 Wenyao Yang
Wenyao Yang Bruce Thompson
Bruce Thompson Faith A. A. Kwa
Faith A. A. Kwa- 1School of Health Sciences, Swinburne University of Technology, Hawthorn, VIC, Australia
- 2Melbourne School of Health Sciences, The University of Melbourne, Parkville, VIC, Australia
Hereditary ataxia (HA) is a diverse group of rare inherited neurological disorders characterised by cerebellar impairment and the progressive degeneration of spinocerebellar tracts and the spinal cord. These conditions manifest predominantly as unsteady gait, speech difficulties, dysphagia and motor skill impairment. The complex genetic causes and varied disease mechanisms underlying HA contribute to the multi-systemic symptoms which pose challenges in developing targeted effective treatments. Currently, available options for HA primarily focus on symptomatic management, highlighting a critical need for complementary therapeutic strategies, such as dietary and lifestyle interventions. This review explains recent findings on dietary and nutraceutical interventions, as well as lifestyle modifications such as exercise and rehabilitation programs for HA. It outlines common types of HA, including Friedreich ataxia, spinocerebellar ataxias, ataxia with vitamin E deficiency, ataxia-telangiectasia, and studies on a mixed cohort of patients with HA. The current management options, therapeutic implications of findings from pre-clinical and clinical data and future directions to advance the treatment of HA will also be discussed. The integration of nutraceuticals and rehabilitation programs with current methods of symptomatic management is encouraged for the holistic treatment of HA. These interventions will complement the use of various technological aids with the support of a multidisciplinary health and medical team to improve monitoring of the health status and disease progression of affected individuals; thus facilitating early treatment and an optimised clinical outcome.
Introduction
Hereditary ataxia (HA) is a group of genetic neurodegenerative disorders characterised by the progressive loss of coordination and balance due to the degeneration of the central nervous system (1). HA can be categorised into various subgroups, with autosomal recessive and autosomal dominant inheritance patterns being the most studied (1). More than 40 types of autosomal dominant HA, often referred to as spinocerebellar ataxias (SCAs), typically exhibit onset during adulthood (2). In contrast, autosomal recessive ataxias generally present in childhood, with common subtypes including Friedreich ataxia (FRDA), ataxia-telangiectasia (AT), and ataxia with vitamin E deficiency (AVED) (1). These ataxias share common symptoms such as unsteady gait, difficulties with speech and swallowing, and a wide range of other neurological symptoms, ultimately impairing the independence and quality of life of affected individuals (3).
To date, amongst all HAs, FRDA is the only subtype with an FDA-approved treatment (i.e., omaveloxolone) (4). Despite not being clinically approved, high-dose vitamin E supplementation has been widely recommended for treating AVED, making it a “dietary treatable condition” (5). Nevertheless, for the remaining types of HA, symptomatic management and supportive care remain the primary treatment options (3). This underscores the pressing need for complementary therapeutic strategies including dietary and lifestyle interventions to bring hope of an improved quality of life for those affected by HA.
This review discusses the pre-clinical and clinical findings related to dietary and nutraceutical interventions, as well as lifestyle modifications, including exercise and rehabilitation programs, for the aforementioned types of HA. In addition, this review highlights the potential benefits and challenges of these interventions in managing HA.
Search strategy and study selection criteria
Due to a small number of articles published on this research topic and in the effort to critique the findings of relatively recent studies, a literature search was conducted to identify peer-reviewed original research articles published within the last 10 years. These articles focused on dietary interventions, nutraceuticals, and lifestyle modifications for the management of hereditary ataxias. Keywords used included “Hereditary ataxia,” “Spinocerebellar Ataxia,” “Friedrich Ataxia,” “Ataxia-telangiectasia,” and “Ataxia with vitamin E deficiency” “physical rehabilitation,” “physical activity,” “dietary interventions,” “nutraceuticals,” “exercise,” and “training.” The search was performed using PubMed, and Web of Science. Full-text articles that met the above criteria were then assessed for their relevance to the objectives of this review.”
Features of hereditary ataxias
Despite sharing the above common clinical manifestations, HA possesses distinct pathogenic pathways and unique disease features. Due to genetic heterogeneity, HA can be a multigenic condition (2, 3). These mutations can impact various aspects of neuronal function, resulting in a wide range of symptoms and differing rates of disease progression (6). While primarily neurological, many HAs also present with non-neurological symptoms that can involve multiple organs, including retinopathy, cardiomyopathy, myopathy, and disorders associated with the endocrine and immune systems (7, 8). For certain types of HA associated with polymorphic tandem repeats, clinical features can vary significantly based on the size of the repeat expansion (9, 10). Depending on their sequence and location, repeat expansions may lead to either a loss- or gain- of-function, affecting gene expression and protein functionality (9). Additionally, specific gene mutations can also result in cellular changes with functional consequences within the nervous systems. These will be discussed in the following subsections.
Autosomal recessive hereditary ataxias
FRDA is caused by the mutation of guanine-adenine-adenine (11) repeat expansion in the frataxin (FXN) gene, which encodes a protein lead to a loss of function that is essential for mitochondrial iron homeostasis and the assembly and transfer of iron–sulfur clusters (11, 12). The mutation results in FXN deficiency, leading to mitochondrial dysfunction and symptoms such as gait ataxia, sensory loss, scoliosis and potential cardiac complications (12). Although symptoms typically reveal around puberty, the age of onset and the disease severity are inversely correlated with the size of the GAA expansion on the smaller of the FXN alleles (13).
A-T is caused by mutations in the Ataxia Telangiectasia Mutated (ATM) gene, which is crucial for the repair of DNA double-strand breaks and maintenance of genomic stability (14). Due to the defects in DNA repair, individuals with A-T have an increased risk of cancer and immunodeficiency (15). It is characterised by progressive cerebellar ataxia, oculomotor apraxia, or uncoordinated eye movements between ages 1 and 4 (1, 3, 15).
AVED is caused by mutations in the TTPA gene, which encodes the alpha-tocopherol transfer protein critical for vitamin E transport (1, 5). This condition is characterised by ataxia and peripheral neuropathy beginning in early childhood due to impaired vitamin E distribution in cells and tissues (1). Symptoms including dysarthria and poor balance in dim lighting result from the early loss of proprioception. As the condition progresses, many individuals become wheelchair-bound due to ataxia or leg weakness between the ages of 11 and 50 (1). While AVED shares phenotypic similarities with FRDA, it has distinct features such as head titubation, and dystonia with reduced frequency of cardiomyopathy (1, 5).
Autosomal dominant hereditary ataxias
The most extensively studied SCAs, characterised by a variable cytosine-adenine-guanine (CAG) expansion within the coding region of their respective genes directly results in the formation of an extended polyQ tract in the encoded proteins, leading to conformational changes that confer a toxic gain of function (6, 16). Uninterrupted (pure CAG) repeat structures increase the risk of expansions whereas CAT interruptions may stabilise these repeats during transmission and enhance meiotic stability (2). The larger the expansion size, the earlier the age of onset and the more severe the disease phenotype are observed in SCA (16). SCA1 is caused by a CAG repeat mutation in the Ataxin-1 (ATXN1) gene, resulting in toxic polyQ expansion that leads to a predominant loss of large populations of neurons in the cerebellum, brainstem, spinal cord, and cerebral cortex, with affected individuals experiencing pyramidal symptoms and muscle atrophy, and respiratory failure being the main cause of death (7, 16, 17). Other SCAs, such as SCA2, SCA3, SCA6, SCA7, and SCA17, are linked to polyQ expansions in distinct proteins, including Ataxin-2 (ATXN2), Ataxin-3 (ATXN3), calcium voltage-gated channel subunit alpha 1 (CACNA1A), Ataxin-7 (ATXN7) and the TATA binding protein (3). Saccadic slowing/saccadic dysmetria, nystagmus, and symptoms related to peripheral neuropathy are frequently seen in affected individuals with SCA2 (16). Notably, repeat sizes greater than 45 are almost always associated with a more aggressive disease course and disease onset before the age of 20 years for SCA2 (7, 18). SCA3, or Machado-Joseph disease, is the most prevalent SCAs worldwide. The protein aggregation leads to the formation of intranuclear inclusions, resulting in neuronal loss and specific symptoms such as pyramidal signs, parkinsonism, dystonia (16, 19, 20). SCA6 is characterised by progressive ataxia and postural instability due to mutations affecting calcium channels in the cerebellum (16). A recent study highlighted that heterozygous loss-of-function variants in the CACNA1A gene not only result in classical ataxia but are also associated with a range of other phenotypes, including epilepsy and intellectual disability (21). SCA7 is associated with vision problems (i.e., slow saccadic eye movement, ophthalmoplegia) due to retinal degeneration in addition to ataxia (16). Mental deterioration, occasional chorea, dystonia, myoclonus, epilepsy are often seen in SCA17 (1, 16). Unlike the above-mentioned common SCA subtypes, SCA38 is caused by missense mutation in the Elongase of Very Long-chain fatty acids 5 (ELOVL5) gene, which is necessary for synthesising omega-3 and omega-6 polyunsaturated fatty acids (PUFAs). It is characterised by a progressive and debilitating condition marked by ataxia, hyposmia, peripheral neuropathy, and cerebellar atrophy. Individuals with SCA38 may lose their ability to walk or may need to rely on assisted feeding within three decades of diagnosis (22).
Current disease management and novel therapeutic options
The pathogenesis underlying HA is complex, addressing the need of a multifaceted therapeutic strategy that can target the genetic cause, regulation of mitochondrial function and metabolism, and are compatible with methods of managing symptoms.
Current therapeutic options for the HA are significantly limited by both availability and the efficacy of available drugs. Many therapies are not widely accessible or approved in all countries or for all age groups. For instance, the first FDA-approved drug for FRDA, Omaveloxolone (SKYCLARYS™), is restricted to individuals under 16 years of age in the countries of US and EU (4). Regarding its efficacy, as a nuclear factor erythroid 2-related factor 2 (Nrf2) activator, it can help improve mitochondrial function, restore redox balance and reduce inflammation; however, it was not designed to target mutation in the FXN gene and improve FXN protein abundance (4).
While potential treatments are currently under investigation, the efficacy of these therapies can vary significantly among individuals due to genetic differences, disease progression, and other comorbidities (3, 23). Various therapeutic approaches, including gene therapy, protein replacement therapy, are being developed (24). However, advancement of these approaches in clinical trials is often hindered by a lack of relevant pre-clinical models (24). For example, while antisense oligonucleotides (ASOs) have demonstrated efficacy in mouse models of SCAs (25, 26). Those models are inadequate to fully recapitulate the full spectrum of disease severity seen in humans, such as neurodegeneration and cardiomyopathy seen in late-stage disease (24). Moreover, despite promising pre-clinical findings associated with molecular therapies, the translation of such data in the clinical setting is questionable due to a small clinical trial community for HA, short follow-up periods, varied outcome measurements and lack of reliable predictors of clinical outcomes. Consequently, these factors can lead to non-reproducible results which challenge the efficacy of new treatments (13, 27).
There is currently no clinically approved pharmacologic therapy for most types of HA. Supportive care and symptomatic treatments, such as physical therapy and medications for muscle spasticity, are the main interventions. Nevertheless, managing ataxic disorders is often difficult due to significant heterogeneity and the presence of symptoms affecting multiple organs. To effectively manage these complex conditions, affected individuals often require a multidisciplinary team comprising neurologists, physiotherapists, and genetic counselors to address the multifaceted symptoms and associated comorbidities (27). For instance, speech therapy is recommended to manage dysarthria and swallowing difficulties, while psychological support plays a crucial role in addressing emotional and cognitive challenges faced by affected individuals. Regular assessments are necessary to monitor disease progression and adjust treatment plans as needed (28).
To address the above concerns, there is an urgent need for the development of safe, accessible and complementary therapeutic strategies such as dietary and lifestyle interventions.
Dietary interventions and nutraceuticals for individuals with hereditary ataxias
Natural products (i.e., nutraceuticals) have had a long-standing reputation as valuable candidates in drug discovery, yielding exciting findings in alleviating symptoms and preventing disease progression in various conditions (29). Extensive evidence from different studies highlights the benefits of nutraceuticals, such as vitamin supplementation and phytochemicals, in treating HA (Table 1) (22, 30–32).
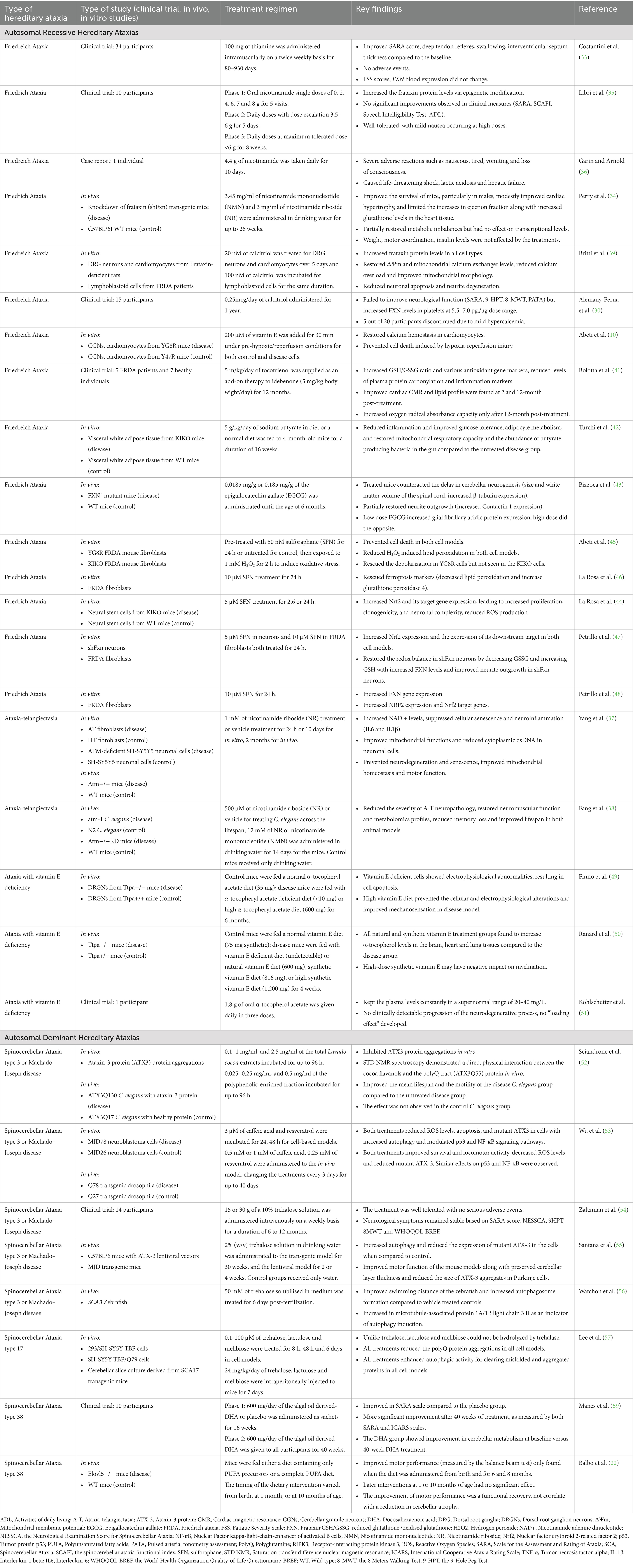
Table 1. Dietary interventions and nutraceuticals for the treatment of hereditary ataxias.
In FRDA, a study involving 34 patients treated with thiamine, also known as vitamin B1, at a dosage of 100 mg twice daily for up to 930 days demonstrated significant improvements in the Scale for the Assessment and Rating of Ataxia (SARA) score, deep tendon reflexes, swallowing, and interventricular septum thickness (33). In contrast, all forms of vitamin B3 consisting of nicotinamide adenine dinucleotide (NAD+) precursors, nicotinamide, nicotinamide mononucleotide (NMN) and nicotinamide riboside (NR), improved the survival, cardiac function and FXN protein levels via epigenetic regulation in FRDA mouse models (34, 35). However, no improvement in motor function was observed in either study (34, 35). Overall, the treatments were well-tolerated; however, one case reported severe side effects in a 40-year-old woman following the administration of 4.4 g of nicotinamide (36). Treatment involving NAD + precursors such as NR and NMN has also been tested in both in vitro and in vivo models of A-T (37, 38). Treated patient fibroblasts were found to have increased NAD + levels as well as decreased levels of mitochondrial reactive oxygen species and senescence markers (37). Similar effects were validated in the C. elegans model where improved locomotion and memory were noted based observational tests (i.e., swimming movement, pharyngeal pumping and chemotaxis assays) (38); Treated mice also found to have enhance survival rate, motor function based on the rotarod analysis, memory through the Y-maze spontaneous alternation test, and mitochondrial function via increased ATP levels and oxygen consumption rate (37, 38). These studies suggest that elevating NAD + levels can benefit motor function, memory and survival. Calcitriol supplementation, the active form of vitamin D, has demonstrated benefits in increasing FXN protein levels in in vitro and clinical studies of FRDA (30, 39). In cell models, calcitriol was found to restore mitochondrial function and enhance neuronal cell viability (39). However, it is notable that no neurological improvements were observed in clinical trials (30). Tocotrienol is a natural form of vitamin E abundant in plant oils, cocoa butter, barley and wheat germ (40). In FRDA cell models, vitamin E was found to help to prevent cell death by restoring calcium homeostasis in cardiomyocytes (10), while in human studies, it demonstrated improvements in cardiac function, lipid profiles and biomarkers related to oxidative stress and inflammation (41). Recently, butyrate supplementation, a short-chain fatty acid produced primarily by the fermentation of dietary fibre, was shown to restore butyrate-producing gut bacteria and improve adipocyte metabolism, mitochondrial respiration and glucose tolerance of the FRDA mice (42). These findings support the application of butyrate supplementation to mitigate diabetes-related symptoms in individuals with FRDA. Epigallocatechin gallate (EGCG), a polyphenol derived from green tea, has shown to restore β-tubulin and Contactin-1 expression, thereby mitigating neuronal developmental delays in FRDA mice and potentially help improve the motor and cognitive issues faced by affected individuals (43). A natural Nrf2 inducer, sulforaphane has been reported to increase FXN expression, elevate Nrf2 levels and the expression of downstream Phase II redox enzymes in cells derived from FRDA mice and FRDA fibroblasts; thus revealing its potential in targeting multiple pathological processes responsible for the disease symptoms (44–48).
There is no doubt that vitamin E has been extensively tested in AVED models. In dorsal root ganglion-derived sensory neurons of vitamin E-deficient mice, high-dose α-tocopheryl acetate supplementation successfully prevented the molecular and functional changes seen in cells and improved mechanosensation (49). In another study, natural and synthetic forms of α-tocopherol were able to increase α-tocopherol levels in the brain, heart, and lung tissues; however, high doses of synthetic α-tocopherol may negatively impact the expression of myelin genes which may impair neural synapses and consequently, motor and cognitive function (50). This underscores the importance of exercising caution with dosage Similarly, in a single case of AVED, daily consumption of 1.8 g of vitamin E successfully maintained plasma α-tocopherol levels within the normal range, with no signs of disease progression detected (51). These findings support the notion that clinical and histological phenotypes can be improved with early supplementation of vitamin E, although further research is needed to fully understand the optimal vitamin E intake.
In SCA3, phytochemicals such as Lavado cocoa extracts, caffeic acid and resveratrol exerted therapeutic benefits in survival and motor function through pleiotropic activities such as enhancing anti-oxidant defenses, downregulating pro-inflammatory markers and supporting mitochondrial functioning (52, 53). Trehalose, a naturally occurring sugar used as a sweetener, has been extensively studied in various models of SCA3 and SCA17 (54–57). Several have evaluated the safety and efficacy of trehalose in healthy subjects and patients with SCAs and in animal models, administered both orally and intravenously (54–57). The formation of misfolded and aggregated proteins in neurons is a hallmark of common types of SCA caused by proteins with polyQ tracts (2). Trehalose supplementation has been shown to effectively reduce the size of these protein aggregates, and improved motor function of the animal models (55–57). Additionally, dietary trehalose has garnered significant attention for its potential to stimulate the growth of health-promoting bacteria in the gastrointestinal tract (58). In SCA38, a diet rich in PUFAs (i.e., omega-3 and/or omega-6), has been shown to significantly enhance motor function in both patients and animal models, this beneficial effect does not appear to be associated with a recovery from cerebellar atrophy, as indicated by morphological measurements (22, 59). These results also highlight the importance of early intervention and diagnosis (22).
Lifestyle interventions in the management of hereditary ataxias
The primary objective of lifestyle interventions in HA is to facilitate recovery of physiological function that enables the performance of daily routine tasks. Table 2 summarises the lifestyle related interventions such as exercise and rehabilitation programs to manage HA.
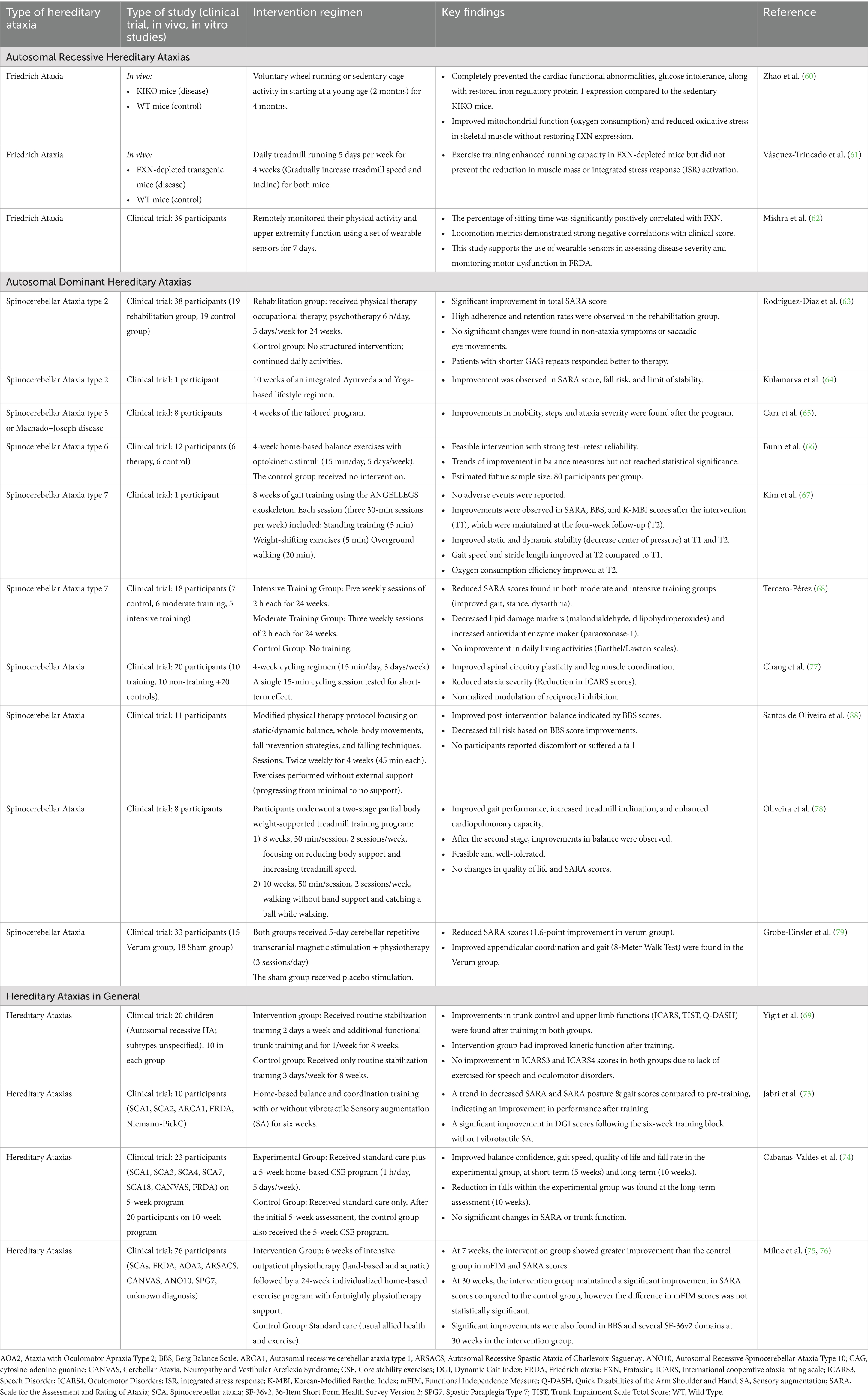
Table 2. Lifestyle interventions for the treatment of hereditary ataxias.
In FRDA, researchers using different mouse models have observed beneficial effects of exercise in preventing cardiac functional abnormalities along with restored iron regulatory protein expression, improving mitochondrial function, reducing oxidative stress in skeletal muscle (60), and enhancing running capacity (61). While in a human study of FRDA, participants used wearable sensors to remotely monitor their physical activity and upper extremity function over a period of 7 days (62). The locomotion metrics demonstrated a strong negative correlation between total walking time/total steps and clinical scores [i.e., modified Friedreich’s Ataxia Rating Scale (mFARS), Friedreich Ataxia Rating Scale Activity of Daily Living (FA-ADL)] (62). These findings indicate that increased walking activity may improve clinical outcomes.
In SCA2, participants who received 6 h of rehabilitation per day, 5 days a week, for 24 weeks focusing on balance, coordination, and muscle strengthening showed improved SARA scores compared to those in the control group (63). However, no improvements were observed in non-ataxia symptoms or saccadic eye movements (63). Notably, the improvement in SARA scores was inversely correlated with the size of the CAG repeat expansion, suggesting that patients with shorter expansions responded better to therapy (63). Furthermore, an integrated Ayurveda and Yoga-based lifestyle regimen applied to a single patient with SCA2 has demonstrated improvements in SARA score, lowered fall risk, and stability (64). In SCA3, implementing a tailored physical activity and lifestyle program for 4 weeks improved mobility and step count while reducing ataxia severity (65). However, a four-week home-based balance training program incorporating opto-kinetic stimuli for individuals with SCA6 did not result in significant improvements in balance measures. Nevertheless, the approach was found to be feasible, with a low dropout rate (66). In a case report of an individual with SCA7, an 8-week gait training program using a robotic exoskeleton demonstrated overall improvements in both motor and cardiac function (67). Moreover, a 24-week physical rehabilitation program, including both moderate and intensive training, led to reductions in SARA scores in participants with SCA7, indicating improvements in cerebellar symptoms such as stance, gait, dysarthria, dysmetria, and tremor (68). Physical training also decreased lipid damage biomarkers (malondialdehyde and lipohydroperoxides) while increasing the activity of the antioxidant enzyme paraoxonase-1, suggesting a beneficial effect on oxidative stress. However, no significant improvements were observed in daily living activities, as measured by the Barthel and Lawton scales (68). Similarly, various studies involving a cohort of participants, each with a different kind of HA, revealed that cycling regimen, physical therapy, treadmill training program, transcranial magnetic stimulation with physiotherapy, trunk training, intensive balance training, core stability exercise, and home-based or goal-directed rehabilitation programs significantly improved motor functions compared to control groups without any interventions (69–79).
Discussion
Due to the lack of clinically approved treatments for HA, supportive care and symptomatic treatments are the main interventions for most of these conditions; To address significant therapeutic gaps, this review aims to explore complementary therapeutic strategies, such as dietary and lifestyle interventions to potentially improve the quality of life in affected individuals.
Natural metabolites exhibit structural diversity and bioactivity, serving as substrates for various transporter systems that facilitate their delivery to targeted intracellular sites of action (80, 81). Phytochemicals such as flavonoids (i.e., Lavado cocoa extracts), polyphenols (i.e., EGCG, caffeic acid and resveratrol) and isothiocyanate (i.e., sulforaphane) are important naturally occurring compounds widely distributed in plants that possess multifaceted effects. A growing amount of evidence is suggestive of their potential therapeutic roles in various types of HA (43, 46, 52, 53). A variety of vitamins, have also been shown to improve certain aspects of pathology in various autosomal recessive ataxias, such as FRDA (30, 33–35, 39), A-T (37, 38) and AVED (49–51). However, a few FRDA studies have yielded inconsistent results (30, 34–36, 39). Potential factors contributing to differences may be due to the use of varied disease models, duration of drug exposure, variations in sample sizes, and the adoption of different biomarkers in pre-clinical or clinical measurements, as listed in Table 1. Furthermore, the results of various animal studies emphasise that individuals should begin treatment as early as possible to achieve optimal improvement in neurological symptoms (22, 34, 82). Meanwhile, caution should be taken when considering the consumption of nutraceuticals and an effective and toxic dose response must be examined rigorously to determine the margin of safety (36, 50).
Recent research suggests NAD + precursors and sulforaphane as promising nutraceuticals for FRDA (34, 46). Pre-clinical studies using NAD + precursors have shown improvements in cardiac function in FRDA mouse models, although effects on motor neurological function remain unclear (34, 35). While sulforaphane also shows promise in extensive pre-clinical models for its anti-oxidant effects, clinical trials are needed to validate its efficacy in FRDA (44–48). It is worth considering integrating lifestyle interventions such as regular exercise or tailored rehabilitation programs into the drug treatment regimen of FRDA for improving the motor function aspects that do not seem to be affected by the nutraceuticals. Additionally, it is not advisable to consume phytochemical supplements without the guidance of a healthcare professional, as individuals may overlook the complex interactions between medications and supplements (83, 84). For instance, dietary polyphenols have been shown to reduce the transport of thiamine and folic acid and to alter the activity of certain drugs by interacting with drug transporters or enzymes involved in metabolic reactions, which can inhibit the absorption of other nutrients (83).
In A-T, NAD + precursors seem to be very promising in mitigating the neuropathology and improving cellular mitochondrial functioning (37, 38). Given its safety profiles observed in FRDA clinical trials and animal models at higher dosages, NAD + precursors should also be trialed in A-T patients. Similarly, mounting evidence has validated the benefits of vitamin E in treating AVED, with the need for more clinical trials to determine the optimal dosages of ɑ-tocopherol acetate in individuals with AVED (49–51).
In studies of common SCAs, trehalose has demonstrated effectiveness in clearing toxic polyQ aggregates and improving motor function in various models of SCA3 and a cell model of SCA17 (55–57). Additionally, dietary trehalose has earned significant attention for its potential to stimulate the growth of health-promoting bacteria in the gastrointestinal tract, thereby promoting beneficial immune response and metabolic homeostasis (58). However, trehalose exhibits reduced efficacy in the brain as it is hydrolsed into glucose by trehalase. A study suggests that trehalose analogs, such as lactulose and melibiose, may serve as alternative strategies to enhance therapeutic effects for their resistance to trehalase hydrolysis (57). In SCA38, foods rich in PUFAs (i.e., fatty fish, olive oil) may support overall brain health and help mitigate some of the neurodegenerative processes based on the outcomes of both animal and human studies (22, 59). Consumption of fat-soluble vitamins and a low fat and carbohydrate diet have been reported to slow the progression of neurological symptoms, which highlights the importance of early diagnosis and dietary modifications (22, 85).
As efforts to source a panacea that involves a drug intervention continue, much research has also focused on rehabilitation methods associated with lifestyle changes to address symptom management (Table 2).
In FRDA, exercise training found to effectively improved the skeletal muscle and cardiac functioning by modulating mitochondrial oxygen consumption levels, glucose tolerance, as well as restoring FXN levels (60–62). However, despite the extensive benefits of exercise reported here, existing literature has limitations in small sample size and lack of sensitive biomarkers to track disease progression and assess less apparent non-motor impairments.
Current treatments for SCAs primarily aims to manage symptoms rather than to halt disease progression. The effectiveness of these treatments may vary based on the disease’s severity and progression rate, which are influenced by repeat length (86). Understanding the relationship between repeat length and disease severity is crucial for predicting outcomes and tailoring interventions for repeat expansion related diseases. A study revealed greater benefits of lifestyle interventions as part of rehabilitation programs in improving SARA scores in patients with smaller CAG repeat expansions (63). Thus, individuals with longer expansions may require more extensive management strategies to address different clinical manifestations (63). While there is evidence indicating that exercise and rehabilitation programs are generally beneficial for individuals with SCAs, there is a need to optimise the nature and scheduling of such activities to be meaningful to individuals and that can be undertaken at their own pace (64, 69–79, 87, 88). Periodic therapy sessions can facilitate customisable home exercise regimens and offer valuable feedback on progress (69, 74). Rehabilitation professionals, including specialists, play a crucial role in assisting individuals in adjusting exercise dosages based on monitored outcomes (65). Furthermore, although high adherence and retention rates were generally observed in most of the rehabilitation programs, the use of technological aids can often further enhance participants’ compliance (63, 66, 89, 90). Activity trackers can help patients to assess their current capabilities and set achievable goals. Smartwatches with safety features such as fall detection and emergency call functionalities can be helpful (91). Wearable sensors are a feasible tool for both assessing and monitoring disease progression (62). A systematic review and meta-analysis suggest that video games, exergames, and apps may be beneficial for ataxia rehabilitation and assessment, but further research is needed to fully establish their health benefits (91). Engaging in exercises like yoga or virtual reality-based activities may be able to motivate individuals to maintain adherence and these have shown improved SARA scores (64, 90). Overall, the limited number of lifestyle intervention studies in HA warrants the need for more studies to investigate the impact of different lifestyle factors (e.g., sleep, specific sport types) alone or in combination with dietary interventions, on the daily function of individuals with HA. Additionally, small sample sizes and the lack of sensitive biomarkers in existing studies limit the capacity to accurately track post-intervention disease progression and detect non-motor impairments (63, 78). Future studies may benefit from involving more participants and incorporating long-term follow-up and objective biomarkers (e.g., neuroimaging, molecular markers).
It is important to note that there are limited studies on lifestyle interventions for HA. Therefore, for the current studies reviewed in this paper, researchers often have to combine cohorts of participants with different kinds of HAs to generate a large enough sample size to increase the statistical power. As such, insufficient data is available to appropriately characterise the efficacy of the various lifestyle interventions according to individuals with different kinds of HAs and disease severity. Consequently, it is difficult to make clinically meaningful conclusions for individual patients suffering from a particular kind of HA.
Conclusion
This review provides an overview of the dietary and lifestyle interventions in the treatment of HAs. NAD + precursors have emerged as the most researched nutraceuticals in FRDA and A-T, demonstrating benefits in cardiac and mitochondrial function, albeit with limited effects on motor and neurological functions. These limitations may be addressed by integrating lifestyle interventions, such as regular physical activity and tailored rehabilitation programs. Trehalose and polyphenols, both of which can inhibit toxic protein aggregation in common SCAs, require clinical trials to validate their efficacy. High PUFA diets and vitamin E diets appear to be promising interventions for individuals with SCA38 and AVED, although further research is needed to determine the optimal dosage. Overall, this review illuminates the great potential of nutraceutical and lifestyle interventions in the mitigation of symptoms, and benefits in promoting health status for individuals with HA.
Author contributions
WY: Conceptualization, Data curation, Formal analysis, Writing – original draft, Writing – review & editing. BT: Supervision, Writing – review & editing. FK: Conceptualization, Data curation, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Gen AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Jayadev, S, and Bird, TD. Hereditary ataxias: overview. Genet Med. (2013) 15:673–83. doi: 10.1038/gim.2013.28
2. Ashizawa, T, Oz, G, and Paulson, HL. Spinocerebellar ataxias: prospects and challenges for therapy development. Nat Rev Neurol. (2018) 14:590–605. doi: 10.1038/s41582-018-0051-6
3. Pilotto, F, Del Bondio, A, and Puccio, H. Hereditary ataxias: from bench to clinic, where do we stand? Cells. (2024) 13:319. doi: 10.3390/cells13040319
4. Pilotto, F, Chellapandi, DM, and Puccio, H. Omaveloxolone: a groundbreaking milestone as the first FDA-approved drug for Friedreich ataxia. Trends Mol Med. (2024) 30:117–25. doi: 10.1016/j.molmed.2023.12.002
5. Thapa, S, Shah, S, Chand, S, Sah, SK, Gyawali, P, Paudel, S, et al. Ataxia due to vitamin E deficiency: a case report and updated review. Clin Case Reports. (2022) 10:e6303. doi: 10.1002/ccr3.6303
6. Mohren, L, Erdlenbruch, F, Leitao, E, Kilpert, F, Hones, GS, Kaya, S, et al. Identification and characterisation of pathogenic and non-pathogenic FGF14 repeat expansions. Nat Commun. (2024) 15:7665. doi: 10.1038/s41467-024-52148-1
7. Velazquez-Perez, LC, Rodriguez-Labrada, R, and Fernandez-Ruiz, J. Spinocerebellar Ataxia type 2: Clinicogenetic aspects, mechanistic insights, and management approaches. Front Neurol. (2017) 8:472. doi: 10.3389/fneur.2017.00472
8. Abramowicz, S, Dentel, A, Chouraqui, M, Bodaghi, B, and Touhami, S. Atypical retinopathy in ataxia with vitamin E deficiency: report of a sibship. Neurogenetics. (2024) 25:33–8. doi: 10.1007/s10048-023-00741-9
9. Leitao, E, Schroder, C, and Depienne, C. Identification and characterization of repeat expansions in neurological disorders: methodologies, tools, and strategies. Rev Neurol (Paris). (2024) 180:383–92. doi: 10.1016/j.neurol.2024.03.005
10. Abeti, R, Brown, AF, Maiolino, M, Patel, S, and Giunti, P. Calcium deregulation: novel insights to understand Friedreich's Ataxia pathophysiology. Front Cell Neurosci. (2018) 12:264. doi: 10.3389/fncel.2018.00264
11. Mueller, A, Paterson, E, McIntosh, A, Praestgaard, J, Bylo, M, Hoefling, H, et al. Digital endpoints for self-administered home-based functional assessment in pediatric Friedreich's ataxia. Ann Clin Transl Neurol. (2021) 8:1845–56. doi: 10.1002/acn3.51438
12. Clark, E, Johnson, J, Dong, YN, Mercado-Ayon, E, Warren, N, Zhai, M, et al. Role of frataxin protein deficiency and metabolic dysfunction in Friedreich ataxia, an autosomal recessive mitochondrial disease. Neuronal Signal. (2018) 2:60. doi: 10.1042/NS20180060
13. Georgiou-Karistianis, N, Corben, LA, Reetz, K, Adanyeguh, IM, Corti, M, Deelchand, DK, et al. A natural history study to track brain and spinal cord changes in individuals with Friedreich's ataxia: TRACK-FA study protocol. PLoS One. (2022) 17:e0269649. doi: 10.1371/journal.pone.0269649
14. Wingard, MC, Dalal, S, Shook, PL, Myers, R, Connelly, BA, Thewke, DP, et al. Deficiency of ataxia-telangiectasia mutated kinase modulates functional and biochemical parameters of the heart in response to Western-type diet. Am J Physiol Heart Circ Physiol. (2021) 320:H2324–38. doi: 10.1152/ajpheart.00990.2020
15. Rothblum-Oviatt, C, Wright, J, Lefton-Greif, MA, McGrath-Morrow, SA, Crawford, TO, and Lederman, HM. Ataxia telangiectasia: a review. Orphanet J Rare Dis. (2016) 11:159. doi: 10.1186/s13023-016-0543-7
16. Buijsen, RAM, Toonen, LJA, Gardiner, SL, and van Roon-Mom, WMC. Genetics, mechanisms, and therapeutic progress in polyglutamine spinocerebellar ataxias. Neurotherapeutics. (2019) 16:263–86. doi: 10.1007/s13311-018-00696-y
17. Kim, JH, Lukowicz, A, Qu, W, Johnson, A, and Cvetanovic, M. Astroglia contribute to the pathogenesis of spinocerebellar ataxia type 1 (SCA1) in a biphasic, stage-of-disease specific manner. Glia. (2018) 66:1972–87. doi: 10.1002/glia.23451
18. Moretti, P, Blazo, M, Garcia, L, Armstrong, D, Lewis, RA, Roa, B, et al. Spinocerebellar ataxia type 2 (SCA2) presenting with ophthalmoplegia and developmental delay in infancy. Am J Med Genet A. (2004) 124A:392–6. doi: 10.1002/ajmg.a.20428
19. Haas, E, Incebacak, RD, Hentrich, T, Huridou, C, Schmidt, T, Casadei, N, et al. A novel SCA3 Knock-in mouse model mimics the human SCA3 disease phenotype including neuropathological, behavioral, and transcriptional abnormalities especially in oligodendrocytes. Mol Neurobiol. (2022) 59:495–522. doi: 10.1007/s12035-021-02610-8
20. Sullivan, R, Yau, WY, O'Connor, E, and Houlden, H. Spinocerebellar ataxia: an update. J Neurol. (2019) 266:533–44. doi: 10.1007/s00415-018-9076-4
21. Hommersom, MP, Doorn, N, Puvogel, S, Lewerissa, EI, Mordelt, A, Ciptasari, U, et al. CACNA1A haploinsufficiency leads to reduced synaptic function and increased intrinsic excitability. Brain (London, England: 1878). (2024) 148:1286–301. doi: 10.1093/brain/awae330
22. Balbo, I, Montarolo, F, Genovese, F, Tempia, F, and Hoxha, E. Effects of the administration of Elovl5-dependent fatty acids on a spino-cerebellar ataxia 38 mouse model. Behav Brain Funct. (2022) 18:8. doi: 10.1186/s12993-022-00194-4
23. Schwab, SM, Spencer, C, Carver, NS, Andrade, V, Dugan, S, Greve, K, et al. Personal factors understood through the ecological-enactive model of disability and implications for rehabilitation research. Front Rehabil Sci. (2022) 3:954061. doi: 10.3389/fresc.2022.954061
24. Mosbach, V, and Puccio, H. A multiple animal and cellular models approach to study frataxin deficiency in Friedreich Ataxia. Biochim Biophys Acta, Mol Cell Res. (2024) 1871:119809. doi: 10.1016/j.bbamcr.2024.119809
25. Schuster, KH, Zalon, AJ, DiFranco, DM, Putka, AF, Stec, NR, Jarrah, SI, et al. ASOs are an effective treatment for disease-associated oligodendrocyte signatures in premanifest and symptomatic SCA3 mice. Mol Ther. (2024) 32:1359–72. doi: 10.1016/j.ymthe.2024.02.033
26. Scoles, DR, Meera, P, Schneider, MD, Paul, S, Dansithong, W, Figueroa, KP, et al. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature. (2017) 544:362–6. doi: 10.1038/nature22044
27. Zesiewicz, TA, Wilmot, G, Kuo, SH, Perlman, S, Greenstein, PE, Ying, SH, et al. Comprehensive systematic review summary: treatment of cerebellar motor dysfunction and ataxia: report of the guideline development, dissemination, and implementation Subcommittee of the American Academy of neurology. Neurology. (2018) 90:464–71. doi: 10.1212/WNL.0000000000005055
28. Stephen, CD, Brizzi, KT, Bouffard, MA, Gomery, P, Sullivan, SL, Mello, J, et al. The Comprehensive Management of Cerebellar Ataxia in adults. Curr Treat Options Neurol. (2019) 21:9. doi: 10.1007/s11940-019-0549-2
29. Harvey, AL, Edrada-Ebel, R, and Quinn, RJ. The re-emergence of natural products for drug discovery in the genomics era. Nat Rev Drug Discov. (2015) 14:111–29. doi: 10.1038/nrd4510
30. Alemany-Perna, B, Tamarit, J, Cabiscol, E, Delaspre, F, Miguela, A, Huertas-Pons, JM, et al. Calcitriol treatment is safe and increases Frataxin levels in Friedreich Ataxia patients. Mov Disord. (2024) 39:1099–108. doi: 10.1002/mds.29808
31. Bawari, S, Tewari, D, Arguelles, S, Sah, AN, Nabavi, SF, Xu, S, et al. Targeting BDNF signaling by natural products: novel synaptic repair therapeutics for neurodegeneration and behavior disorders. Pharmacol Res. (2019) 148:104458. doi: 10.1016/j.phrs.2019.104458
32. Bonanomi, M, Natalello, A, Visentin, C, Pastori, V, Penco, A, Cornelli, G, et al. Epigallocatechin-3-gallate and tetracycline differently affect ataxin-3 fibrillogenesis and reduce toxicity in spinocerebellar ataxia type 3 model. Hum Mol Genet. (2014) 23:6542–52. doi: 10.1093/hmg/ddu373
33. Costantini, A, Laureti, T, Pala, MI, Colangeli, M, Cavalieri, S, Pozzi, E, et al. Long-term treatment with thiamine as possible medical therapy for Friedreich ataxia. J Neurol. (2016) 263:2170–8. doi: 10.1007/s00415-016-8244-7
34. Perry, CE, Halawani, SM, Mukherjee, S, Ngaba, LV, Lieu, M, Lee, WD, et al. NAD+ precursors prolong survival and improve cardiac phenotypes in a mouse model of Friedreich's Ataxia. JCI Insight. (2024) 9:e177152. doi: 10.1172/jci.insight.177152
35. Libri, V, Yandim, C, Athanasopoulos, S, Loyse, N, Natisvili, T, Law, PP, et al. Epigenetic and neurological effects and safety of high-dose nicotinamide in patients with Friedreich's ataxia: an exploratory, open-label, dose-escalation study. Lancet. (2014) 384:504–13. doi: 10.1016/S0140-6736(14)60382-2
36. Garin, N, and Arnold, P. Life-threatening adverse reaction after self-initiated, off-label use of high dose nicotinamide for the treatment of Friedreich’s Ataxia. European journal of case reports. Intern Med. (2015) 2:234. doi: 10.12890/2015_000234
37. Yang, B, Dan, X, Hou, Y, Lee, JH, Wechter, N, Krishnamurthy, S, et al. NAD(+) supplementation prevents STING-induced senescence in ataxia telangiectasia by improving mitophagy. Aging Cell. (2021) 20:e13329. doi: 10.1111/acel.13329
38. Fang, EF, Kassahun, H, Croteau, DL, Scheibye-Knudsen, M, Marosi, K, Lu, H, et al. NAD(+) replenishment improves lifespan and Healthspan in Ataxia telangiectasia models via Mitophagy and DNA repair. Cell Metab. (2016) 24:566–81. doi: 10.1016/j.cmet.2016.09.004
39. Britti, E, Delaspre, F, Sanz-Alcazar, A, Medina-Carbonero, M, Llovera, M, Purroy, R, et al. Calcitriol increases frataxin levels and restores mitochondrial function in cell models of Friedreich Ataxia. Biochem J. (2021) 478:1–20. doi: 10.1042/BCJ20200331
40. Borel, P, Preveraud, D, and Desmarchelier, C. Bioavailability of vitamin E in humans: an update. Nutr Rev. (2013) 71:319–31. doi: 10.1111/nure.12026
41. Bolotta, A, Pini, A, Abruzzo, PM, Ghezzo, A, Modesti, A, Gamberi, T, et al. Effects of tocotrienol supplementation in Friedreich's ataxia: a model of oxidative stress pathology. Exp Biol Med (Maywood). (2020) 245:201–12. doi: 10.1177/1535370219890873
42. Turchi, R, Sciarretta, F, Ceci, V, Tiberi, M, Audano, M, Pedretti, S, et al. Butyrate prevents visceral adipose tissue inflammation and metabolic alterations in a Friedreich's ataxia mouse model. iScience. (2023) 26:107713. doi: 10.1016/j.isci.2023.107713
43. Bizzoca, A, Caracciolo, M, Corsi, P, Magrone, T, Jirillo, E, and Gennarini, G. Molecular and cellular substrates for the Friedreich Ataxia. Significance of Contactin expression and of antioxidant administration. Molecules. (2020) 25:4085. doi: 10.3390/molecules25184085
44. La Rosa, P, Russo, M, D'Amico, J, Petrillo, S, Aquilano, K, Lettieri-Barbato, D, et al. Nrf2 induction re-establishes a proper neuronal differentiation program in Friedreich's Ataxia neural stem cells. Front Cell Neurosci. (2019) 13:356. doi: 10.3389/fncel.2019.00356
45. Abeti, R, Uzun, E, Renganathan, I, Honda, T, Pook, MA, and Giunti, P. Targeting lipid peroxidation and mitochondrial imbalance in Friedreich's ataxia. Pharmacol Res. (2015) 99:344–50. doi: 10.1016/j.phrs.2015.05.015
46. La Rosa, P, Petrillo, S, Turchi, R, Berardinelli, F, Schirinzi, T, Vasco, G, et al. The Nrf2 induction prevents ferroptosis in Friedreich's Ataxia. Redox Biol. (2021) 38:101791. doi: 10.1016/j.redox.2020.101791
47. Petrillo, S, Piermarini, E, Pastore, A, Vasco, G, Schirinzi, T, Carrozzo, R, et al. Nrf2-inducers counteract neurodegeneration in Frataxin-silenced motor neurons: disclosing new therapeutic targets for Friedreich's Ataxia. Int J Mol Sci. (2017) 18:2173. doi: 10.3390/ijms18102173
48. Petrillo, S, D'Amico, J, La Rosa, P, Bertini, ES, and Piemonte, F. Targeting NRF2 for the treatment of Friedreich's Ataxia: a comparison among drugs. Int J Mol Sci. (2019) 20:5211. doi: 10.3390/ijms20205211
49. Finno, CJ, Peterson, J, Kang, M, Park, S, Bordbari, MH, Durbin-Johnson, B, et al. Single-cell RNA-seq reveals profound alterations in mechanosensitive dorsal root ganglion neurons with vitamin E deficiency. iScience. (2019) 21:720–35. doi: 10.1016/j.isci.2019.10.064
50. Ranard, KM, Kuchan, MJ, Bruno, RS, Juraska, JM, and Erdman, JW. Synthetic alpha-tocopherol, compared with natural alpha-tocopherol, downregulates myelin genes in cerebella of adolescent Ttpa-null mice. J Nutr. (2020) 150:1031–40. doi: 10.1093/jn/nxz330
51. Kohlschutter, A, Finckh, B, Nickel, M, Bley, A, and Hubner, C. First recognized patient with genetic vitamin E deficiency stable after 36 years of controlled supplement therapy. Neurodegener Dis. (2020) 20:35–8. doi: 10.1159/000508080
52. Sciandrone, B, Palmioli, A, Ciaramelli, C, Pensotti, R, Colombo, L, Regonesi, ME, et al. Cell-free and in vivo characterization of the inhibitory activity of Lavado cocoa Flavanols on the amyloid protein Ataxin-3: toward new approaches against spinocerebellar Ataxia type 3. ACS Chem Neurosci. (2024) 15:278–89. doi: 10.1021/acschemneuro.3c00560
53. Wu, YL, Chang, JC, Lin, WY, Li, CC, Hsieh, M, Chen, HW, et al. Treatment with Caffeic acid and resveratrol alleviates oxidative stress induced neurotoxicity in cell and Drosophila models of spinocerebellar Ataxia Type3. Sci Rep. (2017) 7:11641. doi: 10.1038/s41598-017-11839-0
54. Zaltzman, R, Elyoseph, Z, Lev, N, and Gordon, CR. Trehalose in Machado-Joseph disease: safety, tolerability, and efficacy. Cerebellum. (2020) 19:672–9. doi: 10.1007/s12311-020-01150-6
55. Santana, MM, Paixao, S, Cunha-Santos, J, Silva, TP, Trevino-Garcia, A, Gaspar, LS, et al. Trehalose alleviates the phenotype of Machado-Joseph disease mouse models. J Transl Med. (2020) 18:161. doi: 10.1186/s12967-020-02302-2
56. Watchon, M, Luu, L, Plenderleith, SK, Yuan, KC, and Laird, AS. Autophagy function and benefits of autophagy induction in models of spinocerebellar Ataxia type 3. Cells. (2023) 12:893. doi: 10.3390/cells12060893
57. Lee, GC, Lin, CH, Tao, YC, Yang, JM, Hsu, KC, Huang, YJ, et al. The potential of lactulose and melibiose, two novel trehalase-indigestible and autophagy-inducing disaccharides, for polyQ-mediated neurodegenerative disease treatment. Neurotoxicology. (2015) 48:120–30. doi: 10.1016/j.neuro.2015.03.009
58. Chen, A, and Gibney, PA. Dietary Trehalose as a bioactive nutrient. Nutrients. (2023) 15:1393. doi: 10.3390/nu15061393
59. Manes, M, Alberici, A, Di Gregorio, E, Boccone, L, Premi, E, Mitro, N, et al. Docosahexaenoic acid is a beneficial replacement treatment for spinocerebellar ataxia 38. Ann Neurol. (2017) 82:615–21. doi: 10.1002/ana.25059
60. Zhao, H, Lewellen, BM, Wilson, RJ, Cui, D, Drake, JC, Zhang, M, et al. Long-term voluntary running prevents the onset of symptomatic Friedreich's ataxia in mice. Sci Rep. (2020) 10:6095. doi: 10.1038/s41598-020-62952-6
61. Vasquez-Trincado, C, Dunn, J, Han, JI, Hymms, B, Tamaroff, J, Patel, M, et al. Frataxin deficiency lowers lean mass and triggers the integrated stress response in skeletal muscle. JCI Insight. (2022) 7:5201. doi: 10.1172/jci.insight.155201
62. Mishra, RK, Nunes, AS, Enriquez, A, Profeta, VR, Wells, M, Lynch, DR, et al. At-home wearable-based monitoring predicts clinical measures and biological biomarkers of disease severity in Friedreich's Ataxia. Commun Med (Lond). (2024) 4:217. doi: 10.1038/s43856-024-00653-1
63. Rodriguez-Diaz, JC, Velazquez-Perez, L, Rodriguez Labrada, R, Aguilera Rodriguez, R, Laffita Perez, D, Canales Ochoa, N, et al. Neurorehabilitation therapy in spinocerebellar ataxia type 2: a 24-week, rater-blinded, randomized, controlled trial. Mov Disord. (2018) 33:1481–7. doi: 10.1002/mds.27437
64. Kulamarva, K, Chikkanna, U, Ramakrishna, KK, Bhargav, H, Velayutham, SG, and Varambally, S. Integrative approach improves fall risk and postural stability in spinocerebellar Ataxia-2 - a case report. Int J Yoga. (2022) 15:168–72. doi: 10.4103/ijoy.ijoy_49_22
65. Carr, J, Lalara, J, Lalara, G, Lalara, G, Daniels, B, Clough, A, et al. Feasibility and impact of a physical activity and lifestyle program for aboriginal families with Machado-Joseph disease in the top end of Australia. Rural Remote Health. (2024) 24:8376. doi: 10.22605/RRH8376
66. Bunn, LM, Marsden, JF, Giunti, P, and Day, BL. Training balance with opto-kinetic stimuli in the home: a randomized controlled feasibility study in people with pure cerebellar disease. Clin Rehabil. (2015) 29:143–53. doi: 10.1177/0269215514539336
67. Kim, SH, Han, JY, Song, MK, Choi, IS, and Park, HK. Effectiveness of robotic exoskeleton-assisted gait training in spinocerebellar Ataxia: a case report. Sensors (Basel). (2021) 21:4874. doi: 10.3390/s21144874
68. Tercero-Perez, K, Cortes, H, Torres-Ramos, Y, Rodriguez-Labrada, R, Cerecedo-Zapata, CM, Hernandez-Hernandez, O, et al. Effects of physical rehabilitation in patients with spinocerebellar Ataxia type 7. Cerebellum. (2019) 18:397–405. doi: 10.1007/s12311-019-1006-1
69. Yigit, S, Usgu, S, Albayrak, HM, Yucel, PP, and Yakut, Y. Effectiveness of functional trunk training on trunk control and upper limb functions in patients with autosomal recessive hereditary ataxia. NeuroRehabilitation. (2022) 51:41–50. doi: 10.3233/NRE-210320
70. Barbuto, S, Kuo, SH, Winterbottom, L, Lee, S, Stern, Y, O'Dell, M, et al. Home aerobic training for cerebellar degenerative diseases: a randomized controlled trial. Cerebellum. (2023) 22:272–81. doi: 10.1007/s12311-022-01394-4
71. Barbuto, S, Martelli, D, Omofuma, IB, Lee, N, Kuo, SH, Agrawal, S, et al. Phase I randomized single-blinded controlled study investigating the potential benefit of aerobic exercise in degenerative cerebellar disease. Clin Rehabil. (2020) 34:584–94. doi: 10.1177/0269215520905073
72. Winser, S, Chan, HK, Chen, WK, Hau, CY, Leung, SH, Leung, YH, et al. Effects of therapeutic exercise on disease severity, balance, and functional Independence among individuals with cerebellar ataxia: a systematic review with meta-analysis. Physiother Theory Pract. (2023) 39:1355–75. doi: 10.1080/09593985.2022.2037115
73. Jabri, S, Bushart, DD, Kinnaird, C, Bao, T, Bu, A, Shakkottai, VG, et al. Preliminary study of Vibrotactile feedback during home-based balance and coordination training in individuals with cerebellar Ataxia. Sensors (Basel). (2022) 22:3512. doi: 10.3390/s22093512
74. Cabanas-Valdes, R, Fernandez-Lago, H, Pelaez-Hervas, S, Serra-Rusinol, L, Lopez-de-Celis, C, and Masbernat-Almenara, M. Effect of a Home-Base Core stability exercises in hereditary Ataxia. A randomized controlled trial. A pilot randomized controlled trial. Mov Disord Clin Pract. (2024) 11:666–75. doi: 10.1002/mdc3.14036
75. Milne, SC, Corben, LA, Roberts, M, Szmulewicz, D, Burns, J, Grobler, AC, et al. Rehabilitation for ataxia study: protocol for a randomised controlled trial of an outpatient and supported home-based physiotherapy programme for people with hereditary cerebellar ataxia. BMJ Open. (2020) 10:e040230. doi: 10.1136/bmjopen-2020-040230
76. Milne, SC, Roberts, M, Williams, S, Chua, J, Grootendorst, AC, Agostinelli, G, et al. Goal-directed rehabilitation versus standard Care for Individuals with hereditary cerebellar Ataxia: a multicenter, single-blind, randomized controlled superiority trial. Ann Neurol. (2024) 97:409–24. doi: 10.1002/ana.27130
77. Chang, YJ, Chou, CC, Huang, WT, Lu, CS, Wong, AM, and Hsu, MJ. Cycling regimen induces spinal circuitry plasticity and improves leg muscle coordination in individuals with spinocerebellar ataxia. Arch Phys Med Rehabil. (2015) 96:1006–13. doi: 10.1016/j.apmr.2015.01.021
78. de Oliveira, LAS, Martins, CP, Horsczaruk, CHR, da Silva, DCL, Vasconcellos, LF, Lopes, AJ, et al. Partial body weight-supported treadmill training in spinocerebellar Ataxia. Rehabil Res Pract. (2018) 2018:1–8. doi: 10.1155/2018/7172686
79. Grobe-Einsler, M, Bork, F, Faikus, A, Hurlemann, R, and Kaut, O. Effects of cerebellar repetitive transcranial magnetic stimulation plus physiotherapy in spinocerebellar ataxias - a randomized clinical trial. CNS Neurosci Ther. (2024) 30:e14797. doi: 10.1111/cns.14797
80. Pohl, F, and Kong Thoo Lin, P. The potential use of plant natural products and plant extracts with antioxidant properties for the prevention/treatment of neurodegenerative diseases: in vitro, in vivo and clinical trials. Molecules. (2018) 23:3283. doi: 10.3390/molecules23123283
81. Zakharenko, AL, Luzina, OA, Chepanova, AA, Dyrkheeva, NS, Salakhutdinov, NF, and Lavrik, OI. Natural products and their derivatives as inhibitors of the DNA repair enzyme Tyrosyl-DNA phosphodiesterase 1. Int J Mol Sci. (2023) 24:5781. doi: 10.3390/ijms24065781
82. Ulatowski, L, Ghelfi, M, West, R, Atkinson, J, Finno, CJ, and Manor, D. The tocopherol transfer protein mediates vitamin E trafficking between cerebellar astrocytes and neurons. J Biol Chem. (2022) 298:101712. doi: 10.1016/j.jbc.2022.101712
83. Cory, H, Passarelli, S, Szeto, J, Tamez, M, and Mattei, J. The role of polyphenols in human health and food systems: a mini-review. Front Nutr. (2018) 5:87. doi: 10.3389/fnut.2018.00087
84. Loughrill, E, Govinden, P, and Zand, N. Vitamins a and E content of commercial infant foods in the UK: a cause for concern? Food Chem. (2016) 210:56–62. doi: 10.1016/j.foodchem.2016.04.014
85. Takahashi, M, Okazaki, H, Ohashi, K, Ogura, M, Ishibashi, S, Okazaki, S, et al. Current diagnosis and Management of Abetalipoproteinemia. J Atheroscler Thromb. (2021) 28:1009–19. doi: 10.5551/jat.RV17056
86. Leotti, VB, de Vries, JJ, Oliveira, CM, de Mattos, EP, Te Meerman, GJ, Brunt, ER, et al. CAG repeat size influences the progression rate of spinocerebellar Ataxia type 3. Ann Neurol. (2021) 89:66–73. doi: 10.1002/ana.25919
87. Carr, JJ, Lalara, J, Lalara, G, Lalara, G, Daniels, B, Clough, AR, et al. Staying strong toolbox: co-design of a physical activity and lifestyle program for aboriginal families with Machado-Joseph disease in the top end of Australia. PLoS One. (2021) 16:e0244311. doi: 10.1371/journal.pone.0244311
88. Santos de Oliveira, LA, Martins, CP, Horsczaruk, CH, Lima da Silva, DC, Martins, JV, Vasconcelos, LF, et al. Decreasing fall risk in spinocerebellar ataxia. J Phys Ther Sci. (2015) 27:1223–5. https://pmc.ncbi.nlm.nih.gov/articles/PMC4434015/
89. Winser, SJ, Kannan, P, Pang, M, Smith, C, and Tsang, WW. Potential benefits and safety of T'ai chi for balance and functional Independence in people with cerebellar Ataxia. J Altern Complement Med. (2018) 24:1221–3. doi: 10.1089/acm.2017.0396
90. Romano, A, Favetta, M, Summa, S, Schirinzi, T, Bertini, ES, Castelli, E, et al. Upper body physical rehabilitation for children with Ataxia through IMU-based Exergame. J Clin Med. (2022) 11:1065. doi: 10.3390/jcm11041065
Keywords: Ataxia telangiectasia, dietary changes, Friedreich Ataxia, hereditary ataxia, lifestyle interventions, nutraceuticals, spinocerebellar ataxia
Citation: Yang W, Thompson B and Kwa FAA (2025) Dietary and lifestyle interventions for the management of hereditary ataxias. Front. Nutr. 12:1548821. doi: 10.3389/fnut.2025.1548821
Edited by:
Jasmina D. Debeljak Martacic, University of Belgrade, SerbiaReviewed by:
Luis E. Almaguer-Mederos, Goethe University, GermanyCopyright © 2025 Yang, Thompson and Kwa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Faith A. A. Kwa, Zmt3YUBzd2luLmVkdS5hdQ==