 Nan Zhang1,2
Nan Zhang1,2 Qiang Zhang
Qiang Zhang Feng Qiu
Feng Qiu- 1School of Chinese Materia Medica, Tianjin University of Traditional Chinese Medicine, Tianjin, China
- 2Tianjin State Key Laboratory of Modern Chinese Medicine, Tianjin University of Traditional Chinese Medicine, Tianjin, China
Phytochemical investigation of Aesculus chinensis Bge. var. chekiangensis (Hu et Fang) Fang obtained 33 triterpenoid saponins, including 14 new ones, aesculiside C–P (1–14). The structure elucidations were performed through comprehensive MS, 1D and 2D-NMR analysis, and their absolute configuration was unambiguously determined by X-ray diffraction analysis as well as Mo2(OAc)4-induced ECD method for the first time. All the substances were examined for their cytotoxic activities against three tumor cell lines, Hep G2, HCT-116, and MGC-803. Of these, compounds 8, 9, 14–16, 18, and 22 exhibited potent cytotoxicities against all cell lines with IC50 of 2–21 μM, while compounds 3, 6, 7, 17–19, 20, 24, and 28 depicted moderate activity (IC50 13 to >40 μM). On these bases, the preliminary structure-activity correlations were also discussed. Meanwhile the neuroprotective properties of triterpenoid saponins from Aesculus genus were evaluated for the first time. Among them, compounds 1, 4, 12, 20, 22, 25, 29, and 31 exhibited moderate activities against COCl2-induced PC12 cell injury.
Introduction
Aesculus chinensis Bge. var. chekiangensis (Hu et Fang) Fang is a shrubby or small tree belonging to the Hippocastanaceae family which is widely distributed in China. The dry seeds of this plant, together with Aesculus chinensis Bge and Aesculus wilsonii Rehd, are the major sources of the traditional Chinese medicine “Suo Luo Zi.” Traditionally, it has been exploited to treat chest and abdominal pain, dysentery and ague (Yang et al., 1999a; Zhang et al., 2006). Earlier phytochemical study of Aesculus chinensis Bge. var. chekiangensis (Hu et Fang) Fang obtained various types of isolates, for example, triterpenoids (Yuan et al., 2013), flavonoids (Kapusta et al., 2007), coumarins (Niu et al., 2015) together with steroids (Zhang et al., 2009). Polyhydroxylated triterpenoid saponins, isolated from Aesculus genus (Wei et al., 2004; Kim et al., 2017) with great structural diversity, have been proved to be the major bioactive principles including anticancer (Patlolla et al., 2006), neuroprotective (Cheng et al., 2016), anti-inflammatory (Matsuda et al., 1997), antioxidative (Küçükkurt et al., 2010), and antiedematous activities (Piller, 1976). As part of our continuous research to screen cytotoxic and neuroprotective compounds of this type, a series of new triterpenoids (1–14) along with 19 reported analogs (15–33) from the seeds of Aesculus chinensis Bge. var. chekiangensis (Hu et Fang) Fang were obtained. Their cytotoxic activity and neuroprotective activity were also examined. Herein, the isolation, structural elucidation, cytotoxic activity, and neuroprotective activities of these isolates are described.
Materials and Methods
General Experimental Procedures
Optical rotations were recorded on a Rudolph (Hackettstown, NJ) Autopol V automatic polarimeter. The UV spectra were acquired on a UNICO 2102PCS spectrophotometer. The IR spectra were obtained in a KBr-disc (cm−1) on a Brucker Tensor II spectrometer. NMR spectra were carried out on a Bruker (Billerica, MA) AM-600 spectrometer at 25°C referencing to the residuals of pyridine-d5. High-Resolution-ESI-MS (HR-ESI-MS) was conducted on a Waters (Milford, MA) Xevo G2-S UPLC-Q/TOF equipped with an ACQUITY UPLC BEH C18 (2.1 × 50 mm, Waters 1.7 μm, USA). Analytical HPLC was performed on a Waters e2695 system equipped with a 2998 PDA detector using a YMC- Pack-ODS-A column (250 × 4.6 mm, 5 μm). Semi-preparative HPLC was performed using a Shimadzu LC-6AD Series instrument equipped with a YMC Packed C18 column (5 μm, 250 × 10.0 mm, YMC Co., Ltd., Kyoto, Japan) and detected with a DAD detector set at 205 and 230 nm. Column chromatography (CC) was done with Sephadex LH-20 (GE Healthcare Co. Ltd., USA), ODS RP-C18 (40–75 μm Merck Darmstadt, Germany), Macroporous resin D101 (Chemical Plant of Nankai University, Tianjin, China), Silica gel (200–400 mesh, Qingdao Haiyang Chemical, China). All reagents used were of analytical grade (Concord Technology Co. Ltd., Tianjin, China).
Plant Material
Seeds of Aesculus chinensis Bge. var. chekiangensis (Hu et Fang) Fang were purchased from the Anguo Chinese medicine market (Hebei Province, P.R. China) in August 2015 and identified by professor Lijuan Zhang (Tianjin University of Traditional Chinese Medicine). The specimen was kept at the School of Chinese Materia Medica, Tianjin University of Traditional Chinese Medicine.
Extraction and Isolation
The dried seeds of A. chinensis Bge. (8.8 kg) were extracted with 70% ethanol (10 L) under reflux for three times (3 h) at 70°C. After the solvent was removed under reduced pressure at <45°C, a dark residue (2,100 g) was obtained. The residue was adsorbed onto D101 resin and then sequentially eluted with H2O, a gradient of EtOH in water to give the corresponding fractions. The 60% EtOH–H2O part was chromatographed on silica gel, eluting with a gradient of 0–100% CH2Cl2/CH3OH to yield four fractions (A–D).
Fraction B (27.0 g) was separated by an RP C18 CC (MeOH–H2O, from 20:80 to 100:0) to give 10 subfractions B1–B10. Subfraction B4 was further purified by an RP-HPLC (MeCN–H2O, 40:60, 3.0 ml/min) to obtain compounds 4 (13.2 mg, tR 21.2 min), 10 (15.6 mg, tR 23.2 min), and 24 (20.6 mg, tR 30.4 min). Further purification of Fr. B6 using preparative RP-HPLC (MeCN–H2O, 43:57, 3.0 ml/min) yielded compounds 1 (48.5 mg, tR 16.5 min) and 3 (10.0 mg, tR 19.2 min). Compounds 22 (11.1 mg, tR 8.9 min), 26 (13.5 mg, tR 14.7 min), 29 (32.0 mg, tR 23.4 min), and 33 (17.6 mg, tR 26.5 min) were obtained from Fr. B7 using a Sephadex LH-20 column and further purified by RP-HPLC (MeCN–H2O 45:55, v/v, 3.0 ml/min). Subfraction B10 was purified by preparative HPLC to afford compounds 2 (12.0 mg, tR 26.5 min), 6–9 (9.7 mg, 21.3 mg, 15.5 mg, 22.7 mg; tR 14.8 min, 16.6 min, 19,4 min, 21.2 min, respectively), and 14–16 (31.1 mg, 12.2 mg, 11.0 mg; tR 31.2 min, 32.3 min, 33.1 min, respectively) using 50% MeCN/H2O.
Fraction C (12.0 g) was subjected to an ODS RP-C18 column (MeOH/H2O, 10:90 to 100:0, v/v) to give six subfractions (C1–C6). Compound 18 (25.3 mg, tR 31.5 min) was purified by preparative HPLC using 30% MeCN/H2O from subfraction C1. Compounds 31 (11.9 mg, tR 26.5 min) and 32 (39.6 mg, tR 28.3 min) were gotten from C2 using the same preparative HPLC procedure with 32% MeCN/H2O. Fraction C4 was subjected to a Sephadex LH-20 column (MeOH) and then purified by recycling preparative HPLC with 33% MeCN/H2O to yield compounds 17 (9.8 mg, tR 22.3 min), 19 (14.5 mg, tR 24.1 min), and 28 (21.4 mg, tR 25.9 min). Fraction C6 was separated using Sephadex LH-20 (MeOH) to obtain four subfractions (C6A–C6D). C6B and C6D were purified using preparative HPLC (35% MeCN/H2O) to yield compounds 20 (59.8 mg, tR 25.4 min), 21 (40.7 mg, tR 27.2 min), and 30 (21.4 mg, tR 16.7 min).
Fraction D (9.0 g) was applied to an RP C18 CC eluting with gradient MeOH–H2O from 10:90 to 100:0 followed by a Sephadex LH-20 column (MeOH) to afford four major subfractions (D1–D4). Subfraction D1 was purified on an RP HPLC (MeCN–H2O 20:80, v/v, 3.0 ml/min) to afford compounds 2 (28.1 mg, tR 19.8 min) and 5 (30.5 mg, tR 22.4 min). Subfraction D2 was purified on an RP HPLC (MeCN–H2O 23: 77, v/v, 3.0 ml/min) to yield compounds 11 (46.0 mg, tR 16.6 min) and 27 (41.8 mg, tR 18.9 min). Compounds 12 (22.6 mg, tR 14.7 min), 13 (16.0 mg, tR 16.6 min), 23 (22.3 mg, tR 17.7 min), and 25 (11.2 mg, tR 18.3 min) were obtained by an RP HPLC (MeCN–H2O 25:75, v/v, 3.0 ml/min) from subfraction D4.
Aesculiside C (1), white amorphous powder; − 6.0 (c 0.10, MeOH); 1H NMR and 13C NMR data (see Tables 1, 3); HR-ESI-MS: m/z 1073.5149 [M–H]− (calcd. for C52H81O23, 1073.5169).
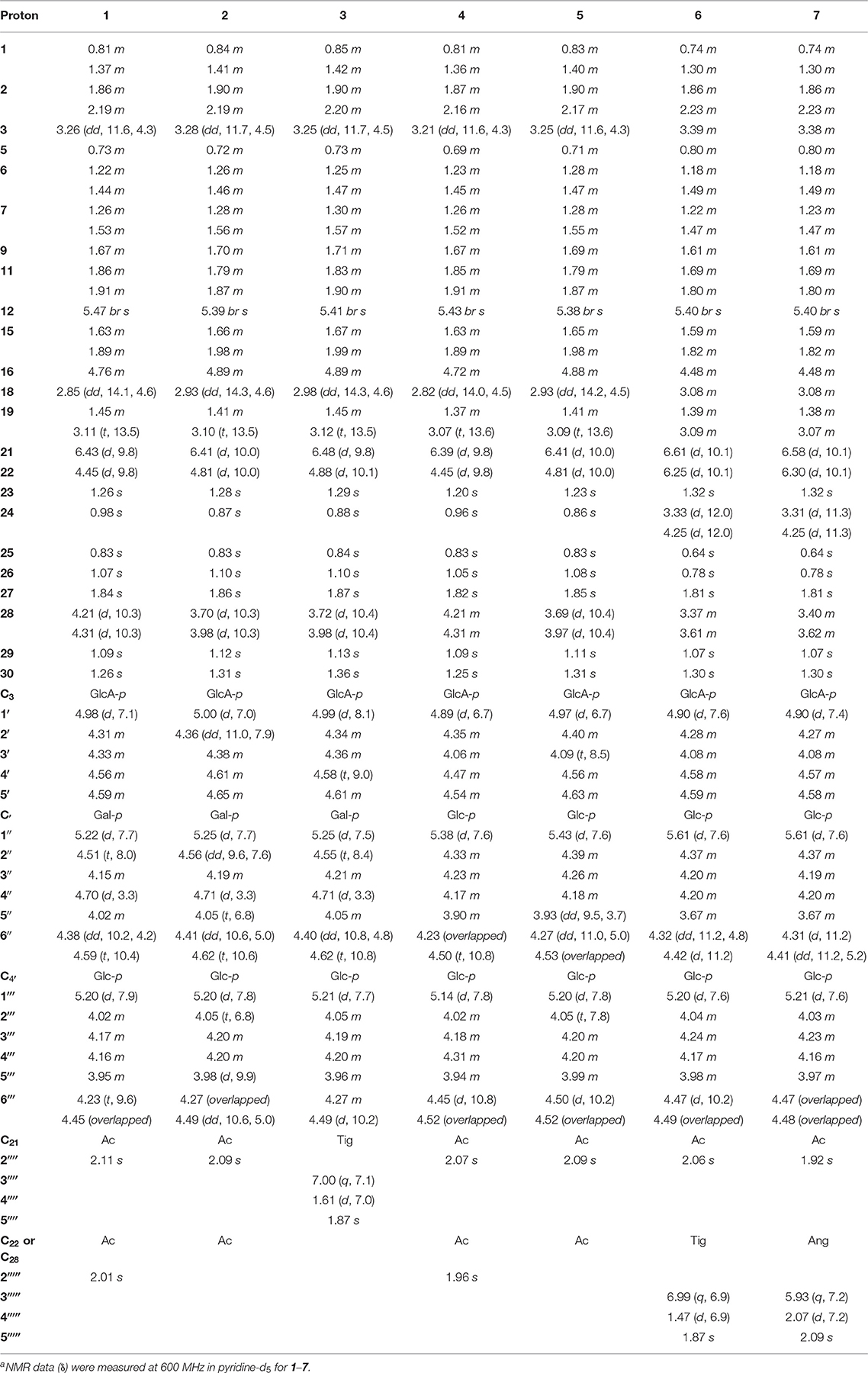
Table 1. 1H NMR spectroscopic data (δ) for compounds 1–7a (δ in ppm, J in Hz).
Aesculiside D (2), white amorphous powder; − 6.0 (c 0.10, MeOH); 1H NMR and 13C NMR data (see Tables 1, 3); HR-ESI-MS: m/z 1031.5067 [M–H]− (calcd. for C50H79O22, 1031.5063).
Aesculiside E (3), white amorphous powder; − 4.0 (c 0.09, MeOH); 1H NMR and 13C NMR data (see Tables 1, 3); HR-ESI-MS: m/z 1071.5376 [M–H]− (calcd. for C53H83O22, 1071.5376).
Aesculiside F (4), white amorphous powder; + 10.0 (c 0.11, MeOH); 1H NMR and 13C NMR data (see Tables 1, 3); HR-ESI-MS: m/z 1073.5153 [M–H]− (calcd. for C52H81O23, 1073.5169).
Aesculiside G (5), white amorphous powder; − 2.0 (c 0.11, MeOH); 1H NMR and 13C NMR data (see Tables 1, 3); HR-ESI-MS: m/z 1031.5051 [M–H]− (calcd. for C50H79O22, 1031.5063).
Aesculiside H (6), white amorphous powder; − 14.0 (c 0.11, MeOH); 1H NMR and 13C NMR data (see Tables 1, 3); HR-ESI-MS: m/z 1131.5586 [M + H]+ (calcd. for C55H87O24, 1131.5587).
Aesculiside I (7), white amorphous powder; − 12.0 (c 0.10, MeOH); 1H NMR and 13C NMR data (see Tables 1, 3); HR-ESI-MS: m/z 1129.5432 [M–H]− (calcd. for C55H85O24, 1129.5431).
Aesculiside J (8), white amorphous powder; − 7.0 (c 0.10, MeOH); 1H NMR and 13C NMR data (see Tables 2, 3); HR-ESI-MS: m/z 1171.5908 [M–H]− (calcd. for C58H91O24, 1171.5900).
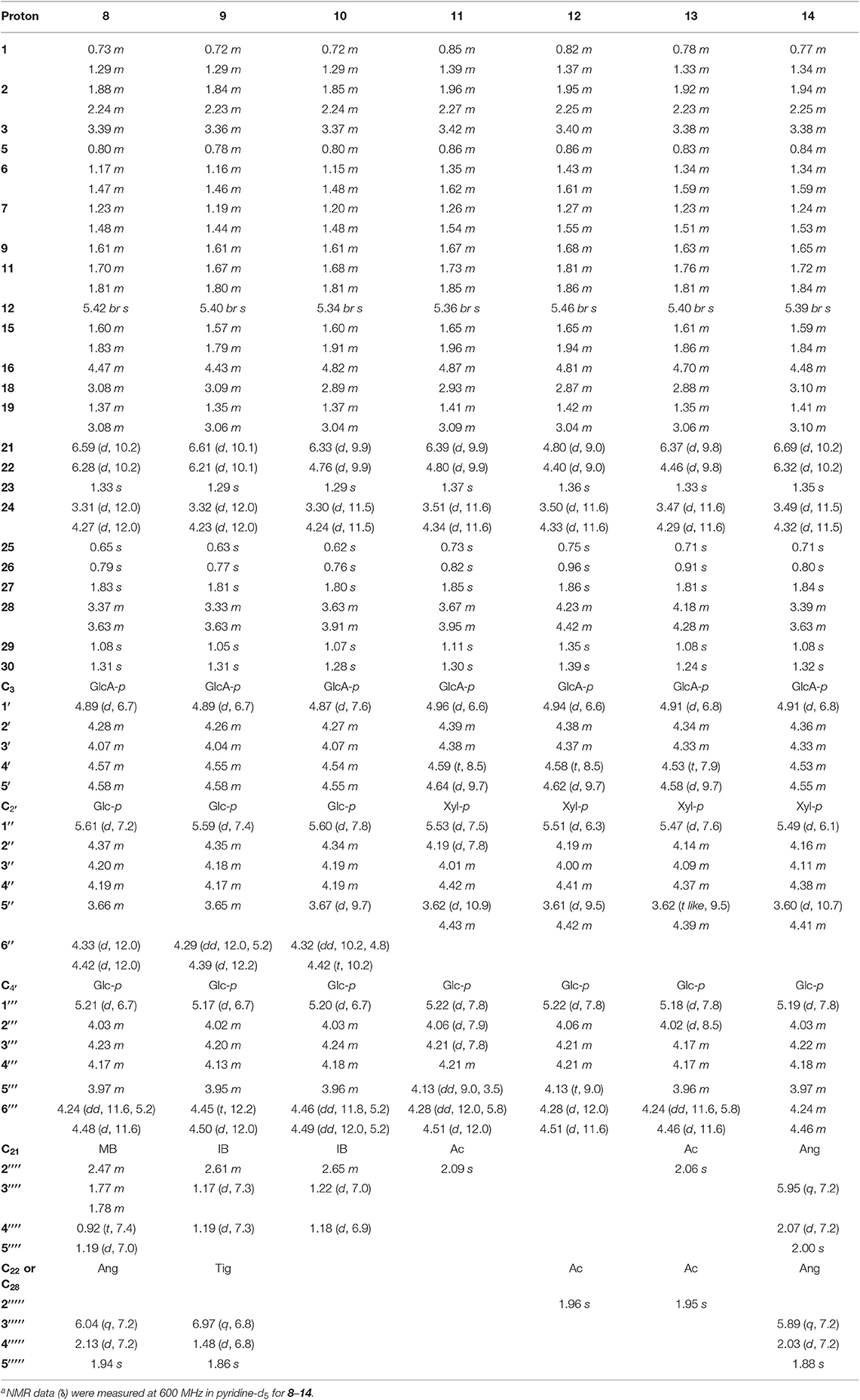
Table 2. 1H NMR spectroscopic data (δ) for compounds 8–14a (δ in ppm, J in Hz).
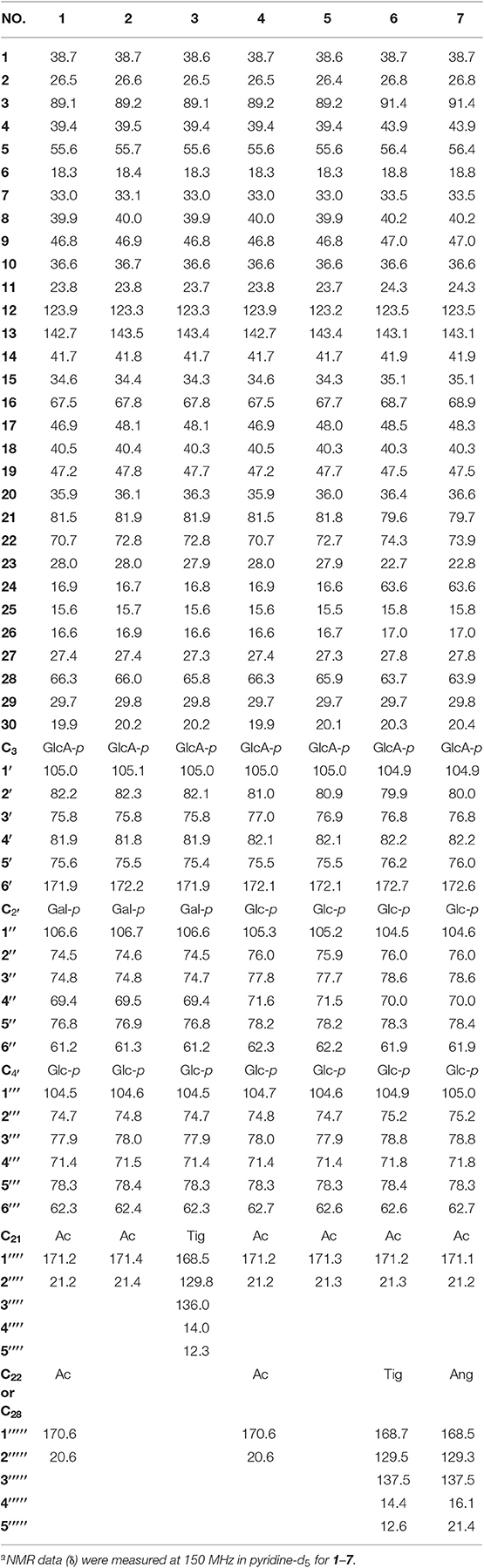
Table 3. 13C NMR spectroscopic data (δ) for compounds 1–7a (δ in ppm).
Aesculiside K (9), white amorphous powder; − 9.0 (c 0.11, MeOH); 1H NMR and 13C NMR data (see Tables 2, 4); HR-ESI-MS: m/z 1159.5879 [M + H]+ (calcd. for C57H91O24, 1159.5900).
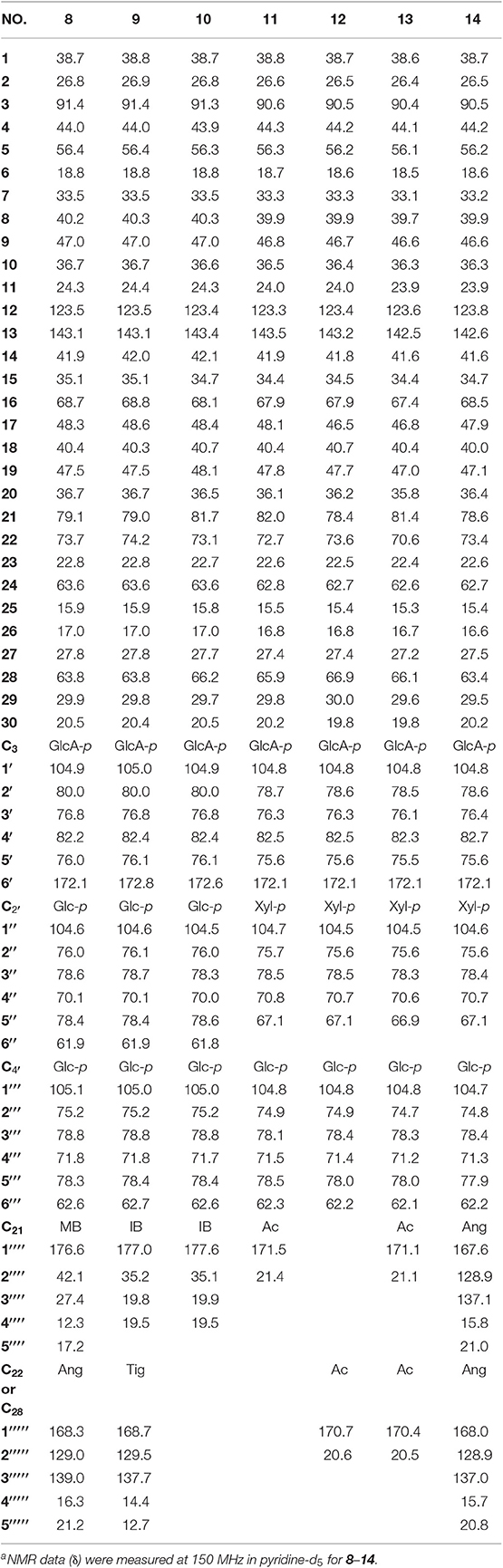
Table 4. 13C NMR spectroscopic data (δ) for compounds 8–14a (δ in ppm).
Aesculiside L (10), white amorphous powder; − 16.0 (c 0.12, MeOH); 1H NMR and 13C NMR data (see Tables 2, 4); HR-ESI-MS: m/z 1077.5471 [M + H]+ (calcd. for C52H85O23, 1077.5482).
Aesculiside M (11), white amorphous powder; + 12.0 (c 0.10, MeOH); 1H NMR and 13C NMR data (see Tables 2, 4); HR-ESI-MS: m/z 1017.4881 [M–H]− (calcd. for C49H77O22, 1017.4906).
Aesculiside N (12), white amorphous powder; + 4.0 (c 0.10, MeOH); 1H NMR and 13C NMR data (see Tables 2, 4); HR-ESI-MS: m/z 1017.4890 [M–H]− (calcd. for C49H77O22, 1017.4906).
Aesculiside O (13), white amorphous powder; + 8.0 (c 0.10, MeOH); 1H NMR and 13C NMR data (see Tables 2, 4); HR-ESI-MS: m/z 1059.4995 [M–H]− (calcd. for C51H79O23, 1059.5012).
Aesculiside P (14), white amorphous powder; − 4.2 (c 0.10, MeOH); 1H NMR and 13C NMR data (see Tables 2, 4); HR-ESI-MS: m/z 1141.5785 [M + H]+ (calcd. for C57H89O23, 1141.5795).
Hydrolysis and Determination of Absolute Configuration of Sugars
A solution of 1–14 (1.0 mg, respectively) in 2 M HCl (4.0 ml) was heated at 90°C for 2 h. The reaction mixture was extracted with EtOAc (2 × 4 ml), and the aqueous phase was evaporated to dryness using a stream of N2. The residues and authentic sugar samples (D/L-galactose, D/L-glucose, D/L-xylose, and D-glucuronic acid) were, respectively, dissolved in pyridine (1.0 ml) containing L-cysteine methyl ester (1.0 mg) and heated at 60°C for 1 h, and then o-tolyisothiocyanate (1.0 ml) was added to the mixture and heated further for 1 h. Then each reaction mixture was analyzed by the Waters e2695 HPLC system using a 2998 PDA detector (at 250 nm). Analytical HPLC was performed on the YMC- Pack-ODS-A column (250 × 4.6 mm, 5 μm) eluting with A (0.1% formic acid): B (acetonitrile) = 80:20 (v/v) at 1.0 ml/min. The absolute configuration of sugars in each compound was established by a comparison of the retention times with the standards where the time differences (Δδ D-L) of one kind of sugar were sufficient to distinguish between D- and L-enantiomers (Tanaka et al., 2007; Zhang N. et al., 2018).
Preparation of the Aglycone of Compound 14
Compound 14 (15.0 mg) in 2 M HCl (10.0 ml) was heated at 50°C for 4 h. The reaction mixture was extracted with EtOAc (2 × 10 ml), and the EtOAc phase was evaporated to dryness using a stream of N2. The residue was dissolved in THF (2.0 ml) and MeOH (1.00 ml), then NaOMe (2.00 mg, 2.2 eq) was added to the solution at 0°C. The mixture was stirred at 25°C for 4 h. The reaction was diluted with H2O (10.0 ml) and the mixture was extracted with ethyl acetate (3 × 3.00 ml). The ethyl acetate fraction was purified by a semipreparative RP HPLC (CH3CN–H2O, 45:65) to gain compound 14a.
Determination of the Absolute Configuration of the 21, 22-Diol Moieties in Compound 14a
First, Mo2(AcO)4 (1.0 mg) dissolved in DMSO (1.0 ml) was subjected to ECD measurement as blank control. Then compound 14a (0.5 mg) and Mo2(AcO)4 (1.0 mg) were added to DMSO (1.0 ml) and scanned directly. The CD spectrum was recorded every 10 min until the Mo2(AcO)4-induced circular dichroism spectrum was stationary. The inherent ECD spectrum of 14a was subtracted. The absolute configuration was elucidated by the diagnostic band at approximately 310–340 nm in the induced ECD spectrum.
X-Ray Crystallographic Analysis of 14a
Single crystals of 14a were obtained from CH3OH and H2O. The intensity data of 14a was collected on a SuperNova, Dual, Cu at zero, AtlasS2 diffractometer at 100.00(11) K. The structure was elucidated with the SHELXS method and refined based on full-matrix least-squares on F2 using SHELXL-2018/3 (Sheldrick, 2015). Crystal data for 14a: orthorhombic, space group P21212 (no. 18), a = 11.7057(7) Å, b = 34.214(3) Å, c = 14.7361(9) Å, V = 5901.7(7) Å3, Z = 8, T = 100.00(11) K, μ(Cu Kα) = 0.617 mm−1, Dcalc = 1.141 g/cm3, 20,468 reflections measured (5.166° ≤ 2Θ ≤ 148.994°), 10,735 unique (Rint = 0.0506, Rsigma = 0.0834) which were used in all calculations. The final R1 was 0.0837 (I > 2σ(I)) and wR2 was 0.2452 (all data). The crystallographic data of 14a have been deposited at the Cambridge Crystallographic Data Centre (CCDC 1957449) and the data can be obtained from supporting information (Data Sheet 1_v1).
Cytotoxicity Assay
The in vitro cytotoxicity of compounds 1–33 was measured by MTT assay (El-Readi et al., 2013; Xia et al., 2015) with 5-fluorouracil as the positive control. The human cancer cell lines, HepG2, HCT-116, and MGC-803 were purchased from ATCC. The tested cell lines were seeded in 96-well plates, and the plates were then incubated in a 37°C incubator containing 5% CO2 for 24 h. Subsequently, the tested compounds in DMSO were added to designated wells at a dosage of 3.125–50 μM. After 24 h, MTT was added to the culture medium and the absorbance at 490 nm was measured using a microplate reader.
Neuroprotective Effect Assay
The neuroprotective effects of compounds 1–33 were tested against COCl2-induced PC12 cell injury (Zou et al., 2002) with MTT method. Rat pheochromocytoma cell line (PC12) was cultured in 96-well plates with RPMI-1640 supplemented with 10% (v/v) inactivated fetal bovine serum and 100 U/ml penicillin/streptomycin. The cells were maintained at 37°C in 5% CO2 and 95% humidified air incubator. Cells were pre-treated for 2 h with or without compounds before incubation in a medium containing 1 mM CoCl2. After 24 h, MTT was added to the culture medium, and the absorbance at 490 nm was measured using a microplate reader.
Results
For the target of isolation of triterpene saponins, the 70% ethanol extracts of air-dried seeds of Aesculus chinensis Bge. var. chekiangensis (Hu et Fang) Fang were chromatographed through a D101 column and eluted with a gradient of EtOH in water. The 60% EtOH part was separated consequently as it contains abundant triterpene saponins under the guidance of UPLC-Q/TOF-MS. Thereafter, 14 undescribed triterpenoid saponins (1–14, aesculiside C–P) (Figure 1) and 19 known analogs (15–33) were afforded and identified (Figures S1–S157). The full assignments of the NMR data of compounds 1–14 are recorded in Tables 1–4.
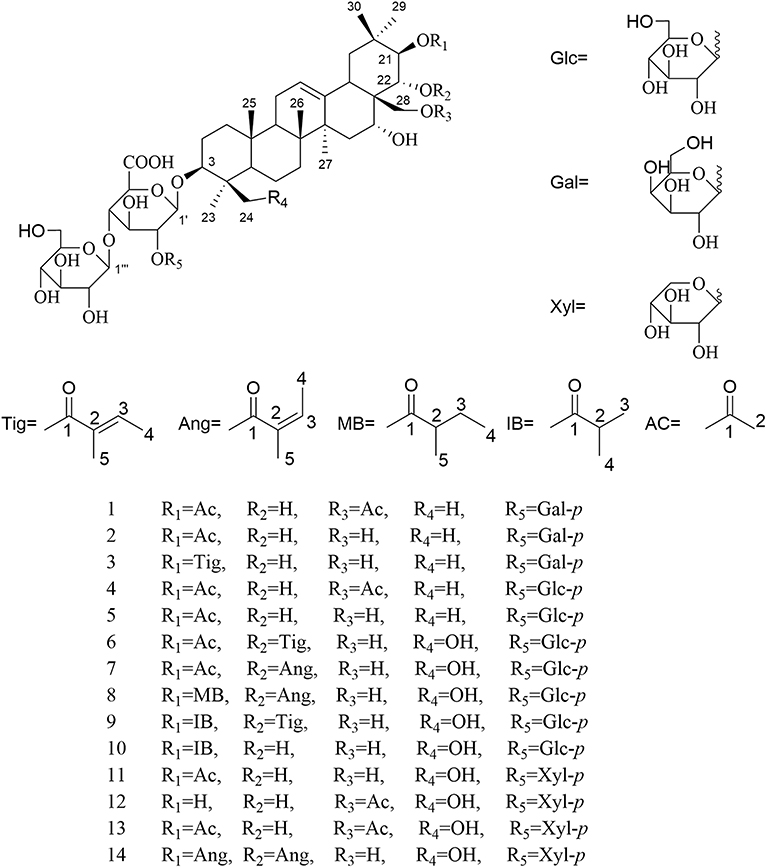
Figure 1. The structures of compounds 1–14.
Aesculiside C (1) was isolated as a white amorphous powder which exhibited an ion peak at m/z 1073.5149 [M–H]− (calcd. 1073.5169). Its molecular formula was confirmed as C52H82O23 based on HR-ESI-MS as well as 13C NMR spectroscopic data. The IR absorptions at 3,414 and 1,732 cm−1 implied the existence of the hydroxyl and carboxyl groups, respectively. The NMR data of 1 exhibited characteristic signals of a triterpenoid saponin.
1H NMR of the aglycone portion indicated the presence of seven methyl protons at δ 0.83, 0.98, 1.07, 1.09, 1.26, 1.84 (each 3H except 1.26 for 6H, s), one olefinic proton at δ 5.47 (1H, br s), and a pair of geminal protons at δ 4.21 and 4.31 (1H each, d, J = 10.3 Hz), indicative of an olean-12-ene skeleton (Zhang S. L. et al., 2018). Four oxymethine proton signals assignable to H-3, H-16, H-21, and H-22 of the aglycone moiety were, respectively, observed at δ 3.26 (1H, dd, J = 11.6, 4.3 Hz), 4.76 (1H, m), 6.43 (1H, d, J = 9.8 Hz), and 4.45 (1H, d, J = 9.8 Hz), which further suggested the aglycon characteristic for 3, 16, 21, 22, 28-pentahydroxyolean-12-ene. As for the relative configurations of C-3, C-16, C-21, C-22, and C-28, the NOESY correlations between H-3/H-5/H3-23, H-21 /H3-29 suggested the H-3 and H-21, while the correlations between H-16 and H2-28, H2-28 and H-22, H-22, and H3-30 H2-28 and H-22, H-22, and H3-30 suggested β-orientations of H-16 and H-22 (Figure 2). On the basis of NOESY correlations and the vicinal coupling constants of the H-21 3β, 16α, 21β, 22α, 22α, 28-pentahydroxyolean-12-ene.
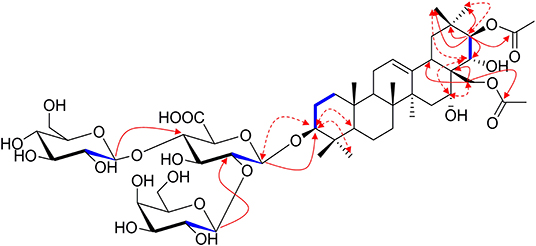
Figure 2. The selected HMBC (H → C), 1H-1H COSY (H — H) and NOESY (H ↔ H) correlations of compound 1.
The 1D NMR spectra of 1 also showed two acetyl signals (δH 2.01, 2.11 and δC 170.6, 171.2, 20.6, 21.2). The cross peak between H-21 (δ 6.43) and C-1′′′′ (δ 171.2) in the HMBC spectrum established one of the acetyl groups was attached to C-21. The other acetyl group was assigned at C-28 according to the HMBC correlation of H2-28 (δ 4.31)/C1′′′′′ (170.6), which was further confirmed by the downfield chemical shifts of H2-28 and C-28 compared with typical oleanane-type triterpenoid (Aki et al., 2004; Zhang and Li, 2007; Yuan et al., 2012) (Figure 2).
The presence of three anomeric protons at δ 4.98 (1H, d, J = 7.1 Hz), 5.20 (1H, d, J = 7.9 Hz), 5.22 (1H, d, J = 7.7 Hz) was correlated with carbons at δ 105.0, 104.5, and 106.6 in HSQC spectrum, respectively, indicating trisaccharide residues. Acid hydrolysis of 1 yielded D-galactose, D-glucose, and D-glucuronic acid, which was established with HPLC analysis by comparing with authentic sugar samples after derivatization. Their relative configuration was determined to be β according to the large coupling constants. The 1H NMR and 13C NMR signals of the trisaccharide group were fully assigned by 2D-NMR spectra and compared with reference data (Yuan et al., 2013). Meanwhile, the upfield shifts of C-3 (δ 89.1) as well as the HMBC correlation between H-1′ (δ 4.98) with C-3 demonstrated that the trisaccharide unit was attached to C-3. The sequence of the sugar chain was further confirmed by the long correlations of H-1′′ (δ 5.22) and C-2′ (δ 82.2), H-1′′′ (δ 5.20) and C-4′ (δ 81.9) (Figure 2). Based on these data, compound 1 was concluded to be 3-O-[β-D-galactopyranosyl-(1→ 2)]-β-D-glucopyranosyl-(1→ 4)-β-D-glucuronopyranosyl-21β, 28-diacetyl-3β, 16α, 21β, 22α, 28-pentahydroxyolean-12-ene, named aesculiside C.
Aesculiside D (2) with the molecular formula of C50H80O22 (m/z 1031.5067 [M–H]−; calcd. for C50H79O22, 1031.5063) was also obtained as a white, amorphous powder. Acid hydrolysis of 2 presented the same sugar moieties as compound 1. The NMR data of 2 are similar to those of 1 except for the absence of an acetyl unit in 2. The essential HMBC correlations of H-21 (δ 6.41)/C-1′′′′ (δC 171.4) indicated the acetyl unit was connected at C-21 (Figure 3). The remaining portion of 2 was superposable to 1 evidenced by careful analysis of their 2D NMR spectra. Thus, compound 2 was established as 3-O-[β-D-galactopyranosyl-(1→ 2)]-β-D-glucopyranosyl-(1→ 4)-β-D- glucuronopyranosyl-21β-acetyl-3β, 16α, 21β, 22α, 28-pentahydroxyolean-12-ene, namely aesculiside D.
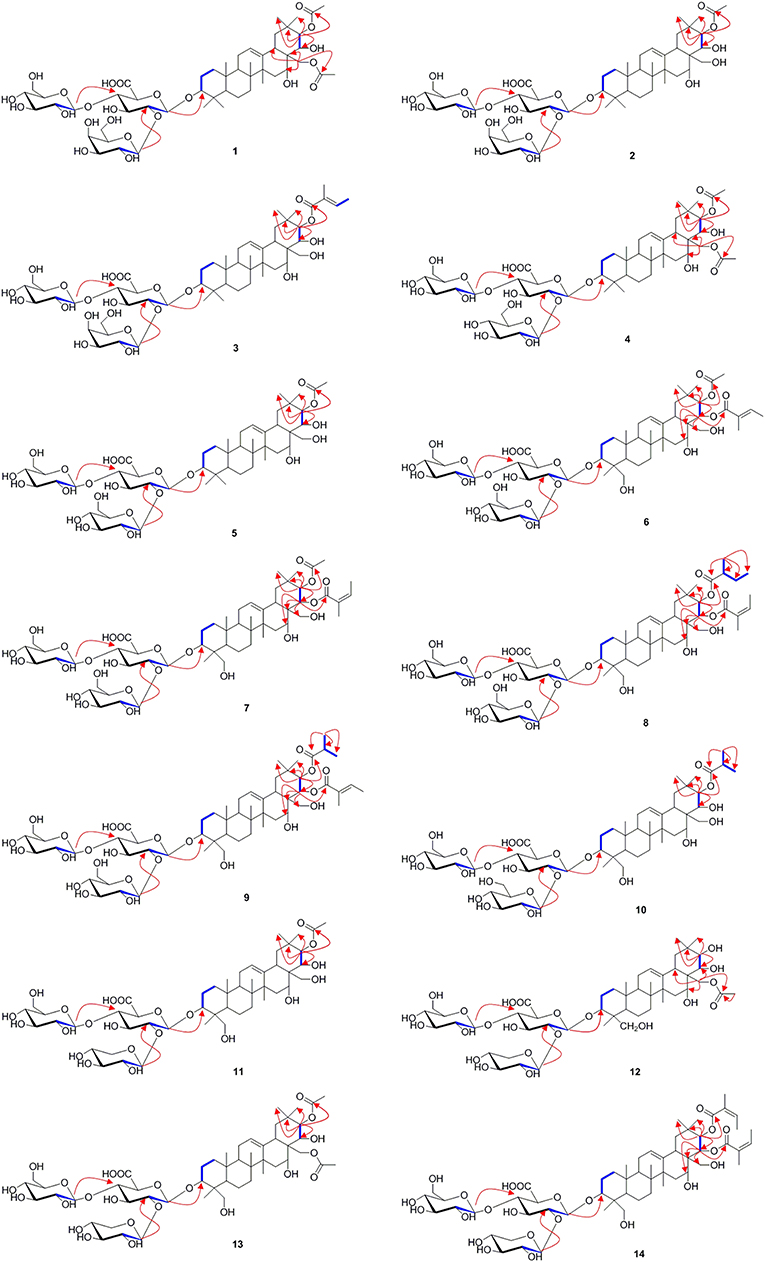
Figure 3. The selected HMBC (H → C) and 1H-1H COSY (H — H) correlations of compounds 1–14.
Aesculiside E (3) was acquired as a white amorphous powder and its molecular formula was determined as C53H84O22 (m/z 1071.5376 [M–H]−; calcd. for C53H83O22, 1071.5376). Acid hydrolysis of 3 yielded D-galactose, D-glucose, and D-glucuronic acid. The NMR data of 3 showed a lot of resemblance with those of 2 except for the presence of a tigloyl moiety instead of an acetyl unit in 3, which was supported by the characteristic olefinic quartet at δ 7.00 in its 1H-NMR spectrum. Moreover, the HSQC correlation signals of δH 1.61 with δC 14.0 and δH 1.87 with δC 12.3 confirmed the existence of a tigloyl group. The aforementioned data, together with the HMBC correlation from H-21 (δ 6.48) to C-1′′′′ (δ 168.5) confirmed the connection between the tigloyl group and C-21. Consequently, it was assigned as 3-O-[β-D-galactopyranosyl-(1→ 2)]-β-D-glucopyranosyl-(1→ 4)-β-D-glucuronopyranosyl-21β-tigloyl-3β, 16α, 21β, 22α, 28-pentahydroxyolean-12-ene.
Aesculiside F (4) and 1 gave the same molecular formula, deduced as C52H82O23 from its HR-ESI-MS and 13C NMR spectroscopic data. Comparison of the NMR data of 4 with those of 1 indicated that both saponins are closely related, differing at trisaccharide moiety where the galactose in 1 was replaced by a glucose in 4, based on the distinction of their 13C NMR data (Table 3) (Yoshikawa et al., 1998), which was further verified by hydrolysis and derivatization as aforementioned. HMBC correlations revealed the position and sequences of the sugar moiety in 4 as described before. Hence, compound 4 was identified and named aesculiside F.
Aesculiside G (5), a white amorphous powder, was established to be an analog of 4 by HRESIMS and NMR spectrum interpretation. Careful analysis of their NMR data suggested that 4 possessed one more acetyl group compared to 5. The key HMBC correlation from H-21 (δ 6.41) to C-1′′′′ (δ 171.3) suggested that the acetyl group was connected to C-21. The sugar residues in 5 was determined to be the same with those in 4 applying the method as before described. Accordingly, aesculiside G (5) was identified as 3-O-[β-D-glucopyranosyl-(1→ 2)]-β-D-glucopyranosyl-(1→ 4)-β-D-glucuronopyranosyl-21β-acetyl-3β, 16α, 21β, 22α, 28-pentahydroxyolean-12-ene.
Aesculiside H (6) and Aesculiside I (7) owned the same molecular formula of C55H86O24, according to their HR-ESI-MS data. The similar NMR spectra of 6 and 7 (Tables 1, 3) to those of 1–5 indicated that 6 and 7 are structural analogs of these compounds. The 1H NMR spectra of the aglycone portion of both compounds exhibited six tertiary methyl groups at δ 0.64 (Me-25), 0.78 (Me-26), 1.07 (Me-29), 1.30 (Me-30), 1.32 (Me-23), and 1.81 (Me-27). The absence of the characteristic singlet at δH 0.86 and δC 16.6 attributable to Me-24 in 5 and the additional resonances at δC 63.6 promoted that Me-24 could be oxygenated. This was corroborated by the key HMBC cross-peaks from H-24 (δ 4.25) to C-3 (δ 91.4), C-5 (δ 56.4). Thus, the structure of the aglycone of 6 and 7 was assigned as 3β, 16α, 21β, 22α, 24, 28-hexahydroxyolean-12-ene. Detailed NMR analysis disclosed that 6 and 7 possessed the same acyl group (acetyl) link to C-21 as that in 5. The 1H and 13C NMR spectra of 6 displayed characteristic signals of a tigeloyl group (Tables 1, 3). The HMBC correlations from H-22 (δ 6.25) to C-1′′′′′ (δ 168.7) and from H-21 (δ 6.61) to C-1′′′′ (δ 171.2) provided definitive evidence that the acetyl was substituted at C-21, the tigeloyl group was substituted at C-22 in 6. The connection of an angeloyl group to C-22 in 7 was validated by the HMBC correlations from H-22 (δ 6.30) to C-1′′′′′ (δ 168.5) and H-21 (δ 6.58) to C-1′′′′ (δ 171.1). The trisaccharide chain of 6 and 7 was the same as that of 5 as determined by the same method as mentioned before. Thus, the chemical structures of compounds 6 and 7 have been elucidated and named aesculiside H (6) and aesculiside I (7).
Aesculiside J (8) was assigned as C58H92O24 based on the [M–H]− ion peak at m/z 1171.5908. Acid hydrolysis suggested that D-glucose and D-glucuronic acid existed in 8. Comparison of the NMR spectroscopic data with those of compound 7 showed many similarities, except for the appearance of a 2-methylbutyryl moiety instead of the acetyl group in 7, which was ascertained by the COSY correlations between H-2′′′′ and H-3′′′′, H-4′′′′ coupled with HMBC correlation of H-21 (δ 6.59)/C-1′′′′ (δ 176.6) and H-22 (δ 6.28)/C-1′′′′′ (δ 168.3). Thus, the structure of aesculiside J (8) was elucidated as 3-O-[β-D-glucopyranosyl-(1→ 2)]-β-D-glucopyranosyl-(1→ 4)-β-D-glucuronopyranosyl-21β-methylbutyryl-22α-angeloyl-3β, 16α, 21β, 22α, 24, 28-hexahydroxyolean-12-ene.
The elemental formula of aesculiside K (9) was confirmed as C57H90O24 by its HRESIMS data. The NMR data of 9 closely resembled those of 6 with the striking difference of the acetyl group signals at δC 171.2 and 21.3 in 6 replaced by an isobutyryl group signals at δC 177.0, 35.2, 19.8 and 19.5. COSY correlations of H-2′′′′/H-3′′′′, H-2′′′′/H-4′′′′ in conjunction with the HMBC cross-peaks of H-3′′′′ with C-1′′′′, C-2′′′′ and C-4′′′′ verified the existence of the isobutyryl group. HMBC correlations from H-21 (δ 6.61) to carbonyl carbon (δ 177.0) of the isobutyryl group, and from H-22 (δ 6.21) to carbonyl carbon (δ 168.7) of the tigeloyl group supported the attachment of the two acyl units to C21 and C22, respectively. Thus, the structure of aesculiside K (9) was fully elucidated as 3-O-[β-D-glucopyranosyl-(1→ 2)]-β-D-glucopyranosyl-(1→ 4)-β-D-glucuronopyranosyl-21β-isobutyryl-22α-tigeloyl-3β, 16α, 21β, 22α, 24, 28-hexahydroxyolean-12-ene.
Aesculiside L (10) had molecular formula of C52H84O23 established through its [M + H]+ ion peak at 1077.5471 and its NMR data. The sugar chain in 10 was same as 9, using the same method as described before. Analysis of the 1H and 13C NMR spectroscopic data (Tables 2, 4) of 10 revealed a close structural resemblance to 9, except for the absence of a tigeloyl group in 9. The key HMBC correlations of H-21 to C-17, C-29, C-30, and C-1′′′′; and of H-22 to C-18, C-20 supported this deduction. Finally, the structure of 10 was proved and named aesculiside L.
According to the [M–H]− ion peak at m/z 1017.4881 and its NMR data, the molecular formula of 11 was established to be C49H78O22, which is 30 mass units less than that of the known compound, aesculusosides C (27). Detailed comparison of the NMR spectroscopic data between 11 and 27 revealed that they shared the same aglycone and C-21 substituent. D-xylose, D-glucose, and D-glucuronic acid were gained from acid hydrolysis of 11. Further NMR analysis of the sugar portion suggested that the β-D-xylopyranose in 11 took the place of the β-D-glucose group substituent at C-2′ in 27. Further confirmation was carried out by the significant cross peak: xyl-H-1 (δH 5.53) with glcA-C-2 (δC78.7) in HMBC spectrum. Accordingly, aesculiside M (11) was unambiguously identified as 3-O-[β-D-xylopyranosyl-(1→ 2)]-β-D-glucopyranosyl-(1→4)-β-D-glucuronopyranosyl-21β-acetyl-3β, 16α, 21β, 22α, 24, 28-hexahydroxyolean-12-ene.
The molecular formula of aesculiside N (12) was calculated as C49H78O22 by virtue of its HR-ESI-MS spectrum. Its 1H NMR spectrum exhibited six singlet methyl protons [δH 0.75, 0.96, 1.35, 1.36, 1.39, 1.86] along with an olefinic proton at δH 5.46. The aforementioned spectroscopic data with its 13C-NMR data (Table 4) for the aglycone portion showed a close resemblance to those of protoaescigenin (Konoshima and Lee, 1986). The relative configuration of C-21 and C-22 was established to be 21β and 22α on the basis of the NOESY correlations of H-21/H3-29, H2-28/H-22/H-30 and the vicinal coupling constants of the H-21 and H-22 (J = 9.0 Hz). We also observed the presence of an acetyl moiety [δH 1.96 (3H, s); δC 20.6, 170.7] which was attached to C-28 due to the HMBC correlation from H-28 to the carbonyl carbon of the acetyl group. The resonances (1D and 2D NMR) of the sugar moieties and the results of hydrolysis of 12 revealed that 12 and 11 possessed the same trisaccharide chain at aglycone C-3. Thus, the structure of aesculiside N (12) was affirmed as 3-O-[β-D-xylopyranosyl-(1→ 2)]-β-D-glucopyranosyl-(1→ 4)-β-D-glucuronopyranosyl-28-acetyl-3β, 16α, 21β, 22α, 24, 28-hexahydroxyolean-12-ene.
Aesculiside O (13) possessed the molecular formula of C51H80O23 based on its HR-ESI-MS data. D-glucuronic acid, D-xylose, and D-glucose were afforded from 13 via the same procedure as before. The side-by-side analysis of the NMR spectroscopic resonances (Tables 2, 4) between 13 and 12 revealed that these two compounds owned similar structural features, with the only difference being due to an additional acetyl group connected with C-21 in 13. HMBC correlations from H-21 (δ 6.37) to ester carbonyl (δ 171.1) of the acetyl unit confirm this proposal. Hence, the structure of 13 was elucidated as 3-O-[β-D-xylopyranosyl-(1→ 2)]-β-D-glucopyranosyl-(1→ 4)-β-D-glucuronopyranosyl-21β, 28-diacetyl-3β, 16α, 21β, 22α, 24, 28-hexahydroxyolean-12-ene.
The HR-ESI-MS of aesculiside P (14) yielded a [M + H] + ion with m/z 1141.5785, consistent with a molecular formula of C57H88O23 (calcd. for C57H89O23, 1141.5795). Analysis of its NMR data (Tables 2, 4) implied the identical trisaccharide chain to 13, which is also established by acid hydrolysis results. The 1H NMR spectrum of its aglycone showed six tertiary methyls: δH 0.71 (Me-25), 0.80 (Me-26), 1.08 (Me-29), 1.32 (Me-30), 1.35 (Me-23), and 1.84 (Me-27); one olefinic proton: δH 5.39 (br s) and a pair of oxygenated methine protons: δH 6.69 and 6.32 (each 1H, d, J = 10.2 Hz). Meanwhile, the 1D-NMR spectra of 14 exhibited typical resonances of two angeloyl groups (Tables 2, 4) and the observed HMBC correlations of H-21/ C-1′′′′ and H-22/ C-1′′′′′ provided definitive evidence of their position. The relative configuration of 14 was established via NOESY experiment. The correlations between H-3/H-5/H3-23, H-21/H3-29 suggested the α-orientations of H-3 and H-21, the correlations between H-16/H2-28/H-22/H3-30 reminded β-orientations of H-16 and H-22. To further confirm its absolute configuration, we made many attempts. Owing to the amount of 14, its aglycone (14a) was easily obtained by hydrolyzation and the absolute configuration of C-21 and C-22 in 14a was determined by Mo2(AcO)4-induced CD. As shown in Figure 4, the ICD exhibited a negative cotton effect at 313 nm, suggesting the R configuration of C-21, according to the Snatzke rule (Snatzke et al., 1981; Di Bari et al., 2001). Fortunately, a single crystal of 14a was obtained and suitable for X-ray crystallographic analysis (Figure 5). The Flack parameter of 0.27 (14) allowed an unambiguous assignment of the absolute configuratihon of 14a. Based on these data, compound 14 was undoubtedly identified as 3-O-[β-D-xylopyranosyl-(1→ 2)]-β-D-glucopyranosyl-(1→ 4)-β-D-glucuronopyranosyl-21R, 22R-diangeloyl-3S, 16R, 21R, 22R, 24, 28-hexahydroxyolean-12-ene.
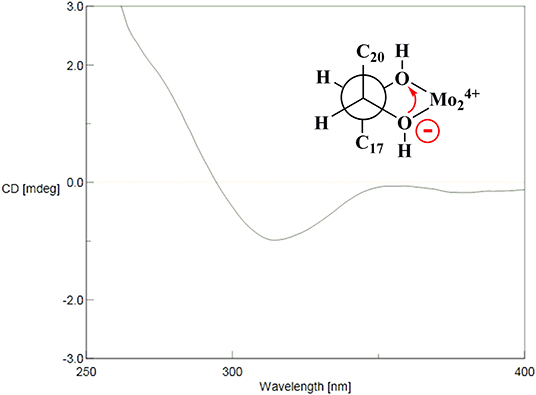
Figure 4. Mo2(OAc)4-induced CD (ICD) spectra of 14a.
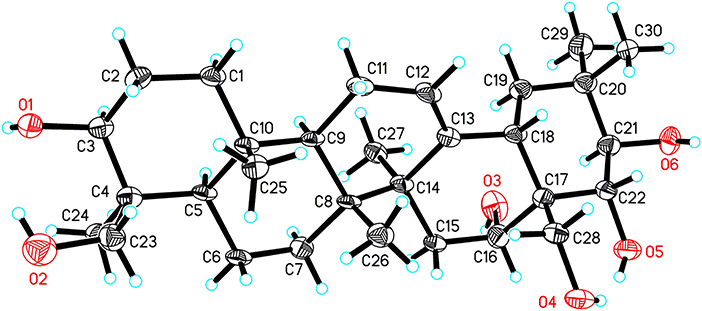
Figure 5. ORTEP drawing of 14a.
The absolute configurations of the aglycones of 1–13 were all deduced to be 3S, 16R, 21R, 22R based on the absolute configuration of 14 and their mutual biogenetic source.
Additionally, the 19 known compounds were identified (Figure S100) as 3-O-[β-D-galactopyranosyl-(1→ 2)]-β-D-glucopyranosyl-(1→ 4)-β-D-glucuronopyranosyl-21β, 22α-ditigeloyl-3β, 16α, 21β, 22α, 22α, 28-pentahydroxyolean-12-ene (15) (Kameyama and Fujimura, 2009), 3-O-[β-D-glucopyranosyl-(1→ 2)]-β-D-glucopyranosyl-(1→ 4)-β-D-glucuronopyranosyl-21β, 22α-diangeloyl-3β, 16α, 21β, 22α, 24, 28-hexahydroxyolean-12-ene (16), escin Ia (17), escin Ib (18), isoescin Ia (18), isoescin Ia (19), isoescin Ib (20) (Zhang et al., 1999), isoescin IIb (21) (Yang et al., 1999b), escin IIIa (22) (Yoshikawa et al., 1996), escin IV (23), escin V (24) (Yoshikawa et al., 1998), aesculusosides A-C (25-27) (Cheng et al., 2018), aesculioside A-B (28, 29) (Zhang et al., 1999), aesculiside A (30) (Cheng et al., 2018), desacylescin I (31) (Cheng et al., 2016), desacylescin II (32) (Yoshikawa et al., 1996), deacetylescinIIb (33) (Kimura et al., 2006) by comparisons of their spectroscopic data with reported values.
The cytotoxic activities against three human cancer cell lines (Hep G2, HCT-116, and MGC-803) of compounds 1–33 were evaluated using the MTT method, with 5-fluorouracil (5-FU) as positive control (Table 5). Among them, compounds 8, 9, 14–16, 18, 22 showed potent cytotoxicity against all the tested human cancer cell lines with IC50 ranging between 2 and 21 μM. Compounds 3, 6, 7, 17–19, 20, 24, 28 were less active (IC50: 13 to >40 μM) whereas the other isolates displayed no toxicity in all cell lines at 50 μM. These results suggested that the compounds with acylations at both C-21 and C-22 exhibited stronger inhibitory activities than those with acylations at C-21 and C-28 or only at C-21. In addition, it seems that the presence of the tigloyl, angeloyl, methylbutyryl, and isobutyryl groups affects the inhibitory activity of these compounds on the tested cell lines positively.
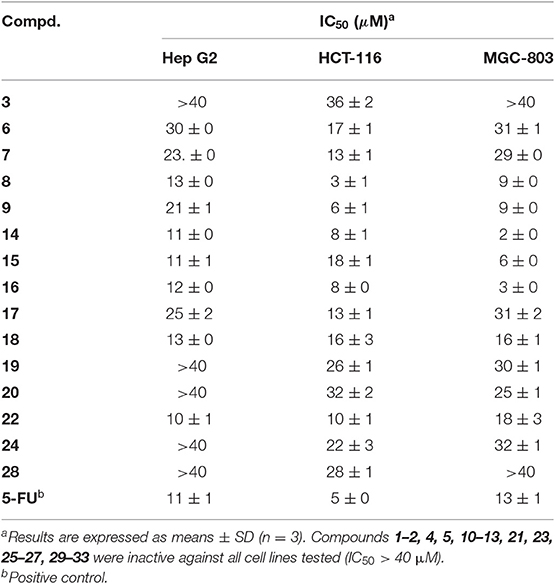
Table 5. Cytotoxic activity of compounds 1–33 by the MTT method.
To examine the neuroprotective effect, the cytotoxic activity of compounds 1–33 against PC12 cell line was first evaluated. Among them, compounds 6–9, 14–16, 18, 22 showed no obvious cytotoxic effects on PC12 cells at a dose of 5 μM, while others at 10 μM. Next, 5 μM compounds 6–9, 14–16, 18, 22 and 10 μM others were tested for their neuroprotective properties against COCl2-induced toxicity in PC12 cells with trolox as the positive control. Among these, compounds 1, 4, 12, 20, 22, 25, 29, 31 exhibited moderate activities against COCl2-induced PC12 cell injury (Figure 6).
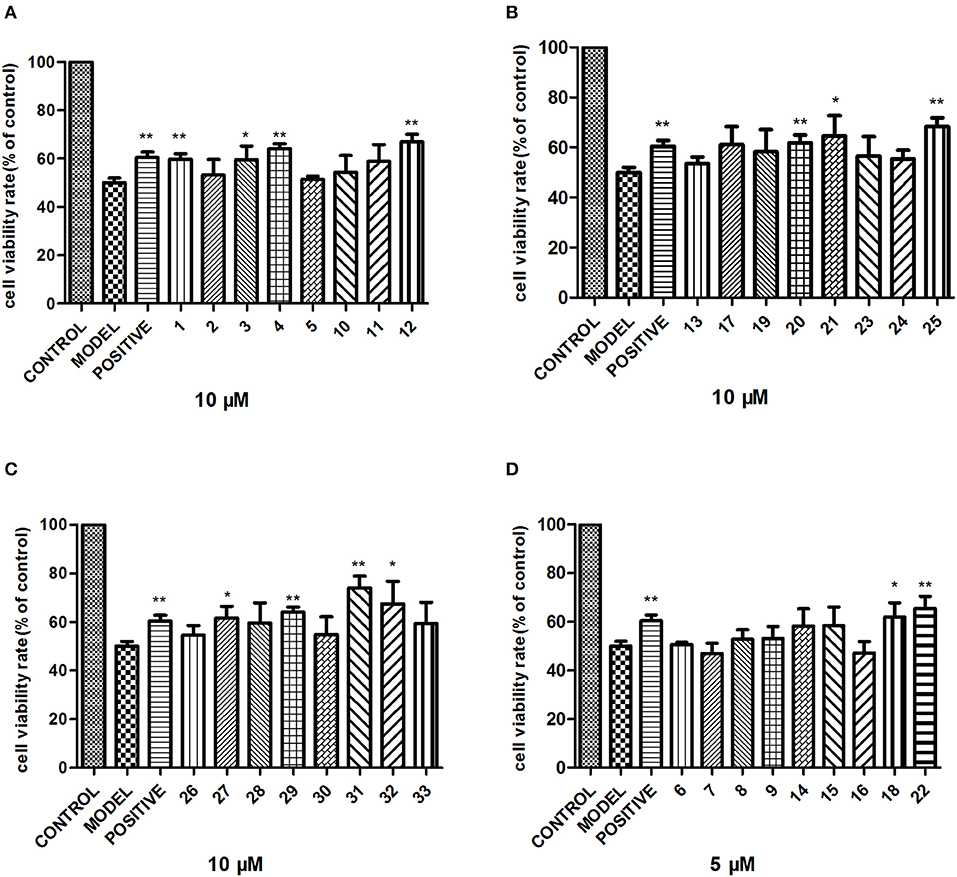
Figure 6. Neuroprotective activity of compounds 1–33. PC12 cells were exposed to 1 mM CoCl2 for 24 h with or without the indicated concentrations of compounds 1–33, and the cell viability was recorded: (A) compounds 1–5, 10–12; (B) compounds 13, 17, 19–21, 23–25; (C) compounds 26–33; (D) compounds 6–9, 14–16, 18, 22. The data are expressed as means ± SEM. Three independent experiments were performed. *P < 0.05, **P < 0.01.
Conclusion
Plants of the genus Aesculus have been proved to be rich in polyhydroxyoleanene triterpenoid saponins which have been characterized more than 100. When compared to the relatively extensive research on other species of Aesculus genus, little is known regarding the chemical constituents and the biological activity of the Aesculus chinensis Bge. var. chekiangensis (Hu et Fang) Fang species. The present paper reports 14 new polyhydroxy oleanene saponins (1–14) along with 19 known analogs from the seeds of A. chinensis Bge. var. chekiangensis. Structure elucidation was achieved via various techniques, and the absolute configuration of the aglycones was undoubtedly defined through X-ray diffraction analysis as well as Mo2(OAc)4-induced ECD method for the first time. Further cytotoxicity evaluation against three human tumor cell lines suggested that compounds 8, 9, 14–16, 18, 22 displayed strong inhibitory activities against all three cell lines; compounds 3, 6, 7, 17–19, 20, 24, 28 exhibited weak activities while the remaining isolates showed no toxicity at 50 μM. These results suggested that isolates with two acylations at C-21 and C-22 might be important for the cytotoxicity, especially substituted by tigloyl, angeloyl, methylbutyryl, and isobutyryl groups. In addition, the first test about the neuroprotective properties of triterpenoid saponins from Aesculus genus found that compounds 1, 4, 12, 20, 22, 25, 29, 31 exhibited moderate activities against CoCl2-induced PC12 cell injury.
Data Availability Statement
The crystallographic dataset generated for this study can be found in the Cambridge Crystallographic Data Centre under the CCDC number 1957449. All other datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
NZ and SW was responsible for the isolation and elucidated of compounds. NZ tested cytotoxicity, neuroprotective effects of the compounds, interpreted the data, and wrote the paper. SC, QZ, and NK revised the manuscript. LD and FQ were the project leaders organizing and guiding the experiment. All authors read and approved the final manuscript.
Funding
This work was financially supported by the State Key Program of National Natural Science of China (Grant No. 81430095).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00908/full#supplementary-material
References
Aki, M., Toshio, M., Hiroshi, N., and David, V. V. (2004). Oleanane saponins from Sanicula elata var. chinensis. J. Nat. Prod. 67, 377–383. doi: 10.1021/np030316v
Cheng, J. T., Chen, S. T., Guo, C., Jiao, M. J., Cui, W. J., Wang, S. H., et al. (2018). Triterpenoid Saponins from the seeds of Aesculus chinensis and their cytotoxicities. Nat. Prod. Bioprospect. 8, 47–56. doi: 10.1007/s13659-017-0148-4
Cheng, P., Kuang, F., and Ju, G. (2016). Aescin reduces oxidative stress and provides neuroprotection in experimental traumatic spinal cord injury. Free Radical Biol. Med. 99, 405–417. doi: 10.1016/j.freeradbiomed.2016.09.002
Di Bari, L., Pescitelli, G., Pratelli, C., Pini, D., and Salvadori, P. (2001). Determination of absolute configuration of acyclic 1,2-Diols with Mo2(OAc)4. 1. Snatzke's Method Revisited. J. Org. Chem. 66, 4819–4825. doi: 10.1021/jo010136v
El-Readi, M. Z., Eid, S., Ashour, M. L., Tahrani, A., and Wink, M. (2013). Modulation of multidrug resistance in cancer cells by chelidonine and Chelidonium majus alkaloids. Phytomedicine 20, 282–294. doi: 10.1016/j.phymed.2012.11.005
Kameyama, A., and Fujimura, T. (2009). Agent Containing Triterpenoid Saponins for Preventing or Ameliorating Skin Aging. Japan patent application. 200980105154.4.
Kapusta, I., Janda, B., Szajwaj, B., Stochmal, A., Piacente, S., Pizza, C., et al. (2007). Flavonoids in horse chestnut (Aesculus hippocastanum) seeds and powdered waste water byproducts. J. Agr. Food. Chem. 55, 8485–8490. doi: 10.1021/jf071709t
Kim, J. W., Ha, T. K., Cho, H., Kim, E., Shim, S. H., Yang, J. L., et al. (2017). Antiviral escin derivatives from the seeds of Aesculus turbinata Blume (Japanese horse chestnut). Bioorg. Med. Chem. Lett. 27, 3019–3025. doi: 10.1016/j.bmcl.2017.05.022
Kimura, H., Ogawa, S., Jisaka, M., Kimura, Y., Katsube, T., and Yokota, K. (2006). Identification of novel saponins from edible seeds of Japanese horse chestnut (Aesculus turbinata Blume) after treatment with wooden ashes and their nutraceutical activity. J. Pharm. Biomed. Anal. 41, 1657–1665. doi: 10.1016/j.jpba.2006.02.031
Konoshima, T., and Lee, K. H. (1986). Antitumor agents, 82. cytotoxic sapogenols from Aesculus hippocastanum. J. Nat. Prod. 49, 650–656. doi: 10.1021/np50046a015
Küçükkurt, I., Ince, S., Keleş, H., Akkol, E. K., Avci, G., Yeşilada, E., et al. (2010). Beneficial effects of Aesculus hippocastanum L. seed extract on the body's own antioxidant defense system on subacute administration. J. Ethnopharmacol. 129, 18–22. doi: 10.1016/j.jep.2010.02.017
Matsuda, H., Li, Y., Murakami, T., Ninomiya, K., Araki, N., Yoshikawa, M., et al. (1997). Antiinflammatory effects of escins Ia Ib IIa and IIb from horse chestnut the seeds of Aesculus hippocastanum L. Bioorg. Med. Chem. Lett. 7, 1611–1616. doi: 10.1016/S0960-894X(97)00275-8
Niu, X., Wang, Y., Li, W., Zhang, H., Wang, X., Mu, Q., et al. (2015). Esculin exhibited anti-inflammatory activities in vivo and regulated TNF-alpha and IL-6 production in LPS-stimulated mouse peritoneal macrophages in vitro through MAPK pathway. Int. Immunopharmacol. 29, 779–786. doi: 10.1016/j.intimp.2015.08.041
Patlolla, J. M., Raju, J., Swamy, M. V., and Rao, C. V. (2006). Beta-escin inhibits colonic aberrant crypt foci formation in rats and regulates the cell cycle growth by inducing p21(waf1/cip1) in colon cancer cells. Mol. Cancer. Ther. 5, 1459–1466. doi: 10.1158/1535-7163.MCT-05-0495
Piller, N. B. (1976). Drug-induced proteolysis: a correlation with oedema-reducing ability. Br. J. Exp. Path. 57, 266–273.
Sheldrick, G. M. (2015). SHELXT - Integrated space-group and crystal-structure determination. Acta Cryst. 71, 3–8. doi: 10.1107/S2053273314026370
Snatzke, G., Wagner, U., and Wolff, H. P. (1981). Circulardichroism—LXXV1: cottonogenic derivatives of chiral bidentate ligands with the complex[Mo2(O2CCH3)4]. Tetrahedron 37, 349–361. doi: 10.1016/S0040-4020(01)92021-6
Tanaka, T., Nakashima, T., Ueda, T., Kenji, T., and Isao, K. (2007). Facile discrimination of aldose enantiomers by reversed-phase HPLC. Chem. Pharm. Bull. 55, 899–901. doi: 10.1248/cpb.55.899
Wei, F., Ma, L.Y., Jin, W. T., Ma, S. C., Han, G. Z., Khan, I. A., et al. (2004). Antiinflammatory triterpenoid saponins from the seeds of Aesculus chinensis. Chem. Pharm. Bull. 52, 1246–1248. doi: 10.1248/cpb.52.1246
Xia, Y. Z., Yang, L., Wang, Z. D., Guo, C., Zhang, C., Geng, Y. D., et al. (2015). Schisandrin A enhances the cytotoxicity of doxorubicin by the inhibition of nuclear factor-kappa B signaling in a doxorubicin-resistant human osteosarcoma cell line. RSC Adv. 5, 13972–13984. doi: 10.1039/C4RA14324H
Yang, X., Zhao, J., Cui, Y., Liu, X., Ma, C., Hattori, M., et al. (1999b). Anti-HIV-1 protease triterpenoid saponins from the seeds of Aesculus chinensis. J. Nat. Prod. 62, 1510–1513. doi: 10.1021/np990180u
Yang, X., Zhao, J., Cui, Y., and Zhang, L. (1999a). A pair of new geometrically isomeric triterpenoid saponins from the seeds of Aesculus chinensis. Chin. Chem. Lett. 10, 925–928.
Yoshikawa, M., Murakami, T., Matsuda, H., Yamahara, J., Murakami, N., and Kitagawa, I. (1996). Bioactive saponins and glycosides. III. Horse chestnut. (1): The structures, inhibitory effects on ethanol absorption, and hypoglycemic activity of escins Ia, Ib, IIa, IIb, and IIIa from the seeds of Aesculus hippocastanum L. Chem. Pharm. Bull. 44, 1454–1464. doi: 10.1248/cpb.44.1454
Yoshikawa, M., Murakami, T., Yamahara, J., and Matsuda, H. (1998). Bioactive saponins and glycosides. XII. Horse Chestnut. (2): structures of Escins IIIb, IV, V, and VI and Isoescins Ia, Ib, and V, acylated polyhydroxyoleanene triterpene oligoglycosides, from the seeds of Horse Chestnut Tree (Aesculus hippocastanum L., Hippocastanaceae). Chem. Pharm. Bull. 46, 1764–1769. doi: 10.1248/cpb.46.1764
Yuan, W., Wang, P., Deng, G., and Li, S. (2012). Cytotoxic triterpenoid saponins from Aesculus glabra Willd. Phytochemistry 75, 67–77. doi: 10.1016/j.phytochem.2011.11.012
Yuan, W., Wang, P., Su, Z. S., Wang, V. S., and Li, S. Y. (2013). Cytotoxic triterpenoid saponins from husks of Aesculus californica (Spach) Nutt. Phytochemistry 90, 95–105. doi: 10.1016/j.phytochem.2013.02.003
Zhang, C. L., Wu, S. Q., and Li, X. S. (2009). Analysis on fatty acids composition in Aesculus chinensis seeds. Seed 28, 53–55. doi: 10.16590/j.cnki.1001-4705.2009.08.056
Zhang, N., Huang, W. X., Xia, G. Y., Oppong, M. B., Ding, L. Q., Li, P., et al. (2018). Methods for determination of absolute configuration of monosaccharides. Chin. Herb. Med. 10, 14–22. doi: 10.1016/j.chmed.2017.12.009
Zhang, S. L., Yang, Z. N., He, C., Liao, H. B., Wang, H. S., Chen, Z. F., et al. (2018). Oleanane-type triterpenoid saponins from Lysimachia fortunei Maxim. Phytochemistry 147, 140–146. doi: 10.1016/j.phytochem.2017.12.022
Zhang, Z., Kazuo, K., Jia, Z., Nikaido, T., Guo, D., and Zheng, J. (1999). New saponins from the seeds of Aesculus chinensis. Chem. Pharm. Bull. 47, 1515–1520. doi: 10.1248/cpb.47.1515
Zhang, Z., and Li, S. (2007). Cytotoxic triterpenoid saponins from the fruits of Aesculus pavia L. Phytochemistry 68, 2075–2086. doi: 10.1016/j.phytochem.2007.05.020
Zhang, Z., Li, S., Zhang, S., and Gorenstein, D. (2006). Triterpenoid saponins from the fruits of Aesculus pavia. Phytochemistry 67, 784–794. doi: 10.1016/j.phytochem.2006.01.017
Keywords: Aesculus chinensis Bge. var. chekiangensis (Hu et Fang) Fang, phytochemistry, triterpenoid saponins, cytotoxic activities, neuroprotective activities
Citation: Zhang N, Wei S, Cao S, Zhang Q, Kang N, Ding L and Qiu F (2020) Bioactive Triterpenoid Saponins From the Seeds of Aesculus chinensis Bge. var. chekiangensis. Front. Chem. 7:908. doi: 10.3389/fchem.2019.00908
Received: 24 October 2019; Accepted: 16 December 2019;
Published: 23 January 2020.
Edited by:
Tara Louise Pukala, University of Adelaide, AustraliaReviewed by:
Qingchao Liu, Northwest University, ChinaCharles Gauthier, Institut National de la Recherche Scientifique (INRS), Canada
Yang Minghua, State Key Laboratory of Natural Medicines, China Pharmaceutical University, China
Copyright © 2020 Zhang, Wei, Cao, Zhang, Kang, Ding and Qiu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liqin Ding, cnVieTcwMzAzQDE2My5jb20=; Feng Qiu, ZmVuZ3FpdTIwMDcwMTE4QDE2My5jb20=