 Hong Xiao
Hong Xiao Tao Hu
Tao Hu Ping Zhou
Ping Zhou Dengfeng Li
Dengfeng Li- School of Science, Chongqing University of Posts and Telecommunications, Chongqing, China
The thermal transport and thermoelectric properties of anti-perovskite M3IO and M4I2O (M = K, Rb) were investigated using first-principles calculations combined with solution of the Boltzmann transport equation. The two-phonon scattering channel was also considered. These structures formed M6O octahedra, accompanied by a rattling motion of the O atoms. They exhibit ultra-low lattice thermal conductivity, ranging from 0.30 to 0.89 W m-1 K−1 at room temperature. M4I2O demonstrates strong anisotropic thermal transport due to weaker bonding interactions along the zz direction, while M3IO shows isotropic thermal conductivity. Specifically, Rb4OI2 has the lowest lattice thermal conductivity of 0.47 W m-1 K−1 along the xx direction and 0.30 W m-1 K−1 along the zz direction. Additionally, M3IO possesses low lattice thermal conductivity of 0.52 W m-1K−1, attributed to the softening behavior of the TA branch at the M and R points. The electronic structure of M3IO and M4OI2 reveals a multi-valley phenomenon in the valence band, resulting in a large Seebeck coefficient under p-type doping. Our results indicate maximum thermoelectric figure of merit (ZT) values of 1.91 for p-type Rb3OI, and 1.41 for p-type Rb4OI2 along the zz direction at 900 K. Rb3OI and Rb4OI2 were proposed as potential p-type thermoelectric materials.
1 Introduction
The energy shortage crisis is intensifying, making the search for sustainable and clean energy sources an urgent priority. Thermoelectric (TE) materials, which can convert thermal energy into electrical energy without generating additional pollution [1–3], show great promise in alleviating energy shortages and environmental pollution. Thermoelectric devices have the advantage of compact, lightweight, durable, and noiseless [4, 5]. The thermoelectric efficiency of materials is typically measured by the dimensionless figure of merit (ZT) [6]. High-performance thermoelectric materials must simultaneously exhibit a high thermoelectric power factor and low thermal conductivity [7]. However, the electronic conductivity and thermal conductivity are inherently coupled, which makes it challenging to achieve high electrical conductivity while maintaining a low electronic thermal conductivity. To overcome this challenge, researchers employ strategies such as introducing defects, alloys, nanostructures, and suppressing the lattice thermal conductivity (κL) [8–14]. Therefore, searching low lattice thermal conductivity materials are important for improving the thermoelectric performance.
The perovskite structure has been classified as a “phonon glass, electron crystal” structure [15]. The octahedral framework vibrations and rattling behavior in anti-perovskite compounds induce strong anharmonic phonon scattering [16, 17], leading to intrinsically low lattice thermal conductivity. Some anti-perovskite derivatives, such as Rb3AuO [18], Rb3ITe [19], Ca6NFSn2 and Sr6NFSn2 [20] demonstrate exceptional thermoelectric performance due to their ultra-low κL. For instance, the room temperature κL of Rb3AuO is 0.73 W m-1 K−1, and the cubic phase of Rb3ITe achieves an ultra-low κL of 0.16 W m-1 K−1 at room temperature. At 800 K, both materials attain ZT > 3, outperforming traditional inorganic thermoelectric materials such as Bi2Te3 [21], SnSe [22], and lead chalcogenide compounds (with typical ZT values greater than 1) [23]. Moreover, Ca6NFSn2 and Sr6NFSn2 exhibit higher electrical conductivity than the halide perovskite CsSnI3 [24]. When coupled with their low lattice thermal conductivity, the calculated ZT values achieve 1.9 and 2.3, respectively.
In this work, we employed first-principles calculations and the Boltzmann transport equation to study the thermal transport and thermoelectric properties of anti-perovskite M3OI and M4OI2(M = K, Rb). Due to their ultra-low lattice thermal conductivity, the dual-phonon channel model was discussed, which includes the contribution of diffuson-like phonon to the lattice thermal conductivity. The calculated κL values are about of 0.30–0.89 W m-1 K−1 at room temperature due to low phonon group velocity and short phonon lifetime. Furthermore, the multi-valley band structure increases the degeneracy of the energy bands, resulting in higher Seebeck coefficients (S) in p-type doping. The p-type anti-perovskite Rb3OI achieves a maximum ZT of 1.91 at 900 K, demonstrating its potential for thermoelectric applications.
2 Computational methods
First-principles calculations were executed within the framework of the Vienna Ab Initio Simulation Package (VASP). It was performed using the Projector Augmented Wave (PAW) method, incorporating the Perdew–Burke–Ernzerhof (PBE) parametrization of the generalized gradient approximation (GGA) for the exchange correlation functional. The electronic properties were investigated based on the hybrid density functional theory (HSE06). The structures of the primitive cells were optimized until the atomic force converged to less than 10–4 eV/Å. The energy cutoff and convergence criterion were set to 520 eV and 10–7 eV, respectively. A 9 × 9 × 9 Γ-centered Monkhorst-Pack k-point mesh was employed to calculate the second- and third-order interatomic force constants (IFCs), which were subsequently utilized to study both harmonic and anharmonic phonon properties. Furthermore, the ShengBTE code was used to compute the lattice thermal conductivity of the materials via the phonon Boltzmann transport equation (BTE), as shown in the following Equation 1:
where Cv is the specific heat, Vg is the group velocity, and λ is the mean free path. To ensure the convergence of the thermal conductivity calculations, a cutoff radius of 0.8 nm was implemented to limit the range of the third-order force constants. For the electrical transport properties, the ab initio scattering and transport (AMSET) software package was used to calculate carrier lifetimes, considering scattering processes due to acoustic deformation potential (ADP), ionized impurities (IMP), and polar-optical phonons (POP). The interpolation parameter ‘interpolation_factor’ is added to 110 to ensure the calculation accuracy and convergence. The final electron scattering time was derived using the following Equation 2, based on Matthiessen’s rule:
3 Results and discussion
3.1 Structure and charge density
M3OI (M = K, Rb) compounds exhibit simple anti-perovskite structures, following the general formula ABX3. As illustrated in Figure 1a, the crystal structures of K3OI and Rb3OI crystallize within the Pm3m space group. In these structures, the alkali metals (M = K, Rb) occupy the X-sites within the unit cell, while the oxygen (O) ions occupy the B-sites. This arrangement results in M6O octahedra formed by the oxygen anions and the alkali metal cations [25, 26]. The iodine (I) atoms are situated at the A-sites, which are located at the corners of the unit cell. Figure 1b depicts the crystal structure of anti-perovskite derivatives K4OI2 and Rb4OI2, which bear resemblance to the (La, Ba)CuO4 structure. These compounds belong to the tetragonal crystal system and crystallize in the space group I4/mmm. In this structure, two distinct alkali metal sites are presented, as noted in previous studies [27]. Supplementary Figure S1 highlights the layered nature of this arrangement, where alternating M3OI and MI layers are stacked along the z-axis(c), mirroring the structural arrangement seen in Na4OI2 [28].
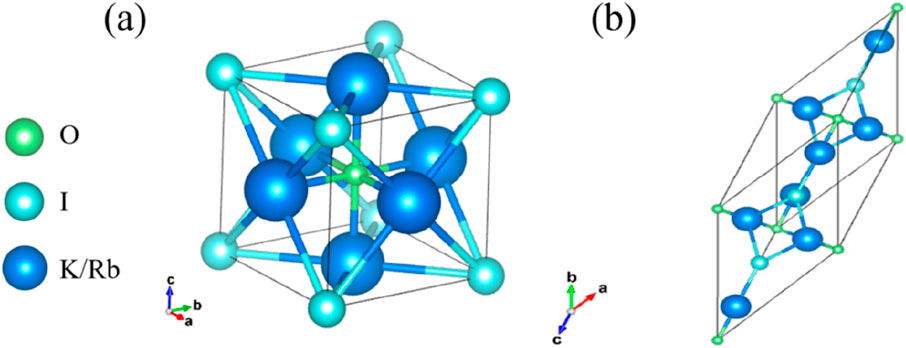
Figure 1. (a) The unit cell of K3OI and Rb3OI, (b) the unit cell of K4OI2 and Rb4OI2.
The optimized lattice constants for these four structures are summarized in Table 1. K3OI has been successfully synthesized in experiments [29]. Additionally, Table 1 provides bond lengths for each structure, along with key mechanical properties such as elastic constants, bulk modulus (B), Young’s modulus (E), shear modulus (G), and the Debye temperature (Θ) [30]. The force constant for M3OI (M = K, Rb) adheres to the mechanical stability criterion in Equation 3, while M4OI2 (M = K, Rb) meets the mechanical stability criterion in Equation 4. Therefore, all four structures are mechanically stable [31].
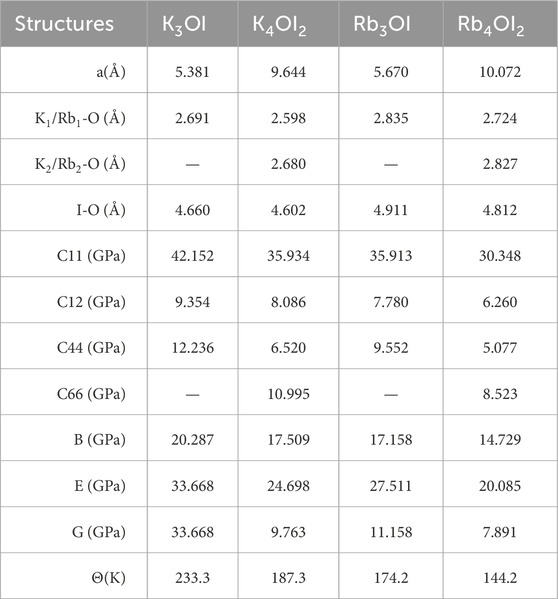
Table 1. Lattice constant (a), bond lengths, elastic constants (C), bulk modulus(B), Young’s modulus(E), shear modulus(G), and Debye temperature(Θ) of the M3OI and M4OI2 (M = K, Rb).
Debye temperature serves as an indicator of the bond strength of materials, with a positive correlation between these two parameters. Among the studied materials, K3OI exhibits the highest Debye temperature of 233.3 K, followed by K4OI2 of 187.3 K and Rb3OI of 174.2 K. The lowest Debye temperature of 144.2 K is observed in Rb4OI2. This trend is consistent with the bond length of them. We find that all the Debye temperatures are significantly below the room temperature of 300 K, suggesting that the M3OI and M4OI2 compounds exhibit pronounced anharmonic behavior.
The charge density in Figure 2 shows a typical covalent bond charge distribution between the alkali metal M and the oxygen (O) element. Specifically, the strong covalent bond between the alkali metal M and O forms a stable M6O octahedral framework structure. In contrast, the iodine (I) element exhibits a strong electronic localization phenomenon, with weaker bonding to other atoms. By comparing the charge densities in Figures 2a,b for K3OI and Rb3OI, and in Figures 2c,d for K4OI2 and Rb4OI2, it can be observed that the Rb-O bond is weaker than the K-O bond, which is consistent with the results of bond length in Table 1.
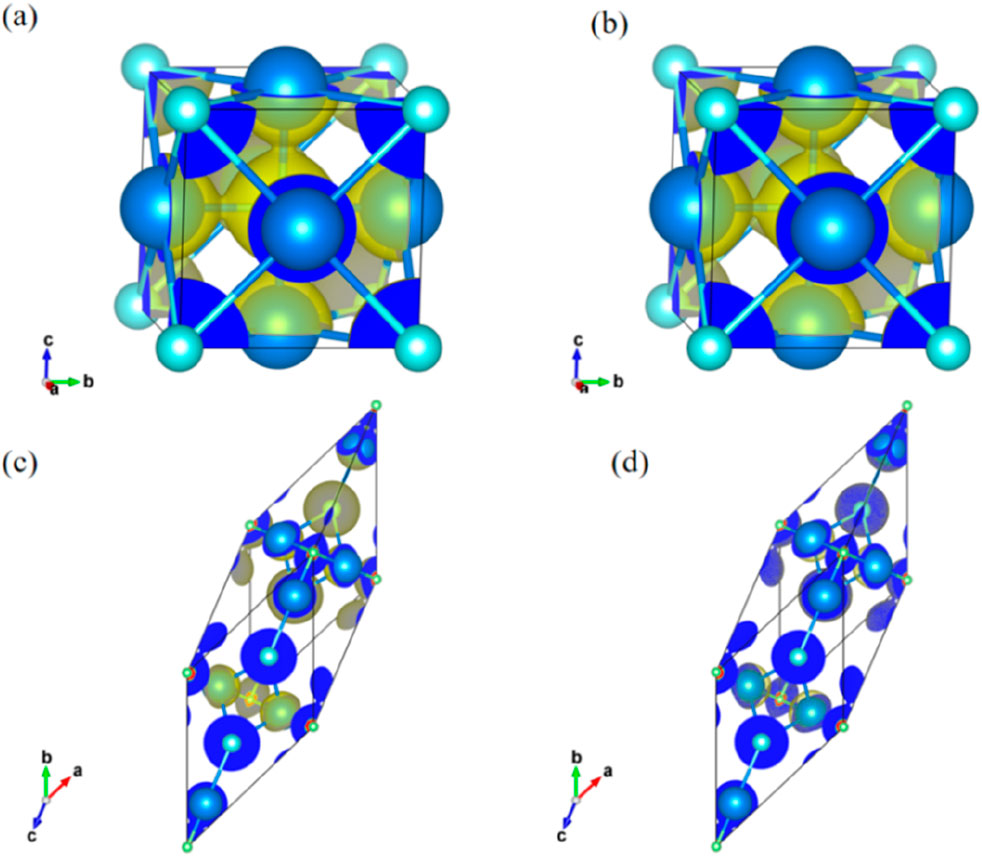
Figure 2. The charge density plot of (a) K3OI, (b) Rb3OI, (c) K4OI2 and (d)Rb4OI2.
3.2 Thermal transport property
The phonon dispersion spectra show that K3OI, K4OI2, Rb3OI, and Rb4OI2 exhibit no imaginary frequencies under the harmonic approximation in Figure 3, indicating that these materials are thermodynamically stable. The high-frequency optical branches are primarily contributed by the vibration modes of the oxygen (O) atoms, while the low-frequency optical and acoustic branches are dominated by the heavier iodine (I) and alkali metal (K, Rb) elements. The phonon dispersions for all four structures exhibit strong phonon-optical coupling within the low-frequency optical and acoustic branch regions, which is typically accompanied by peaks in the phonon density of states (PDOS). A comparison between Figures 3a,c reveals that the highest optical branch frequency for K3OI is 9 THz, which is higher than the 8 THz for Rb3OI. K3OI exhibits stronger phonon dispersion, particularly in the acoustic branch, with the phonon group velocity for K3OI (0–1.44 THz) being significantly higher than for K4OI2 (0–1.19 THz). Similarly, Figures 3b,d show that the highest optical branch frequencies for K4OI2 and Rb4OI2 are 9 THz (K4OI2) and 8 THz (Rb4OI2), respectively, while the highest frequencies for the acoustic branches are 1.11 THz (K4OI2) and 1.00 THz (Rb4OI2). These materials have lower maximum acoustic branch frequencies compared to strong anharmonic materials such as Na2TlSb [32] (∼1.4 THz) and SnSe [33] (∼1.45 THz). Generally, when atoms of a similar structure are replaced with heavier elements, the phonon modes tend to shift to lower frequencies, which reduces the phonon group velocity and consequently suppresses the thermal conductivity. In the M4OI2, the enhanced phonon-optical coupling relative to M3OI provides more phonon scattering channels for the material. Notably, the phonon dispersion for Rb3OI shows a pronounced softening of the TA branch at the M and R points, a phenomenon commonly observed in cubic perovskite structures. This ferroelectric-like vibration behavior is typically attributed to vibrations in the octahedral framework [34, 35]. Figure 4 illustrates the TA branch vibration modes at the high-symmetry M and R points for K3OI and Rb3OI. At the M point, the I atoms in K3OI exhibit in-plane motion along the positive and negative z-axis (c) directions, while in Rb3OI, the I atoms undergo in-plane motion within the xy (ab) plane with some rotational motion, and the O atoms exhibit a rattling behavior similar to that in octahedral structures. Figures 4b,d show that at the R point, the K atoms in K3OI undergo opposing movements around the axis within the xy plane, leading to octahedral distortion, while in Rb3OI, the I atoms exhibit in-plane motion along the positive and negative y-axis directions. In addition to the octahedral distortion [36], the rattling behavior within the octahedral cage is often considered a source of strong anharmonicity in anti-perovskite materials [37].
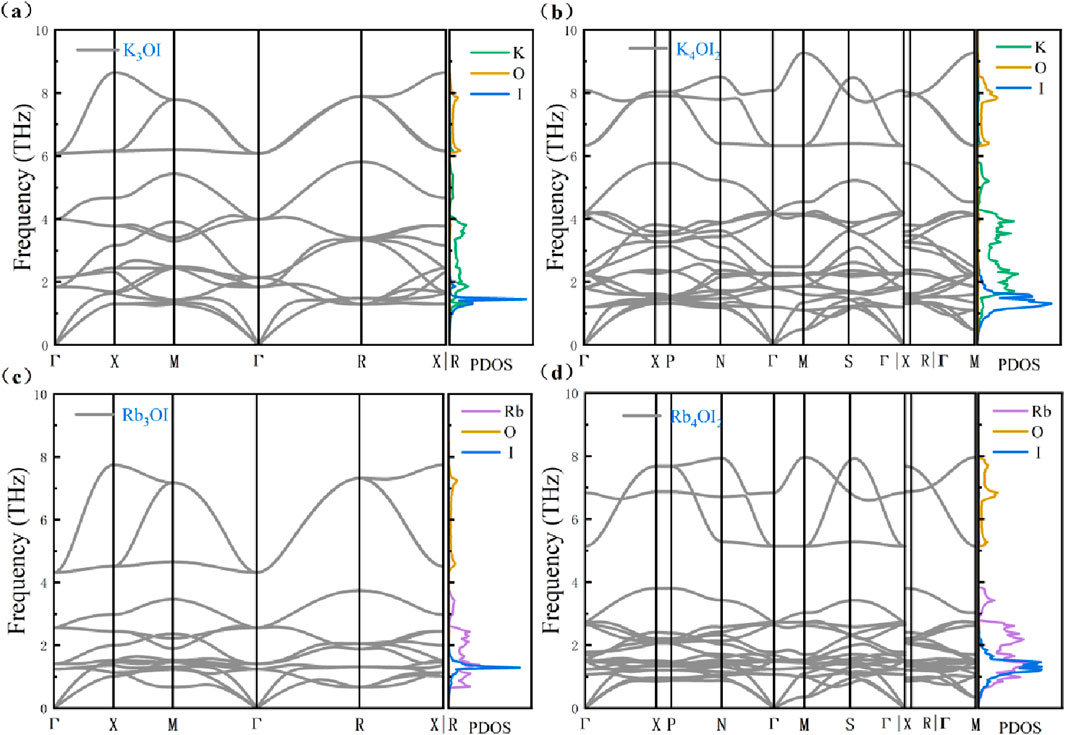
Figure 3. Phonon band and phonon density of states of (a) K3OI, (b) K4OI2, (c) Rb3OI and (d) Rb4OI2.
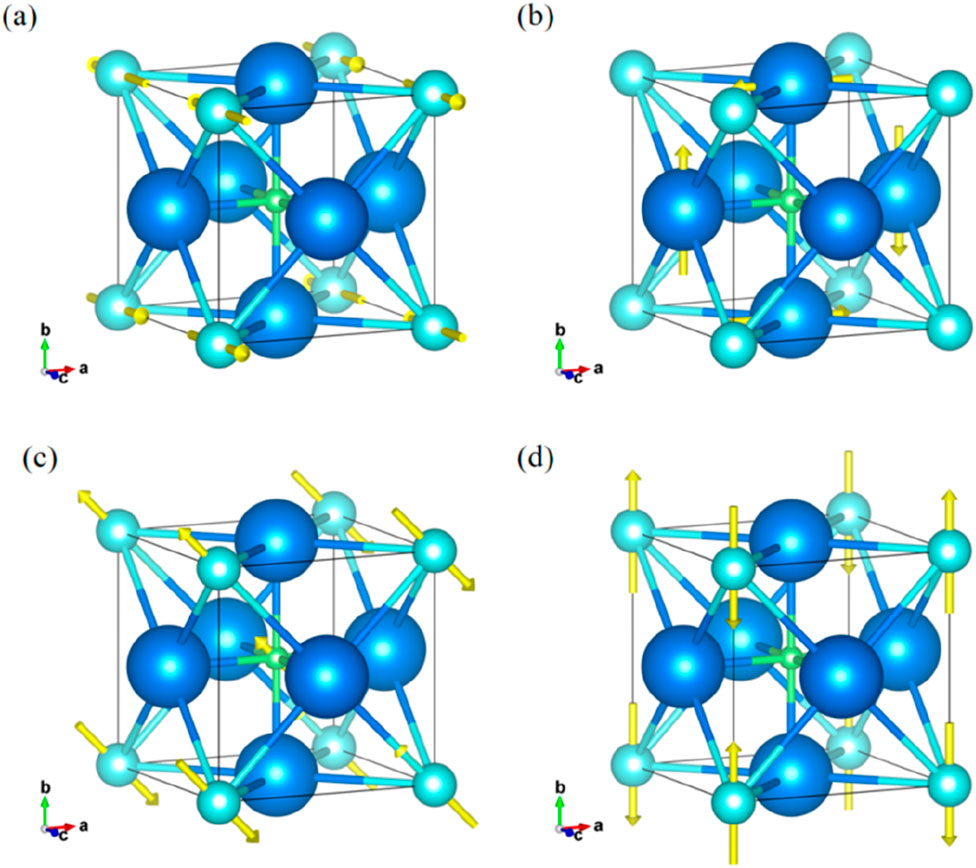
Figure 4. The vibrational modes of the Transverse Acoustic Branch (TA) at high-symmetry points M and R, and yellow arrows indicate the vibration directions, and their lengths represent the vibration amplitudes. (a) The vibrational mode of K3OI at point M; (b) The vibrational mode of K3OI at point R; (c) The vibrational mode of Rb3OI at point M; (d) The vibrational mode of Rb3OI at point R.
We employed the diffusion-like model of dual-phonon [38] with shengBTE to calculate the κL of these materials in the temperature range of 100–900 K. as shown in Figure 5a and Supplementary Figure S2. The lattice thermal conductivity decreases with increasing temperature. We find that Rb4OI2 along the zz (c)direction has the lowest thermal conductivity with and without considering the contribution of the diffuson-like phonon. At 300 K, when the two-phonon channel is neglected, it is 0.18 W m-1 K−1, lower than the 0.20 W m-1 K−1 reported for SnSe [22]. On the other hand, the K3OI and Rb3OI exhibit isotropic thermal conductivity while that for K4OI2 and Rb4OI2 show significant anisotropy. After considering the dual-phonon channel, the lattice thermal conductivity of K3OI and Rb3OI are 0.89 W m-1 K−1 and 0.52 W m-1 K−1, respectively. The lattice thermal conductivity of K4OI2 and Rb4OI2 is 0.71 W m-1 K−1 and 0.47 W m-1 K−1 along the xx direction, and 0.42 W m-1 K−1 and 0.30 W m-1 K−1 along the zz direction, respectively. At room temperature, the κL of these materials is comparable to the industry standard PbTe [39] and significantly lower than most oxide thermoelectric materials [40–43]. At higher temperatures, κL remains below 1 W m-1 K−1, which is a characteristic feature of high-performance thermoelectric materials.
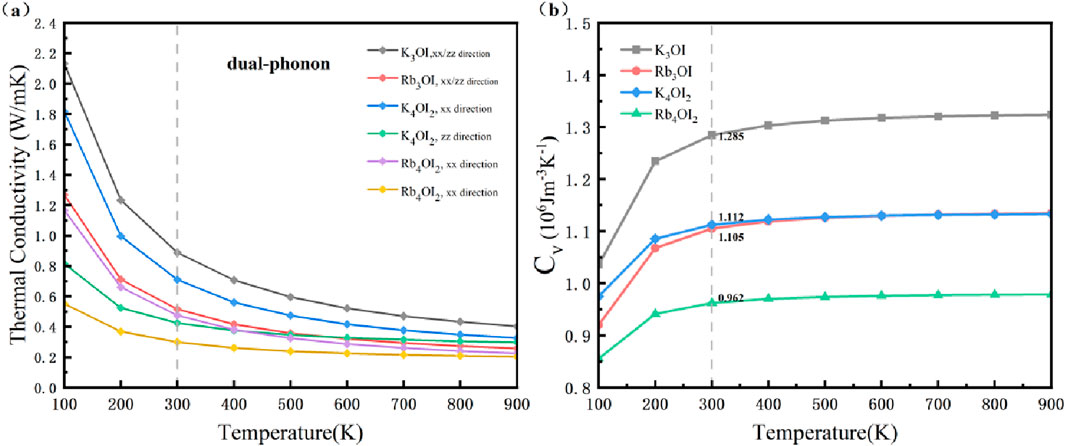
Figure 5. (a) The variation of lattice thermal conductivity (κL) with temperature after considering dual-phonon transport. (b) The variation of specific heat capacity (Cv) with temperature.
Heat capacity curve in Figure 5b shows that the specific heat capacity increases gradually with temperature in the range of 100–400 K. After 400 K, the heat capacities of the materials tend to converge. Over the entire temperature range, K3OI and Rb4OI2 exhibit the highest and the lowest specific heat capacity, respetively. At 300 K, the specific heat capacities of K4OI2 and Rb3OI are 1.112 × 106 Jm-3K−1 and 1.105 × 106 Jm-3K−1, respectively, while the heat capacity curves of the two materials nearly overlap beyond 400 K. Cumulative lattice thermal conductivity, as shown in Supplementary Figure S3, indicates that at 300 K, the lattice thermal conductivity is primarily contributed by phonon modes with an average free path (MFP) below 10 nm. ShengBTE results reveal that the high-frequency optical branches of these materials significantly contribute to the lattice thermal conductivity due to the strong phonon-optical coupling.
To further investigate the physical mechanisms for the thermal transport of these materials, Figure 6 plots the phonon relaxation time, phonon group velocity, scattering phase space, and Grüneisen parameter. As seen in Figure 6b, at 300 K, the phonon group velocity of K3OI and K4OI2 along the xx direction is higher than that for Rb3OI and Rb4OI2 in the acoustic branches and low-frequency optical branches (0-1.64 THz). In addition, the phonon group velocity of the M3OI structure is higher than that of the M4OI2. The difference in the phonon group velocities between K3OI and R3IO or K4OI2, is mainly attributed to the increased effective atomic mass. This result aligns well with the phonon dispersion spectrum in Figure 3. The ultra-low lattice thermal conductivity of K4OI2 and Rb4OI2 in the zz direction, and their remarkable anisotropic structural properties, can be further explained by the softer bonds [44] and the lower phonon group velocity in the zz direction, as seen in Supplementary Figure S4.
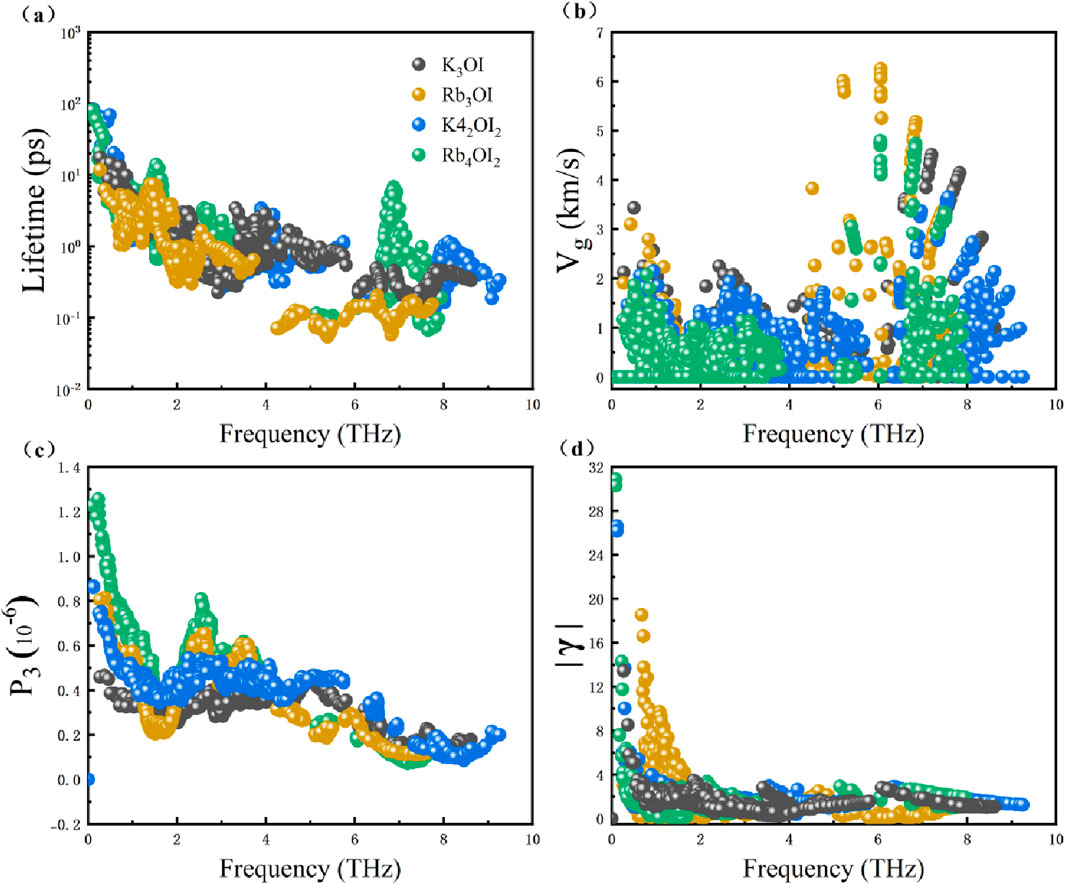
Figure 6. The variation curves of specific phonon thermal transport parameters with phonon frequency at 300 K. (a) Phonon relaxation time, (b) phonon group velocity in the xx direction, (c) P3 scattering phase space, (d) absolute value of the Grüneisen parameter.
Figure 6a presents the phonon relaxation times of these materials. Within the frequency range of the acoustic phonon, K3OI (0-1.64 THz) and K4OI2 (0-1.42 THz) has overall higher phonon relaxation times compared to Rb3OI (0-1.22 THz) and Rb4OI2 (0-1.11 THz), respectively. Moreover, the phonon relaxation times of K4OI2 and Rb4OI2 structures in the acoustic branch frequency range are larger than those of K3OI and Rb3OI structures. Next, we explored the P3 scattering phase space and Grüneisen parameter, as shown in Figures 6c,d. It indicates that K3OI has the smallest scattering phase space, followed by K4OI2, and Rb4OI2 exhibits the largest phase space in the low-frequency acoustic branch frequency range (0-1 THz). This phenomenon is attributed to the phonon dispersion becoming more confined in this frequency range (0-1 THz) as the average relative atomic mass increases, which makes it easier for phonon scattering to satisfy the energy conservation law. In the strong phonon-optical coupling frequency range, K3OI exhibits smaller P3 compared to K4OI2, and Rb3OI is smaller than Rb4OI2 due to the increased number of low-frequency optical branches, which strengthens the phonon-optical coupling and increases the scattering channels. Figure 6d displays the Grüneisen parameter at 300 K. The distribution is concentrated in the acoustic and low-frequency optical branch regions, with Rb3OI showing a distinctly different parameter distribution. Its peak value occurs at ∼1THz, while the other three structures have their peaks near zero frequency. Rb4OI2 has a higher Grüneisen parameter than K4OI2, and K4OI2 is higher than K3OI. Although Rb3OI has the lowest Grüneisen parameter in the acoustic branch (0-0.6 THz), its Grüneisen values in the 0.8–1.8 THz range are significantly largest, leading to the highest overall Grüneisen parameter and strongest phonon anharmonicity, and then the lowest phonon relaxation times. Additionally, the Grüneisen parameter distribution in Rb3OI is influenced by the softening behavior of the transverse acoustic (TA) branch in the corresponding frequency range, which weakens the coupling with the low-frequency optical branch and leads to weaker phonon-optical coupling and a shift of the Grüneisen peak.
3.3 Electrical transport and thermoelectric properties
The electronic band structures were calculated using the hybrid functional HSE06. As shown in Figure 7. The electronic band gaps are 2.18 eV (K3OI), 2.40 eV (K4OI2), 1.61 eV (Rb3OI), and 1.92 eV (Rb4OI2), all of which are direct band gaps. The valence bands are primarily composed of the oxygen (O) orbitals, while the conduction bands are derived from the combined contributions of the alkali metals (K and Rb) and halogen (I) elements. The flat electronic band structure of the valence band indicates that it possesses a larger effective carrier mass, resulting in a lower carrier mobility. On the other hand, the significant band dispersion in the conduction band at the high-symmetry Γ point suggests that carriers near the conduction band exhibit a smaller effective mass and higher mobility. Notably, the valence band demonstrates a distinct multi-valley structure [45], which is often associated with high Seebeck coefficients of thermoelectrics [46].
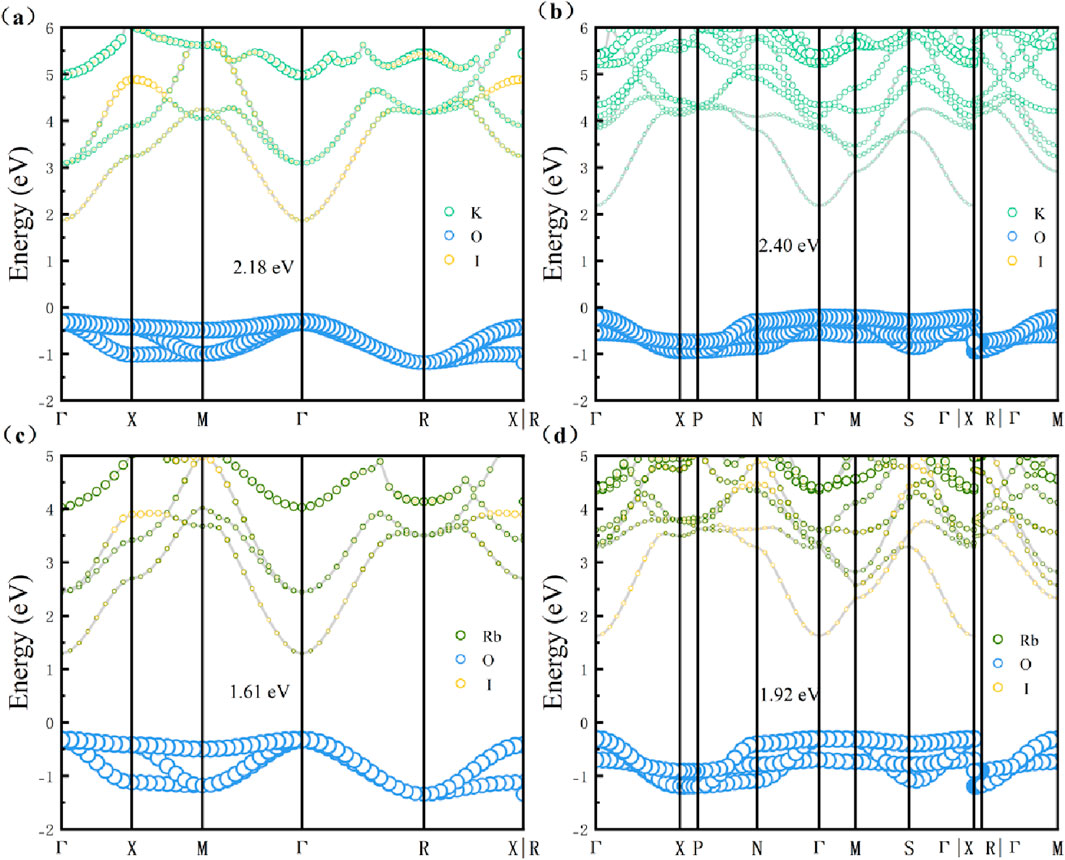
Figure 7. Projected electronic band diagram of (a) K3OI, (b) K4OI2, (c) Rb3OI, (d) Rb4OI2. The diameter of the circle is proportional to their orbital weight.
The electronic transport properties of materials were studied using the AMSET software based on the electronic band structure. Figure 8 presents the computed electrical conductivity (σ), Seebeck coefficient (S), electronic thermal conductivity (κe), and power factor (PF) at different temperatures (300 K, 600 K, and 900 K). From Figures 8a,b, the electrical conductivity of n-type semiconductors is one order of magnitude higher than that of the corresponding p-type semiconductors. It is noteworthy that the electrical conductivity of the anisotropic material along the zz direction is the lowest, with a value ∼1 S/m at a carrier concentration of 1019 cm-3.
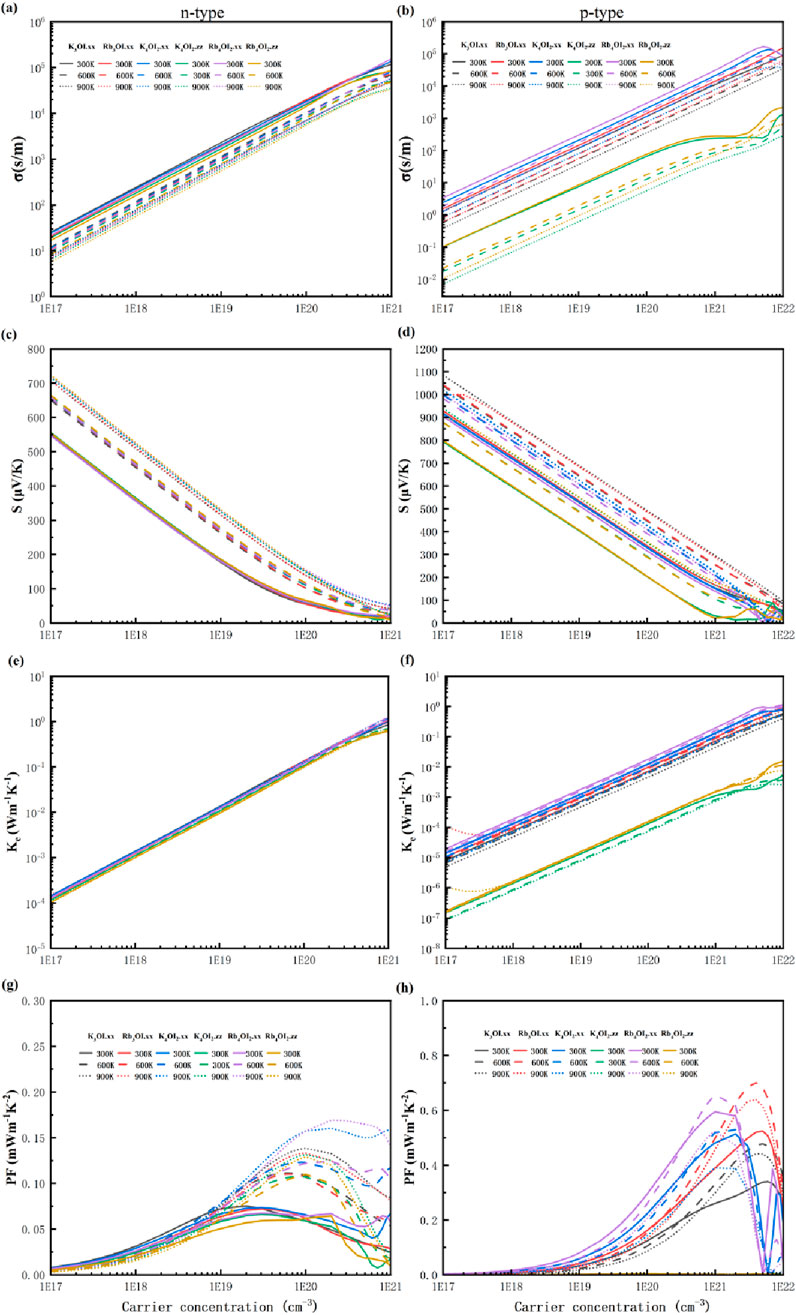
Figure 8. (a) The electronic conductivity (σ), (c) Seebeck coefficient (S), (e) electronic thermal conductivity (κe) and (g) power factor (PF) of n-type materials. (b) The electronic conductivity (σ), (d) Seebeck coefficient (S), (f) electronic thermal conductivity (κe) and (h) power factor (PF) of p-type materials.
The Seebeck coefficient (S) of the p-type semiconductors is significantly higher than that of n-type ones, which can be attributed to the multi-valley phenomenon in the valence band of the electronic structure. For instance, at a carrier concentration of 1019 cm-3, the Seebeck coefficient (S) of Rb3OI ranges from 527 to 684 µVK-1 between 300 K and 900K, which is higher than the 600 µVK-1 of Rb3AuO [7]. And it is notably higher than the typical range (200–300 µVK-1) found in thermoelectric power materials [47]. Additionally, S decreases with increasing temperature as the carrier concentration decreases, which is consistent with the trend for the most semiconductor thermoelectric materials due to carrier excitation by heat.
According to the Wiedemann-Franz law, electrical conductivity is proportional to the electronic thermal conductivity. When doping concentration is low, the electronic thermal conductivity can be neglected. However, at higher doping concentrations, the electronic thermal conductivity becomes comparable to or even greater than the lattice thermal conductivity. The PF of the material is calculated based on the S2σ relationship. Due to the larger Seebeck coefficient (S) of the p-type semiconductors, the power factor of p-type materials is higher than that of n-type semiconductors, with the maximum value reaching 0.7 mWm-1K−2 for Rb3OI at 600 K, with a doping concentration of 4 × 1021 cm-3.
Subsequently, we computed the ZT values for these materials in Figure 9. At 900 K, Rb3OI exhibits the highest ZT value of 1.91 for p-type doping at the carrier concentration of 1 × 1021 cm-3. The carrier scattering rate of R3OI with a hole doping concentration of 1 × 1021 cm-3 were shown in Supplementary Figure S5. We found that the carrier scattering rate increased with the rise in temperature due to the thermal excitation of carriers and the scattering rate of carriers was mainly contributed by POP scattering.
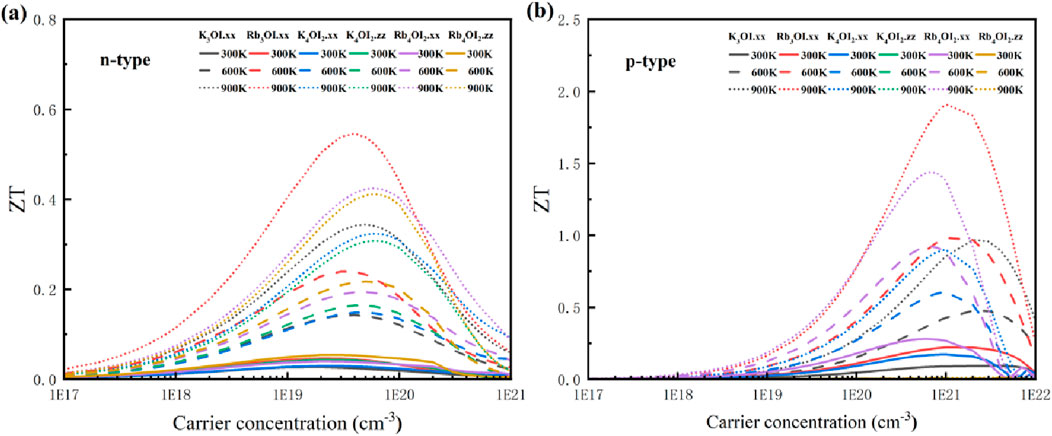
Figure 9. Thermoelectric figure of merit (ZT) of M3OI and M4OI2: (a) n-type, (b) p-type.
Next, Rb4OI2 shows the maximum ZT value of 1.44 in the zz direction, with a p-type doping concentration of 7 × 1020 cm-3. In this system, the thermoelectric performance of the p-type semiconductor is superior to that of the n-type, which is attributed to the higher PF resulting from the larger Seebeck coefficient (S). Comparing with the ZT values of some inorganic halide perovskites of CsPbI3 (0.45), CsSnI3 (0.95), and CsGeI3 (1.05) [48], Rb3OI and Rb4OI2 have better thermoelectric performance. The results indicate that Rb3OI and Rb4OI2 have promising potential for advanced thermoelectric device applications.
4 Conclusion
We investigated the thermal transport and thermoelectric properties of M3IO and M4I2O (M = K, Rb). These structures formed M6O octahedra and rattling behavior of the O atoms existed. The calculated Debye temperatures of these materials are obviously below the room temperature of 300 K. After considering the two-phonon channel, the lattice thermal conductivity ranges from 0.30 to 0.89 W m-1 K−1 at room temperature. Rb4OI2 has the lowest lattice thermal conductivity of 0.47 W m-1 K−1 and 0.30 W m-1 K−1 along the xx direction and the zz direction, respectively. Rb3OI has the lattice thermal conductivity of 0.52 W m-1K−1. M3IO exhibits isotropic lattice thermal conductivity while that for M4I2O shows significant anisotropy. The phonon group velocity and specific heat capacity of the M3OI structure are higher than that of the M4OI2. However, M3OI has lower phonon relaxation times due to the softening behavior of the TA branch at the M and R points. And then, their lattice thermal conductivity is comparable. Due to the multi-valley phenomenon in the valence band of the electronic structure, p-type M3IO and M4OI2 have larger Seebeck coefficient (S). The calculated S of Rb3OI ranges from 527 to 684 µVK-1 at a carrier concentration of 1019 cm-3. We obtained the maximum ZT value of p-type Rb3OI is 1.91 with the carrier concentration of 1 × 1021 cm-3 at 900 K, and that for p-Rb4OI2 is 1.41 in the zz direction at the doping concentration of 7 × 1020 cm-3. We propose that Rb3OI and Rb4OI2 are promising candidates as p-type thermoelectric materials.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author contributions
HX: Visualization, Methodology, Writing – original draft, Formal Analysis, Conceptualization, Data curation, Software, Investigation. TH: Software, Writing – original draft, Investigation, Data curation. PZ: Writing – review and editing, Validation, Supervision. DL: Writing – review and editing, Funding acquisition, Formal Analysis, Validation, Supervision, Project administration.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. We gratefully acknowledge funding supporting from Scientific and Technological Research of Chongqing Municipal Education Commission (KJZD-K202100602).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphy.2025.1619661/full#supplementary-material
References
1. Sootsman J, Chung D, Kanatzidis M. New and old concepts in thermoelectric materials. Angew Chem Int Edition (2009) 48:8616–39. doi:10.1002/anie.200900598
2. Yue T, Baolong X, Yinchang Z, Sheng M, Zhenhong D. Ultra-low lattice thermal conductivity and anisotropic thermoelectric transport properties in Zintl compound β-K2Te2. Phys Chem Chem Phys (2022) 24(7):4666–73. doi:10.1039/D1CP05248A
3. Yinchang Z, Zhenhong D, Chao Z, Chao L, Shuming Z, Geng L, et al. High thermopower and potential thermoelectric properties of crystalline LiH and NaH. Phys Rev B (2017) 95(1):014307. doi:10.1103/PhysRevB.95.014307
4. He J, Kanatzidis MG, Dravid VP. High performance bulk thermoelectrics via a panoscopic approach. Mater Today (2013) 16(5):166–76. doi:10.1016/j.mattod.2013.05.004
5. Fitriani F, Ovik R, Long BD, Barma MC, Riaz M, Sabri MFM A review on nanostructures of high-temperature thermoelectric materials for waste heat recovery. Renew Sustainable Energy Rev (2016) 64:635–59. doi:10.1016/j.rser.2016.06.035
6. Snyder GJ, Toberer ES. Complex thermoelectric materials. Nat Mater (2008) 7(2):105–14. doi:10.1038/nmat2090
7. Zhao Y, Lian C, Zeng S, Dai Z, Meng S, Ni J. Anomalous electronic and thermoelectric transport properties in cubic Rb3AuO antiperovskite. Phys Rev B (2020) 102(9):094314. doi:10.1103/PhysRevB.102.094314
8. Zhou LC, Xi H, ZhuYi X, Xie Y. Defect chemistry for thermoelectric materials. J Am Chem Soc (2016) 138(45):14810–9. doi:10.1021/jacs.6b08748
9. Hu L, Zhu T, Liu X, Zhao X. Point defect engineering of high-performance bismuth-telluride-based thermoelectric materials. Adv Funct Mater (2014) 24:5211–8. doi:10.1002/adfm.201400474
10. Xia Y, Chan MKY. Anharmonic stabilization and lattice heat transport in rocksalt β-GeTe. Appl Phys Lett (2018) 113(19):193902. doi:10.1063/1.5048814
11. Chen Z, Gao B, Tang J, Guo X, Li W, Ang R. Low lattice thermal conductivity by alloying SnTe with AgSbTe2 and CaTe/MnTe. Appl Phys Lett (2019) 115(7):073903. doi:10.1063/1.5109465
12. He J, Girard SN, Kanatzidis MG, Dravid VP. Microstructure-lattice thermal conductivity correlation in nanostructured PbTe0.7S0.3 thermoelectric materials. Adv Funct Mater (2010) 20:764–72. doi:10.1002/adfm.200901905
13. Tadano T, Gohda Y, Tsuneyuki S. Impact of rattlers on thermal conductivity of a thermoelectric clathrate: a first-principles study. Phys Rev Lett (2015) 114(9):095501. doi:10.1103/PhysRevLett.114.095501
14. Nolas GS, Poon J, Kanatzidis M. Recent developments in bulk thermoelectric materials. MRS Bull (2006) 31(3):199–205. doi:10.1557/mrs2006.45
15. Rowe DM. CRC handbook of thermoelectrics. (1995) 1st ed. Boca Raton: CRC Press doi:10.1201/9781420049718
16. Pchelkina ZV, Komleva EV, Irkhina VY, Long Y, Streltsov SV. Rattling phonon modes in quadruple perovskites. JETP Lett (2023) 118(10):738–41. doi:10.1134/S0021364023603202
17. Gu T, Scarbrough T, Yang Y, Íñiguez J, Bellaiche L, Xiang HJ. Cooperative couplings between octahedral rotations and ferroelectricity in perovskites and related materials. Phys Rev Lett (2018) 120(19):197602. doi:10.1103/PhysRevLett.120.197602
18. Zhao Y, Lian C, Zeng S, Dai Z, Meng S, Ni J. Quartic anharmonicity and anomalous thermal conductivity in cubic antiperovskites A3BO (A = K, Rb; B = Br, Au). Phys Rev B (2020) 101(18):184303. doi:10.1103/PhysRevB.101.184303
19. Shuming Z, Qian S, Lina G, Yinchang Z, Hao H, Geng L, et al. Remarkable thermoelectric efficiency of cubic antiperovskites Rb3X (Se and Te) I with strong anharmonicity. J Mater Chem A (2023) 11(44):24047–56. doi:10.1039/D3TA04338J
20. Dan H, Bonan Z, Zenghua C, Kieran BS, Stefan SR, Wolfgang S, et al. Discovery of multi-anion antiperovskites X6NFSn2 (X = Ca, Sr) as promising thermoelectric materials by computational screening. Matter (2024) 7(1):158–74. doi:10.1016/j.matt.2023.10.022
21. Ning W, Chen S, Zhehao S, Haiyan X, Hongbin Z, Zongyou Y, et al. High-temperature thermoelectric monolayer Bi2TeSe2 with high power factor and ultralow thermal conductivity. ACS Appl Energy Mater (2022) 5(2):2564–72. doi:10.1021/acsaem.1c04109
22. Li-Dong Z, Shih-Han L, Yongsheng Z, Hui S, Gangjian T, Ctirad U, et al. Ultralow thermal conductivity and high thermoelectric figure of merit in SnSe crystals. Nature (2014) 508(7496):373–7. doi:10.1038/nature13184
23. Witting IT, Chasapis TC, Ricci F, Peters M, Heinz NA, Hautier G, et al. The thermoelectric properties of bismuth telluride. Adv Electron Mater (2019) 5:1800904. doi:10.1002/aelm.201800904
24. Wudil YS, Peng Q, Alsayoud AQ, Gondal MA. Hydrostatic pressure-tuning of thermoelectric properties of CsSnI3 perovskite by first-principles calculations. Comput Mater Sci (2022) 201:110917. doi:10.1016/j.commatsci.2021.110917
25. Sales BC, Mandrus D, Williams RK. Filled skutterudite antimonides: a new class of thermoelectric materials. Science (1996) 272(5266):1325–8. doi:10.1126/science.272.5266.1325
26. Cohn JL, Nolas GS, Fessatidis V, Metcalf TH, Slack GA. Glasslike heat conduction in high-mobility crystalline semiconductors. Phys Rev Lett (1999) 82(4):779–82. doi:10.1103/PhysRevLett.82.779
27. Rahim W, Skelton JM, Scanlon DO. Ca4Sb2O and Ca4Bi2O: two promising mixed-anion thermoelectrics. J Mater Chem A (2021) 9(36):20417–35. doi:10.1039/D1TA03649A
28. Yonggang W, Ting W, Changyong P, Kenney-Benson C, Pravica M, Wenge Y, et al. Robust high pressure stability and negative thermal expansion in sodium-rich antiperovskites Na3OBr and Na4OI2. J Appl Phys (2016) 119(2):025901. doi:10.1063/1.4940020
29. Sahrowsky H, Vogt P, Hippler K, Sitla S. Crystal structure of K3OI. Z für Kristallographie - Crystalline Mater (1991) 196(1–4), pp. 193–6. doi:10.1524/zkri.1991.196.14.193
30. Yin Y, Yi M, Guo W. High and anomalous thermal conductivity in monolayer MSi2Z4 semiconductors. ACS Appl Mater Inter (2021) 13(38):45907–15. doi:10.1021/acsami.1c14205
31. Mouhat F, François-Xavier C. Necessary and sufficient elastic stability conditions in various crystal systems. Phys Rev B (2014) 90(22):224104. doi:10.1103/PhysRevB.90.224104
32. Yue T, Zhao Y, Ni J, Meng S, Dai Z. Strong quartic anharmonicity, ultralow thermal conductivity, high band degeneracy and good thermoelectric performance in Na2TlSb. npj Comput Mater (2023) 9(1):17. doi:10.1038/s41524-023-00970-4
33. Carrete J, Mingo N, Curtarolo S. Low thermal conductivity and triaxial phononic anisotropy of SnSe. Appl Phys Lett (2014) 105(10):101907. doi:10.1063/1.4895770
34. Wonshik K, Choong HK, Yeong KK, Beomyoung K, Chul K, Woobin J, et al. Electric-field-driven octahedral rotation in perovskite. npj Quan Mater (2021) 6(1):5. doi:10.1038/s41535-020-00306-1
35. Animesh B, Tanmoy G, Koushik P, Kewal SR, Kaushik K, Ajay S, et al. Intrinsically low thermal conductivity in the n-type vacancy-ordered double perovskite Cs2SnI6: octahedral rotation and anharmonic rattling. Chem Mater (2022) 34(7):3301–10. doi:10.1021/acs.chemmater.2c00084
36. Yoonseong J, Wonsik L, Seungbin H, Beom-Soo K, Seung-Jun Y, Hyejin J. Thermal transport properties of phonons in halide perovskites. Adv Mater (2023) 35(43):2204872. doi:10.1002/adma.202204872
37. Akizuki Y, Yamada I, Fujita K, Taga K, Kawakami T, Mizumaki M, et al. Rattling in the quadruple perovskite CuCu3V4O12. Angew Chem Int Ed (2015) 54:10870–4. doi:10.1002/anie.201504784
38. Luo Y, Yang X, Feng T, Wang J, Ruan X. Vibrational hierarchy leads to dual-phonon transport in low thermal conductivity crystals. Nat Commun (2020) 11:2554. doi:10.1038/s41467-020-16371-w
39. El-Sharkawy AA, Abou El-Azm AM, Kenawy MI, Hillal AS. Thermophysical properties of polycrystalline PbS, PbSe, and PbTe in the temperature range 300–700 K. Int J Thermophys (1983) 4(3):261–9. doi:10.1007/BF00502357
40. Fujita K, Mochida T, Nakamura K. High-temperature thermoelectric properties of NaxCoO2-δ single crystals. Jpn J Appl Phys (2001) 40(7R):4644. doi:10.1143/JJAP.40.4644
41. Ohtaki M, Araki K, Yamamoto K. High thermoelectric performance of dually doped ZnO ceramics. J Electron Mater (2009) 38(7):1234–8. doi:10.1007/s11664-009-0816-1
42. Ohta S, Nomura T, Ohta H, Hirano M, Hosono H, Koumoto K. Large thermoelectric performance of heavily Nb-doped SrTiO3 epitaxial film at high temperature. Appl Phys Lett (2005) 87(9):092108. doi:10.1063/1.2035889
43. Kaur T, Sinha MM. Probing thermoelectric properties of high potential Ca3PbO: an ab initio Study. IOP Conf Ser Mater Sci Eng (2021) 1033(1):012080. doi:10.1088/1757-899X/1033/1/012080
44. Chen XK, Zhu J, Qi M, Jia PZ, Xie ZX. Anomalous strain-dependent thermoelectric properties of cubic stuffed-diamond LiCu3TiQ4 (Q= S, Se). Phys Rev Appl (2025) 23(3):034085. doi:10.1103/PhysRevApplied.23.034085
45. Hongwei M, Zhong-Zhen L, Zhigang Z, Mercouri GK. Strategies and prospects for high-performance Te-free thermoelectric materials. Chem Rev (2025) 125(7):3932–75. doi:10.1021/acs.chemrev.4c00786
46. Kumar A, Bano S, Govind B, Bhardwaj A, Bhatt K, Misra DK. A review on fundamentals, design and optimization to high ZT of thermoelectric materials for application to thermoelectric Technology. J Electron Mater (2021) 50(11):6037–59. doi:10.1007/s11664-021-09153-7
47. Wen S, Jianming C, Jinyang X, Dong W, Zhigang S. Search for organic thermoelectric materials with high mobility: the case of 2,7-Dialkyl[1]benzothieno[3,2-b][1]benzothiophene derivatives. Chem Mater (2014) 26(8):2669–77. doi:10.1021/cm500429w
Keywords: first-principles, thermoelectric material, thermal transport, anti-perovskite, multi-valley phenomenon, ultra-low lattice thermal conductivity
Citation: Xiao H, Hu T, Zhou P and Li D (2025) First-principles investigations on the thermal transport and thermoelectric properties of anti-perovskite M3OI and M4OI2 (M = K, Rb). Front. Phys. 13:1619661. doi: 10.3389/fphy.2025.1619661
Received: 28 April 2025; Accepted: 20 May 2025;
Published: 19 June 2025.
Edited by:
San-Dong Guo, Xi’an University of Posts and Telecommunications, ChinaCopyright © 2025 Xiao, Hu, Zhou and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ping Zhou, emhvdXBpbmdAY3F1cHQuZWR1LmNu; Dengfeng Li, bGlkZkBjcXVwdC5lZHUuY24=