1 Introduction
Parkinson’s disease (PD) is among the fastest-growing neurological disorders worldwide, posing a major public health challenge. As of 2021, an estimated 11.77 million individuals were affected globally, with age-standardized incidence and prevalence rates of 15.63 and 138.63 per 100,000 population, respectively. The disability-adjusted life years (DALYs) associated with PD in 2021 reached 7.47 million (95% UI: 6.74–8.14), reflecting a 161.80% increase since 1990. Over this period, the DALYs rose by 179.51% (95% UI: 157.02%–200.85%) for males and 141.37% (95% UI: 123.73%–160.53%) for females [1]. With an aging population, projections indicate that this number could exceed 12 million by 2040 [2]. This escalating disease burden underscores the critical need for comprehensive research into the molecular mechanisms driving PD pathogenesis to facilitate the development of effective therapeutic strategies.
One of the key pathological contributors to PD is blood-brain barrier (BBB) dysfunction, which compromises the brain’s ability to regulate the transport of molecules, thereby accelerating neurodegeneration. In PD, chronic inflammation, oxidative stress, and endothelial dysfunction disrupt BBB integrity, allowing toxic molecules to infiltrate the brain and exacerbate neuronal damage [3, 4]. Beyond its role in disease progression, BBB dysfunction also presents a major challenge for drug delivery, limiting the effectiveness of therapeutic agents. As BBB integrity declines, -synuclein (Syn) aggregation emerges as a central driver of PD pathology. Under normal conditions, Syn functions in synaptic transmission and neurotransmitter release. However, in PD, Syn misfolds and forms insoluble fibrillar aggregates, which accumulate in neurons, disrupt cellular processes, and trigger neuroinflammatory responses [5]. Understanding the mechanisms governing Syn aggregation and its pathological consequences is crucial for developing targeted therapeutic approaches aimed at mitigating its toxic effects. In this context, the presence of divalent metal ions such as and has been shown to significantly influence Syn fibrillation. These ions modulate fibrillation kinetics, polymorphic structure, and cytotoxicity of the resulting fibrils. Notably, ions accelerate aggregation while promoting the formation of shorter, structurally distinct, and less cytotoxic fibrils, as demonstrated by Atarod et al. [6] Moreover, fluctuations in ionic strength influence Syn misfolding, aggregation kinetics, and its interactions with cellular components, exacerbating neurodegenerative processes [7]. Advancing research in this field holds significant potential for improving PD treatments worldwide. The process of Syn aggregation is highly complex and influenced by multiple factors, including genetic mutations, environmental stressors, and post-translational modifications (PTMs) [8–10]. Several familial forms of PD have been linked to point mutations in the SNCA gene, which encodes Syn, such as A53T, A30P, and E46K [11–13]. These mutations accelerate the aggregation process and alter the biophysical properties of Syn, making it more prone to form toxic oligomers and fibrils [14, 15].
Recent studies have explored various strategies to destabilize Syn fibrils as a potential therapeutic approach for PD. Among these, small molecules capable of disrupting key inter-residue interactions within the fibril structure have shown promise. Yun et al. demonstrated that naphthoquinone-dopamine hybrids (NQDA) effectively destabilize Syn fibrils by targeting critical salt bridges (Glu46-Lys80, Lys45-Glu57) and weakening inter-protofibril interactions, thereby promoting fibril disassembly and reducing aggregation [16]. In parallel, computational and experimental studies have identified natural polyphenolic compounds as promising Syn destabilizers. Notably, ellagic acid, a naturally occurring polyphenol, has been shown to interfere with stabilizing interactions within Syn fibrils, particularly by disrupting the Glu46-Lys80 salt bridge and hydrophobic stacking of aromatic residues [17]. These perturbations lead to a reduction in -sheet content and an increase in structural disorder, ultimately impairing fibril stability. Recent research has also explored non-invasive approaches to disrupt Syn fibrils, with external electric fields emerging as a promising strategy [18]. However, as the peptide assembly progresses into trimers and tetramers, their structural stability increases, reducing the effectiveness of the applied electric field. These findings underscore the importance of early intervention, where applying a lower electric field during the early stages of aggregation may effectively disrupt fibril formation and prevent the accumulation of pathogenic Syn structures. Furthermore, MD simulations have demonstrated that electric fields not only induce substantial conformational changes in fibrils but also act on a threshold-dependent manner to disaggregate fibrils by disrupting their hydrophobic core. In particular, oscillating electric fields have been found to be effective in destabilizing polymorphic fibril structures, offering a novel perspective on targeted fibril disaggregation [19]. These findings reinforce the significance of targeting fibril-stabilizing electrostatic and hydrophobic interactions as a means to modulate Syn aggregation. Such an approach complements oxidative stress strategies aimed at destabilizing toxic Syn aggregates, offering a potential therapeutic avenue for PD treatment.
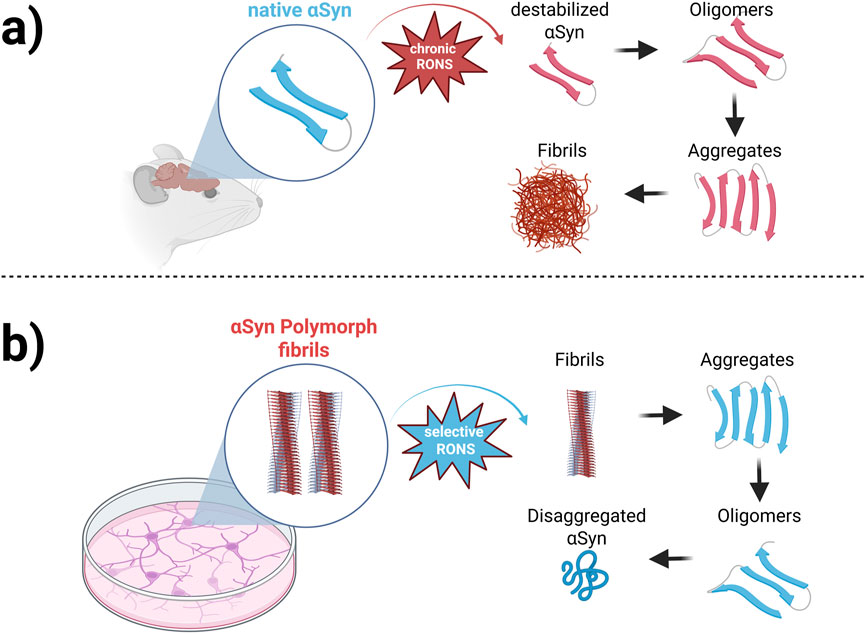
The oxidative stress, driven by excessive production of reactive oxygen and nitrogen species (RONS), is a well-established factor in neurodegenerative diseases [20, 21]. Among its many molecular targets, oxidative stress significantly influences Syn aggregation, altering both its structure and self-assembly behavior. RONS-mediated modifications, such as methionine oxidation, tyrosine nitration, and dityrosine formation, have been shown to impact Syn’s propensity to aggregate and form toxic species [22–24]. However, the effects of oxidation on aggregation are complex and may depend on the specific oxidative modifications and the stage of the aggregation process. Recent studies highlight the dual role of oxidative stress in Syn aggregation. On one hand, oxidative modifications can promote early-stage aggregation by destabilizing native monomeric conformations, making Syn more prone to self-association. In an in vivo study, Won et al. demonstrated that chronic oxidative stress accelerates Syn aggregation in transgenic mouse models, with aggregation being more pronounced in neurons experiencing oxidative damage [25]. This underscores the interplay between oxidative stress and Syn aggregation in idiopathic PD, reinforcing oxidative stress as a major driver of disease progression. On the other hand, oxidative stress may also impair fibril maturation, altering the structural stability of aggregates. Ponzini et al. examined oxidative modifications of Syn, particularly at methionine residues, using dopamine (DA) and epigallocatechin-3-gallate (EGCG) as oxidizing agents [26]. Their findings revealed that oxidized Syn exhibited reduced secondary structure formation, leading to a diminished ability to form stable fibrils. This suggests that oxidation may shift Syn aggregation dynamics toward disordered, less structured aggregates rather than well-defined fibrils.
Taken together, these studies illustrate that RONS-induced modifications influence Syn aggregation in a stage-dependent manner. Oxidation may accelerate early-stage aggregation and oligomer formation, but it can also interfere with fibril elongation and stability, potentially leading to the accumulation of disordered or toxic intermediates. This dual effect underscores oxidative stress as a key modulator of Syn aggregation, with profound implications for PD pathology. As illustrated in Figure 1, oxidative stress modulates Syn aggregation through distinct, stage-dependent mechanisms. Under chronic oxidative conditions, excessive RONS destabilize native Syn, triggering its conversion into misfolded oligomers and promoting fibril formation. Conversely, certain oxidative environments can disrupt mature Syn fibrils, leading to disaggregation into smaller species that may reassemble into aggregates or persist as toxic intermediates. These dynamic interactions emphasize the complex role of oxidative stress in shaping Syn aggregation pathways and structural polymorphism. A deeper understanding of these processes is essential for guiding the development of targeted therapies aimed at modulating oxidative damage or intervening in aggregation at specific stages of disease progression.
Examining the therapeutic potential of controlled oxidation in regulating Syn aggregation presents a promising avenue for addressing neurodegenerative diseases. Controlled oxidative modifications may provide a novel approach for modulating Syn aggregation dynamics. Computational modeling offers valuable insights into how different oxidative modifications influence the structure and aggregation propensity [27]. Such simulations reveal that oxidation at specific residues can have contrasting effects: while some modifications promote the formation of stable aggregates, others destabilize these structures, making them more prone to degradation. This duality underscores the complex role of oxidative stress in Syn polymorphic structure and highlights potential targets for therapeutic intervention. In this context, the primary objective of this study is to investigate the influence of oxidation on the stability of Syn polymorph fibrils, which serve as key intermediates in the aggregation process. Using MD simulations and umbrella sampling techniques, the study explores how varying degrees of oxidation affect the conformational dynamics and dissociation free energy of Syn monomers within fibrils. By providing a detailed molecular-level analysis of oxidation-induced changes in Syn structure and stability, this research aims to advance our understanding of therapeutic strategies to prevent or reverse Syn aggregation. The findings are expected to offer significant insights into the development of therapeutic strategies for neurodegenerative diseases associated with synucleinopathies. The manuscript is organized as follows: Section 2 describes the numerical setup and methodology, including model preparation, force field selection, and simulation parameters. Section 3 presents the results and discussion, encompassing RMSD analysis, secondary structure content, solvent-accessible surface area (SASA), hydrogen bonding, principal component analysis (PCA), and free energy landscape (FEL) comparisons. Finally, Section 4 provides the main conclusions and discusses the implications of oxidation-induced destabilization in Syn fibrils, particularly as it relates to aggregation pathways and potential therapeutic strategies.
2 Numerical setup and methods
MD simulations were carried out using the GROMACS 2023.1 software package [28]. The solid-state NMR structure of the Syn fibril was obtained from the Protein Data Bank (PDB ID: 6H6B), a patient-derived Lewy body fibril, due to its physiological relevance to PD pathology [29]. This structure is stabilized by inter-chain salt bridges (Glu46-Lys80, Lys45-Glu57) and hydrophobic interactions (Phe94 stacking), which play a crucial role in fibril stability. Unlike other reported Syn fibrils, such as 2N0A (recombinant fibrils, see Ref. 30) or 6XYO (alternative polymorphs, see [30], 6H6B represents an in vivo-derived fibril conformation, making it particularly suitable for investigating oxidation-induced destabilization. Moreover, to maintain computational feasibility while preserving key structural interactions, we employed a five-strand fibril model. While larger fibril models (e.g., 10-strand fibrils) may better approximate extended fibril stability, the five-strand system retains essential inter-molecular interactions necessary for studying oxidation-induced effects. Furthermore, the flexibility of this model remains constrained by the core stabilizing interactions, including salt bridges and hydrophobic stacking. However, we acknowledge that extending this approach to larger fibril models-focusing on the internal core to minimize edge effects-could provide additional insights into the impact of oxidative modifications.
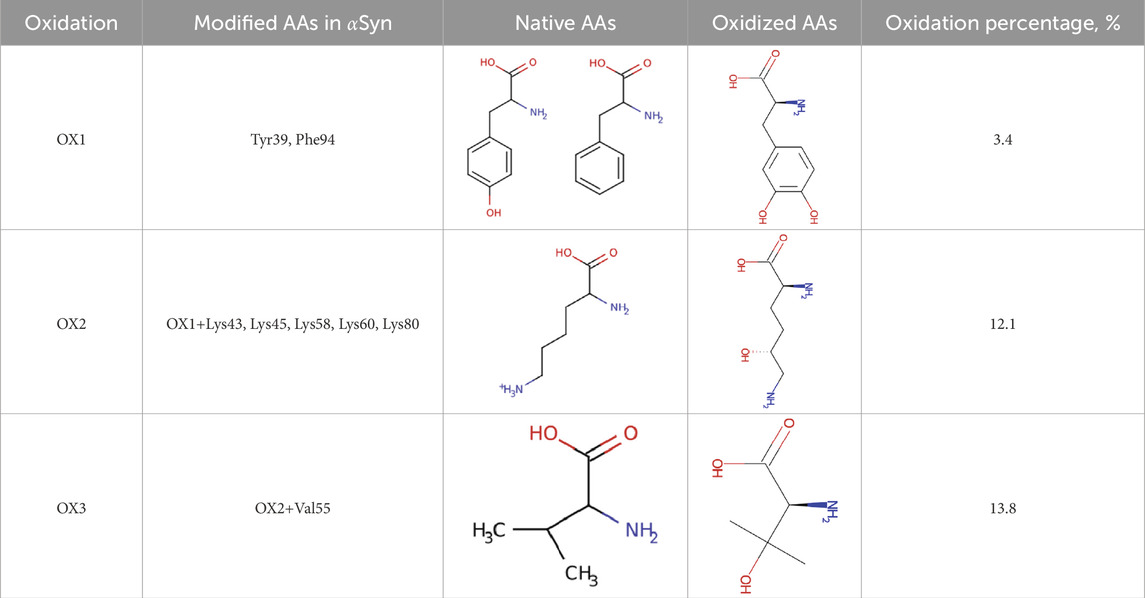
Alongside the native Syn structure, three oxidized models (OX1, OX2, and OX3) were created, featuring oxidation levels of 3.4%, 12.1%, and 13.8%, respectively (see Table 1). These oxidation levels were chosen to reflect progressive oxidative stress conditions, ranging from mild oxidative damage (early-stage PD) to extensive oxidation seen in late-stage neurodegeneration.
The oxidative modifications in our model were selected based on experimental studies showing high reactivity of specific residues under oxidative stress conditions [31–34]. To ensure simulation accuracy, the oxidized variants were generated using CHARMM-GUI [35], which provided force field parameters compatible with CHARMM36 m–a force field specifically optimized for modeling intrinsically disordered proteins (IDPs) like Syn [36]. The CHARMM36 m force field was employed throughout the simulations to accurately capture the conformational flexibility of Syn and the structural effects of oxidation, ensuring a reliable representation of fibril dynamics under oxidative conditions.
• OX1 (3.4%) includes modifications at Tyr39 and Phe94 residues of Syn to 3,4-dihydroxyphenylalanine. Experimental studies have demonstrated that Tyr39 plays a crucial role in the oxidative aggregation of Syn. Ruf et al. [31] showed that when Syn adopts a collapsed conformation, Tyr39 is essential for wild-type-like covalent aggregation. The study found that removing Tyr39 significantly altered the aggregation pattern, emphasizing its importance in the protein’s oxidative behavior. Phe94 is located within the hydrophobic core of Syn, a region critical for fibril formation. Modifications at this site can influence the protein’s folding and aggregation propensity [32]. Although direct experimental evidence of Phe94 oxidation in Syn is limited, its inclusion in our model aims to explore potential structural impacts resulting from oxidative modifications at this position.
•OX2 (12.1%) encompasses the modifications present in OX1 – specifically, Tyr39 and Phe94 – and introduces additional oxidative modifications at Lys43, Lys45, Lys58, Lys60, and Lys80 of Syn to 5-hydroxylysine. The oxidative modification of lysine residues, such as those at positions 43, 45, 58, 60, and 80, can alter the charge and conformation of Syn, potentially influencing its aggregation propensity. By incorporating these specific lysine modifications in the OX2 model, our study aims to elucidate how such oxidative changes affect Syn’s stability and aggregation behavior.
•OX3 (13.8%) encompasses the modifications present in OX2 – specifically, Tyr39, Phe94, and Lys43, Lys45, Lys58, Lys60, and Lys80 – and introduces an additional oxidative modification at Val55 of Syn to 3-hydroxy-L-valine. Valine is a hydrophobic amino acid located within the non-amyloid- component (NAC) region of Syn, which is critical for the protein’s aggregation and fibril formation. These modifications can alter the hydrophobicity and structural conformation of Syn, potentially influencing its aggregation propensity. Although direct experimental evidence of Val55 oxidation in Syn is limited, the strategic location of Val55 within the NAC region suggests that its modification could impact the stability and assembly of Syn fibrils. By incorporating the oxidation of Val55 in the OX3 model, our study aims to explore how such modifications affect Syn’s structural integrity and aggregation behavior.
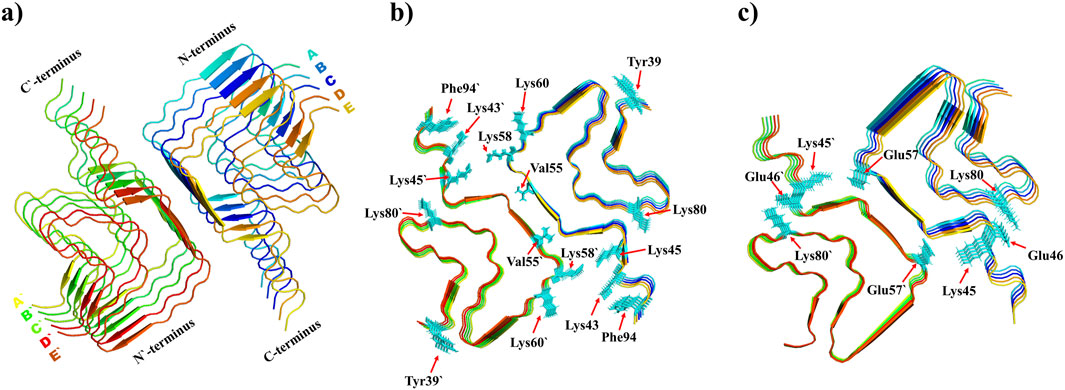
Figure 2 presents a detailed visualization of the Syn polymorph fibril structure, highlighting its key features and molecular interactions. The illustration captures both the intrinsic structural organization and the molecular alterations resulting from oxidation, offering valuable insights into the fibril’s stability and intermolecular dynamics:
Structural overview (see Figure 2a): The polymorph fibril structure is shown as a cartoon representation, where each chain is uniquely color-coded. The C-terminal and N-terminal ends are clearly labeled, providing spatial context for the protein chains.
Oxidation sites (see Figure 2b): Specific amino acid residues subjected to oxidation (Tyr39, Phe94, Lys43, Lys45, Lys58, Lys60, Lys80, and Val55) are displayed in licorice style. These residues, oxidized across all chains (totaling ten instances per residue), are highlighted to show their modification locations within the fibril.
Salt bridge interaction (see Figure 2c): Key residues, Glu46 and Lys80, involved in forming salt bridges within each chain are depicted. These interactions play a significant role in maintaining fibril stability, and their depiction highlights the structural dynamics of the fibril under different conditions.
The four distinct model systems (native, OX1, OX2, and OX3) were placed in a rectangular simulation box, maintaining a minimum distance of 1.0 nm between any atom of the protein and the box boundaries. The box was solvated using the TIP3P water model, and 0.1 M of NaCl was added to mimic physiological conditions [37]. Subsequently, the system underwent a 500 ps equilibration run under the NVT ensemble (constant number of particles, volume, and temperature) with position restraints applied to the heavy atoms of the Syn structure. These steps, including energy minimization and equilibration, were automatically performed following the standard protocol implemented in CHARMM-GUI.
After equilibration, a 600 ns production run was conducted using the NPT ensemble (constant number of particles, pressure, and temperature) without restraints. The simulations were carried out at 300 K and 1 bar, using the V-rescale thermostat with a coupling constant of 1.0 ps and the isotropic C-rescale barostat with a compressibility of and a coupling constant of 5.0 ps. A cut-off distance of 1.2 nm was applied for van der Waals interactions, and long-range electrostatic interactions were handled using the particle mesh Ewald (PME) method.
The production run trajectory was used for data collection, including calculations of the root mean square deviation (RMSD). For assessing secondary structural changes in the Syn fibrils, we used the secondary structure assignment program STRIDE [38], averaging data from the MD trajectory. Visualization and image preparation were done using the PyMol tool [39].
The Free Energy Landscape (FEL) was generated to assess the stability and conformational variability of the fibril structures. FEL calculations were based on the first two principal components (PC1 and PC2), using only the atoms of the protein to reduce noise and highlight collective motions. The FEL was obtained using the gmx sham utility in GROMACS, which calculates the free energy as a function of principal components, allowing identification of stable and metastable states within the simulation.
Further, we utilized the umbrella sampling (US) technique to investigate the interactions between monomers within the Syn fibril structure. This approach enabled us to calculate the potential of mean force (PMF) and estimate the dissociation free energy of a monomer as oxidative stress levels increased. By analyzing these results, we aimed to uncover the molecular mechanisms underlying Syn polymorph fibril destabilization due to oxidative modifications, providing critical insights into the role of oxidative stress in protein aggregation and its implications for neurodegenerative disorders.
3 Results and discussion
3.1 RMSD and conformational changes
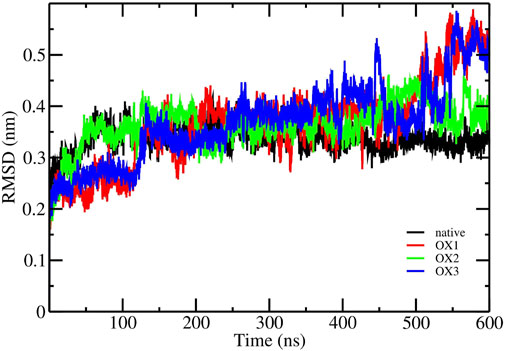
The RMSD analysis was performed to assess the structural stability of the Syn fibrils in its native and oxidized states (OX1, OX2, OX3) over the 600 ns production run. As shown in Figure 3, the RMSD values for the native fibrils, calculated over the last 100 ns trajectories of the simulation, stabilized around nm, indicating a relatively stable structure. In contrast, the oxidized systems exhibited higher RMSD values during the same timeframe, with OX1, OX2, and OX3 showing increased deviations of approximately nm, nm, and nm, respectively. This trend suggests that higher degrees of oxidation lead to greater structural instability in the Syn fibrils. The oxidation of amino acids introduces changes that increase the flexibility of the protein structure. To illustrate, hydrophobic amino acids undergo a conversion into more polar forms, thereby inducing their hydrophilic/solvent environment tendencies and resulting in increased flexibility. The enhanced flexibility of the oxidized protein allows it to explore a greater conformational space during simulations, resulting in elevated RMSD values.
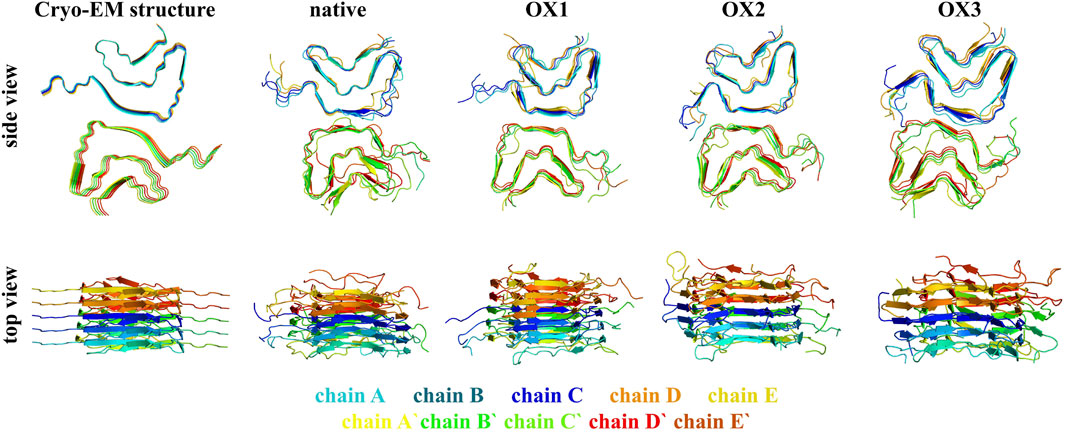
Figure 4 illustrates the structural conformations of native and oxidized Syn fibrils (OX1, OX2, and OX3) based on the final snapshots obtained from 600 ns MD simulation trajectories. Side and top views are presented, highlighting the conformational changes induced by oxidation compared to the native fibril structure. These visualizations provide critical insights into the structural dynamics and stability variations across different oxidation states.
3.2 Secondary structure content
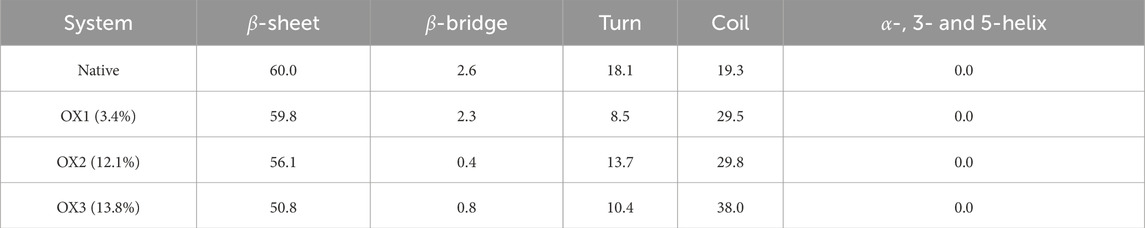
The secondary structure content of Syn fibrils was analyzed using the STRIDE program by examining frames from each model system. The average secondary structure distribution across the native and oxidized systems is summarized in Table 2. The native fibrils maintained a stable -sheet architecture, which is characteristic of amyloid fibrils and critical for their structural integrity. In contrast, the oxidized fibrils displayed a progressive reduction in -sheet content, accompanied by an increase in random coil structures with higher oxidation levels. This trend highlights the destabilizing effect of oxidation, leading to a shift from a more ordered to a less ordered and more flexible conformation. The reduction in -sheet stability under oxidative conditions may stem from the disruption of stabilizing interactions, such as salt bridges and hydrophobic packing. As observed, oxidation of key residues—particularly hydrophobic and charged residues—likely promotes solvent exposure and conformational rearrangements that destabilize the fibril core. Additionally, oxidation-induced changes to critical salt bridges (e.g., Glu46-Lys80 and Lys45-Glu57) and the glutamine ladder (Gln79) contribute to the observed structural changes, further enhancing fibrillar destabilization. These changes in secondary structure align with the higher deviations from the initial fibril conformation, as reflected by the elevated RMSD values across oxidized systems. Together, the reduction in -sheet content, increase in random coils, and enhanced RMSD indicate that oxidation promotes a more dynamic and less stable fibril structure.
3.3 -bond and solvent-accessible surface area (SASA) analysis
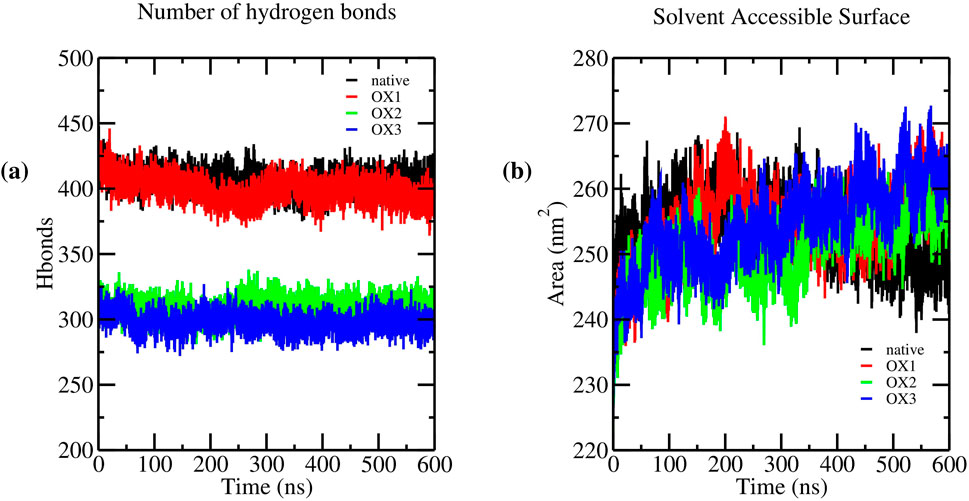
Hydrogen bonding plays a crucial role in stabilizing Syn fibrils, influencing their structural integrity and aggregation behavior. To assess the impact of oxidation on hydrogen bonding, we analyzed the average number of hydrogen bonds in the native and oxidized systems (OX1, OX2, and OX3) over the final 100 ns of molecular dynamics simulations. The results indicate a progressive decline in hydrogen bonding with increasing oxidation levels. The native fibrils maintained an average of hydrogen bonds, whereas the oxidized systems exhibited a gradual reduction: OX1 , OX2 , and OX3 . This decrease suggests that oxidation disrupts inter-peptide interactions, contributing to fibril destabilization. Figure 5a illustrates the number of hydrogen bonds in native and oxidized Syn fibril systems throughout the simulation. The progressive reduction in hydrogen bonding, particularly in OX2 and OX3, highlights the structural destabilization induced by oxidation. Further analysis of hydrogen bond lifetimes and donor-acceptor distributions may provide deeper insights into how oxidation alters fibril stability at the molecular level.
The SASA results, presented in Figure 5b, demonstrate the progressive destabilization of the fibrillar structure with increasing oxidation levels. Throughout the simulation, the native system consistently maintained lower SASA values, reflecting a compact and well-ordered fibril structure. In contrast, oxidized systems displayed higher SASA values that increased with oxidation levels.
The average SASA values calculated over the final 100 ns of the simulation were as follows: native , OX1 , OX2 , and OX3 . This trend indicates enhanced solvent exposure and increased conformational flexibility with oxidation. The observed SASA increase correlates with the degree of oxidation, suggesting that oxidation disrupts fibrillar packing by exposing hydrophobic residues to the solvent and promoting structural instability. These findings are consistent with previous observations of oxidation-induced destabilization in amyloid fibrils.
3.4 Free energy landscape (FEL) analysis
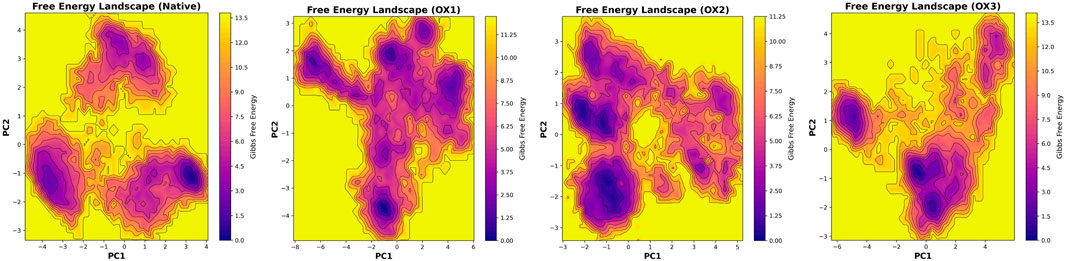
To analyze the influence of oxidation on the conformational ensemble of Syn fibrils, the free energy landscape (FEL) was constructed by plotting the free energy (kcal/mol) as a function of the first two principal components (PC1 and PC2). These principal components correspond to the largest collective motions observed in the fibril structures, providing insights into the stability and conformational flexibility of both the native and oxidized systems (Figure 6). The native structure exhibits well-defined, deep energy basins, indicating the presence of stable conformations. The lowest-energy regions are concentrated within a restricted conformational space, suggesting that the native fibril adopts a highly stable and well-ordered configuration. Upon oxidation, significant alterations in the FEL were observed:
• OX1: The energy landscape undergoes a substantial shift, with the primary deep basin relocating to a different region of the conformational space. This suggests that oxidation induces major structural rearrangements, increasing flexibility. The overall energy distribution appears more dispersed, indicating a loss of well-defined stable states.
•OX2: The energy minima are more widely distributed compared to both the native and OX1 structures, suggesting increased conformational sampling and reduced stability. The shallower energy basins reinforce the notion that oxidation disrupts the native structural integrity, enhancing flexibility.
•OX3: While oxidation alters the energy landscape, the basins in OX3 remain more defined than in OX1 and OX2, suggesting it retains a degree of structural stability. However, the presence of a large high-energy region indicates greater accessibility to higher-energy conformations, which may impact functional dynamics.
3.5 Umbrella sampling and potential of mean force (PMF)
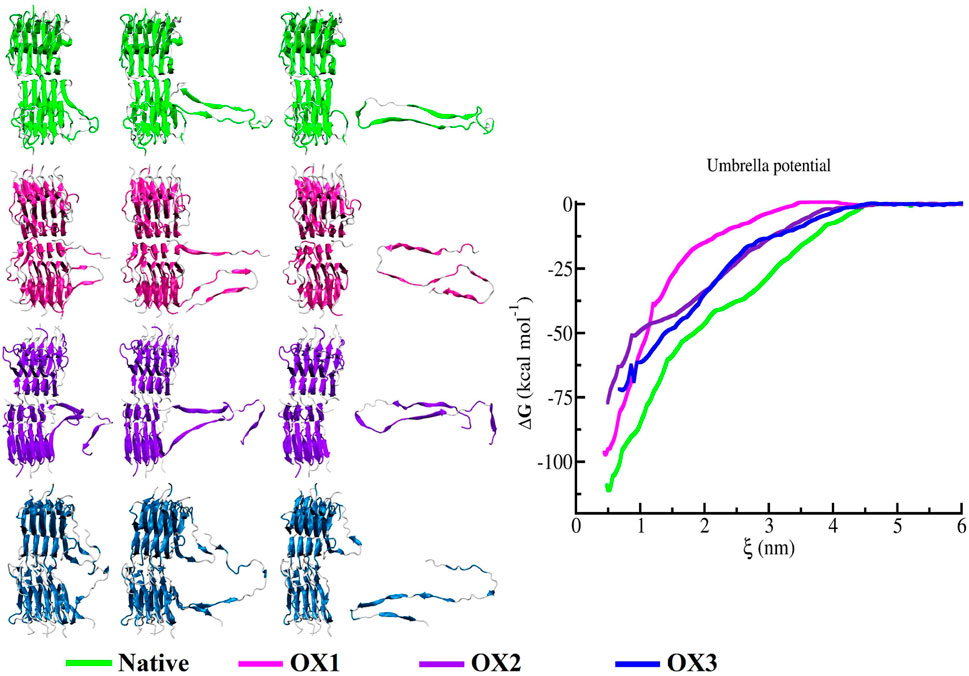
Umbrella sampling was employed to estimate the interaction energies between the monomers of the Syn fibrils. The PMF profiles reveal that the binding free energy of the terminal peptide in the native fibrils is significantly higher than in the oxidized systems. In Figure 7, the snapshots of the pull trajectories of the Syn peptide demonstrate notable conformational alterations throughout the pulling process, as observed across four different model systems (Native, OX1, OX2, and OX3). The PMF profiles exhibit variations, which are represented by green, magenta, violet and blue lines. Snapshots at different moments illustrate significant differences in the conformation of the subunit being pulled away from the protein core, indicating diverse responses during the simulations. Specifically, the dissociation free energy for the native, OX1, OX2, and OX3 systems were registered to be kcal/mol, kcal/mol, kcal/mol and kcal/mol, respectively. This decrease in dissociation free energy with increasing oxidation indicates that oxidation weakens the interactions between the monomers, making the fibrils less stable and more prone to dissociation.
In this study, the free energy profiles derived from umbrella sampling simulations form the basis for defining binding and unbinding events between Syn subunits. We interpret the depth and shape of the PMF wells as indicators of interaction stability rather than imposing a fixed energetic threshold such as the thermal energy scale . This approach is conceptually aligned with criteria used in protein adsorption studies, where critical binding conditions are inferred from system-specific energetics and conformational behavior. For instance, de Carvalho et al. [40] and Caetano et al. [41] employed Monte Carlo simulations to study protein and polyelectrolyte adsorption, defining adsorption transitions through conformational and electrostatic parameters. Their findings emphasize the influence of system geometry, electrostatic complementarity, and solvent conditions on binding phenomena–factors that are equally relevant in the context of Syn fibril destabilization under oxidative stress.
3.6 Discussion
The structural stability of Syn fibrils is maintained by several pivotal interactions, including salt bridges, glutamine ladders, and hydrophobic packing. Among these, the Glu46-Lys80 salt bridges play a crucial role in preserving the characteristic Greek-key topology of the fibril [16]. Additionally, a glutamine ladder involving Gln79 and the hydrophobic packing of aromatic residues, such as Phe94, contribute significantly to fibril integrity, as highlighted by previous studies [42].
Our analysis suggests that oxidative modifications substantially disrupt these critical stabilizing interactions. Oxidation of lysine residues, such as Lys80 and Lys57, removes their positive charge, leading to the weakening or complete disruption of salt bridges with glutamic acid residues (Glu46 and Glu45). The loss of these salt bridges compromises the overall fibril architecture, including the preservation of the Greek-key topology, thereby increasing conformational flexibility and promoting partial unfolding of the fibril structure [43].
The hydrophobic packing of aromatic residues, such as Phe94, is particularly susceptible to oxidation. Oxidative modifications increase the polarity of aromatic residues, enhancing solvent exposure and reducing hydrophobic interactions. This disruption in hydrophobic packing is consistent with the observed increase in solvent-accessible surface area (SASA) and the reduction in -sheet content, both of which contribute to the destabilization of the fibril core [44].
Collectively, these findings underscore the profound impact of oxidative modifications on the molecular interactions that stabilize Syn fibrils. The disruption of salt bridges, glutamine ladders, and hydrophobic packing mechanisms likely drives the observed conformational flexibility, structural destabilization, and fibril destabilization under oxidative conditions. These insights highlight the importance of oxidation-induced alterations in modulating fibril stability and may provide a mechanistic basis for understanding the role of oxidative stress in neurodegenerative diseases like PD.
The destabilization of Syn fibrils upon oxidation is influenced by the specific chemical nature of the oxidized residues. Different types of amino acids exhibit distinct roles in maintaining fibrillar integrity, and oxidation-induced modifications can alter these roles in various ways: 1) Tyrosine (e.g., Tyr39): is an amphipathic amino acid with a hydroxyl group capable of hydrogen bonding and a hydrophobic aromatic ring that participates in fibril packing interactions. Oxidation of tyrosine introduces additional polarity, enhancing solvent exposure and promoting hydrophilic interactions. 2) Similarly, the oxidation of phenylalanine (e.g., Phe94). disrupts the aromatic stacking interactions that are crucial for fibril stabilization. These modifications weaken the structural core of the fibrils. 3) Lysine oxidation, as observed in residues Lys43, Lys45, Lys58, Lys60, and Lys80, significantly impacts fibril stability in multiple ways. Native lysine residues carry a positively charged side chain that is crucial for electrostatic interactions and salt bridges. Upon oxidation, the loss of the positive charge results in an increase in the overall negative charge of Syn fibrils. This charge shift is expected to alter the electrostatic balance within the fibril and weaken key stabilizing interactions. Notably, oxidizing Lys60 disrupts a known stabilizing salt bridge present in the native structure, which could further contribute to fibril destabilization. 4) Valine (e.g., Val55). is a highly hydrophobic amino acid that contributes to the hydrophobic core of the fibril. Oxidation increases the polarity of valine, reducing its hydrophobic effect and leading to a loss of compact packing in the fibril structure. This change enhanced solvent exposure and promoted further destabilization of the fibril.
The structural evidence for destabilization is supported by secondary structure analysis (Table 2), where oxidation reduces the content of -sheets–critical for aggregate stability–while increasing the proportion of random coils, indicating a shift towards less ordered conformations. The loss of -sheet content and the increase in random coil structures further suggest that oxidation promotes fibril loosening and conformational heterogeneity.
Overall, the oxidation of hydrophobic residues primarily promotes increased solvent exposure and hydrophilic interactions, while lysine oxidation alters the fibril’s electrostatic landscape. Both types of modifications work synergistically to destabilize the fibril, favoring partial unfolding or dissociation of fibril subunits. These observations align with our simulation results, where oxidized systems (OX1, OX2, OX3) exhibit increased solvent-accessible surface areas (SASA) and reduced hydrogen bonding compared to the native fibril structure. Further investigations into the specific interactions lost upon oxidation, particularly those involving stabilizing salt bridges and aromatic stacking, are warranted to fully understand how these modifications affect the aggregation behavior and toxicity of Syn fibrils.
The FEL analysis reveals (Figure 6) that oxidation affects both the stability and conformational flexibility of the structure. The native structure maintains distinct deep minima, indicating well-defined stable states. In contrast, the oxidized forms show broader and shallower energy basins, demonstrating increased flexibility and reduced stability. This finding is consistent with RMSD trends and indicates that oxidation increases structural heterogeneity and reduces overall stability. Moreover, a lower -sheet content was observed in the conformations extracted from oxidized Syn fibrils, with values of 59.8% (OX1), 56.1% (OX2), and 50.8% (OX3), compared to the native Syn fibril (60%). This reduction in -sheet content suggests that oxidation disrupts the fibrillar structure, leading to increased conformational flexibility. These observations highlight the potential impact of oxidation on the functional properties of the system, possibly affecting its biological activity and interactions.
Umbrella sampling and PMF profiles (Figure 7) highlight the thermodynamic impact of oxidation. The reduced dissociation free energy of oxidized fibrils ( to kcal/mol compared to kcal/mol for the native fibrils) underscores their weakened monomer interactions. This destabilization suggests that oxidation enhances fibril disassembly tendencies, potentially affecting aggregation dynamics and amyloid propagation.
Oxidative modifications hold critical importance in neurodegenerative diseases, such as PD, where Syn aggregation plays a central role. In our case, we observed that oxidation destabilizes the polymorphic structure of Syn, underscoring the need for a delicate balance to minimize the formation of toxic oligomers while ensuring proper disaggregation. To validate this hypothesis, further experimental investigations are required.
While our simulations provide valuable insights into the oxidative modifications and structural consequences for Syn fibrils, it is important to note that the fibril models used represent a simplified version of the full-length aggregates observed in vivo. Larger fibrils, such as those found in Lewy bodies, may display more complex polymorphic features, additional -sheet stacking, and extensive lateral associations. These features could alter the accessibility of oxidizable residues, impact solvent exposure, and modulate the dynamics of aggregation or disaggregation. Furthermore, the interplay between fibril surface properties and oxidative agents might differ with increased fibril dimensions, potentially influencing how ROS interact with the fibrillar structure. Future studies employing coarse-grained models or multiscale simulations could help bridge this gap and provide a more comprehensive understanding of fibril behavior under physiological conditions.
3.6.1 Comparison with previous studies and novel contributions
Our findings align with and extend several prior studies investigating the influence of oxidation and small molecules on Syn fibril stability. Consistent with the work by Razzokov et al., [27] we observe that oxidative modifications reduce fibril stability, as evidenced by increased RMSD values and reduced PMF well depths. However, our study advances these findings by providing a detailed residue-level interpretation of how oxidation of specific amino acids (Tyr39, Phe94, Lys43, Lys45, Lys58, Lys60, Lys80, and Val55) differentially disrupts hydrophobic and electrostatic interactions.
In contrast to Ponzini et al., [26] who highlighted the role of methionine oxidation in altering fibril morphology, our models - constructed without methionine residues - demonstrate that oxidation of lysine and phenylalanine residues can also induce similar destabilizing effects, thus broadening the understanding of oxidative targets in Syn. Moreover, while Yun et al. [16] and Mankoo et al. [17] investigated the influence of small molecules on disrupting salt bridges like Glu46-Lys80, our PMF and FEL analyses provide new evidence that oxidation alone can similarly impair these critical stabilizing interactions.
Importantly, our simulations are among the first to combine umbrella sampling with principal component-based free energy landscapes to comprehensively capture oxidation-induced conformational shifts in Syn fibrils. This multiscale insight into both thermodynamic and dynamic consequences of oxidation is novel and complements prior purely experimental or monomer-based simulation studies.
Taken together, our results not only validate prior hypotheses but also contribute new computational evidence highlighting the residue-specific mechanisms of oxidative destabilization in Syn fibrils. This comparative synthesis underscores the robustness of our model and its relevance to the broader Syn aggregation literature.
3.6.2 Experimental relevance and comparability
The simulation outcomes presented in this study are supported by and comparable to available experimental data. For example, the observed reduction in -sheet content upon oxidation is consistent with circular dichroism (CD) and solid-state NMR studies, which have reported secondary structure loss in oxidatively modified Syn fibrils [26]. The calculated RMSD and SASA values provide insight into structural stability and solvent exposure, paralleling data from protease accessibility assays and hydrophobicity measurements. Furthermore, the potential of mean force (PMF) profiles generated from umbrella sampling simulations mirror trends seen in calorimetric binding studies such as isothermal titration calorimetry (ITC), offering thermodynamic perspectives on inter-chain dissociation. The disruption of hydrogen bonds and salt bridges noted in our models also aligns with structural observations from high-resolution cryo-EM and NMR structures of Syn polymorphs. These correlations enhance the biological relevance of our simulations and support the interpretation of oxidative effects on fibril stability and aggregation.
4 Conclusion
This study provides a comprehensive molecular-level understanding of how oxidative modifications influence the structural, dynamic, and thermodynamic properties of Syn fibrils, using atomistic MD simulations. Our findings reveal that progressive oxidation significantly compromises fibril stability, alters aggregation dynamics, and induces conformational changes consistent with pathological features observed in PD. Oxidation leads to pronounced structural perturbations, including increased solvent exposure, disrupted hydrogen bonding, and reduced -sheet content. These effects collectively destabilize the fibrillar architecture. Quantitative indicators - elevated RMSD values, broader distributions in FEL, and increased SASA - demonstrate the loss of structural order in oxidized models (OX1, OX2, and OX3) relative to the native fibril. Importantly, this destabilization arises through distinct physicochemical mechanisms that act on different interaction types within the fibril. Oxidation of hydrophobic residues, such as phenylalanine and valine, introduces polarity that weakens core packing and enhances water accessibility - mechanisms reflected in SASA and FEL metrics. Conversely, oxidation of lysine residues neutralizes their positive charge, disrupting key electrostatic interactions such as the Glu46-Lys80 and Lys45-Glu57 salt bridges. These changes reduce inter-subunit cohesion, evidenced by decreased binding free energies and shallower PMF wells.
These two types of interactions - hydrophobic and electrostatic - respond differently to environmental variables. While hydrophobic interactions are generally robust to changes in ionic strength, they are sensitive to oxidation-induced shifts in residue polarity. Electrostatic interactions, on the other hand, are strongly influenced by both oxidation and the dielectric environment, including local salt concentrations. This dual-pathway mechanism of oxidative destabilization underscores the importance of considering both interaction types when designing therapeutic interventions targeting Syn aggregation.
To further build on these findings, future work should extend simulations to larger and more physiologically representative fibril models, potentially incorporating interactions with lipid membranes or molecular chaperones. Multiscale or coarse-grained modeling approaches could capture broader aggregation-disaggregation dynamics, particularly the behavior of toxic oligomeric intermediates. Experimental validation - through structural, biophysical, or cellular assays - will be essential to confirm the therapeutic implications of oxidative modulation of Syn fibrils. Ultimately, this research lays the groundwork for the rational design of oxidation-guided strategies aimed at mitigating fibril-associated toxicity in PD and related synucleinopathies.
Beyond the molecular-level insights into oxidative destabilization mechanisms, it is essential to consider the physiological environment in which Syn aggregation occurs, particularly in aging populations where PD is most prevalent. Aging is associated with a decline in intracellular hydration and an increase in macromolecular crowding (MMC) - a phenomenon that arises from high concentrations of macromolecules in the cellular milieu. MMC has been shown to promote the formation of compact protein-protein complexes by enhancing effective concentrations and reducing conformational entropy costs. These conditions can significantly modulate the relative contributions of hydrophobic and electrostatic interactions to fibril formation. In dehydrated, crowded environments, hydrophobic effects may become more pronounced due to diminished solvent shielding, favoring tighter core packing in protein aggregates. Conversely, electrostatic interactions - particularly salt bridges - may be altered or weakened by shifts in local dielectric properties or ionic strength. Under such circumstances, the oxidative modifications modeled in this study could exert amplified effects. For example, oxidation-induced polarity changes in hydrophobic residues may further disrupt core packing, while the neutralization of positively charged lysine residues could exacerbate the destabilization of key salt bridges.
These results highlight the potential of targeted oxidative modifications, particularly at residues such as tyrosine, lysine, and valine - as a strategy to destabilize Syn fibrils and possibly initiate their disassembly. While the current study focuses on the biophysical and molecular dynamics aspects, the implications of this mechanism extend into therapeutic exploration. Notably, certain naturally derived compounds from traditional Chinese and Arabic medicine such as epigallocatechin gallate (EGCG), ellagic acid, curcumin, and resveratrol have demonstrated inhibitory effects on Syn aggregation or promoted fibril remodeling into less toxic conformations [45–47]. These polyphenolic compounds, widely present in green tea, pomegranate, turmeric, and grapes, exert their actions through redox activity, hydrogen bonding, and hydrophobic interactions with amyloidogenic regions of Syn. Additionally, herbal extracts used in Unani and Ayurvedic medicine have been reported to offer neuroprotective benefits via antioxidant and anti-inflammatory pathways [48, 49]. Our findings therefore offer not only molecular insight into fibril destabilization but also a conceptual foundation for the development of prophylactic or therapeutic strategies aimed at modulating Syn aggregation in neurodegenerative diseases.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
TA: Data curation, Formal Analysis, Methodology, Software, Writing – original draft, Writing – review and editing. PM: Investigation, Supervision, Writing – review and editing. MM: Formal Analysis, Investigation, Methodology, Writing – original draft. AS: Investigation, Methodology, Supervision, Writing – review and editing. RM: Data curation, Formal Analysis, Methodology, Visualization, Writing – original draft. JR: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The authors gratefully acknowledge the financial support from the Innovative Development Agency under the Ministry of Higher Education, Science and Innovations of the Republic of Uzbekistan (grant number AL-4821012320). PM gratefully acknowledges the use of the bioinformatics infrastructure facility supported by Biocenter Finland, Joe, Pentti and Tor Borg Memorial Fund 2021, Juhani Ahon Lääketieteen Tutkimussäätiö sr. for their grant support, CSC-IT Center for Science (Project:2000461) for the computational facility; Dr. Jukka Lehtonen (SBL) for the IT support; and Prof. Outi Salo-Ahen (Pharmacy), Åbo Akademi University, for providing the lab support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. We gratefully acknowledge the assistance of ChatGPT, an advanced AI language model developed by OpenAI, for its valuable support in refining and improving the language and readability of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Luo Y, Qiao L, Li M, Wen X, Zhang W, Li X. Global, regional, national epidemiology and trends of Parkinson’s disease from 1990 to 2021: findings from the Global Burden of Disease Study 2021. Front Aging Neurosci (2025) 16:1498756. doi:10.3389/fnagi.2024.1498756
PubMed Abstract | CrossRef Full Text | Google Scholar
2. Ben-Shlomo Y, Darweesh S, Llibre-Guerra J, Marras C, San Luciano M, Tanner C. The epidemiology of Parkinson’s disease. The Lancet (2024) 403(Issue 10423):283–92. doi:10.1016/S0140-6736(23)01419-8
PubMed Abstract | CrossRef Full Text | Google Scholar
3. Sweeney MD, Sagare AP, Zlokovic BV. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol (2018) 14(3):133–50. doi:10.1038/nrneurol.2017.188
PubMed Abstract | CrossRef Full Text | Google Scholar
4. Upton DH, Ung C, George SM, Tsoli M, Kavallaris M, Ziegler DS. Challenges and opportunities to penetrate the blood-brain barrier for brain cancer therapy. Theranostics (2022) 12(10):4734–52. doi:10.7150/thno.69682
PubMed Abstract | CrossRef Full Text | Google Scholar
6. Atarod D, Mamashli F, Ghasemi A, Moosavi-Movahedi F, Pirhaghi M, Nedaei H, et al. Bivalent metal ions induce formation of α-synuclein fibril polymorphs with different cytotoxicities. Scientific Rep (2022) 12(1):11898. doi:10.1038/s41598-022-15472-4
PubMed Abstract | CrossRef Full Text | Google Scholar
7. Ziaunys M, Sakalauskas A, Mikalauskaite K, Smirnovas V. Polymorphism of alpha-synuclein amyloid fibrils depends on ionic strength and protein concentration. Int J Mol Sci (2021) 22(22):12382. doi:10.3390/ijms222212382
PubMed Abstract | CrossRef Full Text | Google Scholar
11. Polymeropoulos MH, Lavedan C, Leroy E, Dehejia A, Dutra A, Pike B, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science (1997) 276(5321):2045–7. doi:10.1126/science.276.5321.2045
PubMed Abstract | CrossRef Full Text | Google Scholar
12. Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet (1998) 18(2):106–8. doi:10.1038/ng0298-106
PubMed Abstract | CrossRef Full Text | Google Scholar
13. Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, et al. The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann Neurol: Off J American Neurol Assoc Child Neurol Soc (2004) 55(2):164–173. doi:10.1002/ana.10795
PubMed Abstract | CrossRef Full Text | Google Scholar
14. Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc Natl Acad Sci (2011) 108(10):4194–9. doi:10.1073/pnas.1100976108
PubMed Abstract | CrossRef Full Text | Google Scholar
15. Lashuel HA, Overk CR, Oueslati A, Masliah E. The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci (2013) 14(1):38–48. doi:10.1038/nrn3406
PubMed Abstract | CrossRef Full Text | Google Scholar
16. Zhou Y, Yao Y, Yang Z, Tang Y, Wei G. Naphthoquinone-dopamine hybrids disrupt α-synuclein fibrils by their intramolecular synergistic interactions with fibrils and display a better effect on fibril disruption. Phys Chem Chem Phys (2023) 25:14471–83. doi:10.1039/d3cp00340j
PubMed Abstract | CrossRef Full Text | Google Scholar
17. Kaur Mankoo O, Kaur A, Goyal D, Goyal B. Unravelling the destabilization potential of ellagic acid on α-synuclein fibrils using molecular dynamics simulations. Phys Chem Chem Phys (2023) 25:8128–43. doi:10.1039/d2cp06006j
PubMed Abstract | CrossRef Full Text | Google Scholar
18. Khursandov J, Mashalov R, Makhkamov M, Turgunboev F, Sharipov A, Razzokov J. Exploring α-synuclein stability under the external electrostatic field: effect of repeat unit. J Struct Biol (2024) 216(3):108109–8477. doi:10.1016/j.jsb.2024.108109
PubMed Abstract | CrossRef Full Text | Google Scholar
19. Makhkamov M, Baev A, Kurganov E, Razzokov J. Understanding Osaka mutation polymorphic Aβ fibril response to static and oscillating electric fields: insights from computational modeling. Sci Rep (2024) 14:22246. doi:10.1038/s41598-024-72778-1
PubMed Abstract | CrossRef Full Text | Google Scholar
22. Glaser CB, Yamin G, Uversky VN, Fink AL. Methionine oxidation, alpha-synuclein, and Parkinson’s disease. Biochim Biophys Acta (Bba) - Proteins Proteomics (2005) 1703(2):157–69. doi:10.1016/j.bbapap.2004.10.008
PubMed Abstract | CrossRef Full Text | Google Scholar
23. Murray IV, Giasson BI, Quinn SM, Koppaka V, Axelsen PH, Ischiropoulos H, et al. Role of alpha-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry (2003) 42(28):8530–40. doi:10.1021/bi027363r
PubMed Abstract | CrossRef Full Text | Google Scholar
24. Rekas A, Knott RB, Sokolova A, Barnham KJ, Perez KA, Masters CL, et al. The structure of dopamine induced α-synuclein oligomers. Eur Biophys J (2010) 39(10):1407–19. doi:10.1007/S00249-010-0595-X
PubMed Abstract | CrossRef Full Text | Google Scholar
25. Won SJ, Fong R, Butler N, Sanchez J, Zhang Y, Wong C, et al. Neuronal oxidative stress promotes α-synuclein aggregation in vivo. Antioxidants (2022) 11(12):2466. doi:10.3390/antiox11122466
PubMed Abstract | CrossRef Full Text | Google Scholar
26. Ponzini E, De Palma A, Cerboni L, Natalello A, Rossi R, Moons R, et al. Methionine oxidation in α-synuclein inhibits its propensity for ordered secondary structure. J Biol Chem (2019) 294(14):5657–65. doi:10.1074/jbc.RA118.001907
PubMed Abstract | CrossRef Full Text | Google Scholar
30. Schweighauser M, Shi Y, Tarutani A, Kametani F, Murzin AG, Ghetti B, et al. Structures of α-synuclein filaments from multiple system atrophy. Nature (2020) 585:464–9. doi:10.1038/s41586-020-2317-6
PubMed Abstract | CrossRef Full Text | Google Scholar
31. Ruf RA, Lutz EA, Zigoneanu IG, Pielak GJ. Alpha-Synuclein conformation affects its tyrosine-dependent oxidative aggregation. Biochemistry (2008) 47(51):13604–9. doi:10.1021/bi801884z
PubMed Abstract | CrossRef Full Text | Google Scholar
32. Baltazar Gonçalves P, Palhano FL, Cordeiro YAna Carolina Rennó Sodero. How oxidized EGCG remodels α-synuclein fibrils into non-toxic aggregates: insights from computational simulations. Phys Chem Chem Phys (2023) 25:19182–94. doi:10.1039/d3cp02261g
PubMed Abstract | CrossRef Full Text | Google Scholar
34. Takai E, Kitamura T, Kuwabara J, Ikawa S, Yoshizawa S, Shiraki K, et al. Chemical modification of amino acids by atmospheric-pressure cold plasma in aqueous solution. J Phys D: Appl Phys (2014) 47:285403. doi:10.1088/0022-3727/47/28/285403
CrossRef Full Text | Google Scholar
36. Huang J, Rauscher S, Nawrocki G, Ran T, Feig M, de Groot BL, et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat Methods (2017) 14:71–3. doi:10.1038/nmeth.4067
PubMed Abstract | CrossRef Full Text | Google Scholar
37. Berendsen HJC, Postma JPM, van Gunsteren WF, Hermans J. Interaction models for water in relation to protein hydration. In: B Pullman, editor. Intermolecular forces. The Jerusalem Symposia on quantum chemistry and biochemistry, 14. Dordrecht: Springer (1981). p. 331–42. doi:10.1007/978-94-015-7658-1_21
CrossRef Full Text | Google Scholar
42. Tuttle M, Comellas G, Nieuwkoop A, Covell DJ, Berthold DA, Kloepper KD, et al. Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat Struct Mol Biol (2016) 23:409–15. doi:10.1038/nsmb.3194
PubMed Abstract | CrossRef Full Text | Google Scholar
43. Yao Y, Tang Y, Wei G. Epigallocatechin gallate destabilizes α-synuclein fibril by disrupting the E46-K80 salt-bridge and inter-protofibril interface. ACS Chem Neurosci (2020) 11:4351–61. doi:10.1021/acschemneuro.0c00598
PubMed Abstract | CrossRef Full Text | Google Scholar
44. Kaur G, Kaur Mankoo O, Goyal D, Goyal B. Unveiling how hydroxytyrosol destabilizes α-syn oligomers using molecular simulations. J Phys Chem B (2023) 127:5620–32. doi:10.1021/acs.jpcb.3c02434
PubMed Abstract | CrossRef Full Text | Google Scholar
45. Bieschke J, Russ J, Friedrich RP, Ehrnhoefer DE, Wobst H, Neugebauer K, et al. EGCG remodels mature α-synuclein and amyloid-β fibrils and reduces cellular toxicity. Proc Natl Acad Sci (2010) 107(17):7710–5. doi:10.1073/pnas.0910723107
PubMed Abstract | CrossRef Full Text | Google Scholar
46. Kumar S, Kumar R, Kumari M, Kumari R, Saha S, Bhavesh NS, et al. Ellagic acid inhibits α-synuclein aggregation at multiple stages and reduces its cytotoxicity. ACS Chem Neurosci (2021) 12(11):1919–30. doi:10.1021/acschemneuro.1c00001
PubMed Abstract | CrossRef Full Text | Google Scholar
47. Javed H, Meeran MFN, Azimullah S, Adem A, Sadek B, Kumar Ojha S. Plant extracts and phytochemicals targeting α-synuclein aggregation in Parkinson’s disease models. Front Pharmacol (2019) 9:1555. doi:10.3389/fphar.2018.01555
PubMed Abstract | CrossRef Full Text | Google Scholar
48. Wahid M, Ali A, Saqib F, Aleem A, Bibi S, Afzal K, et al. Pharmacological exploration of traditional plants for the treatment of neurodegenerative disorders. Phytotherapy Res (2020) 34(12):3089–112. doi:10.1002/ptr.6742
PubMed Abstract | CrossRef Full Text | Google Scholar
49. Ansari AP, Zaheer Ahmed N, Anwar N. Role of common spices in prevention and management of neurodegenerative diseases. In: Medicinal plants for the management of neurodegenerative diseases. CRC Press (2024). p. 138–49.
Google Scholar
 Tohir Akramov1,2*
Tohir Akramov1,2* Parthiban Marimuthu3,4
Parthiban Marimuthu3,4 Mukhriddin Makhkamov1,5
Mukhriddin Makhkamov1,5 Aamir Shahzad6
Aamir Shahzad6 Rasulbek Mashalov1
Rasulbek Mashalov1 Jamoliddin Razzokov7,8,9
Jamoliddin Razzokov7,8,9