 Yuanyuan Li
Yuanyuan Li Meijun Wu
Meijun Wu Haohua Chen
Haohua Chen Dongdong Xu
Dongdong Xu Lingbo Qu
Lingbo Qu Jing Zhang
Jing Zhang Ruopeng Bai
Ruopeng Bai Yu Lan
Yu Lan- 1Department of Biological and Chemical Engineering, Chongqing University of Education, Chongqing, China
- 2Cooperative Innovation Center of Lipid Resources and Children's Daily Chemicals, Chongqing University of Education, Chongqing, China
- 3College of Chemistry and Molecular Engineering, ZhengZhou University, ZhengZhou, China
- 4School of Chemistry and Chemical Engineering, Chongqing University, Chongqing, China
- 5Department of Chemistry and Chemical Engineering, Jining University, Jining, China
Density functional theory (DFT) calculations have been performed to investigate the mechanism of alkaline-earth-metal-catalyzed hydroboration of pyridines with borane. In this reaction, the active catalytic species is considered to be an alkaline earth metal hydride complex when the corresponding alkaline earth metal is used as the catalyst. The theoretical results reveal that initiation of the catalytic cycle is hydride transfer to generate a magnesium hydride complex when β-diimine alkylmagnesium is used as a pre-catalyst. The magnesium hydride complex can undergo coordination of the pyridine reactant followed by hydride transfer to form a dearomatized magnesium pyridine intermediate. Coordination of borane and hydride transfer from borohydride to magnesium then give the hydroboration product and regenerate the active magnesium hydride catalyst. The rate-determining step of the catalytic cycle is hydride transfer to pyridine with a free energy barrier of 29.7 kcal/mol. Other alkaline earth metal complexes, including calcium and strontium complexes, were also considered. The DFT calculations show that the corresponding activation free energies for the rate-determining step of this reaction with calcium and strontium catalysts are much lower than with the magnesium catalyst. Therefore, calcium and strontium complexes can be used as the catalyst for the reaction, which could allow mild reaction conditions.
Introduction
As an important derivative of pyridine, dihydropyridine is an important raw material to synthesize natural products. For instance, dihydropyridine is extensively used to synthesize nicotinamide adenine dinucleotide, antihypertensive drugs, and anti-inflammatory agents (Karrer et al., 1937; Mauzerall and Westheimer, 1955; Bossert et al., 1981; Schramm et al., 1983; Berg et al., 2002). In addition, because dihydropyridine can provide two hydrogen atoms, it is often used as a mild and efficient reducing agent with high selectivity in catalytic hydrogenation (Adolfsson, 2005; Connon, 2007). Dihydropyridine can be prepared by dearomatization of pyridine and its derivatives. However, application of this reaction is limited by its strict reaction conditions and unstable dearomatized intermediates (Hantzsch, 1881; Stout and Meyers, 1982; Seyferth, 2009; Li Y. Y. et al., 2018). Synthesis of these compounds by homogeneous catalysis has seldom been reported (Harrod et al., 2001; Oshima et al., 2012a,b). In 1998, Harrod's research group (Hao et al., 1998) realized pyridine hydrosilylation and obtained the 1,2-dihydropyridine product with a titanocene derivative as the catalyst. In 2011, Nikonov's group (Gutsulyak et al., 2011) realized hydrosilylation of pyridine with ruthenium as the catalyst at room temperature and obtained a mixture of 1,2- and 1,4-borohydropyridine. However, application of these reactions in synthetic chemistry is greatly limited by the expensive transition metal catalyst (Yaroshevsky, 2006; Dobereiner and Crabtree, 2010; Osakada, 2011; Huang and Xia, 2015; Li et al., 2015; Qi et al., 2016, 2018; Yang et al., 2016; Yu et al., 2016; Xing et al., 2017; Liu et al., 2018; Luo et al., 2018; Zhu et al., 2018).
Alkaline-earth (Ae) metals, which are located in group IIA of the periodic table of elements, have attracted wide attention because of their low cost, availability, and abundant reserves in nature (Green et al., 2007). Ae metals, such as magnesium, calcium, strontium, and barium, are commonly used as homogeneous catalysts. Because the d0 valence electron configuration at the center of the divalent cation (Ae2+) in the Ae metal catalyst shows partial “lanthanide” characteristics, a similar catalytic cycle can be constructed in homogeneous catalysis (Li and Marks, 1996; Westerhausen, 2008; Krieck et al., 2009; Hill et al., 2016; Ma et al., 2016; Rochat et al., 2016; Rossin and Peruzzini, 2016; Xu et al., 2017a).
In the past few decades, many research groups have prepared series of organic Ae metal compounds, which have wide application prospects owing to their low cost and toxicity (Arrowsmith et al., 2011; Liu et al., 2012; Intemann et al., 2014; Schwamm et al., 2014; Hill et al., 2016; Rossin and Peruzzini, 2016). One of the most important applications is the hydrogen transfer reaction, in which Ae metal hydrides are usually used to transfer hydrogen atoms and they show unique catalytic activity (Dunne et al., 2011; Harder et al., 2011; Praneeth et al., 2012; Intemann et al., 2013; Liptrot et al., 2014; Anker et al., 2015; Weetman et al., 2016). The high catalytic activity of Ae metal hydrides can be ascribed to two factors. First, the hydrogen atoms in the Ae metal hydride have high electron density, which means that the hydrogen atoms can easily dissociate in the form of free hydride anions that exhibit Brønsted basicity. Second, Ae metal hydrides also exhibit Lewis acidity owing to the two formal positive charges carried by the metal ions (Rokob et al., 2013; Anker et al., 2014; Stepha, 2015; Hill et al., 2016). For example, the magnesium catalyst can catalyze the hydroboration reaction of borohydride compounds (pinacol—borane (HBpin), 9-borabicyclo [3.3.1]nonane (9-BBN), etc.) with various organic compounds (esters, ketones, amines, pyridines, imines, nitriles, and amides) to construct new C (hetero)–B bonds (Barrett et al., 2007; Hill et al., 2010; Arrowsmith et al., 2012; Butera et al., 2014; Schwamm et al., 2014; Lampland et al., 2015; Liptrot et al., 2015; Liu et al., 2017; Zhao et al., 2017; Jiang et al., 2018).
Hill and co-workers (Arrowsmith et al., 2011) first reported the magnesium-catalyzed hydroboration of Pyridines in 2011. In 2014, Harder's group (Intemann et al., 2014) reported hydroboration of pyridine catalyzed by the Ae metal magnesium and obtained the 1,2-selective addition compound 4 as the major product (Scheme 1). Harder et al. suggested that magnesium hydride species 6 acts as a catalyst in the catalytic cycle. Based on the experimental observations and our previous theoretical studies of Ae catalysis, there are two possible reaction pathways. Raw material pyridine 1 coordinates with magnesium in active catalytic species 6 to give intermediate 7 (Scheme 2). The C = N double bond in pyridine then inserts into the Mg–H bond to give dearomatized magnesium amino intermediate 8, which could coordinate with pinacolborane 2 to give nitrogen–boron compound 9. Finally, magnesium hydride species 6 is regenerated by hydride transfer with the release of 1,2-borohydropyridine derivative 4. Alternatively, intermediate 8 could be isomerized by 1,3-hydrogen migration to give intermediate 10, which then coordinates to pinacolborane 2 to give nitrogen–boron complex 11. After the corresponding hydride transfer, 1,4-borohydropyridine product 5 is obtained and active magnesium hydride species 6 is regenerated.
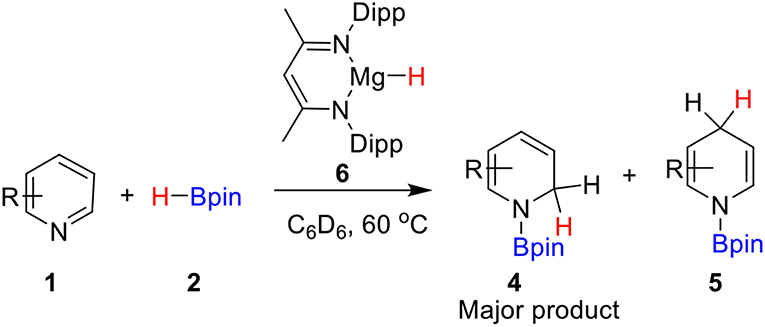
Scheme 1. Magnesium-catalyzed hydroboration of pyridines with borane.
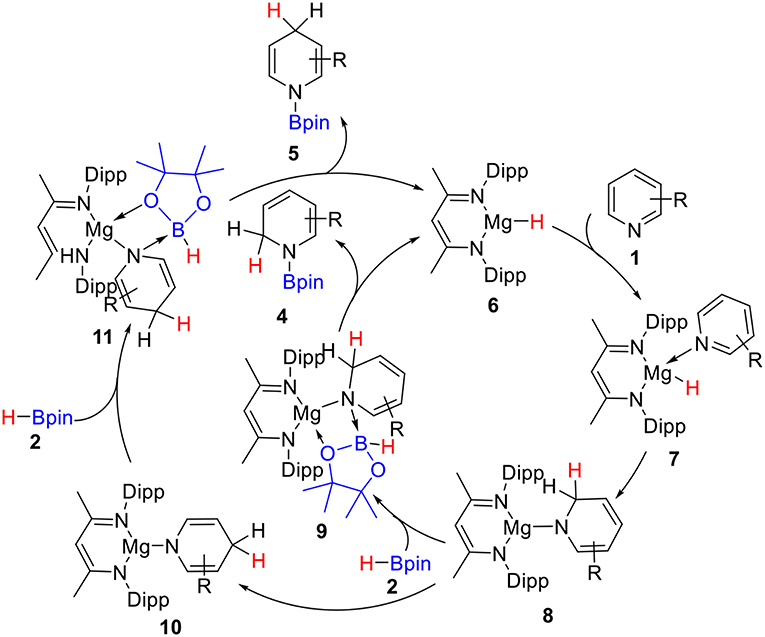
Scheme 2. Plausible mechanism for magnesium-catalyzed hydroboration of pyridines with borane.
Here, we performed a theoretical mechanistic study of Ae catalysis and determined the trend of the reactivates of Ae-catalyzed hydrogenation reactions. The pyridine hydroboration reaction catalyzed by Ae metals reported by Harder's group was used as the template reaction for the theoretical calculations. The calculations were performed based on density functional theory (DFT) and the reaction mechanism is discussed to better understand the reaction process and provide theoretical guidance for subsequent organic reactions catalyzed by Ae metals.
Computational Methods
All of the DFT calculations were performed with the Gaussian 09 software package (Frisch et al., 2013). The B3-LYP (Lee et al., 1988; Becke, 1993; Stephens et al., 1994; Adamo and Barone, 1999) functional with the standard 6-31G(d) (Hehre et al., 1972; Francl et al., 1982) basis set (SDD Dolg et al., 1987 basis set for strontium atoms) was used for the geometry optimizations. Harmonic vibrational frequency calculations were performed for all of the stationary points to determine whether they are local minima or transition structures and to derive the thermochemical corrections for the enthalpies and free energies. The M11 (Peverati and Truhlar, 2011) functional with the 6-311+G(d) basis set (SDD for strontium atoms) was used to calculate the single-point energies, because it is expected that this strategy will provide more accuracy with regard to the energetic information (Peverati and Truhlar, 2012; Zhao et al., 2012; Yu and Lan, 2013; Cui et al., 2019). The solvent effect of benzene was considered by single-point calculations of the gas-phase stationary points with the solvation model based on the density (SMD), which is a continuous model (Cances et al., 1997; Marenich et al., 2009). The reported free energies are the M11-calculated Gibbs free energies in benzene solvent based on the B3-LYP-calculated geometries with thermodynamic corrections. The geometric configurations of the key reaction intermediates and transition states were generated with CYLview software (Legault, 2009). The total energies for all of the calculated structures are listed in Supplementary Material.
Results and Discussion
Mechanism of Pyridine Hydroboration Catalyzed by Magnesium-Hydrogen Species
DFT calculations were performed for hydroboration of pyridines catalyzed by magnesium (Scheme 2). We selected β-diimine magnesium hydride 6 as the relative zero point of the Gibbs free energy (Figure 1). Pyridine 12 coordinates with active magnesium hydride species 6 to form intermediate 13. This process is exergonic by 7.9 kcal/mol, indicating that the magnesium hydride species can be stabilized as the Lewis acid by the corresponding Lewis base. After pyridine is activated by the magnesium hydride species, the C = N double bond in pyridine 12 inserts into the Mg–H bond via transition state 14-ts. the energy barrier of insertion step is 29.7 kcal/mol, indicating that this step is the rate-determining step of the catalytic cycle. The geometric structure of transition state 14-ts is shown in Figure 2. The length of the C–H bond to be formed is 1.58 Å. The length of the Mg–H bond to be ruptured is 1.91 Å. The length of the C–N bond in pyridine involved in the reaction is 1.40 Å. The above data of the geometrical structure show that formation of the C–H bond and rupture of the Mg–H bond simultaneously occur in this reaction. Intermediate 15 is obtained when C = N double insertion occurs. Because the aromaticity of pyridine in intermediate 15 is destroyed, its relative energy is 9.9 kcal/mol higher than that of intermediate 13. In addition, we also considered another route to obtain intermediate 15. With pinacolborane 2 acts as a bridge, the hydrogen atom migrates from magnesium to boron via hexatomic ring transition state 20-ts and then transfers from boron to the ortho carbon of nitrogen to give intermediate 15. The activation energy of this process is 33.5 kcal/mol, which indicates that it is a possible alternative pathway to generate intermediate 15. In intermediate 15, because the Mg–N covalent bond is formed and there is a lone pair of electrons on the nitrogen atom, it can coordinate with pinacolborane 2 to give boron–nitrogen Lewis acid–base complex 17 via low-energy transition state 16-ts. Interestingly, in intermediate 17, there are also two lone pairs of electrons on each oxygen atom in pinacolborane 2, so a coordination bond can form between oxygen and magnesium, increasing the stability of intermediate 17. The pinacolborane 2 moiety of intermediate 17 is activated by the amino group, so hydride can migrate from boron to magnesium. Theoretical calculations show that this process proceeds via transition state 18-ts with an activation free energy of only 5.3 kcal/mol. After this step, 1,2-borohydropyridine product 19 is released and active catalytic species 6 is regenerated. The complete catalytic cycle is only exothermic by 6.2 kcal/mol. This indicates a weak thermodynamic driving force, which can be attributed to the aromaticity of pyridine being destroyed. Nevertheless, a stable N–B covalent bond is generated to drive the reaction.
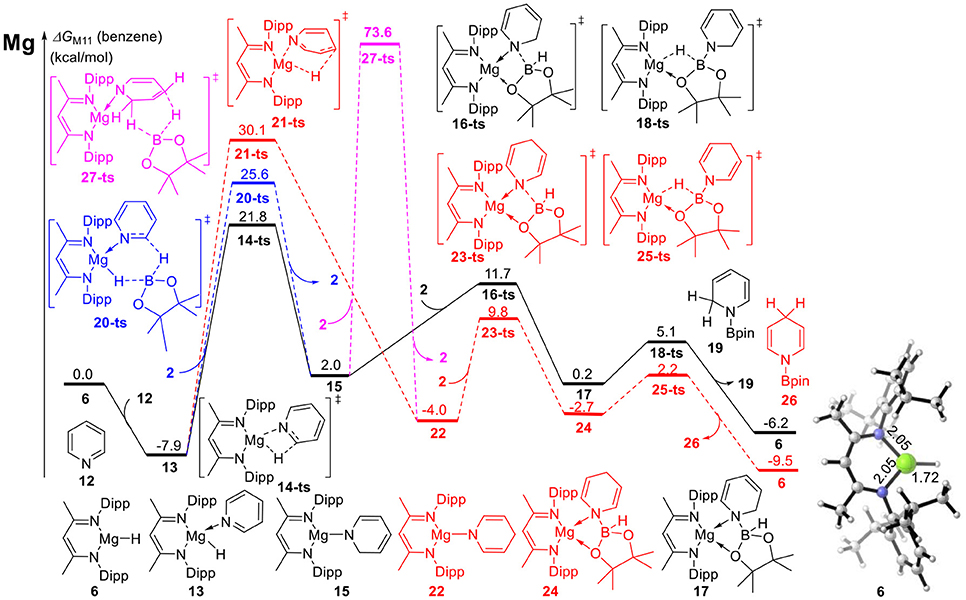
Figure 1. Free energy profile for magnesium-catalyzed hydroboration of pyridines and borane. The relative free energies (ΔG) in benzene are given in kcal/mol.
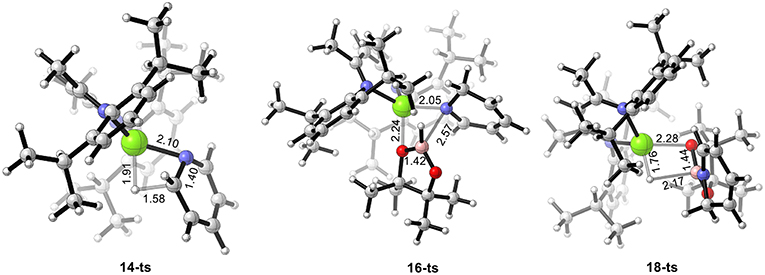
Figure 2. Geometry of transition states 14-ts, 16-ts, and 18-ts. The bond lengths are given in ångstroms.
Mechanism of Pyridine Hydroboration Catalyzed by Other Ae Metals
Harder's group only determined the catalytic activities of magnesium species. Followed these results, Harder and co-workers (Intemann et al., 2015) reported the calcium catalyzed hydrosilylation of pyridine, demonstrating that a beta-deketiminate calcium hydride is capable of faster selective 1,2-dearomatisation in 2015. According to the basic rule of the periodic law of elements and our previous theoretical studies (Gao et al., 2016; Qi et al., 2017; Xu et al., 2017a,b; Li, Y. et al., 2018) elements of the same main group often have similar chemical properties (Cotton et al., 1988). After investigating the mechanism of pyridine hydroboration catalyzed by the magnesium catalyst, we performed M11 calculations to predict the catalytic activities of other Ae metals in the group. In the pyridine hydroboration reaction, the rate-determining step is considered to be pyridine C = N bond insertion with Mg–H bond cleavage. Therefore, a weak metal–H covalent bond is favorable for this reaction. The calculated bond dissociation energies (BDEs) of Ae–H bonds are shown in Figure 3. The BDEs of Be–H, Mg–H, Ca–H, and Sr–H are 96.3, 75.5, 64.6, and 62.2 kcal/mol, respectively. Therefore, beryllium, which is in the same group as magnesium, has a small atomic radius, large steric hindrance, low metallic property, and strong Be–H bond. We believe that hydroboration cannot be catalyzed by beryllium hydride species. Furthermore, calcium and strontium could have higher catalytic activity than magnesium because of the weak metal–H covalent bonds owing to their larger atomic radii and lower electronegativities.
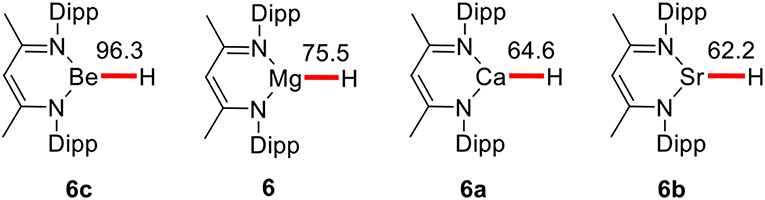
Figure 3. The value of BDEs for corresponding Ae–H bonds given in kcal/mol. Free energies (ΔG) in benzene are given in kcal/mol.
We also calculated the potential energy surfaces of the pyridine hydroboration reaction catalyzed by calcium and strontium hydride (Figure 4). In Figure 4A, active calcium hydride catalyst 6a is selected as the relative zero of the Gibbs free energy. First, calcium hydride species 6a coordinates with pyridine 12 to give intermediate 13a with a relative Gibbs free energy of 1.0 kcal/mol. The pyridine C = N bond then inserts into the Ca–H covalent bond via transition state 14a-ts. The energy barrier of this process is 17.4 kcal/mol, indicating that this step is the rate-determining step of the catalytic cycle. Compared with the corresponding reaction catalyzed by magnesium, the activation energy required for the rate-determining step decreases by 12.3 kcal/mol. Thus, we can conclude that the activity of the calcium catalyst is higher than that of the magnesium catalyst. Intermediate 15a coordinates with pinacolborane 2 via the corresponding low-energy transition state 16a-ts to give boron–nitrogen Lewis acid–base complex 17a. Finally, 1,2-borohydropyridine product 19 is released by hydride transfer with regeneration of active catalytic species 6a. The geometric structures of 14a-ts, 16a-ts, and 18a-ts are shown in Figure 5. The corresponding Ae metal transition states have similar structures.
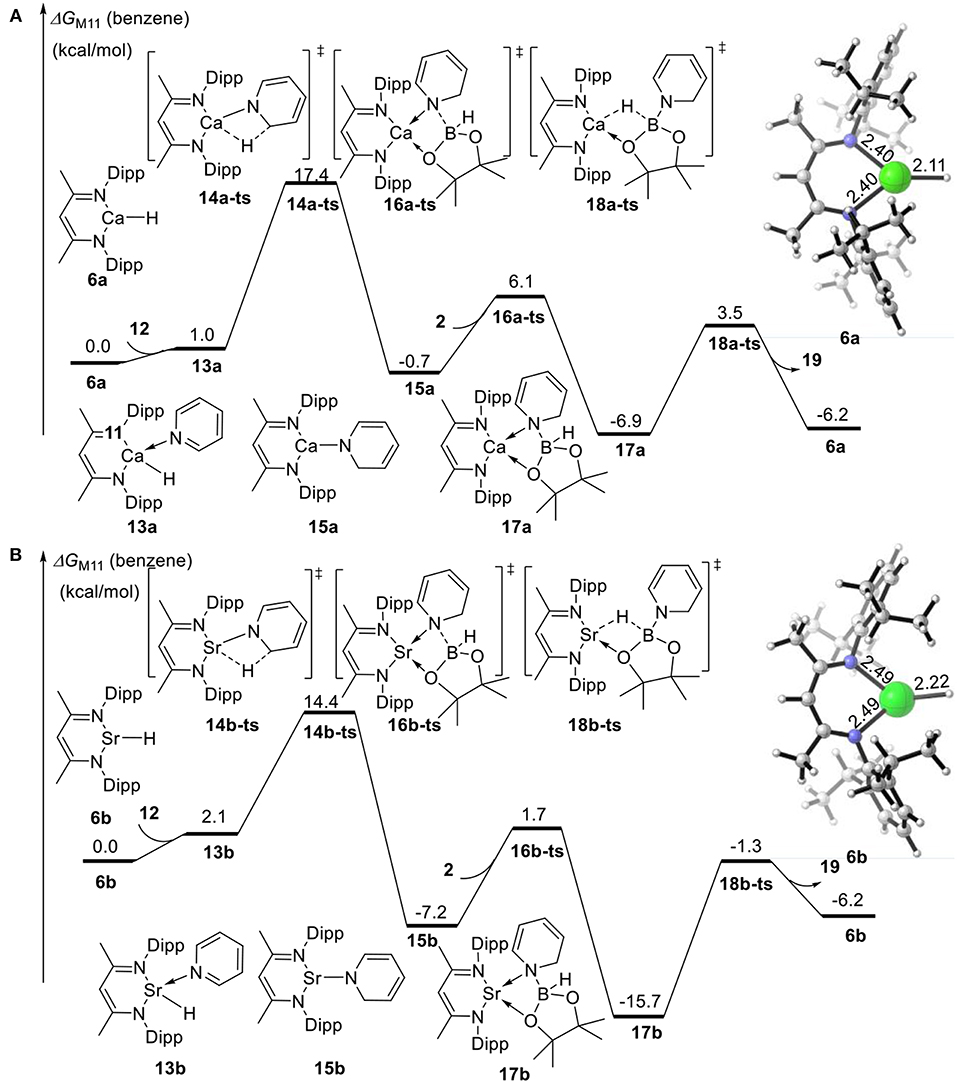
Figure 4. Free energy profile for calcium-catalyzed (A) or strontium-catalyzed (B) hydroboration of pyridines and borane. The relative free energies (ΔG) in benzene are given in kcal/mol.
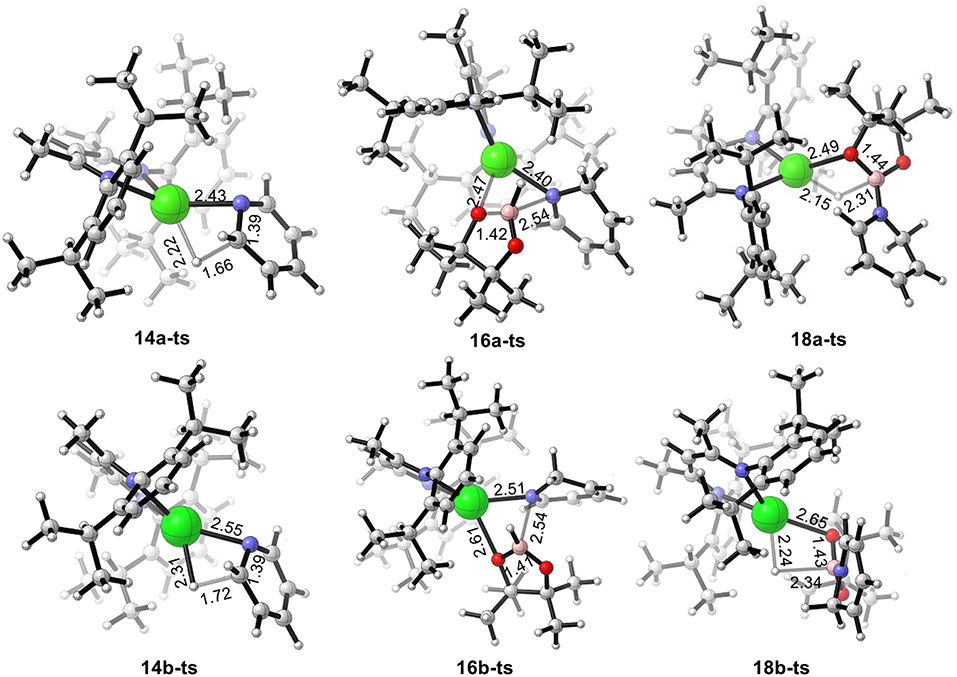
Figure 5. Geometry of transition states 14a-ts, 16a-ts, 18a-ts, 14b-ts, 16b-ts, and 18b-ts. The bond lengths are given in ångstroms.
Strontium hydride species 6b can also coordinate with pyridine 12 to give intermediate 13b, which is endergonic by 2.1 kcal/mol (Figure 4B). Intermediate 13b is unstable and prone to react. The activation energy required for C = N bond insertion into the Sr–H bond via transition state 14b-ts is 14.4 kcal/mol. However, intermediate 17b generated by coordination to pinacolborane 2 is very stable, and its free energy is 9.5 kcal/mol lower than that of the 1,2-borohydropyridine product 19 and regenerated active catalyst 6b. Therefore, the complete activation free energy for strontium catalysis of this reaction is 23.9 kcal/mol, which is 5.8 kcal/mol lower than that for magnesium catalysis, but 5.8 kcal/mol higher than that for calcium catalysis. Therefore, our theoretical calculations predict that the catalytic activities of the Ae metal complexes are calcium > strontium > magnesium.
Conclusion
The mechanism of pyridine hydroboration catalyzed by a series of Ae metals has been systematically investigated by DFT calculations. When a magnesium complex is used as the catalyst, the magnesium hydride intermediate is the active catalytic species for this reaction. According to the theoretical calculations, the reaction mechanism of this reaction is as follows. Pyridine first coordinates with the magnesium atom in the active catalyst, followed by the pyridine C = N double bond inserting into the Mg–H bond, resulting in dearomatization. The magnesium amino intermediate coordinates to pinacolborane and the magnesium hydride intermediate is regenerated by hydride transfer to release the product. The catalytic activities of various Ae metals in the same group were predicted by DFT calculations. The trend of the Ae–H BDEs is Mg–H > Ca–H > Sr–H. The theoretical calculations indicate that when a calcium catalyst is used, there is a much lower activation free energy for C = N double bond insertion into the Ca–H bond, which is the rate-determining step of the catalytic cycle. However, because the boron–nitrogen Lewis acid–base complex with the strontium catalyst is very stable, the apparent activation free energy for the strontium catalyst is higher than that for the calcium catalyst, but lower than that for the magnesium catalyst. Therefore, we believe that milder reaction conditions could be used if a calcium or strontium complex is used as the catalyst.
Author Contributions
The work was completed by cooperation of all authors. YyL, RB, and YL were responsible for the study of concept and design of the project. MW and HC searched the intermediates and transition states, JZ and DX analyzed the data and drew energy profiles. YyL, LQ, and YL drafted and revised the manuscript.
Funding
This project was supported by the Basic and Frontier Research Project of Chongqing science and technology commission (Nos. cstc2018jcyjAX0827, cstc2018jcyjAX0824, cstc2017jcyjAX0371), the Project of Science and technology collaborative innovation platform construction of Chongqing University of Education (No.2017XJPT01), The Project of Scientific and Technological Research Program of Chongqing Municipal Education Commission (No. KJQN201801603) and the National Science Foundation of China (Nos. 21822303, 21772020).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00149/full#supplementary-material
References
Adamo, C., and Barone, V. (1999). Toward reliable density functional methods without adjustable parameters: the PBE0 model. J. Chem. Phys. 110, 6158–6170. doi: 10.1063/1.478522
Adolfsson, H. (2005). Organocatalytic hydride transfers: a new concept in asymmetric hydrogenations. Angew. Chem. Int. Ed. 44, 3340–3342. doi: 10.1002/anie.200500827
Anker, M. D., Arrowsmith, M., Bellham, P., Hill, M. S., Kociok-Köhn, G., Liptrot, D. J., et al. (2014). Selective reduction of CO2 to a methanol equivalent by B(C6F5)3-activated alkaline earth catalysis. Chem. Sci. 5, 2826–2830. doi: 10.1039/C4SC00885E
Anker, M. D., Hill, M. S., Lowe, J. P., and Mahon, M. F. (2015). Alkaline-Earth-Promoted CO homologation and reductive catalysis. Angew. Chem. Int. Ed. 54, 10009–10011. doi: 10.1002/anie.201505851
Arrowsmith, M., Hadlington, T. J., Hill, M. S., and Kociok-Köhn, G. (2012). Magnesium-catalysed hydroboration of aldehydes and ketones. Chem. Commun. 48, 4567–4569. doi: 10.1039/c2cc30565h
Arrowsmith, M., Hill, M. S., and Hadlington, T. G. (2011). Kociok-Köhn, Weetman, C. Magnesium-Catalyzed Hydroboration of Pyridines. Organometallics 30, 5556–5559. doi: 10.1021/om2008138
Barrett, A. G., Crimmin, M. R. S, Hill, M., Hitchcock, P. B., and Procopiou, P. A. (2007). Calcium-Catalyzed Intermolecular Hydrophosphination. Organometallics 26, 4076–4079. doi: 10.1021/om070083t
Becke, A. D. (1993). Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652. doi: 10.1063/1.464913
Berg, J. M., Tymoczko, J. L., and Stryer, L. (2002). Biochemistry, 5th ed.; W. Philadelphia, PA: H. Freeman.
Bossert, F., Meyer, H., and Wehinger, E. (1981). 4-Aryldihydropyridines, a new class of highly active calcium antagonists. Angew. Chem. Int. Ed. 20, 762–769. doi: 10.1002/anie.198107621
Butera, V., Russo, N., and Sicilia, E. (2014). Hydrogen release from dialkylamine–boranes promoted by Mg and Ca complexes: a DFT analysis of the reaction mechanism. Chem. Eur. J. 20, 5967–5976. doi: 10.1002/chem.201304329
Cances, E., Mennucci, B., and Tomasi, J. (1997). A new integral equation formalism for the polarizable continuum model: theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 107, 3032–3041. doi: 10.1063/1.474659
Connon, S. J. (2007). Asymmetric organocatalytic reductions mediated by dihydropyridines. Org. Biomol. Chem. 5, 3407–3417. doi: 10.1039/b711499k
Cotton, F. A., Wilkinson, G., Murillo, C. A., and Bochmann, M. (1988). Advanced Inorganic Chemistry, 6th Edn. New York, NY: A Wiley-interscience publication.
Cui, C. X., Xu, D., Ding, B. W., Qu, L. B., Zhang, Y. P., and Lan, Y. (2019). Benchmark study of popular density functionals for calculating binding energies of three-center two-electron bonds. J. Comput. Chem. 40, 657–670. doi: 10.1002/jcc.25752
Dobereiner, G. E., and Crabtree, R. H. (2010). Dehydrogenation as a substrate-activating strategy in homogeneous transition-metal catalysis. Chem. Rev. 110, 681–703. doi: 10.1021/cr900202j
Dolg, M., Wedig, U., Stoll, H., and Preuss, H. (1987). Energy-adjusted ab initio pseudopotentials for the first row transition elements. J. Chem. Phys. 86, 866–872. doi: 10.1063/1.452288
Dunne, J. F., Neal, S. R., Engelkemier, J., Ellern, A., and Sadow, A. D. (2011). Tris(oxazolinyl)boratomagnesium-Catalyzed Cross-Dehydrocoupling of Organosilanes with amines, hydrazine, and ammonia. J. Am. Chem. Soc. 133, 16782–16785. doi: 10.1021/ja207641b
Francl, M. M., Pietro, W. J., Hehre, W. J., Stephen Binkley, Gordon, M. S., DeFrees, D. J., et al. (1982). Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 77, 3654–3665. doi: 10.1063/1.444267
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2013). Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT: The full author list is shown in the ESI.
Gao, B., Liu, S., Lan, Y., and Huang, H. (2016). Rhodium-catalyzed cyclocarbonylation of ketimines via C-H bond activation. Organometallics 35, 1480–1487. doi: 10.1021/acs.organomet.6b00072
Green, S. P., Jones, C., and Stasch, A. (2007). Stable Magnesium(I) Compounds with Mg-Mg Bonds. Science 318, 1754–1757. doi: 10.1126/science.1150856
Gutsulyak, D. V., van der Est, A., and Nikon, G. I. (2011). Facile catalytic hydrosilylation of pyridines. Angew. Chem. Int. Ed. 50, 1384–1387. doi: 10.1002/anie.201006135
Hantzsch, A. (1881). Condensationsprodukte aus Aldehydammoniak und ketonartigen Verbindungen. Ber. Dtsch. Chem. Ges. 14, 1637–1638. doi: 10.1002/cber.18810140214
Hao, L., Harrod, J. F., Lebuis, A. M., Mu, Y., Shu, R., Samuel, E., et al. (1998). Homogeneous catalytic hydrosilylation of pyridines. Angew. Chem. Int. Ed. 37, 3126–3129. doi: 10.1002/(SICI)1521-3773(19981204)37:22<3126::AID-ANIE3126>3.0.CO;2-Q
Harder, S., Spielmann, J., Intemann, J., and Bandmann, H. (2011). Hydrogen storage in magnesium hydride: the molecular approach. Angew. Chem. Int. Ed. 50, 4156–4160. doi: 10.1002/anie.201101153
Harrod, J. F., Shu, R. G., Woo, H., and Samuel, E. (2001). Titanocene(III) catalyzed homogeneous hydrosilation-hydrogenation of pyridines. Can. J. Chem. 79, 1075–1185. doi: 10.1139/v00-191
Hehre, W. J., Ditchfield, R., and Pople, J. A. (1972). Self-consistent molecular orbital methods. XII. further extensions of Gaussian-type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 56, 2257–2261. doi: 10.1063/1.1677527
Hill, M. S., Liptrot, D. J., and Weetma, C. (2016). Alkaline earths as main group reagents in molecular catalysis. Chem. Soc. Rev. 45, 972–988. doi: 10.1039/C5CS00880H
Hill, M. S., MacDougall, D. J., and Mahon, M. F. (2010). Group 2 promoted hydrogen release from NMe2H·BH3: intermediates and catalysis. Dalton Trans. 39, 11129–11131. doi: 10.1039/c0dt01246g
Huang, G. P., and Xia, Y. Z. (2015). Catalyst-controlled C-C σ bond cleavages in metal halide-catalyzed cycloisomerization of 3-acylcyclopropenes via a formal 1,1-halometalation mechanism: insights from quantum chemical calculations. ACS Catal. 5, 859–868. doi: 10.1021/cs501738p
Intemann, J., Bauer, H., Pahl, J., Maron, L., and Harder, S. (2015). Calcium hydride catalyzed highly 1,2-selective pyridine hydrosilylation. Chem. Eur. J. 21, 11452–11461. doi: 10.1002/chem.201501072
Intemann, J., Lutz, M., and Harder, S. (2014). Multinuclear magnesium hydride clusters: selective reduction and catalytic hydroboration of pyridines. Organometallics 33, 5722–5729. doi: 10.1021/om500469h
Intemann, J., Spielmann, J., Sirsch, P., and Harder, S. (2013). Well-defined molecular magnesium hydride clusters: relationship between size and hydrogen-elimination temperature. Chem. Eur. J. 19, 8478–8489. doi: 10.1002/chem.201300684
Jiang, Y. Y., Liu, T. T., Zhang, R. X., Xu, Z. Y., Sun, X., and Bi, S. (2018). Mechanism and rate-determining factors of amide bond formation through acyl transfer of mixed carboxylic-carbamic anhydrides: a computational study. J. Org. Chem. 83:2676–2685. doi: 10.1021/acs.joc.7b03107
Karrer, P., Ringier, B. H.J, Büchi, Fritzsche, H., and Solmssen, U. (1937). Modellversuche betreffend die wasserstoffübertragenden Wirkungsgruppen der Cofermente. Helv. Chim. Acta., 20, 55–71. doi: 10.1002/hlca.19370200106
Krieck, S., Görls, H., Yu, L., Reiher, M., and Westerhausen, M. (2009). Stable “Inverse” sandwich complex with unprecedented organocalcium(I): crystal structures of [(thf)2Mg(Br)-C6H2-2,4,6-Ph3] and [(thf)3Ca{μ-C6H3-1,3,5-Ph3}Ca(thf)3]. J. Am. Chem. Soc. 131, 2977–2985. doi: 10.1021/ja808524y
Lampland, N. L., Hovey, M., Mukherjee, D., and Sadow, A. D. (2015). Magnesium-catalyzed mild reduction of tertiary and secondary amides to amines. ACS Catal. 5, 4219–4226. doi: 10.1021/acscatal.5b01038
Lee, C., Yang, W., and Robert, G. (1988). Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 37, 785–789. doi: 10.1103/PhysRevB.37.785
Legault, C. Y. (2009). CYLView, 1.0b, Université de Sherbrooke, Canada, Available online at: http://www.cylview.org/
Li, Y., Liu, S., Qi, Z., Qi, X., Li, X., and Lan, Y. (2015). The mechanism of N-O bond cleavage in rhodium-catalyzed C-H bond functionalization of quinoline N-oxides with Alkynes: a computational study. Chem. Eur. J. 21, 10131–10137. doi: 10.1002/chem.201500290
Li, Y., Zou, L., Bai, R., and Lan, Y. (2018). Ni(I)–Ni(III) vs. Ni(II)-Ni(IV): mechanistic study of Ni-catalyzed alkylation of benzamides with alkyl halides. Org. Chem. Front. 5, 615–622. doi: 10.1039/C7QO00850C
Li, Y. W., and Marks, T. J. (1996). Diverse mechanistic pathways and selectivities in organo-F-element-catalyzed hydroamination. Intermolecular Organolanthanide-Catalyzed Alkyne and Alkene Hydroamination. Organometallics 15, 3770–3772. doi: 10.1021/om960293y
Li, Y. Y., Chen, Y., Shan, C. H., Zhang, J., Xu, D. D., Bai, R. P., et al. (2018). Recent advances in alkaline-earth-metal-catalyzed hydrofunctionalization reactions. Chin. J. Org. Chem. 38, 1885–1896. doi: 10.6023/cjoc201804031
Liptrot, D. J., Hill, M. S., and Mahon, M. F. (2014). Accessing the single-electron manifold: magnesium-mediated hydrogen release from silanes. Angew. Chem. Int. Ed. 53, 6224–6227. doi: 10.1002/anie.201403208
Liptrot, D. J., Hill, M. S., Mahon, M. F., and Wilson, A. S. (2015). Alkaline-earth-catalyzed dehydrocoupling of amines and boranes. Angew. Chem. Int. Ed. 54, 13362–13365. doi: 10.1002/anie.201505949
Liu, B., Roisnel, T., Carpentier, J. F., and Sarazin, Y. (2012). When bigger is better: intermolecular hydrofunctionalizations of activated alkenes catalyzed by heteroleptic alkaline earth complexes. Angew. Chem. Int. Ed. 51, 4943–4946. doi: 10.1002/anie.201200364
Liu, S., Qi, X. T., Qu, L. B., Bai, R. P., and Lan, Y. (2018). C-H bond cleavage occurring on a Rh(V) intermediate: a theoretical study of Rh-catalyzed arene azidation. Catal. Sci. Technol. 8, 1645–1651. doi: 10.1039/C7CY02367G
Liu, Y. X., Tang, Y. N., Jiang, Y. Y., Zhang, X. M., Li, P., and Bi, S. W. (2017). Mechanism and Origin of Et2Al(OEt)-induced chemoselectivity of nickel-catalyzed three-component coupling of one diketene and two alkynes. ACS Catal. 7, 1886–1896. doi: 10.1021/acscatal.6b03543
Luo, Y., Liu, S., Xu, D., Qu, L.-B., Luo, X., Bai, R., et al. (2018). Counterion effect and directing group effect in Rh-mediated C-H bond activation processes: a theoretical study. J. Organomet. Chem. 864, 148–153. doi: 10.1016/j.jorganchem.2018.03.016
Ma, M. T., Wang, W. F., and Yao, W. W. (2016). Synthesis of magnesium(I) complexes and their applications. Chin. J. Org. Chem. 36, 72–82. doi: 10.6023/cjoc201507015
Marenich, A. V., Cramer, C. J., and Truhlar, D. G. (2009). Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B. 113, 6378–6396. doi: 10.1021/jp810292n
Mauzerall, D., and Westheimer, F. (1955). H. 1-Benzyldihydronicotinamide-a model for reduced DPN. J. Am. Chem. Soc. 77, 2261–2264. doi: 10.1021/ja01613a070
Osakada, K. (2011). 1,4-hydrosilylation of pyridine by ruthenium catalyst: a new reaction and mechanism. Angew. Chem. Int. Ed. 50, 3845–3846. doi: 10.1002/anie.201008199
Oshima, K., Ohmura, T., and Suginome, M. (2012a). Regioselective synthesis of 1,2-dihydropyridines by rhodium-catalyzed hydroboration of pyridines. J. Am. Chem. Soc., 134, 3699–3702. doi: 10.1021/ja3002953
Oshima, K., Ohmura, T., and Suginome, M. (2012b). Dearomatizing conversion of pyrazines to 1,4-dihydropyrazine derivatives via transition-metal-free diboration, silaboration, and hydroboration. Chem. Commun. 48, 8571–8573. doi: 10.1039/c2cc34086k
Peverati, R., and Truhlar, D. G. (2011). M11-L: a local density functional that provides improved accuracy for electronic structure calculations in chemistry and physics. J. Phys. Chem. Lett. 2, 2810–2817. doi: 10.1021/jz201170d
Peverati, R., and Truhlar, D. G. (2012). Performance of the M11 and M11-L density functionals for calculations of electronic excitation energies by adiabatic time-dependent density functional theory. Phys. Chem. Chem. Phys. 14, 11363–11370. doi: 10.1039/c2cp41295k
Praneeth, V. K., Ringenburg, M. R., and Ward, T. R. (2012). Redox-Active Ligands in Catalysis. Angew. Chem. Int. Ed. 51, 10228–100234. doi: 10.1002/anie.201204100
Qi, X., Bai, R., Zhu, L., Jin, R., Lei, A., and Lan, Y. (2016). Mechanism of synergistic Cu(II)/Cu(I)-mediated alkyne coupling: dinuclear 1,2-reductive elimination after minimum energy crossing point. J. Org. Chem. 81, 1654–1660. doi: 10.1021/acs.joc.5b02797
Qi, X., Li, Y., Bai, R., and Lan, Y. (2017). Mechanism of Rhodium-Catalyzed C-H Functionalization: advances in theoretical investigation. Acc. Chem. Res. 50, 2799–2808. doi: 10.1021/acs.accounts.7b00400
Qi, X. T., Liu, X. F., Qu, L. B., Liu, Q., and Lan, Y. (2018). Mechanistic insight into cobalt-catalyzed stereodivergent semihydrogenation of alkynes: the story of selectivity control. J. Catal. 362, 25–34. doi: 10.1016/j.jcat.2018.03.016
Rochat, R., Lopez, M. J., Tsurugi, H., and Mashima, K. (2016). Recent developments in homogeneous organomagnesium catalysis. Chem. Cat. Chem. 8, 10–20. doi: 10.1002/cctc.201500853
Rokob, T. A., Bakó, I., Stirling, A., Hamza, A., and Pápai, I. (2013). Reactivity models of hydrogen activation by frustrated lewis pairs: synergistic electron transfers or polarization by electric field? J. Am. Chem. Soc. 135, 4425–4437. doi: 10.1021/ja312387q
Rossin, A., and Peruzzini, M. (2016). Ammonia–borane and amine–borane dehydrogenation mediated by complex metal hydrides. Chem. Rev. 116, 8848–8872. doi: 10.1021/acs.chemrev.6b00043
Schramm, M., Thomas, G., Towart, R., and Franckowiak, G. (1983). Novel dihydropyridines with positive inotropic action through activation of Ca2+ channels. Nature 303, 535–537. doi: 10.1038/303535a0
Schwamm, R. J., Day, B. M., Mansfield, N. E., Knowelden, W., Hitchcock, P. B., and Coles, M. P. (2014). Catalytic bond forming reactions promoted by amidinate, guanidinate and phosphaguanidinate compounds of magnesium. Dalton Trans. 43, 14302–14314. doi: 10.1039/C4DT01097C
Stepha, D. W. (2015). Frustrated lewis pairs. J. Am. Chem. Soc. 137, 10018–10032. doi: 10.1021/jacs.5b06794
Stephens, P. J., Devlin, F. J., Chabalowski, C. F., and Frisch, M. J. (1994). Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J Phys. Chem. 98, 11623–11627. doi: 10.1021/j100096a001
Stout, D. M., and Meyers, A. I. (1982). Recent advances in the chemistry of dihydropyridines. Chem. Rev. 82, 223–243. doi: 10.1021/cr00048a004
Weetman, C., Anker, M. D., Arrowsmith, M., Hill, M. S., Kociok-Köhn, G., Liptrot, D. J., et al. (2016). Magnesium-catalysed nitrile hydroboration. Chem. Sci. 7, 628–641. doi: 10.1039/C5SC03114A
Westerhausen, M. (2008). Molecular magnesium(I) compounds: from curiosity to kudos. Angew. Chem. Int. Ed. 47, 2185–2187. doi: 10.1002/anie.200705758
Xing, Y. Y., Liu, J. B., Sheng, X. H., Sun, C. Z., Huang, F., and Chen, D. Z. (2017). Solvent mediating a switch in the mechanism for rhodium(III)-catalyzed carboamination/cyclopropanation reactions between N-enoxyphthalimides and alkenes. Inorg. Chem. 56, 5392–5401. doi: 10.1021/acs.inorgchem.7b00450
Xu, D. D., Shan, C. H., Bai, R. P., and Lan, Y. (2017b). Mechanism of alkaline earth metal catalyzed hydroboration of carbodiimides: a theoretical study. Chin. J. Org. Chem. 37, 1231–1236. doi: 10.6023/cjoc201701033
Xu, D. D., Shan, C. H., Li, Y. Z., Qi, X. T., Luo, X. L., Bai, R. P., et al. (2017a). Bond dissociation energy controlled σ-bond metathesis in alkaline-earth-metal hydride catalyzed dehydrocoupling of amines and boranes: a theoretical study. Inorg. Chem. Front. 4, 1813–1820. doi: 10.1039/C7QI00459A
Yang, Y. F., Houk, K. N., and Wu, Y. D. (2016). Computational exploration of RhIII/RhV and RhIII/RhI catalysis in Rhodium(III)-Catalyzed C-H activation reactions of N-Phenoxyacetamides with Alkynes. J. Am. Chem. Soc. 138, 6861–6868. doi: 10.1021/jacs.6b03424
Yaroshevsky, A. A. (2006). Abundances of chemical elements in the Earth's crust. Geochem. Int. 44, 48–55. doi: 10.1134/S001670290601006X
Yu, Z., and Lan, Y. (2013). Mechanism of rhodium-catalyzed carbon–silicon bond cleavage for the synthesis of benzosilole derivatives: a computational study. J. Org. Chem. 78, 11501–11507. doi: 10.1021/jo402070f
Yu, Z., Qi, X., Li, Y., Liu, S., and Lan, Y. (2016). Mechanism, chemoselectivity and enantioselectivity for the rhodium-catalyzed desymmetric synthesis of hydrobenzofurans: a theoretical study. Org. Chem. Front. 3, 209–216. doi: 10.1039/C5QO00334B
Zhao, Y., Ng, H. T., Peverati, R., and Truhlar, D. G. (2012). Benchmark database for ylidic bond dissociation energies and its use for assessments of electronic structure methods. J. Chem. Theory Comput. 8, 2824–2834. doi: 10.1021/ct300457c
Zhao, Y., Zou, S. L., Jiang, Y. Y., and Bi, S. W. (2017). Theoretical study of the Cl-initiated atmospheric oxidation of methyl isopropenyl ketone. RSC Adv. 7, 52801–52811. doi: 10.1039/C7RA09445K
Keywords: alkaline-earth-metals catalyst, theoretical study, hydroboration, dihydropyridine, metal hydride complex
Citation: Li Y, Wu M, Chen H, Xu D, Qu L, Zhang J, Bai R and Lan Y (2019) Role of Alkaline-Earth Metal-Catalyst: A Theoretical Study of Pyridines Hydroboration. Front. Chem. 7:149. doi: 10.3389/fchem.2019.00149
Received: 22 December 2018; Accepted: 28 February 2019;
Published: 26 March 2019.
Edited by:
Zexing Cao, Xiamen University, ChinaReviewed by:
Zhixiang Wang, University of Chinese Academy of Sciences (UCAS), ChinaHujun Xie, Zhejiang University, China
Copyright © 2019 Li, Wu, Chen, Xu, Qu, Zhang, Bai and Lan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruopeng Bai, cnVvcGVuZ0BjcXUuZWR1LmNu
Yu Lan, bGFueXVAY3F1LmVkdS5jbg==