 Raúl Berenguer
Raúl Berenguer Emilia Morallón
Emilia Morallón- Instituto Universitario de Materiales, Departamento Química Física, Universidad de Alicante (UA), Alicante, Spain
The functionalization of high surface area microporous carbon materials by oxidative treatments receives great interest for multiple applications. The micropore structure of the material and the oxidation method could play an important role in the process. In this work, we analyze and compare the effects of liquid-phase chemical and alternative electrochemical oxidation treatments in the textural and chemical properties of four microporous carbon materials, namely a granular-, cloth-, and powder-like activated carbon (AC) and a powdery zeolite templated carbon (ZTC). Particularly, we provide new data on oxidation kinetics and changes on microporosity and surface chemistry of various microporous carbons. Characterization techniques reveal that the extent, textural changes, and selectivity of the oxidation greatly depend on the type of microporous material and the oxidation method. The incorporation of surface oxygen groups (SOGs) generally causes a more-or-less significant decrease in the measured micropore volume and the specific surface area. In the studied ACs, the extent of oxidation and BET surface area reduction augment with their micropore volume, and the textural changes seem to be governed by the micropore blockage by SOGs. The disordered microporous structure of these materials is then quite robust toward oxidation, but its heterogeneity may contribute to the lack of selectivity during this process. By contrast, the regular micropore framework of the ZTC is rapidly destroyed even under soft oxidizing conditions, but it is proposed to promote certain selectivity during oxidation. The high reactivity and structural fragility of ZTC are assigned to the weak interconnections and large number of exposed edge sites in its structure. Our results demonstrate the fast oxidation rate of chemical treatments under different conditions, especially in the case of ZTC, what is proposed to restrict the control and to limit the oxidizability and selectivity of these functionalization processes. Contrarily, the electrochemical treatments are proved to better control the kinetics of oxidative functionalization, what may explain the observed higher efficiency and selectivity for SOGs introduction and the minimization of degradation in fragile microporous structures.
Introduction
Microporous carbon materials with pores of < 2 nm diameter and high specific surface area (up to 4,000 m2/g) receive great interest for multiple applications, such as gas storage and purification (Bottani and Tascón, 2008; Linares-Solano et al., 2008; Tascón, 2012), catalysis (Rodriguez-Reinoso, 1998; Serp and Figueiredo, 2009), wastewater treatment (Radovic et al., 2000; Bottani and Tascón, 2008; Tascón, 2012), energy storage and conversion (Bottani and Tascón, 2008; Beguin and Frackowiak, 2009; Nishihara and Kyotani, 2012), etc. For these applications, the micropore structure (volume, pore-size distribution, ordered/disordered arrangement, interconnectivity, shape, wall thickness, etc. of micropores) becomes crucial. Moreover, the use of oxidative post-treatments to adjust the number and type of oxygen surface groups (SOGs) in these materials could be very important for their optimization in some of these applications. Nevertheless, the control on the extent and selectivity of the oxidation processes is complex and their possible effects on the textural properties must be considered.
Microporosity features are mainly determined by the precursor and preparation method. Activated carbons (ACs) constitute the most widely known family of microporous carbons. In these materials, microporosity mainly develops as spaces among disordered aromatic sheets and small graphitic domains formed upon heating (Rodríguez-Reinoso and Molina-Sabio, 1998; Molina-Sabio and Rodríguez-Reinoso, 2004) and/or by carbon oxidation to CO or CO2, depending on the activation method (Rodríguez-Reinoso and Molina-Sabio, 1998; Molina-Sabio and Rodríguez-Reinoso, 2004; Linares-Solano et al., 2008). Another family of microporous carbons are the so-called zeolite-templated carbons (ZTCs) obtained by using zeolite as a sacrificial template (Ma et al., 2000; Nishihara and Kyotani, 2018). Unlike ACs, ZTCs exhibit an ordered micropore framework of curved and single-layer graphene coming from the zeolite channels.
The oxidation of microporous ACs has been largely studied in the past. The effect of multiple different oxidation methods and conditions on their properties and applications was analyzed (Shen et al., 2008; Daud and Houshamnd, 2010; Jaramillo et al., 2010; Rivera-Utrilla et al., 2011; Thakur and Thakur, 2016). So far, liquid-phase chemical oxidation by using H2O2, HNO3 or (NH4)2S2O8 has been the most studied oxidative method (Otake and Jenkins, 1993; Moreno-Castilla et al., 1995, 1997, 1998, 2000; De la Puente et al., 1997; Gil et al., 1997; Mangun et al., 1999; Pradhan and Sandle, 1999; El-Sayed and Bandosz, 2001; Shim et al., 2001; Strelko and Malik, 2002; El-Hendawy, 2003; Li et al., 2003, 2011; Houshmand et al., 2011; Ternero-Hidalgo et al., 2016). It has been claimed that the oxidation degree of microporous ACs can be easily controlled by adjusting the oxidation parameters, i.e., treatment time, concentration, and temperature of common oxidants (HNO3 or (NH4)2S2O8) (Moreno-Castilla et al., 1997; Houshmand et al., 2011). These treatments generally cause a decrease in specific surface area and micropore volume, which becomes more significant with an increasing degree of activation in the oxidized material (Moreno-Castilla et al., 1995, 1997, 2000). While some authors suggest that the decrease in textural properties is due to a blockage by SOGs at the entry of the micropores (Moreno-Castilla et al., 1995, 1997; Pradhan and Sandle, 1999; Li et al., 2003; Maroto-Valer et al., 2004; Szymanski et al., 2004; Houshmand et al., 2011), others claim their destruction by the oxidation reaction (Moreno-Castilla et al., 1995, 1997, 1998; Pradhan and Sandle, 1999; Li et al., 2003). These disparities have been attributed to the distinct oxidation method and differences in the micropore texture, thickness of micropore walls, etc. of the oxidized material. And reciprocally, it has been proposed that the porosity of the material plays an important role in the rate of fixation of SOGs (Moreno-Castilla et al., 1997).
Other conventional treatments of microporous ACs are based on gas-phase chemical oxidation at middle/high temperature (García et al., 1998; Figueiredo et al., 1999; Boudou et al., 2000; Gómez-Serrano et al., 2002; Álvarez et al., 2005). Although high oxidation degrees can be reached with these processes, they generally cause significant textural modification. By contrast, electrochemical oxidative treatments at room temperature have been proposed as promising advantageous alternatives (Kinoshita, 1988; Berenguer et al., 2009, 2012, 2013a,b; Tabti et al., 2013; González-Gaitán et al., 2015; Vujković et al., 2018). We previously carried out systematic studies on the electrochemical oxidation of various carbon materials (Berenguer et al., 2009, 2012, 2013a,b; Tabti et al., 2013). For a microporous AC, preliminary results suggested that the kinetics for SOGs generation through electrochemical oxidation are more controllable than with chemical treatments, without significantly affecting its textural properties (Berenguer et al., 2012).
The oxidation of ZTCs, however, remains practically unexplored (Berenguer et al., 2013a,b; Vujković et al., 2018). In a pioneer work, it was found that, compared to chemical treatments, the electrochemical method enabled to better retain the original microporous structure of the ZTC during SOGs incorporation (Berenguer et al., 2013a). Moreover, from variations in CO/CO2 ratios it was proposed that the electrochemical oxidation involves a less aggressive and more controlled oxidation than the chemical one (Berenguer et al., 2013b). Although a better controllability of the oxidation kinetics was proposed to explain these phenomena, no kinetics studies were carried out.
It can be deduced from literature, hence, that not only the oxidative method but also the micropore structure of the oxidized material seem to play an important role on the reactivity and effects of the oxidation processes. Among the vast literature, however, few investigations compare the effect of different oxidation methods on a given carbon material (Otake and Jenkins, 1993; Figueiredo et al., 1999; Li et al., 2003; Houshmand et al., 2011; Berenguer et al., 2012, 2013a) and very few deal with the influence of the different micropore structure of the material when oxidized by a specific method (Moreno-Castilla et al., 1997; El-Sayed and Bandosz, 2001). To the knowledge of the authors, there are no investigations analyzing both effects at the same time. Remarkably, studies on the kinetics and controllability of the different oxidation reactions are lacking. In this work, various carbon materials with disordered (three distinct ACs) and ordered (a ZTC) microporous structure have been oxidized by conventional liquid-phase chemical and alternative electrochemical oxidation methods. The chemical and textural properties of the different materials, before and after oxidation, have been analyzed by various techniques. For these materials, the effects of the different oxidative treatments on the oxidation degree, changes on the microporous texture and the selectivity toward SOGs introduction have been studied. Furthermore, the kinetics of the different oxidative functionalizations have been analyzed. Finally, the effects of the different oxidative methods have been correlated with their kinetics and the microporous structure of the distinct materials.
Materials and Methods
Materials
Four different carbon materials were used for this study: (i) the granular activated carbon (GAC) was supplied by Waterlink Suctliffe Carbons (207 A, pHPZC = 9, Mesh: 12 × 20) and it is referred to as W; (ii) the activated carbon cloth (ACC) was provided by MAST Carbon (C-TEX27, pHPZC = 6.5) and denoted as EX; (iii) the powdery activated carbon (PAC), designated as A, was prepared by chemical activation of an anthracite with KOH as reported elsewhere (Lozano-Castelló et al., 2001). For that, a physical mixture of KOH:anthracite = 3:1 was heated at 5°C/min up to 740°C, and kept at this temperature for 1 h, under a 6,000 mL/min flow of N2; and (iv) the zeolite templated carbon (ZTC) was synthesized by using a zeolite Y template (Na-form, SiO2/Al2O3 = 5.6, from Tosoh Co. Ltd.) as previously described (Ma et al., 2000). The main textural and chemical properties of these materials are collected in Table 1.
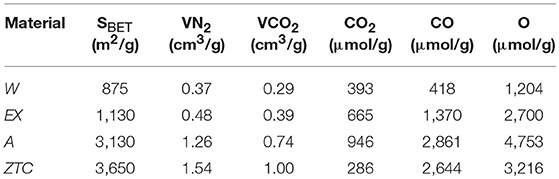
Table 1. Textural and chemical properties of the studied microporous materials.
Oxidation Methods
Chemical Oxidative Functionalization
The chemical oxidation of the different ACs and the ZTC was performed with HNO3, H2O2, and/or (NH4)2S2O8, under different conditions, as reported before (Berenguer et al., 2012, 2013a). The nomenclature of the chemically-oxidized materials includes the name of the carbon material (W, EX, A, ZTC); the letters N, H, or S referring to the used oxidizing agent (HNO3, H2O2, and (NH4)2S2O8, respectively); a number indicating their concentration (%); a number or the letter R (room temperature) related to the temperature of the experiments (°C); and finally, the treatment time (h or min).
Electrochemical Oxidative Functionalization
The electrochemical oxidation of the different ACs was carried out in a filter-press electrochemical cell (Berenguer et al., 2009, 2012; Tabti et al., 2013), whereas the ZTC was assembled into a paste to be electrooxidized in a conventional three-electrode cell (Berenguer et al., 2013a). The different materials were galvanostatically treated at RT in 1 M NaOH, 2 wt% NaCl or 1 M H2SO4 aqueous electrolytes under different conditions. The nomenclature of the electrochemically-oxidized materials comprises the name of the carbon material (W, EX, A, ZTC); a number indicating the applied current; the abbreviations OH−, Cl−, or H+, related to the NaOH, NaCl, and H2SO4 electrolytes, respectively; and finally, the treatment time.
Characterization and Parameters
Prior to their characterization, the functionalized materials were washed with abundant distilled water, vacuum-filtered, and dried at 110°C overnight.
Porous Texture Characterization
The textural properties of the materials before and after oxidation were analyzed by N2 adsorption-desorption at −196°C and CO2 adsorption at 0°C. The different ACs were analyzed in an Autosorb-6 system (Quantrachrome Corporation) after vacuum out-gassing at 250°C for 4 h, while the ZTC samples were characterized in a BEL Japan analyzer (BELSORP-mini) after out-gassing at 150°C under vacuum for 6 h. The apparent surface area (SBET) was determined by the Brunauer–Emmett–Teller (BET) method using the data of N2 adsorption branch in the P/P0 range of 0.01–0.05. The total volumes of micropores (VN2) (d < 2 nm) and narrowest micropores (VCO2) (d < 0.7 nm) were calculated from the Dubinin–Radushkevich (DR) equation applied to N2 (in the range 0.005 < P/P0 < 0.17) and CO2 adsorption isotherms, respectively (Arenillas et al., 2004). The good fitting of the N2 and CO2 adsorption data to the DR equation validated the application of this method for the different microporous materials. Pore size distributions were derived by density functional theory (DFT), applying the QSDFT model to the adsorption branch for the analysis of the geometrically and chemically disordered (slit/cylindrical/sphere) micro-/meso- pores of ACs; and the NLDFT equilibrium model of carbon slit pores for the ZTC samples. Moreover, the changes on ZTC structural order were followed by X-ray diffraction (XRD-6100, Shimadzu) using a Cu-Kα radiation (λ = 1.541 Å) generated at 30 kV and 20 mA.
Particularly, in this work, the change on BET surface area was calculated as:
or, relatively, as the loss (%) of BET surface area:
where, SBET0 and SBETOX are the specific surface area of the material before and after oxidation, respectively. As it is explained below, changes on the SBET of the studied materials are unequivocally attributed to changes on micropore volume.
Surface Chemistry Characterization
The concentration and type of SOGs was characterized by Temperature-Programmed Desorption (TPD) experiments and X-Ray Photoelectron Spectroscopy (XPS). Nevertheless, other techniques, like titration methods, water adsorption, infrared spectroscopy, etc., could have been also used. For the different ACs, the TPDs were performed at 20°C/min up to 950°C under a He flow (100 mL/min) in a SDT 2960 Simultaneous equipment (TA Instruments) coupled to a MSC 200 mass spectrometer (Thermostar, Balzers). The ZTC samples were heated up to 1,000°C (2°C/min), and kept at this temperature for 1 h, under a He flow, and the CO2, and CO evolved gasses were analyzed with a micro gas chromatograph (Varian 490-GC). The XPS analysis was carried out by using a VG-Microtech Multilab electron analyzer and an unmonochromatized Mg-Kα (1253.6 eV) radiation at base pressure of 5 × 10−10 mbar. Photoelectrons were collected at a pass energy of 50 eV and the binding energies (BE) were referenced against the main C1s line at 284.5 eV.
The number of SOGs introduced by a given oxidation method was calculated from TPD measurements by subtraction as:
where, O0 and OOX are the total oxygen evolved (O = 2CO2 + CO) calculated from the TPD-profiles of the material before and after oxidation, respectively. From this parameter, the change on BET surface area relative to SOGs incorporation was calculated as ΔSBET/ΔO.
Results and Discussion
Oxidation Degree and Textural Properties Changes by Chemical Treatments
The studied carbon materials were oxidized by different chemical or electrochemical methods (see experimental section). Figure 1 represents the increment of SOGs (ΔO) caused by different oxidation treatments vs. the variation of specific surface area (ΔSBET) determined for the studied materials. The increment of SOGs (ΔO) can be associated to the oxidizability of the material and/or the efficacy of the oxidative treatment and conditions. Although ΔSBET was arbitrarily represented as positive, a decrease in the measured SBET was found in all cases, so a higher ΔSBET value was characteristic of a larger decrease in specific surface area. Given the lineal dependence of ΔSBET with the change on the micropore volume (ΔVN2) (see section Analysis of Textural Changes), changes on ΔSBET can be directly attributed to variations of the micropore volume determined from N2 adsorption-desorption isotherms.
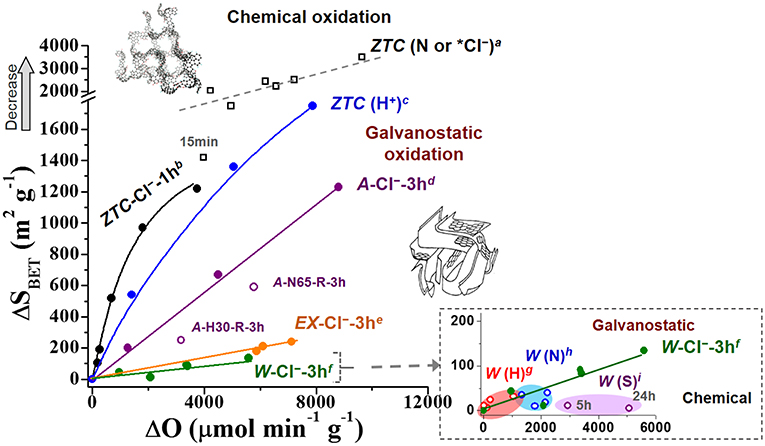
Figure 1. Extent of oxidation (ΔO) and surface area variation (decrease) caused by most common chemical reagents in liquid phase [H2O2, HNO3, and (NH4)2S2O8] and different galvanostatic treatments at different conditions on the studied microporous materials. The carbonaceous structures of the ZTC and the ACs are adapted from Nishihara et al. (2009) and Rodríguez-Reinoso and Molina-Sabio (1998), respectively. Note: (*Cl−) refers to the too strong electrochemical conditions for the oxidation of ZTC (≥ 50 mA in NaCl) at which their oxidizing power approaches those of chemical treatments. Oxidation conditions: a: 30–60 wt% HNO3, R−80°C,15 min-2 h; a*: 50 mA, 3–7 h; b: 2–20 mA; c: 2–5 mA, 1–36 h; d,e,f : 0.2–1.0 A; g: 10–33 wt% H2O2, 2 h; h: 23-65 wt% HNO3, 2 h; i: (NH4)2S2O8-saturated 1 M H2SO4 solution.
Chemical Oxidation of the ACs
As it can be observed in the figure, the extent of both the oxidation degree and decrease in surface area greatly depends on different aspects. The RT-treatment of the W (SBET0 = 875 m2/g) with different oxidizing reagents for 2 h (inlet of Figure 1) enables to introduce a maximum of 3,000 μmolO/g in the case of (NH4)2S2O8, which is even more effective than 65 wt% HNO3 (ΔO = 2,200 μmolO/g) and much than H2O2. When the reaction time is prolonged up to 24 h with (NH4)2S2O8, a maximal increment of 5,000 μmolO/g is observed. These results indicate that the chemical oxidation may be faster at the beginning, so that its rate decreases with time (Figure 2A). Such a kinetic behavior can be observed also in other ACs reported in literature (Moreno-Castilla et al., 1997; Houshmand et al., 2011; Figure 2A) and was associated to the uncontrollability of chemical oxidation (Berenguer et al., 2012). The main consequence is that significantly long times are needed to reach significant oxidation degrees, so that further functionalization of this material by this method and conditions becomes complex and restricted.
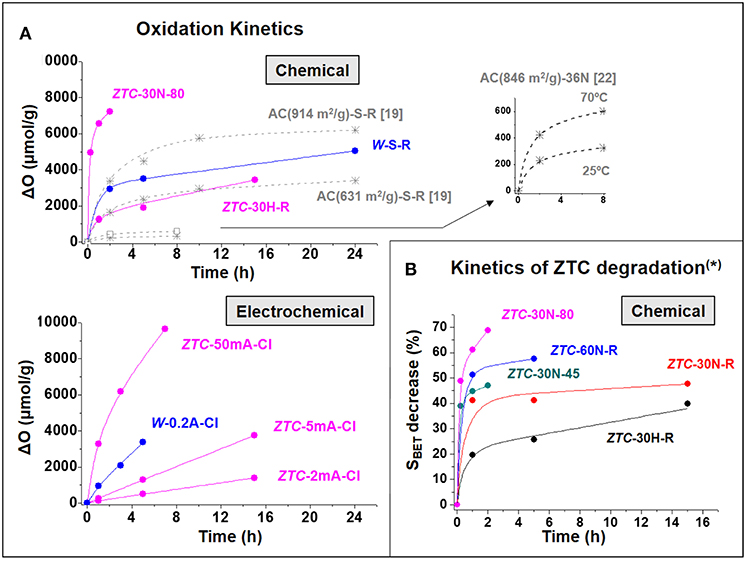
Figure 2. (A) Evolution of the oxidation degree upon time (oxidation kinetics) of various studied and referenced materials functionalized by chemical or electrochemical methods; (B) Evolution upon time of the loss in surface area (associated to structural degradation (*) in section Analysis of Textural Changes) of ZTC functionalized by different chemical methods.
On the other hand, none of these treatments produced significant changes on the surface area (a detailed analysis of the textural properties is presented in section Analysis of Textural Changes). In particular, a maximal 4.5% loss in surface area was observed in the case of 65 wt% HNO3, this being a very low relative decrease of surface area by SOGs gain (0.02 m2/μmolO).
Quite similar results were found for the chemical oxidation of EX under similar conditions (not shown in Figure 1), although in this case both the attained oxidation degrees and the observed changes on surface area were slightly higher (ΔO = 6700 μmolO/g and 13% loss in surface area for EX-S-24 h). This could be explained by the slightly larger micropore volume and surface area of this material (see Table 1), which could result in a greater concentration of reactive edge sites (Arenillas et al., 2004) and larger oxidations (ΔO) (Moreno-Castilla et al., 1995, 1997, 2000; Mangun et al., 1999). For example, in only 1 h a RT-treatment of activated carbon fibers (ACFs) (SBET0 = 1390 m2/g) with concentrated HNO3 resulted in larger oxidation degrees (ΔO = 5420 μmolO/g) and surface area losses (6.8%) (Mangun et al., 1999) than a 2 h-similar-treatment of the studied W. Moreover, such effects of the surface area at RT were also observed and systematically studied, with other reagents [H2O2 and (NH4)2S2O8], by Moreno-Castilla et al. (1995, 1997, 2000).
However, these authors found that the ACs prepared from olive stones display lower oxidative reactivity than those obtained from almond shells (Moreno-Castilla et al., 1995, 1997, 2000). Hence, not only the textural properties but also the nature of the precursor or other features (wettability, dimensions, conformation, mesoporosity, wall thickness, electrical conductivity in the case of electrochemical oxidation, etc.), may affect the reactivity of the material.
The increased reactivity with the development of textural properties was confirmed for the powdery AC, sample A, which presents around triple micropore volume and surface area than previous materials (Table 1). As it can be seen in Figure 1, ca. 5,770 μmolO/g can be introduced after 3 h-oxidation with 65 wt% HNO3 (A-N65-R-3h) and the surface area considerable decreases in a 19.1%. Thus, the relative decrease of surface area by SOGs gain (ΔSBET/ΔO) increases to ca. 0.10 m2/μmolO. Nevertheless, the finer particle size of this powdery material compared to the previous ones, favoring the contact with the oxidizing agents, may also have a strong influence on its greater oxidizability.
It is well-known that the oxidation degree of ACs reached by the chemical method can be further increased in the range ΔO = 5,000–12,000 μmolO/g, respectively by raising the temperature, concentration and/or acidity of the reagent (Moreno-Castilla et al., 1995; Mangun et al., 1999; Ternero-Hidalgo et al., 2016). However, these treatments parallelly caused more severe textural modifications, with a decrease in surface area between 10 and 50%. As it can be deduced from literature data (see Figure 2A the data of Houshmand et al., 2011), for ACs the kinetics of chemical oxidation greatly increases with such stronger oxidizing conditions.
Chemical Oxidation of the ZTC
Unlike the case of ACs, the ZTC experiences a comparable SOGs introduction but a much more marked reduction of surface area when exposed even at much softer oxidizing chemical conditions (see some examples in the upper part of Figure 1). For example, in only 15 min (45°C) or 1 h (room temperature) the oxidation of ZTC with 30 wt% HNO3 introduces 4,000–5,000 μmolO/g, but its surface area decreases in more than 1,400 m2/g. These effects suppose a 39–41% loss in SBET and a considerable enhancement of the relative variation of SBET to 0.30–0.40 m2/μmolO (for 30 wt% HNO3). It can be inferred beforehand (see details on section Oxidation Methods) that this loss in surface area is associated to the destruction of the ordered microporous framework of this material, so it is referred to as structural and/or textural degradation. Hence, chemical treatments at RT produce severe damage in the extraordinary textural properties of ZTC.
Moreover, in this case it is easier to induce further functionalization by simply extending treatment time, temperature or reagent concentration, but any increment in SOGs gives rise to further textural decline. For example, boiling 30 w% HNO3 (80°C) can introduce 5,000–7,200 μmolO/g in only 15 min−2 h of treatment, but it causes a 50–70% loss of SBET. Another interesting feature is that the chemical oxidation reaction of ZTC is very fast, even in the case of 30 wt% H2O2 at RT, the softest conditions studied (Figure 2A). Such oxidation kinetics are greatly accelerated at stronger conditions (see for example the results of boiling 30 wt% HNO3 in Figure 2A). As a result, the degradation of ZTC properties under chemical conditions is remarkably fast. Figure 2B shows that above 40 % of the initial SBET of ZTC is lost within the 1 h of treatment with HNO3 under different conditions. This stresses the extraordinarily high reactivity and fragility of this material toward chemical functionalization, which may be unacceptable for the potential application of these materials with designed unique nanostructure.
Analysis of Textural Changes
The microporosity of the different materials before and after oxidative treatments was analyzed by gas adsorption techniques. Figure 3 compares the N2 adsorption-desorption isotherms of the pristine W, A, and ZTC (see their textural parameters in Table 1) with those of representative oxidized samples. The isotherm of EX (figure not shown) was quite similar to that of W (Zakaria, 2014). For the oxidized ACs, examples of materials obtained by electrochemical treatments have been considered because they cause higher oxidation degrees (see section Electrochemical Oxidation: Greater Controllability at Room Temperature), what may help to discern more clear effects. The isotherms for W and A samples are a combination of type I and IV (Lozano-Castelló et al., 2009). In fact, the good correlation between the volume of micropores and the specific surface area of the many samples oxidized under different conditions (Figure 4A) confirms the microporous nature of all the oxidized materials considered in this work (Mangun et al., 1999; Strelko and Malik, 2002; Szymanski et al., 2004). Nevertheless, the hysteresis loops observed in the isotherms of W and the relatively open elbows at low pressures in the isotherms of A, and their oxidized derivatives, indicates the presence of some mesopores in these (combination of Type I and Type IV isotherms) materials. As explained below, this mesoporosity is also evident in the derived PSDs.
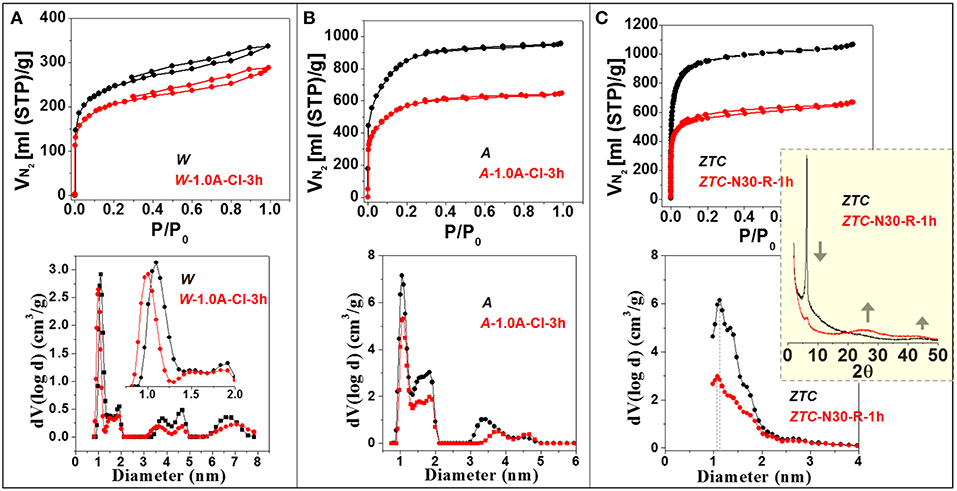
Figure 3. N2 adsorption-desorption isotherms at −196°C and the DFT-derived pore size distributions of the studied W (A), A (B), and ZTC (C) microporous materials compared to those of a representative highly-oxidized material obtained after 3 h-treatment at 1.0 A in 2 wt% NaCl (A,B) or 1 h-oxidation with 30 wt% HNO3 (C) at room temperature. Inlet of (C): X-ray diffractograms of ZTC before and after oxidation.
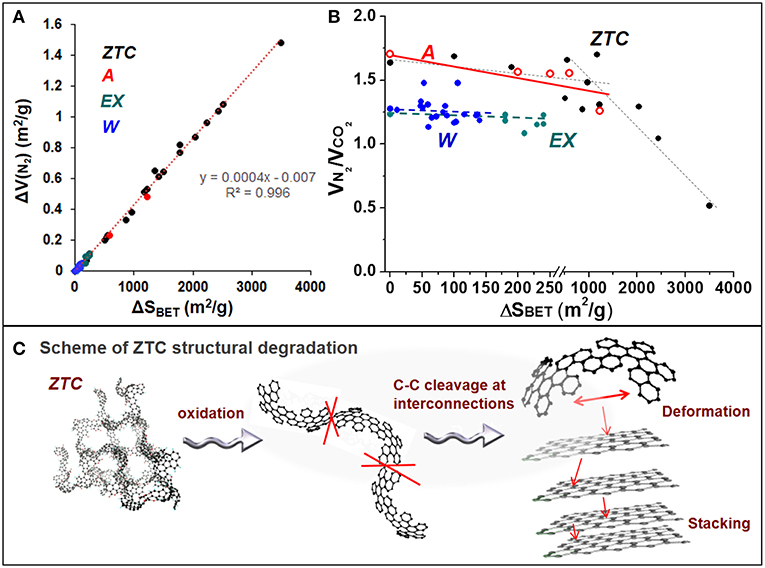
Figure 4. (A) Relation between the change in specific surface area (ΔSBET) and that in the micropore volume (ΔV(N2)); and (B) evolution of the VN2/VCO2 ratio with the change in specific surface area, for the differently oxidized materials prepared for this study. (C) Proposed mechanism for the breakdown of ZTC structure (adapted from Nishihara et al., 2009) after different oxidative treatments.
Particularly, the A material contains ca. 3-fold larger micropore (VN2) and narrow micropore (VCO2) volumes than the W (Table 1), but they show quite similar micropore size distribution. From the derived PSD graphs (Figure 3) it can be inferred that most micropores in W and A materials are 1.10 and 1.05 nm in diameter, respectively, together with a certain concentration of wider micropores and some mesopores, mainly in the case of A, ranging between 1.30 and 2.10 nm. Another difference is that W contains a larger concentration of different mesopores with diameters (maximum population) of 3.80, 4.60, and 6.70 nm, whereas A only contains some mesopores of ca. 3.40 nm. On the other hand, ZTC presents even a higher micropore volume with a narrower PSD, thus most pores ranging between 1.10 and 1.30 nm. Nevertheless, the large micropore volume determined by CO2 adsorption (Table 1) may point out the presence of narrower zones, i.e., neck-like interconnections between successive larger cavities. This texture agrees with the 3D-arranged graphenic framework of interconnected 1.2 nm-micropores coming from the templated zeolite Y (Nishihara et al., 2009).
As shown in Figure 3, the shape of isotherms for the oxidized microporous materials is quite similar to that of the pristine materials. The main difference is the decrease in the volume of N2 adsorbed. The VN2 decrease was generally observed for the different oxidized materials, but its extent depended on the oxidation treatment, like the case of SBET (Figure 1, in agreement with their linear relationship, Figure 4A). Moreover, the shape of the PSDs is also well-conserved after oxidation, although a slight shift of the main peak in the PSD toward smaller diameters (maximum at 1.00 nm) can be observed for the W. These features indicate that, despite the micropore volume decreases, the micropore structure is not significantly affected by the oxidative treatments (at least that of supermicropores, 0.7 < d < 2 nm) (Mangun et al., 1999; El-Sayed and Bandosz, 2001; Shim et al., 2001; Strelko and Malik, 2002; Maroto-Valer et al., 2004). In the same line, the volume of narrower micropores (d < 0.7 nm), deduced from CO2 adsorption isotherms (not shown), also decreased to a different extent after the different oxidative treatments.
Further information was deduced from the changes on both the wider and narrower micropores. Figure 4B depicts the VN2/VCO2 ratio vs. the change on specific surface area. For the W and EX materials, the VN2/VCO2 ratio remains practically constant with the extent of SBET decrease. Contrarily, in the case of A and ZTC the ratio clearly decreases, and, for the later material starts to decrease more rapidly after certain SBET decrease is reached. Particularly, for the most oxidized A sample in this study (A-1.0A-Cl-3h), the VN2/VCO2 ratio is decreased in ca. 25%, and ca 35–70% for the most oxidized ZTC samples.
The similar isotherms and PSDs and nearly constant VN2/VCO2 ratios observed, independently of the extent of SBET decrease, for the differently oxidized W materials suggest that the decrease in their micropore volume after oxidation may be assigned to a pore blockage by the created SOGs and/or split molecules; and/or electrostatic repulsion of N2 probe molecules (Moreno-Castilla et al., 1995; Pradhan and Sandle, 1999; Li et al., 2003; Maroto-Valer et al., 2004; Szymanski et al., 2004; Houshmand et al., 2011). Given the constant VN2/VCO2 ratios, it may equally affect to wider and narrower micropores. The slight narrowing (shift in PSD) of the most abundant micropores in W may be in line with the proposed surface blockage, suggesting that there are N2 molecules that may be able to pass also through the partially blocked (narrower) micropores. Because it is not destroyed, the micropore structure in these materials may be highly tough toward oxidation.
In the case of sample A, the large SBET decrease with the oxidation degree (Figure 1) might be also explained by surface blockage, at least this could be similarly deduced from the results of Figure 3. In this case a shift in the maximum of PSD is not observed probably because these predominant micropores are narrower from the beginning (maximum at 1.05 nm). However, the decrease in the VN2/VCO2 ratio with the extent of oxidation found for A could be indicative of other phenomena, like a favored blockage and/or the destruction of wider micropores (VN2). It has been proposed that the destruction of micropores by breakage of surface C-C bonds can result in the formation of narrower (Gil et al., 1997; El-Sayed and Bandosz, 2001) or wider (Moreno-Castilla et al., 1995, 1997; Pradhan and Sandle, 1999; El-Hendawy, 2003) ones. The much larger relative decrease in VN2 than in VCO2 (for this sample the relative variation of both types of micropores ΔVN2/ΔVCO2 = 4), and the fact that no mesopores are generated (see PSD in Figure 3), seem to neglect the possibility of micropore destruction. Hence, the idea of a favored blockage of wider micropores seems to gain strength. This could be explained by considering that the sample A contains a larger volume and proportion of pores between 1.30 and 2.10 nm than W (compare PSD of samples A and W in Figure 3). Thus, despite blocking the entrance of N2 (of slightly bigger size than CO2 and presenting more interactions with SOGs), some of the more-opened micropores in the sample A could permit the entrance of CO2, thus, observing a decrease in the VN2/VCO2 ratio.
It is noteworthy to mention that other parameters, like the mesoporosity (which may affect the reaction kinetics) and the wall thickness (which can lead to the collapse of the pore structure) could be crucial in the oxidation of microporous carbons. However, the low mesoporosity and the neglected micropore destruction in the studied ACs suggest that these features may not be significant in the observed oxidative behaviors.
The variation of the textural properties of the ZTC upon oxidation deserves a special discussion. A detailed analysis of the effect of different oxidation treatments on ZTC structural properties was previously carried out (Berenguer et al., 2013a,b). Figure 3C shows that, besides a decrease in the adsorption of N2, the oxidation of ZTC (1 h-treatment with 30 wt% HNO3 in this example) entails a marked reduction in the intensity of its characteristic (111) X-ray diffraction peak. Considering that this peak comes from the ZTC regular structure derived from zeolite Y, the decreased intensity or disappearance of this peak evidence the destruction of ZTC structure by oxidative treatments. Another exclusive feature of this degradation is the conversion of the 3D arrangement of graphenic layers into a stacked 2D structure, as confirmed by the evolution of the two broad diffraction peaks around 25 and 45° characteristic of graphitic structures. These results suggest that the breakage of ZTC gives rise to graphenic fragments that can be stacked. Considering the molecular structure of ZTC (Nishihara et al., 2009), it is proposed that the breakage may occur by C-C cleavage at linking/interconnecting points among cavities, where the graphenic layers are thinner, as schematized in Figure 4C. Once fragmented, the wall cavities (the larger graphenic parts in ZTC structure) might be deformed into planar forms able to be stacked.
The destruction of ZTC structure at its very thin interconnecting points among graphenic layers may be the reason explaining the high fragility of this unique microporous structure. In fact, ordered mesoporous carbons (OMCs) with different wall and/or connector thickness and surface area were found differently affected by the chemical treatments (Lu et al., 2005; Vinu et al., 2007; Bazuła et al., 2008). For example, it has been reported that a soft treatment with 30 wt% H2O2 (Lu et al., 2005) or 53 wt% HNO3 (Bazuła et al., 2008) at RT causes, in few minutes, structural collapse of the CMK-5 carbon framework, as deduced from XRD characterization. This high reactivity was attributed to the unique structure of this material, comprised of porous interconnected tubes of very thin carbon walls (2–3 nm) (Lu et al., 2005; Bazuła et al., 2008). Bazula et al. had to work under low HNO3 concentrations (4.5–18 wt %) to functionalize this type of materials (SBET0 = 1,389–1,846 m2/g) (Bazuła et al., 2008). Nevertheless, the temperature was raised to 80°C to generate high oxidation degrees (ΔO = 5,300–10,250 μmolO/g) in 3 h, producing SBET losses of 11–35 % associated to the structure breakdown. On the other hand, unlike tubular OMCs, rod-like CMK-3 carbons with thicker walls and connectors showed increased stability toward oxidative functionalization (Vinu et al., 2007; Bazuła et al., 2008). Such a high fragility exhibited by CMK-5 and CMK-3 OMCs resembles that of the ZTC and it was attributed to C-C cleavage on the links between adjacent tubular or rod-like structures, respectively (Lu et al., 2005; Bazuła et al., 2008). Moreover, these studies showed that the oxidation of these OMCs leads to the formation of small organic molecules and a substantial weight loss; and, whenever the whole structure is not collapsed, the N2 isotherms of the materials were quite similar shape to those of pristine materials. All these similarities between the oxidative behavior of OMCs and that of ZTC support the proposed degradation mechanism and fragility reasons.
Nevertheless, the high reactivity of ZTC may not be exclusively related to a weak interconnectivity in its 3D microporous structure. In this sense, the preparation of a mesoporous graphene-based framework, by replication of the outer surface of Al2O3 nanoparticles, resulted in a continuous 3D structure with a minimal number of edge sites (Nishihara et al., 2016). This unique material realized both a large surface area (1,940 m2/g) and an extraordinarily high stability against oxidation. Hence, another crucial aspect involved in the reactivity and fragility of the studied ZTC may be the large number of exposed edge sites, associated to the very small graphenic layers and/or incomplete replication of the Y zeolite channels in this material (Nishihara et al., 2009), as well as its thin (single layer) wall thickness (Vinu et al., 2007; Bazuła et al., 2008).
Electrochemical Oxidation: Greater Controllability at Room Temperature
Sections Chemical Oxidation of the ACs and Chemical Oxidation of the ZTC have evidenced the limitations of chemical treatments to control the oxidation of carbon materials. Figure 1 compiles the effects of various electrochemical oxidative treatments on the properties of the studied carbon materials. As it can be observed in this figure, the different materials exhibit variable fragility toward electrochemical oxidation, but remarkable differences can be observed when compared with chemical treatments.
The anodic treatment in NaCl at enough-high currents enables, at room temperature and in short time, to greatly enhance the oxidation degree of the ACs. Particularly, strong electrochemical oxidizing conditions, polarization at 1.0 A, introduced in 3 h a larger amount of SOGs (ΔO = 5,600 μmolO/g for W and 7,150 μmolO/g for EX) than chemical oxidation with (NH4)2S2O8 in 24 h (ca. 5,000 μmolO/g for W and 6,700 μmolO/g for EX). Similar results are obtained when considered other ACs, like A (see for example CO2-like TPD profiles of A-oxidized samples in Figure 5A) or other reagents (see for example CO-like TPD profiles of W-oxidized samples in Figure 5B).
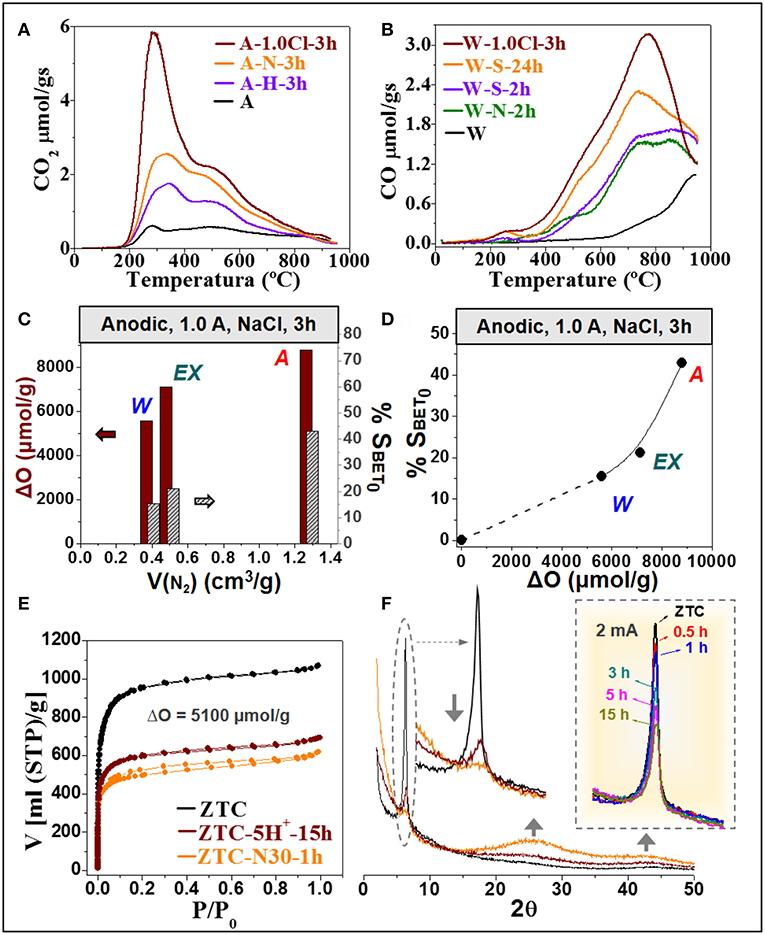
Figure 5. (A,B) Comparison of CO2-like (for A sample) (A), and CO-like (for W sample) (B) TPD profiles of samples oxidized under different chemical or electrochemical conditions; (C,D) Influence of the micropore volume of the studied ACs (V(N2)) in the SOGs gain (ΔO) and SBET decrease (%SBET0) (C), and relation between this SOGs gain and SBET decrease (D) for a given anodic oxidation treatment (1.0 A, in NaCl, 3 h); (E,F) Comparison of N2 adsorption-desorption isotherms (−196°C) (E), and X-ray diffractograms (F) of the ZTC before and after chemical and electrochemical oxidation treatments with similar efficiency (ΔO = 5100 μmolO/g). Inlet of (F) example of the high control on the structural degradation (111-X-ray characteristic peak of the ZTC) achieved by electrochemical oxidation in NaCl at low current densities.
It was previously demonstrated that the oxidation degree introduced on ACs by electrochemical methods can be easily adjusted, without the need of working at higher temperatures or using highly corrosive reagents, by the suitable choice of the applied current and treatment time (Berenguer et al., 2009, 2012). For example, it was found that similar oxidation degrees can be reached when the W is treated at 500 mA for 3 h than when it is oxidized at 200 mA for 5 h (Berenguer et al., 2012). The choice of these parameters may be strongly affected by the reactivity of the material, but also by its dimensions, morphology, and configuration, which may determine their effective polarization in the electrochemical cells and the contribution of direct and/or indirect oxidation mechanisms (Berenguer et al., 2013a). Furthermore, apart from current and time, the electrolyte also plays a key role. For instance, in Cl− solutions, the anodic generation of Cl2 and other oxidants can produce, through the indirect mechanism, larger functionalization than in NaOH or H2SO4. Thus, the concentration of oxidants during treatments in NaCl can be so high that the oxidizing conditions become even harder than those of chemical treatments, explaining the observed greater oxidation efficiency (Figure 5A). Considering all these facts, enough high anodic currents (usually above 200 mA) are necessary to cause similar or larger oxidation degrees than chemical treatments, and specifically in the case of ACs in the form of particles or cloths, which are not completely fixed to/contacting the current collector (i.e., when direct electrooxidation is less important).
Figure 5C represents the SOGs gain and SBET decrease, after similar electro-oxidation treatments, as a function of micropore volume of the studied ACs. It can be observed that the extent of oxidation and induced textural changes in the studied ACs augment with their micropore volume. This trend observed for the electrochemical method agrees with those found in literature and hereby presented for the chemical oxidation of ACs. Respect to the textural properties of the ACs, the extent of SBET-decrease with the oxidation degree seems to be independent of the oxidative method. Thus, the decrease in surface area may be better correlated with the extent of oxidation (Figure 5D), so a larger SBET decrease is generally observed for the more efficient electrochemical treatments (inlet of Figure 1). This fact is in line with the blockage of micropores as the main mechanism affecting the textural properties of oxidized ACs (see section Analysis of Textural Changes).
In the case of the ZTC, both the higher reactivity and the optimum polarization that is achieved by using ZTC-pastes (see experimental section) involves the use of lower currents for electrooxidation experiments. Figure 1 shows that under different conditions the electrochemical oxidation allows to fill the gap of properties that are difficult to obtain by uncontrolled chemical treatments. For example, short (1 h) oxidation times in NaCl at current between 2 and 20 mA (ZTC-Cl- curve) permit to reach lower oxidation degrees that are rapidly overcome during fast chemical treatments. On the other hand, long treatments (15–36 h) in H2SO4 at low current (2–5 mA) (ZTC-H+- curve) enable to reach similar or larger oxidation degrees to those of most oxidizing chemicals but with considerably lower SBET losses. The effect on the textural properties is shown in the Figure 5E. The figure shows that for a similar oxidation degree of 5,100 μmolO/g, the ZTC oxidized at 5 mA for 15 in H2SO4 maintains a larger volume of micropores (larger volume of N2 adsorbed at low pressures in the isotherm) than the sample treated with 30 wt% HNO3 for 1 h, both at RT. This greater microporosity in the electro-oxidized sample agrees with the higher intensity of the X-ray diffraction peak related to the long-range microporous ordering from the Y zeolite template. In this sense, Figure 5F demonstrates that at low currents, the extent of structural degradation of ZTC (decrease in the intensity of 111-X-ray peak) can be gradually controlled just by adjusting the treatment time. Hence, the considerably higher oxidation degrees with lesser structural degradation in ZTC reached by extending the treatment time of low-current anodic oxidations in H2SO4 suggests that these are the most suitable conditions to functionalize ZTC. Similar conclusions were reached from the analysis of the modification of CO/CO2 ratios after the different oxidative treatments (Berenguer et al., 2012, 2013a,b).
Apart from all these effects, to talk about the controllability of the oxidative treatments is necessary to analyze the kinetics of the process. As it can be observed in Figure 2A, the linear or almost linear (gradual) evolutions of the oxidation degree with time, for different microporous materials and under quite different electrochemical conditions, are indicative of a controlled oxidation process. The slope of these linear trends is related to the kinetics of the oxidation processes. One of the main consequences of this behavior is that the oxidation kinetics remain approximately constant for a given set of experimental conditions (at least during the analyzed periods of time). Moreover, Figure 2A also shows that the modification of these electrochemical parameters (current, time, electrolyte) enables to adjust the slope of the linear trend, i.e., the rate of the oxidation reaction. By contrast, chemical oxidation treatments show all (more exaggerated in the case of the ZTC) a fast-initial oxidation (high slope) that rapidly slows down (low slope), i.e., a highly variable reaction rate difficult to control. For the ZTC, the consequence of this fast reaction rate is that above 40% of its initial BET surface area is lost just in few minutes, i.e., within the 1 h of chemical oxidation (Figure 2B). This indicates that the collapse of ZTC structure during chemical oxidation is remarkably fast.
Interestingly, even at strong electrochemical conditions for ZTC (ZTC-50 mA-Cl), where the oxidizing conditions are comparable to those of most oxidizing chemical treatments, the rate of ZTC electro-oxidation is slower (more controlled) than chemical oxidation with 30 wt% HNO3 at 80°C (Figure 2A). This may be explained by considering that the concentration of reactive species is initially zero and increases in a controlled manner for the NaCl anodic treatment, whereas it is high from the beginning for any chemical oxidation (Berenguer et al., 2012). Another possible consequence of the less controllability of oxidizing conditions is that the chemical oxidation may occur rapidly on the outer surface of the carbon material, just after physical interaction, so that the introduced SOGs could hamper the diffusion of reagent into the inner pores. By contrast, in indirect anodic treatments the reactants may be homogeneously generated in any effectively-polarized micropore impregnated with NaCl. This effect could also contribute to explain the comparatively higher oxidation degrees and greater micropore blockage (during N2 adsorption-desorption analysis) when the ACs are anodically oxidized in NaCl.
Selectivity
As exposed in the introduction section, among the spectrum of different functionalities, only some specific groups are desired for certain applications. For these applications, hence, the selectivity of the functionalization becomes important. Figure 6A compiles the CO2-evolving TPD profiles of various carbon materials after different effective oxidative treatments (ΔO > 3,500 μmolO/g). The chemical oxidation of the W with (NH4)2S2O8 (W-S-5 h) mainly increases the CO2-evolving SOGs with lower thermal stability, i.e., those decomposing at lower temperatures. Particularly, the profile is characterized by two peaks, the most intense with maximum at 275°C and a wider overlapped contribution with maximum at 450°C. These evolutions have been assigned to carboxylic- and lactone-like groups, respectively, on carbon surfaces (Figueiredo et al., 1999). As it can be seen in the figure, the electrochemical oxidation of the GAC in NaCl produces exactly the same CO2-TPD profile, and the same was observed also for the chemical treatment with 65 wt% HNO3 (figure not shown). Furthermore, exactly the same CO2-TPD profiles were also obtained for A after electrochemical oxidation in NaCl (Figure 6A) and chemical treatments with 65 wt% HNO3 or H2O2 (figures not shown), as well as many other ACs in literature (Otake and Jenkins, 1993; Román-Martínez et al., 1993; Moreno-Castilla et al., 1995, 1997; De la Puente et al., 1997; Figueiredo et al., 1999; Pradhan and Sandle, 1999; Strelko and Malik, 2002; Li et al., 2003; Bleda-Martínez et al., 2006). In particular, different studies on the chemical oxidation of ACs point out that, compared to HNO3, (NH4)2S2O8 favors the generation of carboxylic groups (Moreno-Castilla et al., 1995, 1997; Vinu et al., 2007). On the other hand, quite similar CO-TPD profiles, with a general increase in the whole temperature range and a shift of the maximum toward lower temperatures, were also obtained for the different materials treated under these conditions.
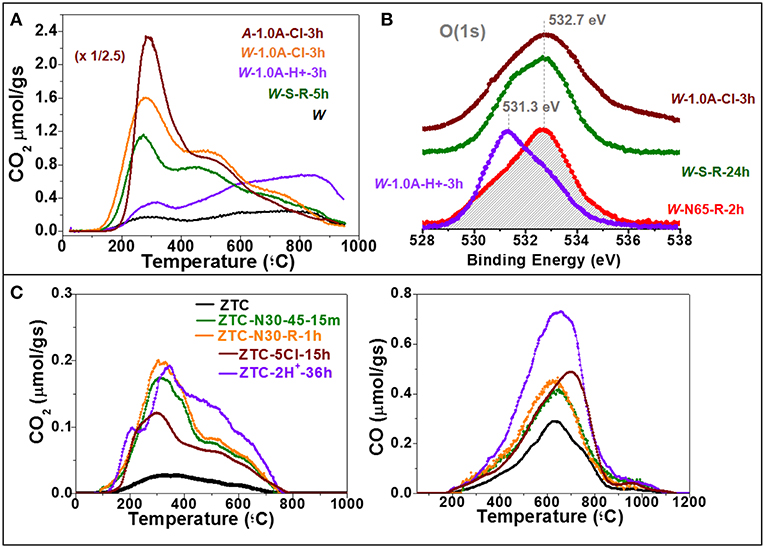
Figure 6. (A) CO2 evolution from TPD experiments for the original and the chemically- and anodically-treated W and A; (B) O(1s) XPS spectra of the W samples oxidized by different chemical and electrochemical methods; (C) CO2 and CO evolution from TPD experiments for the original and the chemically- and anodically-oxidized ZTC.
The results observed by TPD were confirmed by XPS. Figure 6B shows the O(1s) XPS spectra of the W samples oxidized in 65 wt% HNO3, (NH4)2S2O8, or anodically in NaCl. As observed, the spectra of the three oxidized samples are centered at the same binding energy (532.7 eV). This suggests that the same type of oxygen bonds prevail in these samples. All these results indicate that, though different in concentration, the SOGs introduced by the chemical reagents and the anodic treatment in NaCl are of the same nature, stressing the lack of selectivity by these oxidative treatments.
By stark contrast, the GAC samples electrochemically oxidized in H2SO4 and NaOH electrolytes, where the electrochemical oxidation may be governed by direct polarization, displayed TPD profiles and XPS spectra with different shape. For example, the anodic oxidation in H2SO4 leads to an increase in the most stable CO2-evolving SOGs (Figure 6A) and, accordingly, a remarkable shift in the O(1s) XPS binding energy toward lower binding energies (centered at 531.3 eV) (Figure 6B). Hence, by changing the electrolyte the electrochemical oxidation method enables to reach a different selectivity.
In the case of highly-reactive ZTC, the oxidation by chemicals or anodically in NaCl at high currents produced a general increase in the whole TPDs, confirming the lack of selectivity. As it can be observed in Figure 6C, the relative increase in CO2-evolving groups was more significant after chemical oxidation (CO/CO2 = 2.80–2.90) than after electro-oxidations at softer conditions in NaCl or H2SO4 (CO/CO2 = 3.60–5.60), the later exhibiting a remarkably higher relative increase in CO-evolving groups (sample ZTC-2H+-36 h). In addition, this sample treated in H2SO4 electrolyte shows a relative larger generation of the most stable CO2-evolving groups, thus bringing a different selectivity.
The remarkable increase in the CO-profile after electrochemical oxidation in H2SO4 was associated to a large generation of quinone-like SOGs (Itoi et al., 2014; Nueangnoraj et al., 2015). Indeed, by controlling the parameters of the electrochemical method, oxidized ZTC-derived carbons with ca. 80–90% selectivity toward quinone-like SOGs generation have been obtained. Since such a significant selectivity cannot be achieved in ACs by using similar electrochemical methods, the nature of the material may play also a crucial role. Accordingly, it is hypothesized that the characteristic regular structure of ZTC may somehow influence in the incorporation of quinones.
Conclusions
The results presented in this work clearly show that the efficiency, selectivity and effects on microporous texture associated to the oxidative functionalization depends on the nature of the carbon material and the oxidation method. Independently of the oxidative method, the incorporation of SOGs on ACs decreases the specific surface area by blocking and/or constricting the entry of micropores, while it causes the collapse of the ordered microporous framework of the ZTC. The kinetic analysis indicates that this oxidation reaction for chemical treatments is quite fast and variable, restricting the control of the process. By contrast, by using proper electrolytes, low current and/or extending the treatment time, the electrochemical oxidative method permits at room temperature not only a greater control of the oxidation degree of the microporous carbons, but also to minimize the degradation of the fragile microporous structure of ZTC. Moreover, the electrochemical oxidation enables different selectivity to that provided by chemical methods.
Data Availability
All datasets generated for this study are included in the manuscript.
Author Contributions
RB performed the different oxidative experiments, materials characterization and devised the subject of the manuscript. EM greatly contributed to data analyses, results discussion and manuscript preparation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Financial support from the Spanish Ministerio de Economía y Competitividad and FEDER funds (RYC-2017-23618, MAT2016-76595-R) is gratefully acknowledged.
References
Álvarez, P. M., García-Araya, J. F., Beltrán, F. J., Masa, F. J., and Medina, F. (2005). Ozonation of activated carbons: effect on the adsorption of selected phenolic compounds from aqueous solutions. J. Colloid. Interf. Sci. 283, 503–512. doi: 10.1016/j.jcis.2004.09.014
Arenillas, A., Rubiera, F., Pevida, C., Ania, C. O., and Pis, J. J. (2004). Relationship between structure and reactivity of carbonaceous materials. J. Therm. Anal. Cal. 76, 593–602. doi: 10.1023/B:JTAN.0000028038.34976.83
Bazuła, P. A., Lu, A. H., Nitz, J. J., and Schüth, F. (2008). Surface and pore structure modification of ordered mesoporous carbons via a chemical oxidation approach. Micropor. Mesopor. Mater. 108, 266–275. doi: 10.1016/j.micromeso.2007.04.008
Beguin, F., and Frackowiak, E. (2009). Carbons for Electrochemical Energy Storage and Conversion Systems. Boca Raton, FL: CRC Press.
Berenguer, R., Marco-Lozar, J. P., Quijada, C., Cazorla-Amorós, D., and Morallón, E. (2009). Effect of electrochemical treatments on the surface chemistry of activated carbon. Carbon 47, 1018–1027. doi: 10.1016/j.carbon.2008.12.022
Berenguer, R., Marco-Lozar, J. P., Quijada, C., Cazorla-Amorós, D., and Morallón, E. (2012). A comparison between oxidation of activated carbon by electrochemical and chemical treatments. Carbon 50, 1123–1134. doi: 10.1016/j.carbon.2011.10.025
Berenguer, R., Morallón, E., Cazorla-Amorós, D., Nishihara, H., Itoi, H., Ishii, T., et al. (2013b). Tailoring the surface chemistry of zeolite templated carbon by electrochemical methods. Boletin Del Grupo Espanol Del Carbon 28, 10–14. Available online at: http://www.gecarbon.org/boletines/articulos/boletinGEC_028-A2.pdf
Berenguer, R., Nishihara, H., Itoi, H., Ishii, T., Morallón, E., Cazorla-Amorós, D., et al. (2013a). Electrochemical generation of O-containing groups in an ordered microporous zeolite-templated carbon. Carbon 54, 94–104. doi: 10.1016/j.carbon.2012.11.007
Bleda-Martínez, M. J., Lozano-Castelló, D., Morallón, E., Cazorla-Amorós, D., and Linares-Solano, A. (2006). Chemical and electrochemical characterization of porous carbon materials. Carbon 44 2642–2651. doi: 10.1016/j.carbon.2006.04.017
Boudou, J. P., Martínez-Alonso, A., and Tascón, J. M. D. (2000). Introduction of acidic groups at the surface of activated carbon by microwave-induced oxygen plasma at low pressure. Carbon 38, 1021–1029. doi: 10.1016/S0008-6223(99)00209-2
Daud, W. M. A. W., and Houshamnd, A. H. (2010). Textural characteristics, surface chemistry and oxidation of activated carbon. J. Nat. Gas Chem. 19, 267–279. doi: 10.1016/S1003-9953(09)60066-9
De la Puente, G., Pis, J. J., Menéndez, J. A., and Grange, P. (1997). Thermal stability of oxygenated functions in activated carbons. J. Anal. Appl. Pyrolysis 43, 125–138. doi: 10.1016/S0165-2370(97)00060-0
El-Hendawy, A. N. A. (2003). Influence of HNO3 oxidation on the structure and adsorptive properties of corncob-based activated carbon. Carbon 41, 713–722. doi: 10.1016/S0008-6223(03)00029-0
El-Sayed, Y., and Bandosz, T. J. (2001). A study of acetaldehyde adsorption on ACs. J. Colloid Interface Sci. 242, 44–51. doi: 10.1006/jcis.2001.7772
Figueiredo, J. L., Pereira, M. F. R., Freitas, M. M. A., and Órfäo, J. J. M. (1999). Modification of the surface chemistry of activated carbons. Carbon 37, 1379–1389. doi: 10.1016/S0008-6223(98)00333-9
García, A. B., Martínez-Alonso, A., León y León, C. A., and Tascón, J. M. D. (1998). Modification of the surface properties of an activated carbon by oxygen plasma treatment. Fuel 77, 61–624. doi: 10.1016/S0016-2361(97)00111-7
Gil, A., de la Puente, G., and Grange, P. (1997). Evidence of textural modifications of an activated carbon on liquid-phase oxidation treatments. Microporous Mater. 12, 51–61. doi: 10.1016/S0927-6513(97)00057-6
Gómez-Serrano, V., Álvarez, P. M., Jaramillo, J., and Beltrán, F. J. (2002). Formation of oxygen structures by ozonation of carbonaceous materials prepared from cherry stones. II. kinetic study. Carbon 40, 523–529. doi: 10.1016/S0008-6223(01)00142-7
González-Gaitán, C., Ruíz-Rosas, R., Morallón, E., and Cazorla-Amorós, D. (2015). “Electrochemical methods to functionalize carbon materials”, in Chemical Functionalization of Carbon Nanomaterials: Chemistry and Applications, eds V. K. Thakur and M. K. Thakur (Boca Raton, FL: CRC Press), 231–262.
Houshmand, A., Daud, W. M. A. W., and Shafeeyan, M. S. (2011). Tailoring the surface chemistry of activated carbon by nitric acid: study using response surface method. Bull. Chem. Soc. Jpn. 84, 1251–1260. doi: 10.1246/bcsj.20110145
Itoi, H., Nishihara, H., Ishii, T., Nueangnoraj, K., Berenguer, R., and Kyotani, T. (2014). Large pseudocapacitance in quinone-functionalized zeolite templated carbon. Bull. Chem. Soc. Jpn. 87, 250–257. doi: 10.1246/bcsj.20130292
Jaramillo, J., Álvarez, P. M., and Gómez-Serrano, V. (2010). Oxidation of activated carbon by dry and wet methods surface chemistry and textural modifications. Fuel Process. Technol. 91, 1768–1775. doi: 10.1016/j.fuproc.2010.07.018
Li, N., Ma, X., Zha, Q., Kim, K., Chen, Y., and Song, C. (2011). Maximizing the number of oxygen-containing functional groups on activated carbon by using ammonium persulfate and improving the temperature-programmed desorption characterization of carbon surface chemistry. Carbon 49, 5002–5013. doi: 10.1016/j.carbon.2011.07.015
Li, Y. H., Lee, C. W., and Gullett, B. K. (2003). Importance of activated carbon's oxygen surface functional groups on elemental mercury adsorption. Fuel 82, 451–457. doi: 10.1016/S0016-2361(02)00307-1
Linares-Solano, A., Lozano-Castelló, D., Lillo-Ródenas, M. A., and Cazorla-Amorós, D. (2008). “Carbon activation by alkaline hydroxides preparation and reactions, porosity and performance,” in Chemistry and Physics of Carbon, Vol. 30, ed L. R. Radovic (Boca Raton, FL: CRC Press), 1–62.
Lozano-Castelló, D., Lillo-Ródenas, M. A., Cazorla-Amorós, D., and Linares-Solano, A. (2001). Preparation of activated carbons from Spanish anthracite. I. activation by KOH. Carbon 39, 741–749. doi: 10.1016/S0008-6223(00)00185-8
Lozano-Castelló, D., Suárez-García, F., Cazorla-Amorós, D., and Linares-Solano, A. (2009). “Porous texture of carbons,” in Carbons for Electrochemical Energy Storage and Conversion Systems, eds F. Beguin and E. Frackowiak (Boca Raton, FL: CRC Press), 115–162.
Lu, A. H., Li, W. C., Muratova, N., Spliethoff, B., and Schüth, F. (2005). Evidence for C–C bond cleavage by H2O2 in a mesoporous CMK-5 type carbon at room temperature. Chem. Commun. 41, 5184–5186. doi: 10.1039/b509300g
Ma, Z. X., Kyotani, T., and Tomita, A. (2000). Preparation of a high surface area microporous carbon having the structural regularity of Y zeolite. Chem. Commun. 2, 2365–2366. doi: 10.1039/b006295m
Mangun, C. L., Benak, K. R., Daley, M. A., and Economy, J. (1999). Oxidation of activated carbon fibers: effect on pore size, surface chemistry, and adsorption properties. Chem. Mater. 11, 3476–3483. doi: 10.1021/cm990123m
Maroto-Valer, M. M., Dranca, I., Lupascu, T., and Nastas, R. (2004). Effect of adsorbate polarity on thermodesorption profiles from oxidized and metal-impregnated activated carbons. Carbon 42, 2655–2659. doi: 10.1016/j.carbon.2004.06.007
Molina-Sabio, M., and Rodríguez-Reinoso, F. (2004). Role of chemical activation in the development of carbon porosity. colloids surf. a physicochem. Eng. Asp. 241, 15–25. doi: 10.1016/j.colsurfa.2004.04.007
Moreno-Castilla, C., Carrasco-Marín, F., and Mueden, A. (1997). The creation of acid carbon surfaces by treatment with (NH4)2S2O8. Carbon 35, 1619–1626. doi: 10.1016/S0008-6223(97)00121-8
Moreno-Castilla, C., Carrasco-Martín, F., Maldonado-Hodar, F. J., and Rivera-Utrilla, J. (1998). Effects of non-oxidant and oxidant acid treatments on the surface properties of an activated carbon with very low ash content. Carbon 36, 145–151. doi: 10.1016/S0008-6223(97)00171-1
Moreno-Castilla, C., Ferro-García, M. A., Joly, J. P., Bautista-Toledo, I., Carrasco-Marín, F., and Rivera-Utrilla, J. (1995). Activated carbon Surface modifications by nitric acid, hydrogen peroxide, and ammonium peroxydisulfate treatments. Langmuir 11, 4386–4392. doi: 10.1021/la00011a035
Moreno-Castilla, C., López-Ramón, M. V., and Carrasco-Marín, F. (2000). Changes in surface chemistry of activated carbons by wet oxidation. Carbon 38, 1995–2001. doi: 10.1016/S0008-6223(00)00048-8
Nishihara, H., and Kyotani, T. (2012). Templated nanocarbons for energy storage. Adv. Mater. 24, 4473–4498. doi: 10.1002/adma.201201715
Nishihara, H., and Kyotani, T. (2018). Zeolite-templated carbons – three-dimensional microporous graphene frameworks. Chem. Commun. 54, 5648–5673. doi: 10.1039/C8CC01932K
Nishihara, H., Simura, T., Kobayashi, S., Nomura, K., Berenguer, R., Ito, M., et al. (2016). Stability oxidation-resistant and elastic mesoporous carbon with single-layer graphene walls. Adv. Funct. Mater. 26, 6418–6427. doi: 10.1002/adfm.201602459
Nishihara, H., Yang, Q. H., Hou, P. X., Unno, M., Yamauchi, S., Saito, R., et al. (2009). A possible buckybowl-like structure of zeolite templated carbon. Carbon 47, 1220–1230. doi: 10.1016/j.carbon.2008.12.040
Nueangnoraj, K., Nishihara, H., Ishii, T., Yamamoto, N., Itoi, H., Berenguer, R., et al. (2015). Pseudocapacitance of zeolite-templated carbon in organic electrolytes. Energy Storage Mater. 1, 35–41. doi: 10.1016/j.ensm.2015.08.003
Otake, Y., and Jenkins, R. G. (1993). Characterization of oxygen-containing surface complexes created on a microporous carbon by air and nitric acid treatment. Carbon 31, 109–121. doi: 10.1016/0008-6223(93)90163-5
Pradhan, B. K., and Sandle, N. K. (1999). Effect of different oxidizing agent treatments on the surface properties of activated carbons. Carbon 37 1323–1332. doi: 10.1016/S0008-6223(98)00328-5
Radovic, L. R., Moreno-Castilla, C., and Rivera-Utrilla, J. (2000). “Carbon materials as adsorbents in aqueous solutions,” in Chemistry and Physics of Carbon, Vol. 27, ed L. R. Radovic (Boca Raton, FL: CRC Press), 227–405.
Rivera-Utrilla, J., Sánchez-Polo, M., Gómez-Serrano, V., Álvarez, P. M., Alvim-Ferraz, M. C. M., and Dias, J. M. (2011). Activated carbon modifications to enhance its water treatment applications. an overview. J. Haz. Mater. 187, 1–23. doi: 10.1016/j.jhazmat.2011.01.033
Rodriguez-Reinoso, F. (1998). The role of carbon materials in heterogeneous catalysis. Carbon 36, 159–175. doi: 10.1016/S0008-6223(97)00173-5
Rodríguez-Reinoso, F., and Molina-Sabio, M. (1998). Textural and chemical characterization of microporous carbons. Adv. Colloid Interface Sci. 76–77, 271–294. doi: 10.1016/S0001-8686(98)00049-9
Román-Martínez, M. C., Cazorla-Amorós, D., Linares-Solano, A., and Salinas-Martínez de Lecea, C. (1993). TPD and TPR characterization of carbonaceous supports and Pt/C catalysts. Carbon 31, 895–902. doi: 10.1016/0008-6223(93)90190-L
Serp, P., and Figueiredo, J. L. (2009). Carbon Materials for Catalysis. Hoboken, NJ: John Wiley & Sons, Inc.
Shen, W., Li, Z., and Liu, Y. (2008). Surface chemical functional groups modification of porous carbon. Recent Pat. Chem. Eng. 1, 27–40. doi: 10.2174/2211334710801010027
Shim, J. W., Park, S. J., and Ryu, S. K. (2001). Effect of modification with HNO3 and NaOH on metal adsorption by pitch-based activated carbon fibers. Carbon 39, 1635–1642. doi: 10.1016/S0008-6223(00)00290-6
Strelko, V., and Malik, D. J. (2002). Characterization and metal sorptive properties of oxidized active carbon. J. Colloid Interface Sci. 250, 213–220. doi: 10.1006/jcis.2002.8313
Szymanski, G. S., Grzybek, T., and Papp, H. (2004). Influence of nitrogen surface functionalities on the catalytic activity of activated carbon in low temperature SCR of NOx with NH3. Catal. Today 90, 51–59. doi: 10.1016/j.cattod.2004.04.008
Tabti, Z., Berenguer, R., Ruiz-Rosas, R., Quijada, C., Morallón, E., and Cazorla-Amorós, D. (2013). Electrooxidation methods to produce pseudocapacitance-containing porous carbons. Electrochemistry 81, 833–839. doi: 10.5796/electrochemistry.81.833
Ternero-Hidalgo, J. J., Rosas, J. M., Palomo, J., Valero-Romero, M. J., Rodríguez-Mirasol, J., and Cordero, T. (2016). Functionalization of activated carbons by HNO3 treatment: Influence of phosphorus surface groups. Carbon 101, 409–419. doi: 10.1016/j.carbon.2016.02.015
Thakur, V. K., and Thakur, M. K. (2016). Chemical Functionalization of Carbon Nanomaterials: Chemistry and Applications. Boca Raton, FL: CRC Press.
Vinu, A., Hossian, K. Z., Srinivasu, P., Miyahara, M., Anandan, S., Gokulakrishnan, N., et al. (2007). Carboxy-mesoporous carbon and its excellent adsorption capability for proteins. J. Mater. Chem. 17, 1819–1825. doi: 10.1039/b613899c
Vujković, M., Bajuk-Bogdanović, D., Matović, L, Stojmenović, M., and Mentus, S. (2018). Mild electrochemical oxidation of zeolite templated carbon in acidic solutions, as a way to boost its charge storage properties in alkaline solutions. Carbon 138, 369–378. doi: 10.1016/j.carbon.2018.07.053
Zakaria, T. (2014). Electroadsorción de Plomo Sobre Carbones Activados en Diferentes Conformaciones: Modificación de la Química Superficial por Métodos Electroquímicos (dissertation thesis). University of Alicante. Available online at: http://hdl.handle.net/10045/41922
Keywords: microporous carbons, chemical oxidation, electrochemical oxidation, functionalization, oxidation kinetics, selectivity
Citation: Berenguer R and Morallón E (2019) Oxidation of Different Microporous Carbons by Chemical and Electrochemical Methods. Front. Mater. 6:130. doi: 10.3389/fmats.2019.00130
Received: 15 March 2019; Accepted: 16 May 2019;
Published: 04 June 2019.
Edited by:
Jie-Sheng Chen, Shanghai Jiao Tong University, ChinaReviewed by:
Fotios K. Katsaros, National Centre of Scientific Research Demokritos, GreeceStanisław Biniak, Nicolaus Copernicus University in Torun, Poland
Copyright © 2019 Berenguer and Morallón. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Raúl Berenguer, cmF1bC5iZXJlbmd1ZXJAdWEuZXM=