Infection of animals and humans by these bacteria induces the production of antibodies that can cross-react with the Brucella O-polysaccharide, leading to false positive test results.
In addition, the labelling of O-polysaccharide with fluorescein is poorly reproducible from the point of view of the amount of the attached fluorescein residues and their location along the polysaccharide chain. The overall disadvantages of natural O-polysaccharides led to the idea of using synthetic oligosaccharides, which mimic the M epitope of the polysaccharide chain, as diagnostic antigens. The presence of the (1→3)-bond is a unique feature of the Brucella O-polysaccharide, as this structural element has not been found in the O-polysaccharides of other bacteria. Thus, Bundle et al. synthesized a series of oligosaccharides representing various structural motifs within the O-polysaccharide chain of Brucella (Bundle and McGiven, 2017; Ganesh et al., 2014; Guiard et al., 2013). Their BSA conjugates were employed as diagnostic antigens in ELISA. Notably, short oligosaccharides demonstrated greater specificity compared to natural O-antigens (Duncombe et al., 2022). Based on this observation, the authors proposed that the epitope formed by the terminal monosaccharide unit provides higher diagnostic specificity than that formed by internal monosaccharide residues within native O-antigens.
Recently, we reported the synthesis of a series of spacered oligosaccharides featuring the presence of an α-(1→3)-glycoside bond and related to the M epitope of the Brucella O-polysaccharide (Tsvetkov et al., 2024; Tsvetkov and Nifantiev, 2023). Fluorescein conjugates of these compounds were used in the development of brucellosis diagnosis based on FPA (Mukhametova et al., 2024a). Fluorescein-based dyes are the most popular due to their unique spectral properties (high quantum yield of 92% and fluorescence lifetime of 4.05 ns). Fluorescein dyes are chemically stable, inexpensive and commercially available. In addition, most FP readers are equipped with filters for measuring fluorescence of fluorescein. As demonstrated, the FPA method has proven to be effective for detecting antibodies against Brucella in various physiological fluids – such as serum, whole blood, and milk – across different animal species and in humans (Dong et al., 2021; Ibarra et al., 2023; Olsen et al., 2025; Sotolongo-Rodríguez et al., 2022). Here we describe the synthesis of α-(1→2)-linked D-perosamine oligomers (1a–4a), corresponding to the A epitope of the Brucella O-polysaccharide (Figure 1), the preparation of their fluorescein-labeled conjugates (1b–4b) and the investigation of their applicability as tracers in FPA to evaluate anti-Brucella serum immunoglobulins and to determine the optimal tracer size for effective diagnosis of brucellosis in animals.
Results and discussion
Synthesis of oligosaccharides 1a-4a and fluorescein labeled conjugates 1b-4b thereof
The known 1,2-diacetate 5 (Bundle et al., 1988) was employed to prepare the key synthetic blocks 6, 7, and 9, which were then used to assemble all protected oligosaccharides (Scheme 1). Treatment of diacetate 5 with 5-(tert-butyl)-2-methylthiophenol in the presence of BF3⋅Et2O smoothly produced thioglycoside 6, which was O-deacetylated to give 2-OH derivative 7 almost quantitatively. The use of azide groups as precursors to amides is a well-established strategy in the synthesis of perosamine and structurally related anthrose oligomers (Bundle et al., 1988; Bundle and McGiven, 2017; Saksena et al., 2005; Saksena et al., 2008). The preference for azide over amide protecting groups arises from the observation that amide at the C-4 position of glycosyl donors significantly decreased the stereoselectivity of glycosylation (Adamo and Kováč, 2007). Acetyl group at C-2 atom of glycosyl donor 8 provided the required α-selectivity in glycosylation and served as a convenient temporary protecting group. Orthogonal glycosylation of acceptor 7 with imidate 8 (Ariosa-Alvarez et al., 1998) afforded disaccharide thioglycoside 9 in high yield. The latter was used for the block-wise assembly of the target (1→2)-linked oligosaccharides.
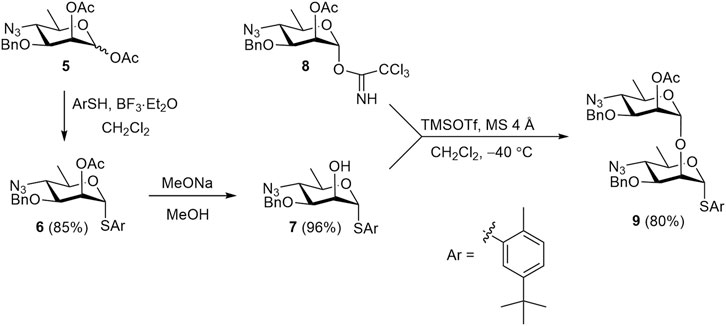
Scheme 1. Synthesis of synthetic blocks 6, 7, and 9.
The published data evidenced (Hou and Kovác, 2010; Ogawa et al., 1996; Peters and Bundle, 1989; Saksena et al., 2005) that glycosyl donors derived from 4-azido-4,6-dideoxy-d-mannose are able to glycosylate secondary hydroxyl groups highly α-stereoselectively, even if these donors bear non-participating substituents (for example, monosaccharide residue) at O-2. At the same time, glycosylation of much more reactive primary aliphatic acceptors can lead to the formation of considerable amounts of β-anomers (Ogawa and Kovac, 1997).
Indeed, TMSOTf–NIS promoted glycosylation of acceptor 10 with donor 9 under the conditions commonly used for the thioglycoside activation (at −45 °C) resulted in a mixture of glycosides 11α and 11β in a ratio of 2:1 (Scheme 2, route A). To improve the selectivity of glycosylation, the reaction was carried out in toluene at elevated temperature (Scheme 2, route B). This led to a significant increase in the α-stereoselectivity of the reaction, resulting in an 11α:11β ratio of 11:1. The anomeric configuration of the products was confirmed by 1JC1,H1 values (170.1 Hz for 11α and 155.7 Hz for 11β).
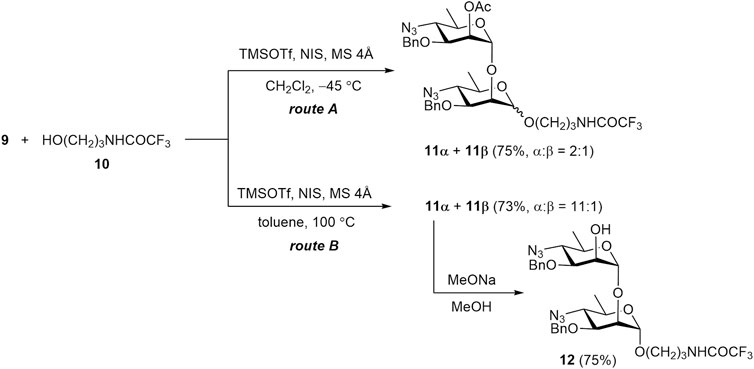
Scheme 2. Glycosylation of 3-trifluoroacetamidopropanol 10 with donor 9 under different conditions.
The preferential formation of the thermodynamically more stable α-anomer at elevated temperature was reported by Hou and Kovác (2010) and termed “thermodynamically controlled glycosylation”. However, a detailed mechanistic explanation for this phenomenon has not been clearly addressed in the literature. The term “thermodynamic control” conventionally implies that equilibrium is established among reaction products during the course of the reaction. Therefore, we investigated the possibility of interconversion between α- and β-glycosides under the reported reaction conditions.
Although anomerization of a glycosidic bond without a leaving group is rare, several examples of such a transformation have been described in the literature (Manabe et al., 2014). In this connection, we examined whether equilibration between 11α and 11β really occurs under the conditions used for “thermodynamically controlled” glycosylation (i.e., TMSOTf, toluene, 100 °C) (Hou and Kovác, 2010). However, when β-glycoside 11β was thus treated with TMSOTf the expected transformation to a mixture of 11α and 11β was not observed. This result suggests that the concept of “thermodynamic control” may refer to the equilibrium between some intermediate products rather than the final glycosides.
Conventional deacetylation of 11α with sodium methoxide yielded 2-OH derivative 12. It was then used for transformation into final disaccharide 1, and also served as a glycosyl acceptor in the synthesis of tetrasaccharide 12.
NIS–TMSOTf promoted glycosylation of known acceptor 13 (Tsvetkov et al., 2024) with thioglycoside 9 smoothly gave trisaccharide 14 (Scheme 3). As expected, glycosylation of the less reactive secondary OH group in 13 exclusively produced the α-linked product 14. Its O-deacetylation gave alcohol 15, the precursor of target trisaccharide 2.
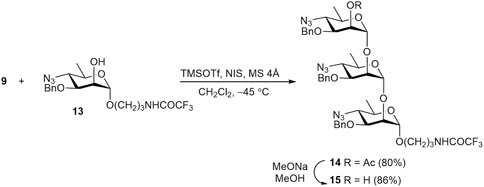
Scheme 3. Synthesis of protected trisaccharide 14.
Similarly, glycosylation of disaccharide acceptor 12 with thioglycoside 9 resulted in α-linked tetrasaccharide 16, with no detectable formation of the corresponding β-anomer (Scheme 4). Subsequent O-deacetylation of 16 gave rise to 2-OH derivative 17, which was then converted into tetrasaccharide 3.
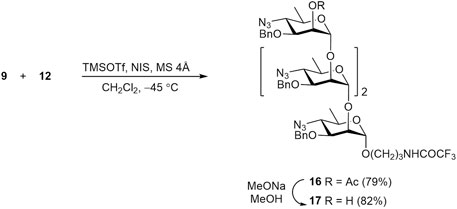
Scheme 4. Synthesis of protected tetrasaccharide 16.
Finally, tetrasaccharide acceptor 17 was subjected to glycosylation with thioglycoside donor 6 to produce protected pentasaccharide 18 in high yield. Its O-deacetylation led to the formation of alcohol 19, which was then transformed to free pentasaccharide 4 (Scheme 5). The structure of protected oligosaccharides 14–19, particularly the configuration of all anomeric centers was confirmed by characteristic values of corresponding JH1,H2 constants and other data of fully assigned 1H and 13C NMR spectra (see Supplementary Material) and high-resolution mass spectra (HRMS).
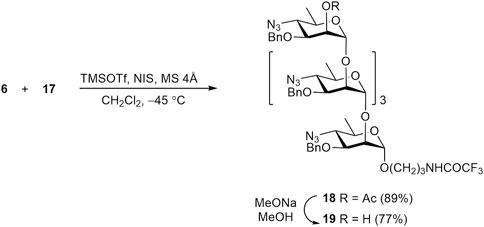
Scheme 5. Synthesis of protected pentasaccharide 18.
The transformation of protected oligomers 12, 15, 17, and 19 into final products 1a-4a was accomplished by using the reaction sequence described in our previous publication (Tsvetkov et al., 2024). It included reduction of azido groups by catalytic hydrogenation, N-formylation, debenzylation and removal of the N-trifluoroacetyl group. The details of this transformation are shown in Scheme 6, using disaccharide 12 as an example. Thus, catalytic reduction of diazide 12 over Pd(OH)2/C was accompanied with only minor affecting benzyl groups (Ariosa-Alvarez et al., 1998) and produced, after chromatographic purification, diamine 20 in 78% yield. N-Formylation of the latter with formic anhydride generated in situ from formic acid and DCC resulted in the formation of bis(formamide) 21 (86%). Following catalytic debenzylation of 21 afforded triol 22 (94%); final basic removal of the N-trifluoroacetyl group from 22 gave target 3-aminopropyl glycoside 1a in 85% yield after purification by size-exclusion chromatography.
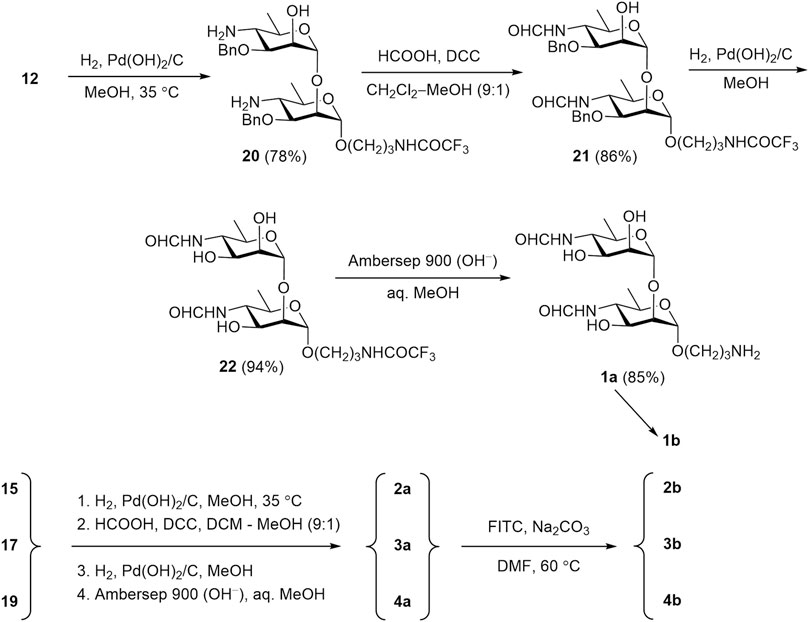
Scheme 6. Synthesis of free oligosaccharides 1a-4a and their conversion in fluorescein labeled conjugates 1b-4b.
Intermediate products 20–22 were characterized as follows. The structure of compound 20 was confirmed by 1H and 13C NMR, and HRMS data, while the composition of N-formyl derivatives 21 and 22 was characterized by only HRMS data, since their NMR spectra were poorly informative due to the Z/E-isomerism of the N-formyl groups (Peters et al., 1990).
Protected oligomers 15, 17, and 19 underwent the same transformations to produce desired free 3-aminopropyl glycosides 2a-4a (Scheme 6). The existence of compounds 2a-4a as mixtures of 2n isomers (where n is the number of the N-formyl monosaccharide units in the molecule) (Peters et al., 1990) strongly complicates NMR spectra, thus making their full assignment virtually impossible. Nevertheless, the presence of key structural features, such as N-formyl groups (both Z- and E-isomers), anomeric centers, the 3-aminopropyl spacer moiety, and some others, can be inferred from 1H and 13C NMR spectra of compounds 1a-4a. Further introduction of the fluorescein tag was conducted with FITC in the presence of base (Na2CO3) in DMF. The primary amino group in the spacer reacts rapidly and regioselectively with the isothiocyanate group of FITC, leading to fluorescein-labeled tracers with good yields (80–88%).
Assaying of serum immunoglobulins with the use of fluorescein labeled conjugates 1b-4b
Brucella-positive (+) (N = 19) and Brucella-negative (−) (N = 20) serum samples, which were validated through conventional serological methods (the Rose Bengal test (RBT), the complement fixation test (CFT) and ELISA), were used to evaluate the diagnostic performance of FPA employing the fluorescent tracers 1b–4b synthesized in this work. Also, the previously reported monosaccharide tracer (compound 23) (Mukhametova et al., 2024a) was included in this study.
The FPA method is based on the increase in the FP signal caused by the interaction of fluorescence oligosaccharide probes with oligosaccharide-specific antibodies (Eremin et al., 2024). Accordingly, all tracers tested (23, 1b–4b) produced a significant increase in FP signal in the Brucella-positive group compared to the Brucella-negative group (see Figure 2; Table 1). Notably, in Brucella-negative samples, a slight gradual increase in FP signal was observed with enlargement of oligosaccharide length–median FP values ranged from 47.0 for the monosaccharide tracer 23 to 58.3 for the pentasaccharide tracer 4b. This trend may be attributed to nonspecific interactions between the larger oligosaccharides and various serum immunoglobulins and other proteins.
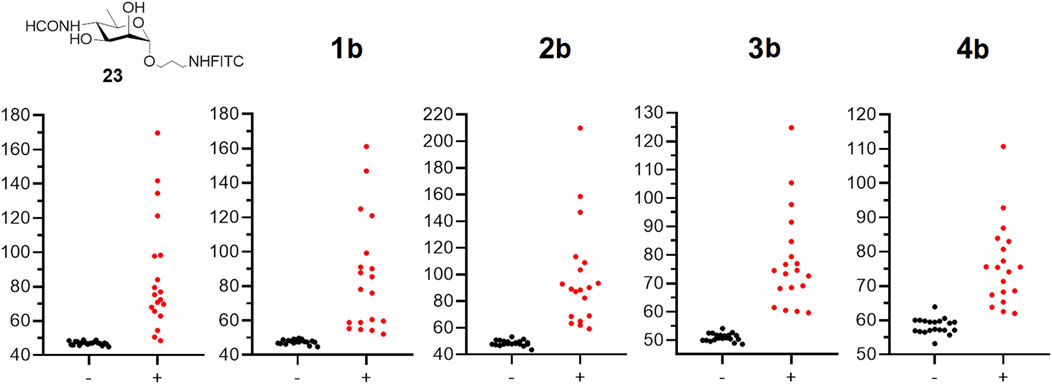
Figure 2. The FPA results for negative (−) (N = 20) and positive (+) (N = 19) bovine serum samples on brucellosis using tracers: 23 and 1b–4b. All experiments were repeated three times and the mean values and standard deviations were calculated. For all samples the experimental error did not exceed 10%.
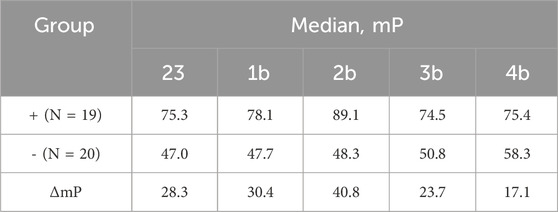
Table 1. Median FP signal values obtained by binding fluorescently labeled conjugates to negative (−) sera and Brucella-positive (+) bovine sera samples using tracers 23 and 1b–4b.
To determine the specificity and sensitivity of the FPA method when using fluorescently labeled saccharides of different lengths, ROC analysis of the obtained data was performed. The results are presented in Table 2. As can be seen, the diagnostic sensitivity of the method for all synthesized tracers is comparable. However, the specificity is higher for conjugates containing from 2 to 4 perosamine units. While testing, Brucella-positive sera with fluorescent glycoconjugate tracers, a bell-shaped dependence was observed with a maximum increase in the FP signal in the case of trisaccharide conjugate 2b (Table 1). It is possible that the decrease in the FP signal for 3b and 4b containing four and five perosamine units, respectively, can be explained by the so-called “propeller effect” (Nasir and Jolley, 2003). It is generally assumed that specific anti-Brucella antibodies primarily recognize epitopes located at the non-reducing end of the oligosaccharide chain. Consequently, the addition of extra sugar residues beyond the antibody binding site may increase the rotational mobility of the part bearing the fluorescent label and of the fluorescein itself (Figure 3).
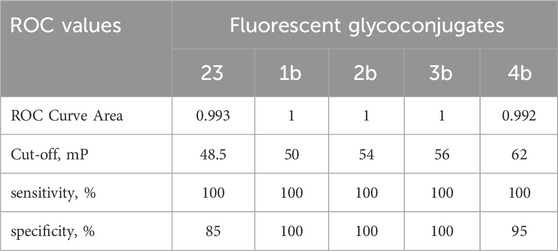
Table 2. Receiver operating characteristic (ROC) analysis of sensitivity and specificity of FPA with conjugates 1b-4b and 23.
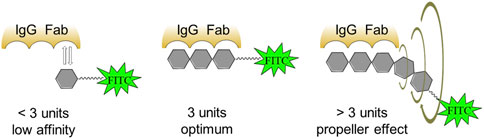
Figure 3. Occurrence of the propeller effect upon binding of fluorescently labeled glycoconjugates to antibodies.
To interpret this phenomenon, a “wobbling-in-a-cone” model has been proposed (Kinosita et al., 1982; Palchowdhury et al., 2024), which suggests that the fluorophore undergoes restricted rotational motion within an imaginary cone. It is known that FP measures the alignment of emitted light relative to polarized excitation, which depends on the tracer’s rotation during its fluorescence lifetime (τ). Rotation angle (θ) is the angular displacement of the tracer during τ. Increase in θ leads to a faster rotation and lower polarization (P). In FP, the rotation angle of the tracer depends on the rotational diffusion, flexibility of the tracer and the hydrodynamic radius of the complex (tracer-antibody). When the tracer binds to the antibody, the FP signal increases. However, the signal increase is also affected by the size of the tracer itself. As can be seen, binding of a short tracer (2–3 saccharides) to the antibody leads to a sharp slowdown in its rotation and an increase in the FP signal is observed. Growth in numbers of monosaccharide units in the tracer glycoconjugates increases θ, since the distance between the fluorophore and the antibody-antigen complex expands, thereby multiplying the freedom of movement of the fluorophore and leading to a corresponding decrease in the FP signal. A similar effect was described in works on the design of tracers for the detection of low-molecular toxicants by FPIA (Dong et al., 2019).
Thus, as shown in Table 1, the maximum difference in median FP signals between Brucella-positive and Brucella-negative sera was observed for conjugate 2b, which contains three monosaccharide units. Varying of the oligosaccharide length influenced the FP signal difference. Therefore, the trisaccharide represents the most efficient size for use as a fluorescent tracer in FPA for the detection of anti-Brucella antibodies. It should be mentioned that the advantage of synthetic oligosaccharide antigens, which permits better distinguishing of anti-Brucella and anti-Y. enterocolitica O:9, was reported (Duncombe et al., 2022). Overall the obtained results can form the basis for the elaboration of advantageous serological tests for brucellosis.
Conclusion
In conclusion, 3-aminopropyl glycosides of α-(1→2)-linked oligomers of 4,6-dideoxy-4-formamido-d-mannopyranose up to the pentasaccharide, which represent the A epitope of the Brucella O-polysaccharide, have been successfully synthesized using a block-wise approach. The α-(1→2)-linked disaccharide thioglycoside 9 was employed as the key donor block for the assembly of the required oligomers. Despite the presence of a non-participating glycosyl substituent at O-2 of the donor molecule, glycosylation of the 2-OH groups in mono- and oligosaccharide acceptors demonstrated high α-stereoselectivity. On the contrary, glycosylation of the more reactive primary aliphatic alcohol, 3-trifluroacetamidopropanol, with the same donor resulted in a significant decrease in α-stereoselectivity. It was considerably improved by performing the reaction under the conditions of “thermodynamic control”, i.e., at elevated (100 °C) temperature. However, a control experiment showed that interconversion of the anomeric glycosides did not occur under these conditions, and thus the improvement in α-stereoselectivity is a more complex process than simply achieving an equilibrium state.
The synthesized α-(1→2)-linked oligomers were transformed into corresponding fluorescein labelled conjugates, which were applied as tracers for assaying anti-Brucella immunoglobulins by fluorescence polarization. This study demonstrated that the trisaccharide derivative is the most efficient for detecting antibodies against Brucella by the FPA. It is important to point out that the use of longer oligosaccharides gave a smaller fluorescence polarization signal that is explained by us by the existence of known “propeller-effect.” This finding shows again the advantage of synthetic fluorescein-labelled oligosaccharide tracers of distinct structure when compared with fluorescein conjugates of Brucella O-polysaccharide, which are used often today as tracers for FPA but contain poorly characterized amounts of label attached along the polysaccharide chains. Thus, fluorescently labeled α-(1→2)-linked trisaccharide exhibits the best properties among the tested compounds and permits the most efficient detecting of antibodies to Brucella. Thus, this tracer can be recommended for the development of diagnostic tools to identify brucellosis in animals and humans.
Experimental section
General methods
NMR spectra were recorded on a Bruker Fourier 300 HD and Bruker Avance 600 NMR spectrometers. Protected oligosaccharides were measured in CDCl3, and 1H NMR chemical shifts were referenced to the solvent residual signal (δH 7.27). 13C chemical shifts were referenced to the central resonance of CDCl3 (δC 77.0). Free oligosaccharides were measured in deuterium oxide (D2O) with suppression of the HOD signal. Acetone (δH 2.225, δC 31.45) was used as an internal standard. Signal assignment was made using COSY, TOCSY, HSQC, and ROESY experiments. In the presentation of NMR data, monosaccharide residues in oligosaccharides are denoted by the capital letters (A, B, C, etc.) starting from the reducing end. NMR spectra of synthesized compounds are presented in the Supplementary Material. High resolution mass spectrometry (HRMS) with electrospray ionization (ESI) was performed on a MicrOTOF II (Bruker Daltonics) instrument. Optical rotations were measured using a JASCO P-2000 polarimeter at ambient temperature (20 °C–25 °C) in chloroform (protected oligosaccharides) or water (free oligosaccharides). TLC was performed on Silica Gel 60 F254 plates (Merck) and visualization was accomplished using UV light or by charring at ∼150 °C with orcinol–phosphoric acid [180 mg of orcinol in a mixture of 85% H3PO4 (10 mL), ethanol (5 mL), and water (85 mL)]. Column chromatography was carried out using Silica Gel 60 (40–63 μm; Merck Millipore). Gel-permeation chromatography of free oligosaccharides was performed on a Toyopearl TSK HW-40(S) column (2.8 × 80 cm) in 0.1 M acetic acid. A K-2401 refractive index detector (Knauer) was used to monitor gel-permeation chromatography. All moisture-sensitive reactions were carried out using dry solvents under dry argon. Powdered molecular sieve 4 Å was activated at 300 °C under vacuum (∼1 mbar) for 30 min directly prior to the reaction.
5-tert-Butyl-2-methylphenyl 2-O-acetyl-4-azido-3-O-benzyl-4,6-dideoxy-1-thio-α-d-mannopyranoside (6). 5-tert-Butyl-2-methylthiophenol (0.89 g, 4.92 mmol) and BF3⋅Et2O (0.49 mL, 3.93 mmol) were added to a chilled (ice bath) solution of 1,2-diacetate 5 (1.22 g, 3.28 mmol) in CH2Cl2 (30 mL), and the mixture was stirred for 10 min with chilling and then for 2.5 h at room temperature. The mixture was diluted with CHCl3 (70 mL), washed with water (80 mL) and aqueous saturated NaHCO3. The organic solution was concentrated, and residue was purified by column chromatography (petroleum ether–EtOAc, 0→5%) to yield thioglycoside 6 (1.38 g, 85%) as a colorless syrup, [α]D +116.2 (c 1.0, CHCl3); the product contained 8%–9% of the β-anomer. 1H NMR (CDCl3, 300 MHz): δ 7.56–7.12 (m, 8 H, Ar), 5.65 (dd, 1 H, J2,1 = 1.6 Hz, J2.3 = 3.1 Hz, H-2), 5.37 (d, 1 H, J1,2 = 1.6 Hz, H-1), 4.76 (d, 1 H, J = 11.0 Hz, PhCHaHb), 4.61 (d, 1 H, J = 11.0 Hz, PhCHaHb), 4.11 (dq, 1 H, J5,6 = 6.2 Hz, J5,4 = 10.0 Hz, H-5), 3.87 (dd, 1 H, J3,2 = 3.1 Hz, J3,4 = 10.1 Hz, H-3), 3.54 (t, 1 H, J = 10.0 Hz, H-4), 2.42 (s, 3 H, Ar-CH3), 2.16 (s, 3H, CH3CO), 1.38 (d, 3 H, J6,5 = 6.2 Hz, H-6), 1.33 (s, 9 H, C(CH3)3). 13C NMR (76 MHz, CDCl3): δ 170.2 (CH3CO), 149.9, 137.0, 136.7, 132.2, 130.1, 129.8, 128.5, 128.4, 128.1, 125.2 (Ar), 85.7 (C-1), 71.8 (PhCH2), 69.4 (C-2), 68.3 (C-5), 64.2 (C-4), 34.5 (C(CH3)3), 31.3 (C(CH3)3), 21.0 (CH3CO), 20.3 (Ar-CH3), 18.5 (C-6). HRMS (ESI): calcd. for C26H33N3O4S [M + K]+ m/z 522.1819; found m/z 522.1823.
5-tert-Butyl-2-methylphenyl 4-azido-3-O-benzyl-4,6-dideoxy-1-thio-α-d-mannopyranoside (7). 1 M Sodium methoxide in MeOH (0.47 mL) was added to a solution of 2-acetate 6 (229 mg, 0.47 mmol) in MeOH (5 mL), and the mixture was stirred for 16 h at room temperature. The mixture was neutralized with AcOH, and the solvent was evaporated. The residue was dissolved in CHCl3 (25 mL), and the solution was washed with water (20 mL). The aqueous phase was extracted with CHCl3 (25 mL), and the combined organic solutions were concentrated. Column chromatography (petroleum ether–EtOAc, 5→10%) produced title product 7 (201 mg, 96%) as a colorless syrup, [α]D +201.6 (с 1, CHCl3). 1H NMR (CDCl3, 300 MHz): δ 7.61–7.13 (m, 8 H, Ar), 5.53 (br. s, 1H, H-1), 4.83–4.72 (m, 2 H, PhCH2), 4.30 (br. s, 1 H, H-2), 4.09 (dq, 1 H, J5,6 = 6.2 Hz, J5,4 = 10.1 Hz, H-5), 3.73 (dd, 1H, J3,4 = 9.7 Hz, J3,2 = 3.1 Hz, H-3), 3.54 (t, 1 H, J = 9.9 Hz, H-4), 2.67 (br. s, 1 H, 2-OH), 2.39 (s, 1 H, Ar-CH3), 1.36 (d, 3 H J6,5 = 6.2 Hz, H-6), 1.32 (s, 9 H, C(CH3)3). 13C NMR (76 MHz, CDCl3): δ 149.9, 137.0, 136.2, 132.5, 130.0, 129.1, 128.7, 128.4, 128.2, 124.8 (Ar) 86.4 (C-1), 78.7 (C-3), 72.3 (PhCH2), 69.1 (C-2), 68.0 (C-5), 64.3 (C-4), 34.5 (C(CH3)3), 31.3 (C(CH3)3), 20.2 (Ar-CH3), 18.4 (C-6). HRMS (ESI): calcd. for C24H31N3O3S [M + Na]+ m/z 464.1966; found m/z 464.1978.
5-tert-Butyl-2-methylphenyl 2-O-acetyl-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-1-thio-α-d-mannopyranoside (9). A mixture of imidate 8 (181 mg, 0.389 mmol) and thioglycoside acceptor 7 (159 mg, 0.353 mmol) was dried by co-evaporation with toluene, dried under vacuum for 1 h, and dissolved in CH2Cl2 (5 mL). Powdered molecular sieve 4 Å (350 mg) was added to the solution, and the mixture was stirred for 30 min at room temperature and cooled to −40 °C. TMSOTf (14 μL, 0.078 mmol) was added and the mixture was stirred for 30 min, while the temperature was gradually increased to −10 °C. Stirring was continued at this temperature for another 1 h and then the reaction was quenched by adding Et3N (20 μL). The solids were filtered off through a Celite layer, washed with CHCl3 (4 × 5 mL), and the filtrate was washed with water (20 mL). The solvent was evaporated and the residue was purified by column chromatography (petroleum ether–EtOAc, 0→7%) to afford disaccharide 9 (212 mg, 80%) as a colorless syrup, [α]D +130.8 (с 1, CHCl3). 1H NMR (CDCl3, 300 MHz): δ 7.53–7.13 (m, 13 H, Ar), 5.41 (dd, 1 H, J2,1 = 1.8 Hz, J2,3 = 3.2 Hz, H-2B), 5.31 (d, 1 H, J1,2 = 1.6 Hz, H-1A), 4.84 (d, 1 H, J1,2 = 1.6 Hz, H-1B), 4.85–4.53 (m, 4 H, 2 PhCH2), 4.12 (poorly resolved t, 1 H, H-2A), 4.02 (dq, 1 H, J5,4 = 10.1 Hz, J5,6 = 6.2 Hz, H-5B), 3.83–3.76 (m, 2 H, H-3A, H-3B), 3.61 (dq, 1 H, J5,4 = 10.1 Hz, J5,6 = 6.2 Hz, H-5A), 3.48–3.36 (m, 2 H, H-4A, H-4B), 2.38 (s, 3 H, Ar-CH3), 2.13 (s, 3 H, CH3CO), 1.35–1.30 (m, 12 H, H-6B, C(CH3)3), 1.23 (d, 3 H, J6,5 = 6.2 Hz, H-6A). 13C NMR (76 MHz, CDCl3): δ 169.9 (CH3CO), 149.9, 137.4, 137.1, 136.5, 132.6, 130.1, 129.7, 128.6, 128.5, 128.1, 125.2 (Ar), 99.7 (C-1A), 86.9 (C-1B), 78.2 (C-3B), 76.4 (C-2A), 75.3 (C-3A), 72.2, 71.6 (2 PhCH2), 68.4 (C-5B), 67.7 (C-5A), 67.2 (C-2B), 64.3 (C-4B), 63.8 (C-4A), 31.3 (C(CH3)3), 21.0 (CH3CO), 20.2 (Ar-CH3), 18.5 (C-6B), 18.4 (C-6A). HRMS (ESI): calcd. for C39H48N6O7S [M + Na]+ m/z 767.3197; found m/z 767.3200.
3-Trifluoroacetamidopropyl 2-O-acetyl-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α- and β-d-mannopyranosides (11α, 11β).
1. A mixture of donor 9 (100 mg, 0.134 mmol), alcohol 10 (34.5 mg, 0.202 mmol) and mol. sieve 4 Å (150 mg) in CH2Cl2 (3 mL) was stirred for 30 min at room temperature, and then was cooled to −45 °C. NIS (60.3 mg, 0.268 mmol) was added, the mixture was stirred for 30 min, and then TMSOTf (5 μL, 0.027 mmol) was added. The mixture was stirred for 40 min, after which more TMSOTf (7 μL, 0.038 mmol) was added. The resulting mixture was stirred for 4 h, gradually increasing the temperature to −10 °C. The reaction was quenched by adding pyridine (30 μL), the mixture was diluted with CHCl3 (10 mL), the mol. sieve was filtered off through a Celite layer, and washed with CHCl3 (3 × 5 mL). The filtrate was washed with 0.5 M Na2S2O3 solution (20 mL) and water (30 mL), and concentrated. The solvent was evaporated and the residue was subjected to column chromatography (toluene – EtOAc, 0→15%) to provide a mixture of glycosides 11α and 11β (70 mg, 75%) in a ratio ∼2:1 (NMR data).
2. A solution of donor 9 (996 mg, 1.34 mmol) in toluene (45 mL) was added to alcohol 10 (404 mg, 2.36 mmol) and the mixture was heated to 100 °C. Mol. sieve 4 Å (1.5 g), NIS (608 mg, 2.70 mmol), and a solution of TMSOTf (49 μL) in toluene (5 mL) were added and the mixture was stirred at 100 °C. Additional portions of the promoting reagents were added after 2 h (304 mg (1.35 mmol) of NIS; 25 μL (0.14 mmol) of TMSOTf) and 4 h (152 mg of NIS; 12 μL of TMSOTf). Stirring was continued at 100 °C for another 1 h and then the mixture was cooled to room temperature, and quenched by adding pyridine (200 μL). The mol. sieve was filtered off through a Celite layer and washed with toluene (3 × 10 mL). The filtrate was washed with 0.5 M Na2S2O3 solution (50 mL) and water (50 mL), and concentrated. Column chromatography (petroleum ether–EtOAc, 10→25%) of the residue gave α-glycoside 11α (472 mg), β-glycoside 11β (24 mg), and 310 mg of their mixture. The latter mixture was rechromatographed to produce more 11α (186 mg) and 11β (47 mg). Total yields of 11α and 11β were 658 mg (67%) and 71 mg (6%), respectively.
α-Anomer 11α, white crystals, m. p. 111°C–113°C, [α]D +78.3 (с 1, CHCl3). 1H NMR (CDCl3, 300 MHz): δ 7.44–7.26 (m, 10 H, Ar), 6.68 (br. s, 1 H, NH), 5.42 (dd, 1 H, J2,1 = 1. 9 Hz, J2,3 = 3.3 Hz, H-2B), 4.89 (d, 1 H, J1,2 = 1.9 Hz, H-1B), 4.78–4.68 (m, 3 H, H-1A, 2 PhCHaHb), 4.63 (d, 1 H, J = 11.5 Hz, PhCHaHb), 4.55 (d, 1 H, J = 11.1 Hz, PhCHaHb), 3.88 (poorly resolved dd, 1 H, H-2A), 3.83–3.73 (m, 2 H, H-3B, OCHaHbCH2CH2N), 3.70 (dd, 1 H, J3,2 = 2.9 Hz, J3,4 = 9.3 Hz, H-3A), 3.65–3.33 (m, 7 H, H-4A, H-4B, H-5A, H-5B, OCHaHbCH2CH2N, OCH2CH2CH2N), 2.10 (s, 3 H, CH3CO), 1.94–1.83 (m, 2 H, OCH2CH2CH2N), 1.36–1.29 (m, 6 H, H-6A, H-6B). 13C NMR (76 MHz, CDCl3): δ 169.8 (CH3CO), 137.4, 137.1, 128.5, 128.5, 128.1, 128.0, 127.8 (Ar), 99.4 (C-1B), 98.9 (C-1A), 77.8 (C-3A), 75.3 (C-3B), 73.6 (C-2A), 72.1, 71.6 (2 PhCH2), 67.7 (C-5B), 67.4 (C-5A), 67.1 (C-2B), 65.8 (OCH2CH2CH2N), 64.0 (C-4B), 63.8 (C-4A), 38.1 (OCH2CH2CH2N), 28.4 (OCH2CH2CH2N), 21.0 (CH3CO) 18.6 (C-6A), 18.5 (C-6B). HRMS (ESI): calcd. for C33H40F3N7O9 [M + K]+ m/z 774.2471; found m/z 774.2472.
β-Anomer 11β, colorless syrup, [α]D +9.9 (с 1, CHCl3). 1H NMR (CDCl3, 300 MHz): δ 7.42–7.27 (m, 10 H, Ar), 7.13 (br. s, 1 H, NH), 5.49 (dd, 1 H, J2,1 = 1.9 Hz, J2,3 = 3.3 Hz, H-2B), 5.09 (d, 1 H, J1,2 = 1.9 Hz, H-1B), 4.77–4.65 (m, 3 H, 2 PhCHaHb, PhCHaHb), 4.51 (d, 1 H, J = 11.1 Hz, PhCHaHb), 4.33 (br. s, 1 H, H-1A), 4.13 (poorly resolved d, 1 H, H-2A), 4.00 (dq, 1 H, J5,4 = 10.3 Hz, J5,6 = 6.2 Hz, H-5B), 3.87–3.78 (m, 2 H, H-3B, OCHaHbCH2CH2N), 3.66–3.58 (m, 1 H, OCHaHbCH2CH2N), 3.52–3.26 (m, 5 H, H-3A, H-4A, H-4B, OCH2CH2CH2N), 3.21–3.11 (m, 1 H, H-5A), 2.10 (s, 3 H, CH3CO), 1.96–1.71 (m, 2 H, OCH2CH2CH2N), 1.39 (d, 3H, J6,5 = 6.1 Hz, H-6A), 1.30 (d, 3 H, J6,5 = 6.2 Hz, H-6B). 13C NMR (76 MHz, CDCl3): δ 169.9 (CH3CO), 137.2, 136.9, 128.6, 128.4, 128.1, 128.0, 127.8 (Ar) 99.5 (C-1A), 98.1 (C-1B), 80.5 (C-3A), 75.4 C-3B), 71.9 (PhCH2), 71.4 (×2) (C-5A, PhCH2), 70.7 (C-2A), 67.0 (C-2B), 66.9 (C-5B), 66.6 (OCH2CH2CH2N), 64.0 (C-4B), 63.9 (C-4A), 37.0 (OCH2CH2CH2N), 28.6 (OCH2CH2CH2N), 21.0 (CH3CO), 18.4 (×2) (C-6A, C-6B).). HRMS (ESI): calcd. for C33H40F3N7O9 [M + Na]+ m/z 758.2753; found m/z 758.2732.
An attempt to transform 11β into an equilibrium mixture of 11β and 11α. A solution of TMSOTf in toluene (0.03 M, 43 μL) was added at 100 °C to a stirred mixture of glycoside 11β (19 mg, 0.026 mmol) in toluene (1 mL) and mol. sieve 4 Å (43 mg). Stirring was continued at 100 °C, and the reaction was monitored by TLC in petroleum ether–EtOAc (7:3). Starting glycoside 11β had Rf = 0.29 in the specified solvent system. Comparison with an authentic sample of 11α (Rf = 0.36) showed that the reaction mixture contained only 11β after 5.5 h of heating, with no formation of 11α.
3-Trifluoroacetamidopropyl 4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranoside (12). 1 M Sodium methoxide in MeOH (157 μL) was added to a solution of acetylated disaccharide 11α (576 mg, 0.84 mmol) in MeOH (16 mL), the mixture was stirred for 4 h at room temperature, and then made neutral by adding Amberlite IR-120 (H+). The resin was filtered off, washed with MeOH (3 × 5 mL), and the combined filtrate and washings were concentrated. Column chromatography of the residue (toluene – EtOAc, 0→20%) yielded deacetylated product 12 (438 mg, 75%) as a colorless syrup, [α]D +84.1 (с 1, CHCl3). 1H NMR (CDCl3, 300 MHz): δ 7.46–7.16 (m, 10 H, Ar), 6.66 (br. s, 1 H, NH), 4.96 (d, 1 H, J1,2 = 1.3 Hz, H-1B), 4.78–4.58 (m, 5 H, H-1A, 2 PhCH2), 4.00 (br. s, 1 H, H-2B), 3.91 (poorly resolved t, 1 H, H-2A), 3.82–3.29 (m, 10 H, H-3A, H-3B, H-4A, H-4B, H-5A, H-5B, OCH2CH2CH2N, OCH2CH2CH2N), 2.35 (br. s, 1 H, OH), 1.95–1.82 (m, 2 H, OCH2CH2CH2N), 1.38–1.25 (m, 6 H, H-6B, H-6A). 13C NMR (76 MHz, CDCl3): δ 137.1, 128.7, 128.6, 128.4, 128.3, 128.1, 128.0 (Ar) 101.0 (C-1B), 99.1 (C-1A), 77.9 (C-3A), 77.5 (C-3B), 73.7 (C-2A), 72.2, 72.1 (2 PhCH2), 67.4 (C-5A), 67.4 (C-5B), 67.1 (C-2B), 65.9 (OCH2CH2CH2N), 64.1 (C-4A), 63.8 (C-4B), 38.1 (OCH2CH2CH2N), 28.4 (OCH2CH2CH2N), 18.6 (C-6A), 18.4 (C-6B). HRMS (ESI): calcd. for C31H38F3N7O8 [M + K]+ m/z 732.2366; found m/z 732.2364.
3-Trifluoroacetamidopropyl 2-O-acetyl-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranoside (14). A mixture of donor 9 (76 mg, 0.102 mmol), acceptor 13 (40 mg, 0.093 mmol), and mol. sieve 4 Å (140 mg) in CH2Cl2 (4 mL) was stirred for 30 min. at room temperature, and then cooled to −35 °C. NIS (42 mg, 0.186 mmol) and a solution of TMSOTf (3.4 μL, 0.019 mmol) in CH2Cl2 (0.1 mL) were added and the mixture was stirred for 40 min, while the temperature was gradually increased to −10 °C. The reaction was quenched with pyridine (20 μL), the mixture was diluted with CHCl3 (20 mL), the solids were filtered off through a Celite layer and washed with CHCl3 (3 × 5 mL). The filtrate was washed with 0.5 M Na2S2O3 solution (20 mL) and water (20 mL), and concentrated. Column chromatography of the residue (petroleum ether–EtOAc, 5→25%) produced trisaccharide 14 (74 mg, 80%) as a colorless syrup, [α]D +85.1 (с 1, CHCl3). 1H NMR (600 MHz, CDCl3): δ 7.41–7.34 (m, 15 H, Ar), 6.63 (br. s, 1 H, NH), 5.41 (dd, 1 H, J2,1 = 1.7 Hz, J2,3 = 3.1 Hz, H-2C), 4.97 (d, 1 H, J1,2 = 1.8 Hz, H-1B), 4.82 (d, 1 H, J1,2 = 1.7 Hz, H-1C), 4.74–4.53 (m, 7 H, 3 PhCH2, H-1A), 3.87 (poorly resolved t, 1 H, H-2B), 3.84 (poorly resolved t, 1 H, H-2A), 3.79–3.71 (m, 3 H, H-3C, OCHaHbCH2CH2N, H-3B), 3.66 (dd, 1 H, J3,4 = 9.9 Hz, J3,2 = 2.9 Hz H-3A), 3.56–3.35 (m, 8 H, H-4B, H-4C, H-5A, H-5B, H-5C, OCHaHbCH2CH2N, OCH2CH2CH2N), 3.26 (t, 1 H, J = 10.0 Hz, H-4A), 2.13 (s, 3 H, CH3CO), 1.90–1.84 (m, 2 H, OCH2CH2CH2N)), 1.33–1.26 (m, 6 H, H-6A, H-6B), 1.20 (d, 3 H, J6,5 = 6.2 Hz, H-6C). 13C NMR (151 MHz, CDCl3): δ 169.8 (CH3CO), 137.4, 137.2, 137.1, 128.6, 128.5, 128.5, 128.2, 128.1, 128.0 (Ar), 100.4 (C-1B), 99.2 (C-1C), 99.0 (C-1A), 77.6 (C-3A), 76.7 (C-3B), 75.4 (C-3C), 73.8 (C-2B, C-2A), 72.2, 72.0, 71.6 (3 PhCH2), 67.9 (C-5B), 67.7 (C-5C), 67.5 (C-5A), 67.2 (C-2C), 65.8 (OCH2CH2CH2N), 64.2 (C-4A), 64.0 (C-4B), 63.8 (C-4C), 38.2 (OCH2CH2CH2N), 28.4 (OCH2CH2CH2N), 21.0 (CH3CO), 18.6 (C-6A), 18.6 (C-6B), 18.4 (C-6C). HRMS (ESI): calcd. for C46H55F3N10O12 [M + Na]+ m/z 1019.3845; found m/z 1019.3839.
3-Trifluoroacetamidopropyl 4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranoside (15). 1 M Sodium methoxide in MeOH (48 μL) was added to a solution of 2-acetate 14 (239 mg, 0.24 mmol) in MeOH (4.8 mL), the mixture was stirred at room temperature for 5 h, and then made neutral by adding Amberlite IR-120 (H+). The resin was filtered off, washed with MeOH (3 × 5 mL), and the filtrate was concentrated. Column chromatography of the residue (toluene–EtOAc, 0→15%) gave deacetylated trisaccharide 15 (196 mg, 86%) as a colorless foam, [α]D +85.4 (с 1, CHCl3). 1H NMR (400 MHz, CDCl3): δ 7.42–7.26 (m, 15 H, Ar), 6.55 (br. s, 1 H, NH), 4.96 (d, 1 H, J1,2 = 1.7 Hz, H-1B), 4.92 (d, 1 H, J1,2 = 1.5 Hz, H-1C), 4.74–4.58 (m, 7 H, 3 PhCH2, H-1A), 3.97 (poorly resolved t, 1 H, H-2C), 3.90 (poorly resolved t, 1 H, H-2B), 3.81 (poorly resolved t, 1 H, H-2A), 3.77–3.62 (m, 4 H, OCHaHbCH2CH2N, H-3B, H-3C, H-3A), 3.55–3.34 (m, 7 H, H-4C, H-5A, H-5B, H-5C, OCHaHbCH2CH2N, OCH2CH2CH2N), 3.31 (t, 1 H, J = 10.0 Hz, H-4B), 3.24 (t, 1 H, J = 9.8 Hz, H-4A), 2.28 (br. s, 1 H, OH) 1.88–1.80 (m, 2 H, OCH2CH2CH2N), 1.31–1.25 (m, 6 H, H-6A, H-6B), 1.18 (d, 3 H, J6,5 = 6.1 Hz, H-6C). 13C NMR (101 MHz, CDCl3): δ 137.3, 137.2, 129.0, 128.6, 128.3, 128.2, 128.1, 128.1 (Ar) 100.6 (×2) (C-1B, C-1C), 99.0 (C-1A), 77.6 (C-3A), 77.5 (C-3C), 76.8 (C-3B), 73.7 (C-2A), 73.4 (C-2B), 72.2, 72.14, 72.07 (3 PhCH2), 67.9 (C-5B), 67.5 (C-5A), 67.4 (C-5C), 67.2 (C-2C), 65.8 (OCH2CH2CH2N), 64.4 (C-4A), 64.2 (C-4B), 63.8 (C-4C), 38.0 (OCH2CH2CH2N), 28.4 (OCH2CH2CH2N), 18.6 (C-6A), 18.5 (C-6C), 18.3 (C-6B). HRMS (ESI): calcd. for C44H53F3N10O11 [M + K]+ m/z 993.3479; found m/z 993.3475.
3-Trifluoroacetamidopropyl 2-O-acetyl-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranoside (16). A mixture of thioglycoside 9 (298 mg, 0.401 mmol), acceptor 12 (242 mg, 0.349 mmol) and mol. sieve 4 Å (550 mg) in CH2Cl2 (20 mL) was stirred at room temperature for 30 min, and then cooled to −35 °C. NIS (157 mg, 0.698 mmol) and a solution of TMSOTf (13 μL, 0.07 mmol) in CH2Cl2 (0.35 mL), and the resulting mixture was stirred for 1.5 h, while the temperature was gradually increased to −10 °C. The mixture was neutralized by adding pyridine (50 μL), diluted with CHCl3 (20 mL) and filtered through a Celite layer. The solids were washed with CHCl3 (3 × 10 mL), and the filtrate was washed with 0.5 M Na2S2O3 solution (20 mL) and water (20 mL). The organic solution was concentrated, and the residue was purified by column chromatography (toluene–EtOAc, 0→10%) to produce tetrasaccharide 16 (346 mg, 79%) as a colorless syrup, [α]D +81.6 (с 1, CHCl3). 1H NMR (600 MHz, CDCl3): δ 7.46–7.26 (m, 20 H, Ar), 6.61 (br. s, 1 H, NH), 5.43 (dd, 1 H, J2,1 = 1.8 Hz, J2,3 = 3.1 Hz, H-2D), 4.95 (d, 1 H, J1,2 = 1.6 Hz, H-1C), 4.91 (d, 1 H, J1,2 = 1.6 Hz, H-1B), 4.87 (d, 1 H, J1,2 = 1.8 Hz, H-1d), 4.76–4.72 (m, 2 H, 2 benzylic H), 4.68–4.60 (m, 6 H, 5 benzylic H, H-1A), 4.56 (d, 1 H, J = 11.2 Hz, benzylic H) 3.90 (poorly resolved t, 1 H, H-2C), 3.85 (poorly resolved t, 1 H, H-2B), 3.82 (poorly resolved t, 1 H, H-2A), 3.80 (dd, 1 H, J3,2 = 3.2 Hz, J3,4 = 9.9 Hz, H-3D), 3.78–3.73 (OCHaHbCH2CH2N), 3.71 (dd, 1 H, J3,2 = 2.9 Hz, J3,4 = 9.9 Hz, H-3C), 3.69 (dd, 1 H, J3,2 = 2.8 Hz, J3,4 = 9.9 Hz, H-3B), 3.66 (dd, 1 H, J3,2 = 3.1 Hz, J3,4 = 9.9 Hz, H-3A), 3.57–3.37 (m, 8 H, H-4D, H-5A, H-5B, H-5C, H-5D, OCHaHbCH2CH2N, OCH2CH2CH2N), 3.35 (t, 1 H, J = 10.1 Hz, H-4C), 3.26 (t, 1 H, J = 9.9 Hz, H-4B), 3.24 (t, 1 H, J = 9.9 Hz, H-4A), 2.13 (s, 3 H, CH3CO), 1.90–1.84 (m, 2 H, OCH2CH2CH2N), 1.34–1.27 (m, 6 H, H-6A, H-6B), 1.23 (d, 3 H, J6,5 = 6.2 Hz, H-6D), 1.18 (d, 3 H, J6,5 = 6.1 Hz, H-6C). 13C NMR (101 MHz, CDCl3): δ 170.0 (CH3CO) 157.2 (q, 2JC,F = 36.3 Hz, CF3CO), 137.6, 137.4, 137.3, 129.2, 129.1, 128.8, 128.7, 128.6, 128.4, 128.3, 128.2 (Ar) 116.0 (q, 1JC,F = 288 Hz, CF3CO) 100.7 (C-1B), 100.3 (C-1C), 99.3 (C-1D), 99.1 (C-1A), 77.7 (C-3A), 76.9 (C-3C), 76.7 (C-3B), 75.6 (C-3D), 73.9 (C-2A), 73.7 (C-2B), 73.6 (C-2C), 72.4 (×2), 72.2, 71.7 (4 PhCH2), 68.1 (C-5B), 68.0 (C-5C), 67.9 (C-5D), 67.6 (C-2D), 67.3 (C-5A), 66.0 (OCH2CH2CH2N), 64.4 (×2) (C-4B, C-4A), 64.2 (C-4C), 64.0 (C-4D), 38.2 (OCH2CH2CH2N), 28.6 (OCH2CH2CH2N), 21.2 (CH3CO), 18.8 (C-6A), 18.7 (C-6B), 18.6 (C-6C), 18.5 (C-6D). HRMS (ESI): calcd. for C59H70F3N13O15 [M + Na]+ m/z 1280.4959; found m/z 1280.4963.
3-Trifluoroacetamidopropyl 4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranoside (17). A solution of 2-acetate 16 (408 mg, 0.324 mmol) in MeOH (6.5 mL) was treated with methanolic sodium methoxide (65 μL) at room temperature for 5 h. The reaction mixture was worked up as described above for compound 15. The title product 17 (324 mg, 82%) was isolated by column chromatography (toluene–EtOAc, 0→15%); a colorless foam, [α]D +90.4 (с 1, CHCl3). 1H NMR (400 MHz, CDCl3): δ 7.43–7.28 (m, 20 H, Ar), 6.54 (br. s, 1 H, NH), 4.97 (d, 1 H, J1,2 = 1.6 Hz, H-1D), 4.93 (d, 1 H, J1,2 = 1.8 Hz, H-1С), 4.89 (d, 1 H, J1,2 = 1.8 Hz, H-1B), 4.76–4.57 (m, 9 H, H-1A, 4 PhCH2), 3.95 (poorly resolved t, 1 H, H-2D), 3.93 (poorly resolved t, 1 H, H-2C), 3.81 (poorly resolved t, 1 H, H-2B), 3.79 (poorly resolved t, 1 H, H-2A), 3.76–3.61 (m, 5 H, OCHaHbCH2CH2N, H-3A, H-3B, H-3C, H-3D), 3.56–3.33 (m, 8H, H-4D, H-5A, H-5B, H-5C, H-5D, OCHaHbCH2CH2N, OCH2CH2CH2N), 3.29 (t, 1 H, J = 10.0 Hz, H-4C), 3.24 (t, 1 H, J = 10.0 Hz, H-4B 3.21 (t, 1 H, J = 10.0 Hz, H-4A), 2.29 (br. s, 1 H, OH) 1.88–1.81 (m, 2 H, OCH2CH2CH2N), 1.30–1.25 (m, 6 H, H-6A, H-6B), 1.20 (d, 3 H, J6,5 = 6.1 Hz, H-6D), 1.16 (d, 3 H, J6,5 = 6.1 Hz, H-6C). 13C NMR (101 MHz, CDCl3): δ 137.3, 137.1, 128.6, 128.4, 128.3, 128.2, 128.0 (Ar), 100.5 (×2) (C-1B, C-1D), 100.3 (C-1C), 98.9 (C-1A), 77.7 (C-3D), 77.5 (C-3A), 76.9 (C-3B), 76.5 (C-3C), 73.7 (C-2A), 73.6 (C-2B), 73.3 (C-2C), 72.19 (×2), 72.16, 72.1 (4 PhCH2), 67.91 (C-5B), 67.85 (C-5С), 67.5 (C-5A), 67.4 (C-5D), 67.2 (C-2D), 65.8 (OCH2CH2CH2N), 64.2 (×3) (C-4A, C-4B, C-4D), 63.9 (C-4C), 38.0 (OCH2CH2CH2N), 28.4 (OCH2CH2CH2N), 18.6 (×2) (C-6A, C-6B) 18.5 (C-6D), 18.3 (C-6C). HRMS (ESI): calcd. for C57H68F3N13O14 [M + Na]+ m/z 1238.4853; found m/z 1238.4848.
3-Trifluoroacetamidopropyl 2-O-acetyl-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranoside (18). A mixture of donor 6 (58 mg, 0.118 mmol), acceptor 17 (120 mg, 0.099 mmol) and mol sieve 4 Å (200 mg) in CH2Cl2 (9 mL) was stirred at room temperature for 30 min, and then cooled to −40 °C. NIS (45 mg, 0.198 mmol) and a solution of TMSOTf (4 μL, 0.02 mmol) in CH2Cl2 (24 μL), and the mixture was stirred for 10 min, gradually increasing the temperature to −30 °C. The mixture was neutralized by adding pyridine (20 μL), diluted with CHCl3 (10 mL), and filtered through a Celite layer. The solids were washed with CHCl3 (3 × 5 mL), and the filtrate was washed with 0.5 M Na2S2O3 solution (10 mL) and water (20 mL), and concentrated. Column chromatography of the residue (toluene–EtOAc, 0→10%) yielded pentasaccharide 18 (134 mg, 89%) as a colorless foam, [α]D +92.3 (с 1, CHCl3). 1H NMR (400 MHz, CDCl3): δ 7.41–7.28 (m, 25 H, Ar), 6.52 (br. s, 1 H, NH), 5.40 (poorly resolved t, 1 H, H-2E), 4.95 (d, 1 H, J1,2 = 1.5 Hz, H-1D), 4.88–4.83 (m, 3 H, H-1B, H-1E, H-1C), 4.74–4.69 (m, 2 H, 2 benzylic H), 4.67–4.51 (m, 9 H, H-1A, 8 benzylic H), 3.87 (poorly resolved t, 1 H, H-2D), 3.83 (poorly resolved t, 1 H, H-2C), 3.80–3.60 (m, 8 H, H-2A, H-2B, H-3A, H-3B, H-3C, H-3E, H-3D, OCHaHbCH2CH2N), 3.56–3.30 (m, 10 H, H-4D, H-4E, H-5A, H-5B, H-5C, H-5D, H-5E, OCHaHbCH2CH2N, OCH2CH2CH2N), 3.24–3.16 (m, 3 H, H-4A, H-4B, H-4C), 2.10 (s, 3 H, CH3CO), 1.88–1.80 (m, 2 H, OCH2CH2CH2N), 1.27 (d, 3 H, J6,5 = 6.2 Hz, H-6A), 1.25 (d, 3 H, J6,5 = 6.4 Hz, H-6D), 1.21 (d, 3 H, J6,5 = 6.2 Hz, H-6E), 1.18 (d, 3 H, J6,5 = 6.0 Hz, H-6B), 1.14 (d, 3 H, J6,5 = 6.0 Hz, H-6C). 13C NMR (101 MHz, CDCl3): δ 169.8 (CH3CO), 128.6, 128.4, 128.3, 128.1, 128.0 (Ar), 100.5 (C-1B), 100.2 (C-1D), 100.1 (C-1E), 99.1 (C-1С), 98.9 (C-1A), 77.5 (C-3A), 76.8 (C-3B), 76.6 (C-3D), 76.4 (C-3C), 75.4 (C-3E), 73.8 (C-2A), 73.6 (C-2B), 73.5 (C-2D), 73.4 (C-2C), 72.2 (×3), 72.0, 71.6 (5 PhCH2), 67.9 (×3) (C-5B, C-5D, C-5E), 67.7 (C-5C), 67.5 (C-5A), 67.2 (C-2E), 65.8 (OCH2CH2CH2N), 64.2 (×3) (C-4A, C-4A, C-4C), 64.1 (C-4D), 63.9 (C-4E), 38.0 (OCH2CH2CH2N), 28.4 (OCH2CH2CH2N), 21.0 (CH3CO), 18.6 (×2), 18.5 (×2), 18.4 (C-6A, C-6B, C-6C, C-6D, C-6E). HRMS (ESI): calcd. for C72H85F3N16O18 [M + Na]+ m/z 1541.6065; found m/z 1541.6072.
3-Trifluoroacetamidopropyl 4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-azido-3-O-benzyl-4,6-dideoxy-α-d-mannopyranoside (19). 1 M sodium methoxide (20 μL) was added to a solution of 2-acetate 18 (162 mg, 0.107 mmol) in MeOH (2.2 mL), the mixture was stirred for 4.5 h at room temperature, and then worked up as described above for compound 15. Column chromatography (toluene–EtOAc, 0→15%) produced title product 19 (122 mg, 77%) as a colorless foam, [α]D +98.7 (с 1, CHCl3). 1H NMR (600 MHz, CDCl3): δ 7.42–7.29 (m, 25 H, Ar), 6.54 (br. s, 1 H, NH), 4.97 (d, 1 H, J1,2 = 1.7 Hz, H-1B), 4.96 (d, 1 H, J1,2 = 1.8 Hz, H-1C), 4.86 (d, 1 H, J1,2 = 1.7 Hz, H-1E), 4.85 (d, 1 H, J1,2 = 1.7 Hz, H-1D), 4.74–4.57 (m, 11 H, H-1A, 10 benzylic H), 3.99 (br. s, 1 H, H-2E), 3.94–3.93 (poorly resolved t, 1 H, H-2C), 3.82 ((poorly resolved t, 1 H, H-2D), 3.79–3.77 (m, 2 H, H-2A, H-2B), 3.75–3.69 (m, 3 H, H-3B, H-3C, OCHaHbCH2CH2N), 3.66–3.61 (m, 3 H, H-3A, H-3D, H-3E), 3.55–3.34 (m, 9 H, H-4c, H-5a, H-5b, H-5c, H-5e, H-5d, OCHaHbCH2CH2N, OCH2CH2CH2N), 3.31 (t, 1 H, J = 10.1 Hz, H-4B) 3.24–3.17 (m, 3 H, H-4A, H-4D, H-4E), 2.29 (br. s, 1 H, OH), 1.87–1.81 (m, 2 H, OCH2CH2CH2N), 1.27 (d, 3 H, J6,5 = 6.2 Hz, H-6), 1.25 (d, 3 H, J6,5 = 6.2 Hz, H-6), 1.20 (d, 3 H, J6,5 = 6.2 Hz, H-6), 1.18 (d, 3 H, J6,5 = 6.2 Hz, H-6), 1.14 (d, 3 H, J6,5 = 6.2 Hz, H-6). 13C NMR (151 MHz, CDCl3): δ 128.7, 128.6, 128.4, 128.4, 128.3, 128.3, 128.1 (Ar), 100.5 (×2) (C-1B, C-1C), 100.2 (×2) (C-1D, C-1E), 98.9 (C-1A), 77.7 (C-3E), 77.5 (C-3C), 77.0 (C-3B), 76.6 (C-3D), 76.4 (C-3A), 73.7 (C-2E), 73.6 (C-2A), 73.4 (C-2D), 73.3 (C-2C), 72.2 (×4) 72.1 (5 PhCH2), 67.9 (×2) 67.8 67.5, 67.4, (C-5A, C-5B, C-5C, C-5D, C-5E), 67.2 (C-2E), 65.8 (OCH2CH2CH2N), 64.2 (×3), 64.1, 63.8 (5 C-4), 38.0 (OCH2CH2CH2N), 28.4 (OCH2CH2CH2N), 18.6 (×2),18.5 (×2), 18.3 (5 C-6). HRMS (ESI): calcd. for C70H83F3N16O17 [M + Na]+ m/z 1499.5965; found m/z 1499.5966.
3-Trifluoroacetamidopropyl 4-amino-3-O-benzyl-4,6-dideoxy-α-d-mannopyranosyl-(1→2)-4-amino-3-O-benzyl-4,6-dideoxy-α-d-mannopyranoside (20). A mixture of diazide 11α (85 mg, 0.123 mmol) and Pd(OH)2/C (31 mg) in MeOH (3 mL) was vigorously stirred at 35 °C under hydrogen for 30 min. Then the catalyst was filtered off through a Celite layer, thoroughly washed with MeOH (4 × 5 mL), and the filtrate was concentrated. Column chromatography of the residue (CH2Cl2 – MeOH, 0→15%) yielded diamine 20 (62 mg, 78%) as a colorless syrup. 1H NMR (600 MHz, CD3OD): δ 7.44–7.27 (m, 10 H, Ar), 4.91 (d, 1 H, J1,2 = 1.9 Hz, H-1B), 4.79 (d, 1 H, J1,2 = 1.9 Hz, H-1A), 4.72 (d, 1 H, J = 11.7 Hz, PhCHaHb), 4.68 (d, 1 H, J = 11.4 Hz, PhCHaHb′), 4.58 (d, 1 H, J = 11.4 Hz, PhCHaHb′), 4.51 (d, 1 H, J = 11.7 Hz, PhCHaHb), 4.08 (poorly resolved t, 1 H, H-2B), 3.95 (poorly resolved t, 1 H, H-2A), 3.78 (dq, 1 H, J5,6 = 6.2 Hz, J5,4 = 10.1 Hz, H-5B), 3.71–3.66 (m, 2 H, H-3A, OCHaHbCH2CH2N), 3.65–3.59 (m, 2 H, H-5A, H-3B), 3.42–3.37 (m, 3 H, OCHaHbCH2CH2N, OCH2CH2CH2N), 3.02 (t, 1 H, J = 10.1 Hz, H-4B), 2.85 (t, 1 H, J = 9.9 Hz, H-4A), 1.87–1.81 (m, 2 H, OCH2CH2CH2N), 1.25 (d, 3 H, J6,5 = 6.2 Hz, H-6B), 1.23 (d, 3 H, J6,5 = 6.2 Hz, H-6A). 13C NMR (151 MHz, CD3OD): δ 139.2, 139.0, 129.9, 129.8, 129.7, 129.7, 129.3, 129.2 (Ar), 103.4 (C-1B), 100.5 (C-1A), 78.9 (C-3A), 77.0 (C-3B), 74.6 (C-2A), 72.7, 71.3 (2 PhCH2), 69.3 (C-5A), 69.2 (C-5B), 66.6 (C-2B), 65.9 (OCH2CH2CH2N), 54.9 (C-4A), 54.2 (C-4B), 38.0 (OCH2CH2CH2N), 29.9 (OCH2CH2CH2N), 18.5 (C-6A), 18.3 (C-6B). HRMS (ESI): calcd. for C31H42F3N3O8 [M + Na]+ m/z 664.2817; found m/z 664.2816.
3-Trifluoroacetamidopropyl 3-O-benzyl-4,6-dideoxy-4-formamido-α-d-mannopyranosyl-(1→2)-3-O-benzyl-4,6-dideoxy-4-formamido-α-d-mannopyranoside (21). Formic acid (22 μL, 0.57 mmol) and DCC (59 mg, 0.285 mmol) were added to a solution of diamine 20 (61 mg, 0.095 mmol) in CH2Cl2 and MeOH (9:1, 3 mL), and the mixture was stirred at room temperature for 1.5 h. Then more formic acid (5 μL, 0.130 mmol) and DCC (16 mg, 0.077 mmol) were added, and stirring was continued for the next 1.5 h. The solvents were evaporated, the residue was suspended in CH2Cl2 (3 mL), and the precipitate of dicyclohexylurea was filtered off and washed with CH2Cl2 (3 × 2 mL). The filtrate was concentrated to a volume of ∼2 mL, and the resulting solution was subjected to column chromatography (CH2Cl2 – MeOH, 0→10%) to produce bis(formamide) 21 (57 mg, 87%) as a colorless amorphous solid. HRMS (ESI): calcd. for C33H42F3N3O10 [M + Na]+ m/z 720.2709; found m/z 720.2715.
3-Trifluoroacetamidopropyl 4,6-dideoxy-4-formamido-α-d-mannopyranosyl-(1→2)-4,6-dideoxy-4-formamido-α-d-mannopyranoside (22). Pd(OH)2/C (20 mg) was added to a solution of disaccharide 21(57 mg, 0.082 mmol) in MeOH (3 mL) and the mixture was vigorously stirred under hydrogen at room temperature for 24 h. More catalyst (7 mg) was added and stirring was continued for the next 1.5 h. The catalyst was filtered of through a Celite layer, washed with MeOH (4 × 4 mL), and the filtrate was concentrated. The residue was purified by column chromatography (CH2Cl2 – MeOH, 15→25%) to give debenzylated product 22 (34 mg, 94%) as a colorless amorphous solid. HRMS (ESI): calcd. for C19H30F3N3O10 [M + H]+ m/z 518.1953; found m/z 518.1956.
3-Aminopropyl 4,6-dideoxy-4-formamido-α-d-mannopyranosyl-(1→2)-4,6-dideoxy-4-formamido-α-d-mannopyranoside (1a). Ambersep 900 (OH−) (2 mL) was added to a solution of disaccharide 22 (33 mg, 0.064 mmol) in 50% aqueous MeOH (3 mL). The mixture was kept for 1 h with periodic shaking, and then the resin was filtered off and washed with 50% aqueous MeOH (6 × 3 mL). The filtrate was concentrated, and the residue was subjected to gel chromatography to afford 3-aminopropyl glycoside 1a (25 mg, 85%) as a white amorphous solid; contained a non-stoichiometric amount (∼0.40 equiv.) of AcOH; [α]D +31.0 (с 0.25, water). 1H NMR (600 MHz, D2O): δ 8.20, 8.19 (2 s, 1.6 H, HZC(O)NH), 8.03, 8.02 (2 s, 0.4 H, HEC(O)NH), 5.03 (br. s, 0.2 H, HE-1B) 5.02 (d, 0.8 H, J1,2 = 1.7 Hz, HZ-1B), 4.94 (br. s, 0.8 H, HZ-1A), 4.92 (br. s, 0.2 H, HE-1A), 4.12–4.0 (m, 1 H, H-2B), 4.02–3.77 (m, 7.6 H, H-2A, H-3A, H-3B, HZ-4A, HZ-4B, H-5A, H-5B, OCHaHbCH2CH2N), 3.60–3.55 (m, 1 H, OCHaHbCH2CH2N), 3.40 (t, 0.2 H, J = 10.3 Hz, HE-4), 3.38 (t, 0.2 H, HE-4), 3.16–3.07 (m, 2 H, OCH2CH2CH2N), 2.02–1.95 (m, 2 H, OCH2CH2CH2N), 1.90 (s, 1.2 H, CH3COO−), 1.28–1.20 (m, 6 H, H-6A, H-6B). 13C NMR (151 MHz, D2O): δ 167.9, 167.8 (HECONH), 164.92 (HZCONH), 102.2 (CE-1B), 102.1 (CZ-1B), 98.4 (C-1A), 78.0 (CZ-2A), 77.9 (CE-2A), 69.8 (H-2B), 68.0, 67.9, 67.6, 67.5 (С-3A, C-3B, C-5A, C-5B), 65.2 (OCH2CH2CH2N), 56.9, 56.7 (CE-4A, CE-4B), 52.1, 51.9, 51.7 (CZ-4A, CZ-4B), 37.4 (OCH2CH2CH2N), 26.7 (OCH2CH2CH2N), 16.9, 16.7 (C-6A, C-6B). HRMS (ESI): calcd. for C17H32N3O9 [M + Na]+ m/z 444.1952; found m/z 444.1953.
3-Aminopropyl 4,6-dideoxy-4-formamido-α-d-mannopyranosyl-(1→2)-4,6-dideoxy-4-formamido-α-d-mannopyranosyl-(1→2)-4,6-dideoxy-4-formamido-α-d-mannopyranoside (2a). Compound 2a was synthesized starting from trisaccharide 15 (103 mg, 0.108 mmol) according to the procedure used for transformation of disaccharide 12 into target compound 1a to give 39.8 mg (66%) of trisaccharide 2a as a white fluffy solid; contained a non-stoichiometric amount (∼0.40 equiv.) of AcOH, [α]D +36.5 (с 1, water). 1H NMR (600 MHz, D2O): δ 8.21–8.18 (m, 2.4 H, HZCON), 8.04–8.02 (m, 0.6 H, HECON), 5.21–4.87 (m, 3 H, 3 H-1), 4.18–3.78 (m, 12.4 H, 3 H-2, 3 H-3, 3 H-4Z, 3 H-5, OCHaHbCH2CH2N), 3.60–3.55 (m, 1 H, OCHaHbCH2CH2N), 3.45–3.35 (m, 0.6 H, 3 H-4E), 3.16–3.07 (m, 2 H, OCH2CH2CH2N), 2.01–1.95 (m, 2 H, OCH2CH2CH2N), 1.29–1.18 (m, 9 H, 3 H-6). 13C NMR (151 MHz, D2O): δ 169.0, 168.9 (HECON), 166.0 (HZCON), 103.2, 103.1, 103.0, 101.8, 99.6 (3 C-1), 78.8, 78.7, 78.5, 78.4 (C-2A, C-2B), 70.1, 69.4, 69.3, 69.1, 69.0, 68.8, 68.6, 68.5, 68.4, 68.3 (C-3A, C-5A, C-3B, C-5B, C-2C, C-3C, C-5C), 66.3 (OCH2CH2CH2N), 58.0, 57.8 (3 C-4E), 53.2, 53.1, 53.0, 52.9 (3 C-4Z), 38.5 (OCH2CH2CH2N), 27.8 (OCH2CH2CH2N), 24.4 (CH3COO−), 18.1, 18.0, 17.9, 17.8 (3 C-6). HRMS (ESI): calcd. for C24H42F3N4O13 [M + Na]+ m/z 617.2646; found m/z 617.2641.
3-Aminopropyl 4,6-dideoxy-4-formamido-α-d-mannopyranosyl-(1→2)-4,6-dideoxy-4-formamido-α-d-mannopyranosyl-(1→2)-4,6-dideoxy-4-formamido-α-d-mannopyranosyl-(1→2)-4,6-dideoxy-4-formamido-α-d-mannopyranoside (3a). Compound 3a was synthesized starting from tetrasaccharide 17 (85 mg, 0.071 mmol) according to the procedure used for transformation of disaccharide 12 into target compound 1a to give 27.7 mg (53%) of tetrasaccharide 3a as a white fluffy solid; contained a non-stoichiometric amount (∼0.35 equiv.) of AcOH, [α]D +39.4 (с 1, water). 1H NMR (600 MHz, D2O): δ 8.22–8.18 (m, 3.2 H, HZCON), 8.05–8.01 (m, 0.8 H, HECON), 5.23–4.87 (m, 4 H, 4 H-1), 4.19–3.78 (m, 16.2 H, 4 H-2, 4 H-3, 4 H-4Z, OCHaHbCH2CH2N), 3.61–3.55 (m, 1 H, OCHaHbCH2CH2N), 3.46–3.35 (m, 0.8 H, 4 H-4E), 3.16–3.07 (m, 2 H, OCH2CH2CH2N), 2.02–1.94 (m, 2 H, OCH2CH2CH2N), 1.90 (s, 1.1 H, CH3COO−), 1.29–1.17 (m, 12 H, 4 H-6). 13C NMR (151 MHz, D2O): δ 168.9 (HECON), 166.0 (HZCON), 103.1, 103.0, 101.7, 101.6, 99.5 (4 C-1), 78.7, 78.4, 78.3, 78.1 (C-2A, C-2B, C-2C), 70.0, 69.4, 69.3, 69.1, 69.0, 68.7, 68.5, 68.2 (4 C-3, 4 C-5, C-2D), 66.2 (OCH2CH2CH2N), 58.0, 57.8 (C-4E), 53.2, 53.1, 53.0, 52.8 (C-4Z), 38.5 (OCH2CH2CH2N), 27.8 (OCH2CH2CH2N), 24.4 (CH3COO−), 18.0, 17.9, 17.8 (4 C-6). HRMS (ESI): calcd. for C31H53N5O17 [M + Na]+ m/z 790.3327; found m/z 790.3329.
3-Aminopropyl 4,6-dideoxy-4-formamido-α-d-mannopyranosyl-(1→2)-4,6-dideoxy-4-formamido-α-d-mannopyranosyl-(1→2)-4,6-dideoxy-4-formamido-α-d-mannopyranosyl-(1→2)-4,6-dideoxy-4-formamido-α-d-mannopyranosyl-(1→2)-4,6-dideoxy-4-formamido-α-d-mannopyranoside (4a). Compound 4a was synthesized starting from pentaasaccharide 19 (47 mg, 0.032 mmol) according to the procedure used for transformation of disaccharide 12 into target compound 1a to give 10.0 mg (39%) of pentaasaccharide 4a as a white fluffy solid; contained a non-stoichiometric amount (∼0.30 equiv.) of AcOH, [α]D +42.5 (с 0.5, water). 1H NMR (600 MHz, D2O): δ 8.18 (br s, 1.5 H, HZCON), 8.03, 8.01 (2 s, 0.7 H, HECON), 5.21–4.85 (m, 5 H, 5 H-1), 4.19–3.76 (m, 17.3 H, 5 H-2, 5 H-3, 5 H-4Z, OCHaHbCH2CH2N), 3.60–3.53 (m, 1 H, OCHaHbCH2CH2N), 3.45–3.33 (m, 0.8 H, 5 H-4E), 3.16–3.05 (m, 2 H, OCH2CH2CH2N), 2.01–1.93 (m, 2 H, OCH2CH2CH2N), 1.89 (s, 1.2 H, CH3COO−), 1.33–1.15 (m, 15 H, 5 H-6). 13C NMR (151 MHz, D2O) δ 167.9 (HECON), 165.0 (HZCON), 102.0, 100.7, 100.6, 98.5 (5 C-1), 77.6, 77.2, 77.0 (C-2A, C-2B, C-2C, C-2D), 69.0, 68.3, 68.0, 67.9, 67.7, 67.5 (5 C-3, 5 C-5, C-2E), 65.2 (OCH2CH2CH2N), 56.9 (C-4E), 52.2, 52.0, 51.7 (C-4Z), 37.4 (OCH2CH2CH2N), 26.8 (OCH2CH2CH2N), 17.0, 16.9, 16.8, 16.7 (5 C-6). HRMS (ESI): calcd. for C38H64N6O21 [M + H]+ m/z 941.4190; found m/z 941.4197.
Preparation of fluorescein labelled glycoconjugates 1b-4b. General procedure: FITC was conjugated to synthetic oligosaccharides 1a–4a as described previously (Mukhametova et al., 2024b). Briefly, to aminopropyl glycoside 1a–4a (1 equiv.) and Na2CO3 (3 equiv.) water solution fluorescein isothiocyanate (FITC) (1.2 eq.) in DMF was added. The obtained mixture was vigorously mixed and kept at 60 °C for 2 h. The reaction mixture was concentrated in vacuo, dissolved in water (300 μL) and the mixture was loaded onto a Sep-Pak C-18 cartridge, which was preliminarily washed with methanol and then with excess water. The preliminarily washed cartridge was eluted with 2 mL portions of methanol–water mixture (from 0 to 60 vol% of methanol) with the concentration increasing in increments of 5 vol%. Product 1b was collected at eluent concentrations between 10 and 20 vol%, products 2b between 25 and 40 vol%, 3b between 30 and 40 vol%, 4b between 15 and 30 vol%. After evaporation and lyophilization, the products 1b–4b were obtained as light orange fluffy solids. The purity of the products was confirmed by thin-layer chromatography.
Synthesis of disaccharide tracer 1b. The fluorescent labelling of aminoethyl glycoside 1a (1.07 mg, 2.54 μmol) as described in general procedure, gave a light orange product (1.81 mg, 88%). HRMS (ESI) calcd. for C38H42N4O14S [M + Na]+ 833.2310 was found to be 833.2306.
Synthesis of trisaccharide tracer 2b. The fluorescent labelling of aminoethyl glycoside 2a (1.00 mg, 1.69 μmol) as described in general procedure, gave a light orange product (1.31 mg, 80%). HRMS (ESI) calcd. for calcd. for 3b C45H53N5O18S [M + Na]+ 1006.2999 found 1006.2987.
Synthesis of tetrasaccharide tracer 3b. The fluorescent labelling of aminoethyl glycoside 3a (1.54 mg, 2.00 μmol) as described in general procedure, gave a light orange product (1.57 mg, 83%). HRMS (ESI) calcd. for C52H64N6O22S [M + Na]+ 1179.3687 was found to be 1179.3683.
Synthesis of pentasaccharide tracer 4b. The fluorescent labelling of aminoethyl glycoside 4a (0.68 mg, 0.723 μmol) as described in general procedure, gave a light orange product (0.8234 mg, 86%). HRMS (ESI) calcd. for C59H75N7O26S [M + Na]+ 1352.4375 was found to be 1352.4373.
Serum samples
Positive serum samples from multiple brucellosis-unfavorable farms (N = 19) were provided by Federal state budgetary institution «The Russian state center for animal feed and drug standardization and quality» (Moscow, Russia). These Brucella-positive sera were confirmed by at least two serological assays, including RBT, CFT, and ELISA. Brucellosis negative (N = 20) samples were provided from brucellosis-free farms and the reaction to all serological tests for brucellosis was negative.
Fluorescence polarization assay
FITC-labeled glycoconjugates 23, 1b–4b tracer working solutions (2.5 nM) in 10 mM phosphate buffer with 0.15 M NaCl, pH 7.4, were prepared so that the fluorescence intensity of the solutions was 10 times more than the buffer background signal, or roughly 200,000 U. The tracer working solution (1.0 mL) was then mixed with 10 µL of the tested serum, then the tube was vigorously shaken and after 1–2 min incubation time at 20°C. The intensity and polarization of fluorescence were measured using a portable device Sentry-200 (Ellie LLC, Germantown, WI, USA) λex = 485 nm and λem = 535 nm). Every measurement was carried out three times. Analysis and statistical evaluation of the experimental data was performed using SigmaPlot 11 (Systat Software Inc., Palo Alto, CA, United States) software.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was approved by Russian State Centre of Quality and Standardization of Veterinary Drugs and Feeds. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
TV: Investigation, Writing – original draft. YT: Writing – original draft, Investigation. DY: Investigation, Writing – original draft. AK: Investigation, Writing – original draft. OS: Writing – original draft, Investigation. OB: Investigation, Writing – original draft. DZ: Investigation, Writing – original draft. LM: Writing – original draft, Investigation. SE: Investigation, Writing – original draft. VK: Writing – original draft, Formal Analysis, Investigation. NN: Supervision, Data curation, Writing – original draft, Writing – review and editing, Methodology, Conceptualization, Funding acquisition, Resources, Validation, Formal Analysis, Project administration.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Russian Science Foundation (Grant 19-73-30017-P). This work was performed using the equipment in the Shared Research Center (Department of Structural Studies) of N.D. Zelinsky Institute of Organic Chemistry RAS, Moscow.
Acknowledgments
The authors are grateful to Dr. A. S. Dmitrenok and Dr. A. O. Chizhov for recording NMR and mass spectra of synthesized compounds, respectively.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2025.1662885/full#supplementary-material
References
Adamo, R., and Kováč, P. (2007). Glycosylation under thermodynamic control: synthesis of the di- and the hexasaccharide fragments of the O-sp of Vibrio cholerae O:1 serotype Ogawa from fully Functionalized Building blocks. Eur. J. Org. Chem. 2007, 988–1000. doi:10.1002/ejoc.200600851
Andrade, R. S., Oliveira, M. M. de, Bueno Filho, J. S. de S., Ferreira, F., Godfroid, J., Lage, A. P., et al. (2024). Accuracy of serological tests for bovine brucellosis: a systematic review and meta-analysis. Prev. Vet. Med. 222, 106079. doi:10.1016/j.prevetmed.2023.106079
Ariosa-Alvarez, A., Arencibia-Mohar, A., Madrazo-Alonso, O., Garcia-Imia, L., Sierra-Gonzalez, G., and Verez-Bencomo, V. (1998). Synthesis of the Vibrio cholerae O1 Ogawa and Inaba terminal disaccharides with Dioxolane-type spacers and their Coupling to Proteins1. J. Carbohydr. Chem. 17, 1307–1320. doi:10.1080/07328309808002355
Bundle, D. R., and McGiven, J. (2017). Brucellosis: improved diagnostics and vaccine Insights from synthetic Glycans. Acc. Chem. Res. 50, 2958–2967. doi:10.1021/acs.accounts.7b00445
Bundle, D. R., Gerken, M., and Peters, T. (1988). Synthesis of antigenic determinants of the Brucella A antigen, utilizing methyl 4-azido-4,6-dideoxy-alpha-D-mannopyranoside efficiently derived from D-mannose. Carbohydr. Res. 174, 239–251. doi:10.1016/0008-6215(88)85094-8
Caroff, M., Bundle, D. R., and Perry, M. B. (1984). Structure of the O-chain of the phenol-phase soluble cellular lipopolysaccharide of Yersinia enterocolitica serotype O:9. Eur. J. Biochem. 139, 195–200. doi:10.1111/j.1432-1033.1984.tb07994.x
Dong, B., Zhao, S., Li, H., Wen, K., Ke, Y., Shen, J., et al. (2019). Design, synthesis and characterization of tracers and development of a fluorescence polarization immunoassay for the rapid detection of ractopamine in pork. Food Chem. 271, 9–17. doi:10.1016/j.foodchem.2018.07.147
Dong, S.-B., Xiao, D., Liu, J.-Y., Bi, H.-M., Zheng, Z.-R., Wang, L.-D., et al. (2021). Fluorescence polarization assay improves the rapid detection of human brucellosis in China. Infect. Dis. Poverty 10, 46. doi:10.1186/s40249-021-00834-3
Duncombe, L., Howells, L., Haughey, A., Taylor, A. V., Kaveh, D., Erdenliğ Gϋrbilek, S., et al. (2022). The Tip of Brucella O-polysaccharide is a potent epitope in Response to brucellosis infection and Enables short synthetic antigens to Be Superior diagnostic reagents. Microorganisms 10, 708. doi:10.3390/microorganisms10040708
El Ayoubi, L. W., Challita, C., and Kanj, S. S. (2024). The many faces of brucellosis: diagnostic and management approach. Curr. Opin. Infect. Dis. 37, 474–484. doi:10.1097/QCO.0000000000001045
Eremin, S. A., Mukhametova, L. I., Krylov, V. B., and Nifantiev, N. E. (2024). Fluorescence polarization assay for infection diagnostics: a review. Molecules 29, 4712. doi:10.3390/molecules29194712
Franco, M. P., Mulder, M., Gilman, R. H., and Smits, H. L. (2007). Human brucellosis. Lancet Infect. Dis. 7, 775–786. doi:10.1016/S1473-3099(07)70286-4
Ganesh, N. V., Sadowska, J. M., Sarkar, S., Howells, L., McGiven, J., and Bundle, D. R. (2014). Molecular recognition of Brucella A and M antigens dissected by synthetic oligosaccharide glycoconjugates leads to a disaccharide diagnostic for brucellosis. J. Am. Chem. Soc. 136, 16260–16269. doi:10.1021/ja5081184
Godfroid, J., Nielsen, K., and Saegerman, C. (2010). Diagnosis of brucellosis in livestock and wildlife. Croat. Med. J. 51, 296–305. doi:10.3325/cmj.2010.51.296
Guiard, J., Paszkiewicz, E., Sadowska, J., and Bundle, D. R. (2013). Design and synthesis of a universal antigen to detect brucellosis. Angew. Chem. Int. Ed. Engl. 52, 7181–7185. doi:10.1002/anie.201302303
Hou, S., and Kovác, P. (2010). Enhanced stereoselectivity of alpha-mannosylation under thermodynamic control using trichloroacetimidates. Carbohydr. Res. 345, 999–1007. doi:10.1016/j.carres.2010.03.025
Ibarra, M., Campos, M., Hernán, B., Loor-Giler, A., Chamorro, A., and Nuñez, L. (2023). Comparison of diagnostic tests for detecting bovine brucellosis in animals vaccinated with S19 and RB51 strain vaccines. Vet. World 16, 2080–2085. doi:10.14202/vetworld.2023.2080-2085
Jolley, M. E., and Nasir, M. S. (2003). The use of fluorescence polarization assays for the detection of infectious diseases. Comb. Chem. High. Throughput Screen. 6, 235–244. doi:10.2174/138620703106298419
Kenne, L., Lindberg, B., Unger, P., Gustafsson, B., and Holme, T. (1982). Structural studies of the Vibrio cholerae O-antigen. Carbohydr. Res. 100, 341–349. doi:10.1016/s0008-6215(00)81047-2
Khurana, S. K., Sehrawat, A., Tiwari, R., Prasad, M., Gulati, B., Shabbir, M. Z., et al. (2021). Bovine brucellosis - a comprehensive review. Vet. Q. 41, 61–88. doi:10.1080/01652176.2020.1868616
Kinosita, K., Ikegami, A., and Kawato, S. (1982). On the wobbling-in-cone analysis of fluorescence anisotropy decay. Biophys. J. 37, 461–464. doi:10.1016/S0006-3495(82)84692-4
Kubler-Kielb, J., and Vinogradov, E. (2013). Reinvestigation of the structure of Brucella O-antigens. Carbohydr. Res. 378, 144–147. doi:10.1016/j.carres.2013.03.021
Kuznetsov, A. N., Gerbst, A. G., Tsvetkov, Y. E., Khodzhibekov, R. R., Dalgatova, A. A., Burgasova, O. A., et al. (2025). Synthesis and immunochemical studies of linear oligoglucosides structurally related to the cyclic β-(1→2)-D-glucan of Brucella. Commun. Chem. 8, 172. doi:10.1038/s42004-025-01570-7
Manabe, S., Satoh, H., Hutter, J., Lüthi, H. P., Laino, T., and Ito, Y. (2014). Significant substituent effect on the anomerization of pyranosides: mechanism of anomerization and synthesis of a 1,2-cis glucosamine oligomer from the 1,2-trans anomer. Chem. – Eur. J. 20, 124–132. doi:10.1002/chem.201303474
Mukhametova, L. I., Zherdev, D. O., Eremin, S. A., Kuznetsov, A. N., Yudin, V. I., Sclyarov, O. D., et al. (2024a). Applying a fluorescence polarization assay for detection of brucellosis in animals using the fluorescently labeled synthetic oligosaccharides as Biosensing tracer. Biosensors 14, 404. doi:10.3390/bios14080404
Mukhametova, L. I., Zherdev, D. O., Kuznetsov, A. N., Yudina, O. N., Tsvetkov, Y. E., Eremin, S. A., et al. (2024b). Fluorescence-polarization-based assaying of Lysozyme with Chitooligosaccharide tracers. Biomolecules 14, 170. doi:10.3390/biom14020170
Nasir, M. S., and Jolley, M. E. (2003). Fluorescence polarization (FP) assays for the determination of grain mycotoxins (fumonisins, DON vomitoxin and aflatoxins). Comb. Chem. High. Throughput Screen. 6, 267–273. doi:10.2174/138620703106298310
Ogawa, Y., and Kovac, P. (1997). Synthesis of the dodecasaccharide fragment representing the O-polysaccharide of Vibrio cholerae O:1, serotype Ogawa, bearing an aglycon offering flexibility for chemical linking to proteins. Glycoconj. J. 14, 433–438. doi:10.1023/a:1018591132723
Ogawa, Y., Lei, P. S., and Kovác, P. (1996). Synthesis of eight glycosides of hexasaccharide fragments representing the terminus of the O-polysaccharide of Vibrio cholerae O:1, serotype Inaba and Ogawa, bearing aglycons suitable for linking to proteins. Carbohydr. Res. 293, 173–194. doi:10.1016/0008-6215(96)00202-9
Olsen, S. C., Putz, E., and Boggiatto, P. M. (2025). Mucosal and systemic humoral responses after experimental Brucella abortus challenge in non-vaccinated or RB51-vaccinated elk. Res. Vet. Sci. 187, 105591. doi:10.1016/j.rvsc.2025.105591
Palchowdhury, S., Mondal, S., Kwak, K., and Cho, M. (2024). Recasting the wobbling-in-a-cone model for the rotational anisotropy of phenylselenocyanate in poly(methyl methacrylate): effect of internal bond rotation and polymer segmental motion. J. Chem. Phys. 161, 214112. doi:10.1063/5.0239896
Perry, M. B., and Bundle, D. R. (1990). Antigenic relationships of the lipopolysaccharides of Escherichia hermannii strains with those of Escherichia coli O157:H7, Brucella melitensis, and Brucella abortus. Infect. Immun. 58, 1391–1395. doi:10.1128/iai.58.5.1391-1395.1990
Peters, T., and Bundle, D. R. (1989). Block synthesis of two pentasaccharide determinants of the Brucella M antigen using thioglycoside methodologies. Can. J. Chem. 67, 497–502. doi:10.1139/v89-077
Peters, T., Brisson, J.-R., and Bundle, D. R. (1990). Conformational analysis of key disaccharide components of Brucella A and M antigens. Can. J. Chem. 68, 979–988. doi:10.1139/v90-154
Saksena, R., Zhang, J., and Kováč, P. (2005). Immunogens from a synthetic hexasaccharide fragment of the O-SP of Vibrio cholerae O:1, serotype Ogawa. Tetrahedron Asymmetry 16, 187–197. doi:10.1016/j.tetasy.2004.11.021
Saksena, R., Chernyak, A., and Kovác, P. (2008). Synthesis of spacer-equipped di-tri-and the tetrasaccharide fragments of the deacetylated O-PS1 of Citrobacter gillenii O9a,9b, and a related pentasaccharide. Carbohydr. Res. 343, 1693–1706. doi:10.1016/j.carres.2008.03.037
Sotolongo-Rodríguez, D., Gomez-Flores, R., Navarro-Soto, M. C., Arellano-Reynoso, B., Tamez-Guerra, P., and Ramírez-Pfeiffer, C. (2022). Evaluation of the fluorescence polarization assay for the diagnosis of brucellosis in Goat milk. Vet. Sci. 9, 303. doi:10.3390/vetsci9060303
Tian, M., Song, M., Yin, Y., Lian, Z., Li, Z., Hu, H., et al. (2020). Characterization of the main immunogenic proteins in Brucella infection for their application in diagnosis of brucellosis. Comp. Immunol. Microbiol. Infect. Dis. 70, 101462. doi:10.1016/j.cimid.2020.101462
Tsvetkov, Y.E., and Nifantiev, N.E. (2023). Synthesis of a spacer-armed disaccharide structurally related to the M antigenic fragment of Brucella O-polysaccharides. Russ. Chem. Bull. 72, 2731–2737. doi:10.1007/s11172-023-4079-4
Keywords: Brucella, O-antigen, N-formyl-D-perosamine, antibodies detection, fluorescence polarization assay
Citation: Volkov TM, Tsvetkov YE, Yashunsky DV, Kuznetsov AN, Sclyarov OD, Babicheva OV, Zherdev DO, Mukhametova LI, Eremin SA, Krylov VB and Nifantiev NE (2025) Synthesis of oligo-α-(1→2)-4,6-dideoxy-4-formamido-d-mannopyranosides related to the A epitope of the Brucella O-polysaccharide and their use for assaying of serum immunoglobulins. Front. Chem. 13:1662885. doi: 10.3389/fchem.2025.1662885
Received: 09 July 2025; Accepted: 05 August 2025;
Published: 29 August 2025.
Edited by:
Bing Yang, Nantong University, ChinaReviewed by:
Yu-Shun Yang, Nanjing University, ChinaRamesh Maruthi Chingle, National Institutes of Health (NIH), United States
Daniel Varon Silva, University of Applied Sciences and Arts Northwestern Switzerland, Switzerland
Copyright © 2025 Volkov, Tsvetkov, Yashunsky, Kuznetsov, Sclyarov, Babicheva, Zherdev, Mukhametova, Eremin, Krylov and Nifantiev. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nikolay E. Nifantiev, bmVuQGlvYy5hYy5ydQ==