 Xiuqin Hu1Disha Wang1Yi Tong1Linjiang Tong2Xia Wang1Lili Zhu1Hua Xie2
Xiuqin Hu1Disha Wang1Yi Tong1Linjiang Tong2Xia Wang1Lili Zhu1Hua Xie2 Shiliang Li1
Shiliang Li1 You Yang1*Yufang Xu1*
You Yang1*Yufang Xu1*- 1Shanghai Key Laboratory of New Drug Design, School of Pharmacy, East China University of Science and Technology, Shanghai, China
- 2Division of Anti-tumor Pharmacology, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, China
The synthesis of a series of ribose-modified anilinopyrimidine derivatives was efficiently achieved by utilizing DBU or tBuOLi-promoted coupling of ribosyl alcohols with 2,4,5-trichloropyrimidine as key step. Preliminary biological evaluation of this type of compounds as new EGFR tyrosine kinase inhibitors for combating EGFR L858R/T790M mutant associated with drug resistance in the treatment of non-small cell lung cancer revealed that 3-N-acryloyl-5-O-anilinopyrimidine ribose derivative 1a possessed potent and specific inhibitory activity against EGFR L858R/T790M over WT EGFR. Based upon molecular docking studies of the binding mode between compound 1a and EGFR, the distance between the Michael receptor and the pyrimidine scaffold is considered as an important factor for the inhibitory potency and future design of selective EGFR tyrosine kinase inhibitors against EGFR L858R/T790M mutants.
Introduction
Epidermal growth factor receptor (EGFR), a transmembrane protein with tyrosine kinase activity, is essential for cell growth, differentiation, migration, adhesion, and proliferation under normal physiological conditions (Gschwind et al., 2004). However, overexpression of EGFR has been associated with tumor growth and progression in a variety of cancers including non-small cell lung cancer (NSCLC), head and neck squamous cell carcinoma, and pancreatic cancer (Huang and Harari, 1999; Kris et al., 2003; Moore et al., 2007; Harrington et al., 2009). Therefore, regulation of EGFR has been deemed as an important strategy for the development of cancer therapy (Huang and Harari, 1999; Gschwind et al., 2004; Steuer et al., 2015).
First-generation EGFR tyrosine kinase inhibitors (TKIs) such as gefitinib and erlotinib that possess a 4-anilinoquinazoline scaffold, reversibly inhibit EGFR mutants (L858R and delE746_A750) as well as wild-type (WT) EGFR, resulting in significant disease control of patients with NSCLC (Figure 1) (Cohen et al., 2005; Cheng et al., 2016). However, drug resistance driven by activating mutation of the gatekeeper T790M residue in which the threonine group is replaced with the methionine moiety, greatly counteracted the clinical efficiency of first-generation TKIs against NSCLC (Ozvegy-Laczka et al., 2005; Balak et al., 2006; De Luca et al., 2008; Pao and Chmielecki, 2010). To address this issue, the irreversible EGFR TKIs (afatinib, osimertinib, WZ4002, and CO-1686) which contain a Michael acceptor moiety for binding covalently to the thiol group of Cys797 in the ATP binding domain of EGFR, were developed to treat NSCLC via the efficient inhibition of EGFR mutants (Figure 1; Castellanos and Horn, 2015). Among them, second-generation TKIs such as afatinib potently inhibited both EGFR mutants (L858R/T790M) and WT-EGFR without mutant selectivity, thereby leading to side effects such as rash and diarrhea (Dungo and Keating, 2013). In contrast, third-generation TKIs such as osimertinib, WZ4002, and CO-1686 bearing an anilinopyrimidine core, showed high potency and selectivity for EGFR L858R/T790M over WT EGFR, therefore serving as mutant-selective TKIs targeting EGFR mutants involved in NSCLC (Zhou et al., 2009; Walter et al., 2013; Cross et al., 2014; Finlay et al., 2014; Gray and Haura, 2014).
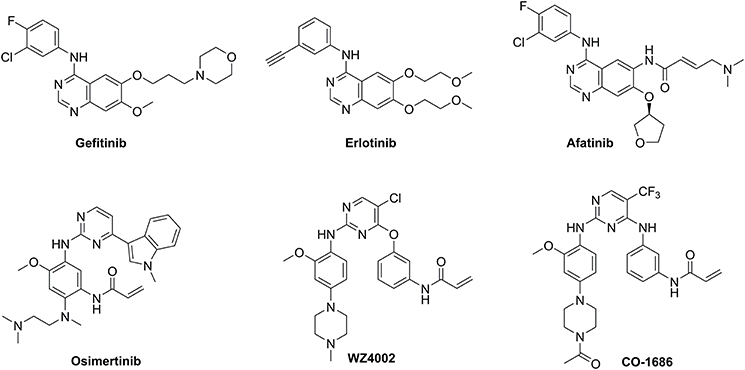
Figure 1. Structures of three generations of EGFR tyrosine kinase inhibitors.
Considering the drug resistance is rapidly emerging for third-generation TKIs (Eberlein et al., 2015; Niederst et al., 2015; Piotrowska et al., 2015; Thress et al., 2015), design of EGFR inhibitors with new structural skeletons could lead to the discovery of novel types of TKIs against EGFR mutants such as the triple mutant L858R/T790M/C797S (Günther et al., 2016, 2017; Jia et al., 2016; Juchum et al., 2017; Park et al., 2017). Based on the fact that most commercially available TKIs are ATP-competitive inhibitors for binding at the catalytic domain of the EGFR tyrosine kinase (Traxler and Furet, 1999; Grünwald and Hidalgo, 2003; Normanno et al., 2003), we envisioned that replacement of the phenyl ring on the right side of WZ4002 with a chiral ribosyl moiety would provide compound 1 as a novel type of carbohydrate-based EGFR TKI against the drug resistance involved in NSCLC (Figure 2). Here we report the synthesis, preliminary biological evaluation and molecular docking studies of ribose-containing anilinopyrimidine derivatives as EGFR TKIs against EGFR L858R/T790M.
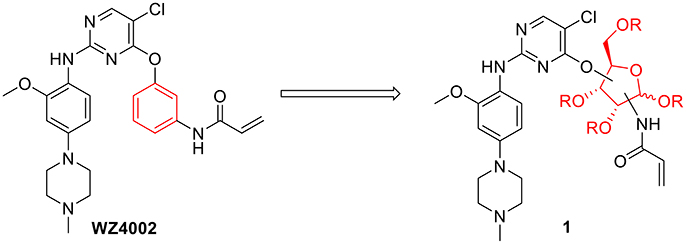
Figure 2. Design of novel EGFR tyrosine kinase inhibitors.
Materials and Methods
Commercial reagents were used without further purification except where noted. Solvents were dried and redistilled prior to use in the usual way. All reactions were performed in oven-dried glassware with magnetic stirring under an inert atmosphere unless noted otherwise. Analytical thin layer chromatography (TLC) was performed on precoated plates of Silica Gel (0.25–0.3 mm, Shanghai, China). The TLC plates were visualized with UV light and by staining with sulfuric acid-ethanol solution. Silica gel column chromatography was performed on Silica Gel AR (100–200 mesh, Shanghai, China). NMR spectra were measured with a Bruker Avance III 400 or Bruker Avance III 500 spectrometer. The 1H and 13C NMR spectra were calibrated against the residual proton and carbon signals of the solvents as internal references (CDCl3: δH = 7.26 ppm and δC = 77.2 ppm; CD3OD: δH = 3.31 ppm and δC = 49.0 ppm). Multiplicities are quoted as singlet (s), broad singlet (br s), doublet (d), doublet of doublets (dd), triplet (t), or multiplet (m). All NMR chemical shifts (δ) were recorded in ppm and coupling constants (J) were reported in Hz. Mass spectra were recorded on an Agilent Technologies 6120 or LCT Premier XE FTMS instrument.
1,2-O-Isopropylidene-3-N-acryloyl-3-deoxy-5-O-(2,5-dichloropyrimidin-4-yl)-α-D-ribofuranoside 7
To a solution of compound 5 (0.60 g, 2.47 mmol) in anhydrous CH2Cl2 (30 mL) at room temperature, was added DBU (1.48 mL, 5.94 mmol) and 2,4,5-trichloropyrimidine 6 (0.57 mL, 4.94 mmol). After stirring at room temperature for 2 h, the reaction mixture was diluted with saturated aqueous NH4Cl, and extracted with CH2Cl2. The organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by silica gel chromatography (petroleum ether/EtOAc: 80/1) to give 7 (0.88 g, 92%) as a pale yellow syrup: 1H NMR (400 MHz, CDCl3) δ 8.30 (s, 1 H), 6.29 (dd, J = 1.2, 17.2 Hz, 1 H), 6.12 (dd, J = 10.4, 17.2 Hz, 1 H), 6.07 (d, J = 8.8 Hz, 1 H, NH), 5.90 (d, J = 3.6 Hz, 1 H, H-1), 5.69 (dd, J = 1.2, 10.4 Hz, 1 H), 4.73 (dd, J = 2.4, 12.0 Hz, 1 H), 4.66 (t, J = 4.0 Hz, 1 H), 4.61–4.55 (m, 2 H), 4.16 (m, 1 H), 1.57 (s, 3 H), 1.35 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 165.4, 165.3, 157.4, 157.2, 130.1, 128.0, 117.0, 113.0, 104.7, 79.1, 78.2, 67.3, 52.1, 26.8, 26.5; ESI-MS (ESI) m/z calcd for C15H17O5N3Cl2Na [M + Na]+ 412.0, found 412.0.
1,2-O-Isopropylidene-3-N-acryloyl-3-deoxy-5-O-[5-chloro-2-N-(2-methoxy-4-(4-methylpiperazin-1-yl)phenyl)pyrimidin-4-yl]-α-D-ribofuranoside 1a
To a solution of compound 7 (80 mg, 0.21 mmol) and aniline derivative 8 (91 mg, 0.41 mmol) in isobutanol (3 mL), was added TFA (0.12 mL, 1.55 mmol). The mixture was heated to 100°C and stirred for 5 h. After cooling down to room temperature, the mixture was quenched with Et3N (3 mL) and concentrated in vacuo to give a residue, which was purified by silica gel column chromatography (CH2Cl2/MeOH: 30/1) to give 1a (82 mg, 69%) as a pale yellow powder: 1H NMR (400 MHz, CDCl3) δ 8.10 (d, J = 8.8 Hz, 1 H), 8.08 (s, 1 H), 7.35 (br s, 1 H), 6.56 (dd, J = 2.4, 8.8 Hz, 1 H), 6.52 (d-like, J = 2.4 Hz, 1 H), 6.30 (dd, J = 1.2, 17.2 Hz, 1 H), 6.11 (m, 2 H), 5.91 (d, J = 3.6 Hz, 1 H, H-1), 5.67 (dd, J = 1.2, 10.4 Hz, 1 H), 4.66 (m, 2 H), 4.59–4.49 (m, 3 H), 4.22 (m, 1 H), 3.86 (s, 3 H), 3.16 (t-like, J = 5.2 Hz, 4 H), 2.58 (t-like, J = 5.2 Hz, 4 H), 2.34 (s, 3 H), 1.58 (s, 3 H), 1.35 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 165.4, 164.1, 157.9, 156.5, 149.2, 147.5, 130.2, 127.8, 122.0, 120.0, 112.9, 108.3, 106.2, 104.8, 100.6, 79.1, 78.2, 66.6, 55.8, 55.3, 52.7, 50.2, 46.2, 26.8, 26.5; HRMS (ESI) m/z calcd for C27H36O6N6ClNa [M + H]+ 575.2385, found 575.2385.
3-N-Acryloyl-3-deoxy-5-O-[5-chloro-2-N-(2-methoxy-4-(4-methylpiperazin-1-yl)phenyl)pyrimidin-4-yl]-D-ribofuranose 1b
A solution of compound 1a (58 mg, 0.10 mmol) in TFA/acetic acid/water (1/15/4, v/v/v, 5 mL) was stirred at 70°C for 7 h. Concentration in vacuo and elution through reverse phase C-18 column (H2O/MeOH: 2/3) provided 1b (44 mg, 83%) as a pale yellow syrup: 1H NMR (400 MHz, CD3OD) δ 8.07 (s, 1 H), 8.05 (s, 2.4 H), 7.92 (m, 3.4 H), 6.67 (d-like, J = 2.4 Hz, 3.4 H), 6.58 (dd, J = 2.8, 8.8 Hz, 3.4 H), 6.35 (m, 3.4 H), 6.21 (m, 3.4 H), 5.67 (m, 3.4 H), 5.38 (d, J = 4.0 Hz, 1 H), 5.22 (s, 2.4 H), 4.67 (m, 2.4 H), 4.58 (m, 3.4 H), 4.49 (m, 4.4 H), 4.34–4.24 (m, 4.4 H), 4.00 (d-like, J = 4.4 Hz, 2.4 H), 3.87 (s, 10.2 H), 3.78 (m, 6.8 H), 3.58 (m, 6.8 H), 3.26 (m, 6.8 H), 3.03 (m, 6.8 H), 2.96 (s, 10.2 H); HRMS (ESI) m/z calcd for C24H32O6N6ClNa [M + H]+ 535.2072, found 535.2079.
2,3-O-Isopropylidene-5-O-(2,5-dichloropyrimidin-4-yl)-α-D-ribofuranosyl acrylamide 14
To a solution of compound 13 (147 mg, 0.60 mmol) in anhydrous CH2Cl2 (25 mL) at room temperature, was added DBU (0.21 mL, 1.42 mmol) and 2,4,5-trichloropyrimidine 6 (0.12 mL, 1.07 mmol). After stirring at room temperature for 2 h, the reaction mixture was diluted with saturated aqueous NH4Cl, and extracted with CH2Cl2. The organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by silica gel chromatography (petroleum ether/EtOAc: 5/1) to give 14 (217 mg, 93%) as a colorless syrup: 1H NMR (400 MHz, CDCl3) δ 8.36 (s, 1 H), 6.58 (d, J = 9.2 Hz, 1 H, NH), 6.32 (dd, J = 1.2, 17.2 Hz, 1 H), 6.15 (dd, J = 10.4, 17.2 Hz, 1 H), 6.08 (dd, J = 4.0, 9.2 Hz, 1 H, H-1), 5.72 (dd, J = 1.2, 10.4 Hz, 1 H), 4.86 (d-like, J = 6.0 Hz, 1 H), 4.82 (dd, J = 4.4, 6.0 Hz, 1 H), 4.55 (m, 2 H), 4.45 (t, J = 2.8 Hz, 1 H), 1.57 (s, 3 H), 1.38 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 165.1, 165.0, 157.7, 157.4, 130.6, 128.2, 116.8, 113.4, 82.3, 81.6, 79.7, 79.4, 70.4, 26.4, 24.8; HRMS (ESI) m/z calcd for C15H17O5N3Cl2Na [M + Na]+ 412.0443, found 412.0448.
5-O-[5-Chloro-2-N-(2-methoxy-4-(4-methylpiperazin-1-yl)phenyl)pyrimidin-4-yl]-D-ribofuranosyl acrylamide 1c
To a solution of compound 14 (58 mg, 0.15 mmol) and aniline derivative 8 (66 mg, 0.30 mmol) in isobutanol (3 mL), was added TFA (0.084 mL, 1.13 mmol). The mixture was heated to 100°C and stirred for 5 h. After cooling down to room temperature, the mixture was quenched with Et3N (3 mL) and concentrated in vacuo to give a residue, which was purified by silica gel column chromatography (CH2Cl2/MeOH: 30/1) to give 15 (60 mg, 70%) as a pale yellow oil: HRMS (ESI) m/z calcd for C27H36O6N6Cl [M + H]+ 575.2385, found 575.2383. A solution of compound 15 (85 mg, 0.15 mmol) in TFA/acetic acid/water (1/20/4, v/v/v, 4 mL) was stirred at 50°C for 5 h. Concentration in vacuo and elution through reverse phase C-18 column (H2O/MeOH: 2/3) provided 1c (64 mg, 80%) as a pale yellow syrup. 1c (α): 1H NMR (400 MHz, CD3OD) δ 8.06 (s, 1 H), 7.88 (d, J = 8.8 Hz, 1 H), 6.63 (d-like, J = 2.4 Hz, 1 H), 6.53 (dd, J = 2.8, 8.8 Hz, 1 H), 6.34 (dd, J = 10.0, 17.2 Hz, 1 H), 6.27 (dd, J = 2.0, 17.2 Hz, 1 H), 5.83 (d, J = 4.4 Hz, 1 H, H-1), 5.70 (dd, J = 2.0, 10.0 Hz, 1 H), 4.57 (dd, J = 3.2, 12.0 Hz, 1 H), 4.40 (dd, J = 4.0, 11.6 Hz, 1 H), 4.25 (m, 3 H), 3.85 (s, 3 H), 3.16 (t, J = 4.8 Hz, 4 H), 2.61 (t, J = 4.8 Hz, 4 H), 2.34 (s, 3 H); 13C NMR (100 MHz, CD3OD) δ 168.0, 165.6, 159.5, 157.3, 151.6, 148.8, 132.1, 128.1, 123.0, 122.3, 109.3, 106.7, 101.9, 81.9 (C-1), 81.5, 73.3, 71.8, 68.2, 56.4, 55.8, 50.2, 45.6; 1c (β): 1H NMR (400 MHz, CD3OD) δ 8.05 (s, 1 H), 7.87 (d, J = 8.4 Hz, 1 H), 6.65 (d-like, J = 2.4 Hz, 1 H), 6.54 (dd, J = 2.4, 8.8 Hz, 1 H), 6.23 (m, 2 H), 5.67 (dd, J = 4.8, 6.8 Hz, 1 H), 5.48 (d, J = 4.4 Hz, 1 H, H-1), 4.58 (dd, J = 3.6, 12.0 Hz, 1 H), 4.41 (dd, J = 4.8, 12.0 Hz, 1 H), 4.24–4.14 (m, 2 H), 4.03 (t, J = 4.8 Hz, 1 H), 3.84 (s, 3 H), 3.76 (m, 2 H), 3.57 (m, 2 H), 3.23 (m, 2 H), 3.00 (m, 2 H), 2.93 (s, 3 H); HRMS (ESI) m/z calcd for C24H32O6N6Cl [M + H]+ 535.2072, found 535.2087.
5-O-tert-Butyldiphenylsilyl-3-O-(2,5-dichloropyrimidin-4-yl)-2-O-tert-butyldimethylsilyl-β-D-ribofuranosyl azide 21
To a solution of compound 19 (0.86 g, 1.63 mmol) in anhydrous CH2Cl2 (25 mL) at room temperature, was added tBuOLi (1.83 g, 22.82 mmol) and 2,4,5-trichloropyrimidine 6 (0.37 mL, 3.26 mmol). After stirring under reflux for 36 h, the reaction mixture was diluted with saturated aqueous NH4Cl, and extracted with CH2Cl2. The organic layer was washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by silica gel chromatography (petroleum ether/EtOAc: 80/1) to give 21 (0.88 g, 80%) as a pale yellow syrup: 1H NMR (400 MHz, CDCl3) δ 8.34 (s, 1 H), 7.71–7.67 (m, 4 H), 7.45–7.34 (m, 6 H), 5.66 (t, J = 4.8 Hz, 1 H, H-3), 5.25 (d, J = 3.6 Hz, 1 H, H-1), 4.40 (dd, J = 3.6, 7.6 Hz, 1 H), 4.33 (t, J = 4.4 Hz, 1 H), 3.91 (dd, J = 4.0, 11.6 Hz, 1 H), 3.84 (dd, J = 3.6, 11.6 Hz, 1 H), 1.09 (s, 9 H), 0.75 (s, 9 H), 0.05 (s, 3 H), −0.15 (s, 3 H); HRMS (ESI) m/z calcd for C31H42O4N5Cl2Si2 [M + H]+ 674.2152, found 674.2155.
5-O-tert-Butyldiphenylsilyl-3-O-[5-chloro-2-N-(2-methoxy-4-(4-methylpiperazin-1-yl)phenyl)pyrimidin-4-yl]-2-O-tert-butyldimethylsilyl-β-D-ribofuranosyl azide 22
To a solution of compound 21 (0.53 g, 0.79 mmol) and aniline derivative 8 (0.70 g, 3.16 mmol) in isobutanol (12 mL), was added TFA (1.47 mL, 19.75 mmol). The mixture was heated to 100°C and stirred for 5 h. After cooling down to room temperature, the mixture was quenched with Et3N (8 mL) and concentrated in vacuo to give a residue, which was purified by silica gel column chromatography (CH2Cl2/MeOH: 30/1) to give 22 (0.47 g, 69%) as a white powder: 1H NMR (400 MHz, CDCl3) δ 8.12 (s, 1 H), 8.08 (d, J = 9.2 Hz, 1 H), 7.69 (dd, J = 1.6, 7.6 Hz, 2 H), 7.63 (dd, J = 1.6, 8.0 Hz, 2 H), 7.43–7.25 (m, 6 H), 6.54 (m, 2 H), 5.58 (dd, J = 4.4, 7.2 Hz, 1 H, H-3), 5.29 (d, J = 1.6 Hz, 1 H, H-1), 4.47 (m, 1 H), 4.38 (dd, J = 2.0, 4.4 Hz, 1 H), 4.02 (dd, J = 2.8, 11.6 Hz, 1 H), 3.88 (s, 3 H), 3.83 (dd, J = 3.6, 12.0 Hz, 1 H), 3.17 (t, J = 5.2 Hz, 4 H), 2.61 (t, J = 4.8 Hz, 4 H), 2.37 (s, 3 H), 1.06 (s, 9 H), 0.78 (s, 9 H), −0.07 (s, 3 H), −0.25 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 163.6, 157.9, 156.9, 149.3, 147.6, 135.8, 135.7, 133.1, 133.0, 129.9, 127.9, 127.8, 121.8, 120.1, 108.4, 106.1, 100.6, 95.8, 81.6, 74.7, 74.2, 62.8, 55.8, 55.3, 50.1, 46.3, 26.9, 25.6, 19.3, 18.0, −4.9, −5.4; HRMS (ESI) m/z calcd for C43H60O5N8ClSi2 [M + H]+ 859.3914, found 859.3920.
5-O-tert-Butyldiphenylsilyl-3-O-[5-chloro-2-N-(2-methoxy-4-(4-methylpiperazin-1-yl)phenyl)pyrimidin-4-yl]-2-O-tert-butyldimethylsilyl-α-D-ribofuranosyl acrylamide 23
A mixture of compound 22 (190 mg, 0.22 mmol) and Pd/C (50 mg, 10%) in EtOH (7 mL) was stirred under an atmosphere of H2 at room temperature for overnight. The mixture was filtered through celite, washed with EtOH and concentrated in vacuo to afford the corresponding amine for the next step without further purification. To a solution of the resulting amine in CH2Cl2 (7 mL) at room temperature, was added DCC (69 mg, 0.33 mmol), DMAP (41 mg, 0.33 mmol), and acrylic acid (0.061 mL, 0.89 mmol). After stirring at room temperature for 4 h, the mixture was concentrated in vacuo to give a residue, which was purified by silica gel column chromatography (CH2Cl2/MeOH: 30/1) to afford 23 (76 mg, 39% over two steps) as a pale yellow syrup: 1H NMR (400 MHz, CDCl3) δ 8.17 (s, 1 H), 8.09 (d, J = 8.8 Hz, 1 H), 7.71 (m, 4 H), 7.43–7.36 (m, 6 H), 7.12 (d, J = 9.2 Hz, 1 H), 6.53 (d, J = 2.4 Hz, 1 H), 6.42 (m, 1 H), 6.36 (dd, J = 1.2, 16.8 Hz, 1 H), 6.14 (dd, J = 10.4, 17.2 Hz, 1 H), 6.02 (d-like, J = 4.8 Hz, 1 H), 5.98 (dd, J = 6.0, 9.2 Hz, 1 H), 5.70 (dd, J = 1.2, 10.4 Hz, 1 H), 4.72 (dd, J = 5.2 Hz, 1 H), 4.36 (br s, 1 H), 3.86 (m, 5 H), 3.07 (br s, 4 H), 2.54 (br s, 4 H), 2.35 (s, 3 H), 1.11 (s, 9 H), 0.68 (s, 9 H), −0.02 (s, 3 H), −0.09 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 165.9, 164.0, 157.8, 157.0, 149.3, 147.6, 135.8, 135.5, 133.4, 132.5, 131.0, 130.1, 130.0, 128.9, 128.0, 127.4, 121.5, 120.0, 108.1, 105.8, 100.4, 82.2, 80.4, 77.4, 71.0, 64.2, 55.7, 55.2, 49.9, 46.2, 27.0, 25.4, 19.5, 17.7, −5.1, −5.3; HRMS (ESI) m/z calcd for C46H64O6N6ClSi2 [M + H]+ 887.4114, found 887.4116.
3-O-[5-Chloro-2-N-(2-methoxy-4-(4-methylpiperazin-1-yl)phenyl)pyrimidin-4-yl]-2-O-tert-butyldimethylsilyl-α-D-ribofuranosyl acrylamide 1d
To a solution of compound 23 (91 mg, 0.11 mmol) in pyridine (3 mL) at room temperature, was added HF·pyridine (0.19 mL). After stirring at room temperature for overnight, the mixture was poured into saturated aqueous NaHCO3 and extracted with CH2Cl2. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (CH2Cl2/MeOH: 20/1) to afford 1d (40 mg, 68%) as a pale yellow syrup: 1H NMR (400 MHz, CDCl3) δ 8.15 (s, 1 H), 8.02 (d, J = 8.4 Hz, 1 H), 7.71 (m, 4 H), 7.30 (br s, 1 H), 7.06 (d, J = 8.4 Hz, 1 H), 6.54 (m, 2 H), 6.33 (d-like, J = 17.2 Hz, 1 H), 6.12 (dd, J = 10.0, 16.8 Hz, 1 H), 5.90 (dd, J = 6.0, 8.4 Hz, 1 H), 5.73 (m, 2 H), 4.51 (t, J = 5.6 Hz, 1 H), 4.30 (br s, 1 H), 3.87 (s, 3 H), 3.82 (d-like, J = 12.4 Hz, 1 H), 3.71 (d-like, J = 11.2 Hz, 1 H), 3.18 (br s, 4 H), 2.61 (br s, 4 H), 2.37 (s, 3 H), 0.72 (s, 9 H), −0.02 (s, 3 H), −0.10 (s, 3 H); 13C NMR (100 MHz, CDCl3) δ 166.1, 163.8, 157.9, 157.0, 149.6, 147.5, 130.9, 127.7, 121.9, 120.5, 108.6, 106.1, 100.8, 82.1, 80.6, 76.3, 70.9, 62.5, 55.8, 55.0, 49.6, 45.7, 25.5, 17.9, −5.0, −5.2; HRMS (ESI) m/z calcd for C30H46O6N6ClSi [M + H]+ 649.2937, found 649.2940.
Kinase Assay
Kinases domain of EGFR WT and EGFR L858R/T790M were expressed using the Bac-to-Bac™ baculo virus expression system (Invitrogen, Carlsbad, CA, USA) and purified in Ni-NTA columns (QIAGEN Inc., Valencia, CA, USA). The kinase activity was evaluated with enzyme-linked immunosorbent assay (ELISA). Briefly, 20 μg/mL Poly (Glu, Tyr) 4:1 (Sigma, St. Louis, MO) was precoated in 96-well ELISA plates as substrate. After adding 50 μL of 10 μmol/L ATP solution which was diluted in kinase reaction buffer (50 mM HEPES pH 7.4, 20 mM MgCl2, 0.1 mM MnCl2, 0.2 mM Na3VO4, 1 mM DTT), the plate was treated with 1 μL of indicated concentrations of compounds (dissolved in DMSO) per well. Experiments at each concentration were performed in duplicate. Reaction was initiated by adding tyrosine kinase diluted in kinase reaction buffer. After incubation at 37°C for 1 h, the wells were washed three times with phosphate buffered saline (PBS) containing 0.1% Tween 20 (T-PBS). One hundred microliters of anti-phosphotyrosine (PY99) antibody (1:1,000, Santa Cruz Biotechnology, Santa Cruz, CA) diluted in T-PBS containing 5 mg/mL BSA was added and the plate was incubated at 37°C for 30 min. After the plate was washed three times, 100 μL horseradish peroxidase-conjugated goat anti-mouse IgG (1:2,000, Calbiochem, SanDiego, CA) was added and the plate was incubated at 37°C for 30 min. The plate was washed, added with 100 μL citrate buffer (0.1 M, pH 5.5) containing 0.03% H2O2. Then 2 mg/mL o-phenylenediamine was added, and samples were incubated at room temperature until color emerged. The reaction was terminated immediately by adding 50 μL of 2 M H2SO4. Plate was read using a multiwell spectrophotometer (VERSAmax™, Molecular Devices, Sunnyvale, CA, USA) at 492 nm. The inhibitory rate (%) was calculated with the formula: [1 − (A492 treated/A492 control)] × 100%. IC50 values were calculated from the inhibitory curves.
Molecular Docking
EGFRT790M/L858R structure (PDB: 3IKA) was retrieved from the Protein Data Bank and covalent docking was performed with maestro (Schrödinger, Inc., version 10.2). Compound 1a was docked into the EGFR protein as an irreversible inhibitor using Covalent Docking module. The docking procedure was validated by re-docking the co-crystallized ligand WZ4002 into the ATP binding site of EGFRT790M/L858R structure. The details of the docking workflow are listed below:
(1) Protein was prepared using the “Protein Preparation Wizard” workflow. All water molecules were removed from the structure of the complex. Hydrogen atoms and charges were added during a brief relaxation. After optimizing the hydrogen bond network, the crystal structure was minimized using the OPLS_2005 force field with the maximum root mean square deviation (RMSD) value of 0.3 Å.
(2) The ligand was prepared with LigPrep module in Maestro, including adding hydrogen atoms, ionizing at a pH range from 5.0 to 9.0, and producing the corresponding low-energy 3D structure.
(3) Pose prediction mode of Covalent Docking module was adopted to dock the molecules into the ATP-binding site with the default parameters. The center of the grid box was defined with the intrinsic ligand and Michael addition reaction type was chosen. The top-ranking poses of molecule 1a were retained.
Result and Discussion
The synthesis of 3-N-acryloyl-5-O-anilinopyrimidine ribose derivatives 1a and 1b commenced with 3-amino ribose derivative 3 that can be readily prepared from D-xylose 2 in 41% yield over six steps (Scheme 1; Shie et al., 2007). Protection of the primary hydroxyl group in 3 with TBDPSCl followed by condensation with acryloyl chloride using Et3N as base gave ribose derivative 4 in 92% yield (two steps). Treatment of 4 with HF·pyridine afforded alcohol 5 in 71% yield. When 5 was reacted with 2,4,5-trichloropyrimidine 6 in the presence of K2CO3 or DIPEA, almost no desired product was observed. Exhilaratingly, nucleophilic reaction of 5 with 6 employing stronger base (DBU) as promoter proceeded smoothly to provide ribose derivative 7 in excellent yield (92%). Subjection of 7 to the known aniline derivative 8 (Han et al., 2014) under the promotion of TFA in isobutanol at 100°C delivered 1a in 69% yield. Removal of the 1,2-O-isopropylidene group in 1a with TFA in acetic acid and water at 70°C produced 1b in 83% yield.
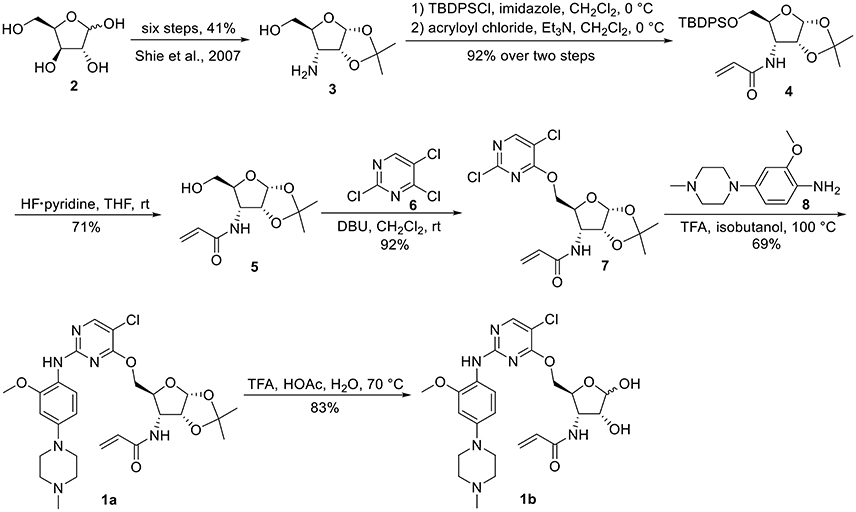
Scheme 1. Synthesis of 3-N-acryloyl-5-O-anilinopyrimidine ribose derivatives 1a and 1b.
For the synthesis of 1-N-acryloyl-5-O-anilinopyrimidine ribose derivative 1c, β-ribosyl azide 10 was conveniently prepared from D-ribose 9 in three steps and 53% yield according to the procedures described in the literature (Scheme 2; Bonache et al., 2009). The acetyl group in 10 was then replaced with TBS group to give compound 11 in 85% yield. Hydrogenolysis of the azide group in 11 over Pd/C followed by condensation with acrylic acid in the presence of DCC and DMAP afforded a mixture of ribose derivative 12 in 43% yield (α/β = 1:1; α-anomer: δH = 6.00 ppm, δC = 81.4 ppm; β-anomer: δH = 5.95 ppm, δC = 87.2 ppm), which were easily separated by silica gel column chromatography (Numao et al., 1981; Bonache et al., 2009). After removal of the TBS group in 12α, the resulting alcohol 13 reacted with 6 in the presence of DBU to produce ribose derivative 14 in an excellent 93% yield. TFA-promoted reaction of 14 with aniline 8 led to 15 in 70% yield as a mixture of α/β anomers probably arising from the anomerization of the 1-N-acryloyl ribose derivative under strong acidic conditions (Boschelli et al., 1989). Finally, acidic cleavage of the isopropylidene group of 15 in the mixture of TFA/HOAc/water provided 1c in 80% yield.
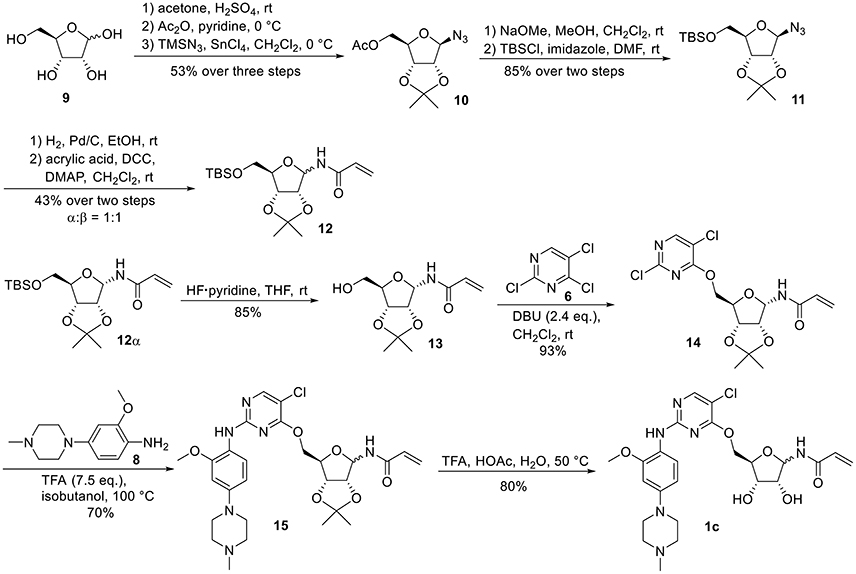
Scheme 2. Synthesis of 1-N-acryloyl-5-O-anilinopyrimidine ribose derivative 1c.
Synthetic work toward 1-N-acryloyl-3-O-anilinopyrimidine ribose derivative 1d started from replacement of the 2,3-isopropylidene group of azide 10 with 2,3-orthoester group by treatment with TFA and subsequent protection with triethyl orthoacetate under the catalysis of TsOH·H2O, affording azide 16 in 76% yield over two steps (Scheme 3). Substitution of the acetyl group in 16 with TBDPS group and subsequent acidic cleavage of the orthoester group led to an inseparable mixture of 2-acetyl and 3-acetyl ribose derivatives 17 and 18 (77% yield over three steps). Treatment of the mixture of 17 and 18 with TBSCl followed by removal of the acetyl groups gave alcohols 19 and 20 in 83% yield, allowing for the separation of 3-hydroxyl ribose derivative 19 from 2-hydroxyl ribose derivative 20 (19:20 = 3:2). Nucleophilic attack of 19 on 6 required stronger basic conditions to promote the reaction due to the steric hindrance of the silyl groups on 19. As such, excess tBuOLi in dichloromethane under reflux was employed for this conversion, providing ribose derivative 21 in 80% yield. TFA-promoted coupling of 21 with aniline 8 generated ribose derivative 22 (69%), which was then subjected to hydrogenolysis over Pd/C and subsequent condensation with acrylic acid to afford ribose derivative 23 as single anomer in moderate yield (39% over two steps). Exposure of 23 to HF·pyridine in pyridine resulted in cleavage of the TBDPS group without affecting the TBS group, providing 1d in 68% yield.
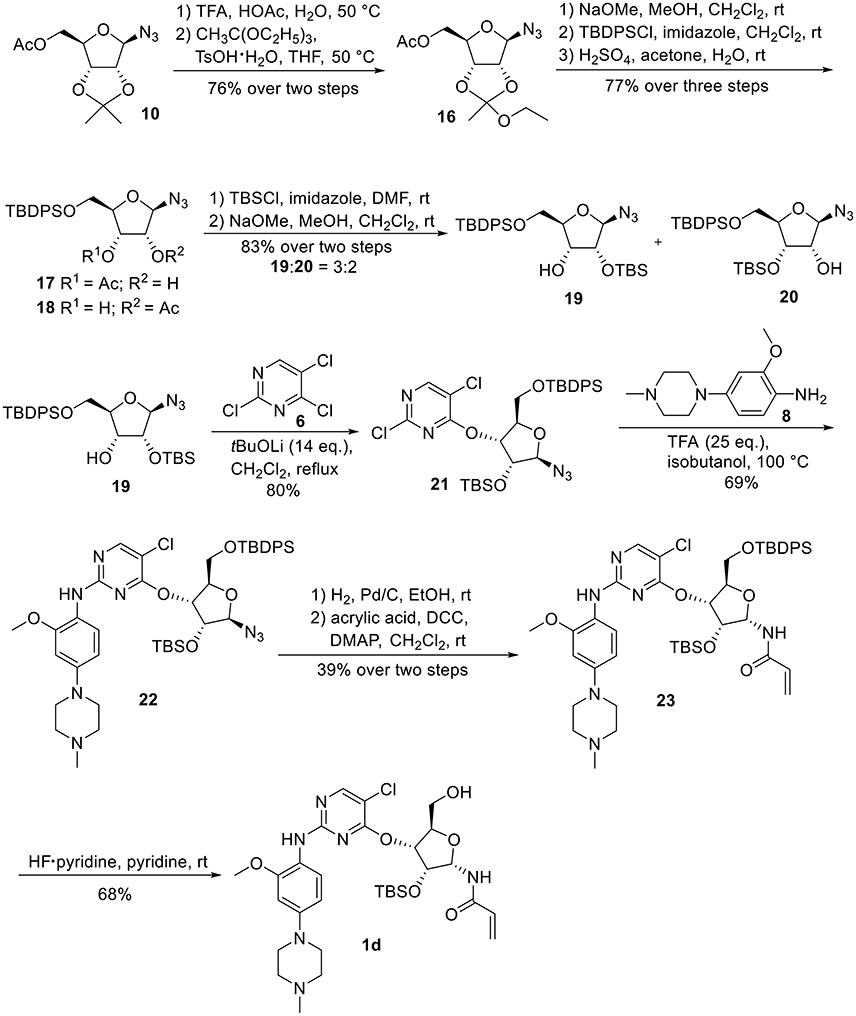
Scheme 3. Synthesis of 1-N-acryloyl-3-O-anilinopyrimidine ribose derivative 1d.
To determine whether the Michael acceptor played a significant role in the inhibitory activity of ribose-modified pyrimidine derivatives against EGFR tyrosine kinase, 1-azide-5-O-anilinopyrimidine ribose derivative 24, 1-azide-3-O-anilinopyrimidine ribose derivative 25, and 5-O-anilinopyrimidine ribose derivative 26 were readily synthesized following the similar procedures described for 1a-1d (Table 1; see Supplementary Material for details).
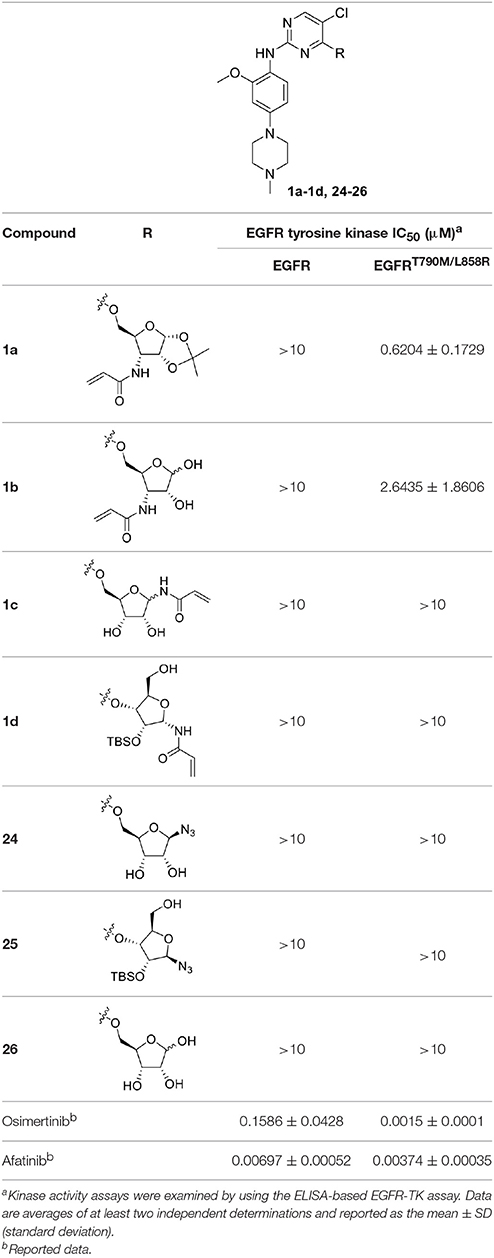
Table 1. In vitro inhibitory activities of ribose-modified pyrimidine derivatives 1a–1d and 24–26 against EGFR tyrosine kinase.
As shown in Table 1, compounds 1a and 1b containing 3-N-acryloyl-5-O-anilinopyrimidine ribosyl moiety potently inhibited EGFR L858R/T790M mutant with IC50 values of 0.62 and 2.64 μM, revealing specific inhibitory activity for EGFR L858R/T790M over WT EGFR, although they are not comparable to the positive controls osimertinib (IC50 = 1.5 nM for EGFR L858R/T790M) and afatinib (IC50 = 3.7 nM for EGFR L858R/T790M). In contrast, other compounds (1c, 1d, and 24–26) bearing 5-O-anilinopyrimidine ribosyl moiety, 1-N-acryloyl-5-O-anilinopyrimidine ribosyl moiety, 1-N-acryloyl-3-O-anilinopyrimidine ribosyl moiety, or their 1-azide counterparts, showed no inhibitory activities against EGFR tyrosine kinases.
In order to better understand the mechanism of this type of compounds binding to EGFR T790M, molecular docking was adopted to predict the binding mode of the representative compound 1a. The docking procedure was validated in advance by re-docking the co-crystallized ligand WZ4002 (Zhou et al., 2009) into the ATP binding site of EGFR L858R/T790M structure (PDB ID: 3IKA). The root mean square deviation (RMSD) between the crystallographic and docked conformation of WZ4002 is 0.57 Å (Figure S1), demonstrating that the present docking procedure was feasible in generating the binding conformation accurately. As expected based upon co-crystal structure of the anilinopyrimidine-derived inhibitor WZ4002, the anilinopyrimidine core of compound 1a forms a bidentate hydrogen bonding interaction with the “hinge” residue Met793 (Figure 3). The chlorine substituent on the pyrimidine ring could form hydrophobic contact with the mutant gatekeeper residue, Met790. The aniline ring is oriented to form hydrophobic interactions with Leu792 and Pro794 in the hinge region. Moreover, the acrylamide group attached to the sugar ring of compound 1a could form a covalent bond with Cys797 to achieve irreversible binding. The sugar ring acts like a linker to tune the orientation of the electrophilic acrylamide moiety that can covalently alkylate the conserved cysteine residue Cys797. For the 1,2-O-isopropylidene moiety in compound 1a, it could form favorable vdW interactions with residues ARG841, ASN842, and Thr854. Therefore, compound 1b without that protecting group on the sugar ring, displayed less potent bioactivity against EGFR T790M/L858R compared with compound 1a. Lacking of the Michael receptor, compounds 24–26 are unable to form covalent bond with Cys797 and thus displayed sharply decreased inhibitory activity against EGFR T790M/L858R. Compound 1c displayed no inhibitory activity of EGFR probably because of the long distance between the Michael receptor and Cys797. Although compound 1d also has an acrylamide group attached to the sugar ring, it showed no inhibitory activity probably due to the conformational alteration of compound 1d caused by the TBS protecting group. Briefly, it could be concluded that the distance between the Michael receptor and the pyrimidine scaffold has a significant effect on the inhibitory potency of this type of compounds. Employing the ribosyl moiety as a chiral building block for modulating the distance between the Michael receptor and the pyrimidine scaffold could pave a new avenue for future design of EGFR inhibitors against EGFR mutants.
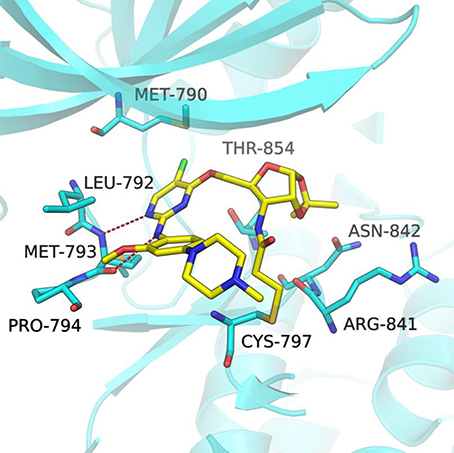
Figure 3. Docking pose of compound 1a in complex with the EGFRT790M/L858R (PDB ID 3IKA). The EGFR kinase is shown as cartoon (cyan) with the bound inhibitor in a stick representation (yellow). Key residues are represented as cyan sticks. The expected hydrogen bonds are indicated by red dashes.
Conclusion
In summary, we have described a DBU- or tBuOLi-promoted coupling of ribosyl alcohols with 2,4,5-trichloropyrimidine as key step for the synthesis of a series of ribose-modified anilinopyrimidine derivatives as EGFR TKIs. Preliminary biological evaluation indicated that compound 1a displayed potent inhibitory activity against EGFR L858R/T790M with an IC50 value of 0.62 μM, and good selectivity for EGFR L858R/T790M over WT EGFR. Molecular docking studies revealed that the inhibitory activities of this type of compounds are largely influenced by the distance between the Michael receptor and the pyrimidine scaffold. As a novel type of EGFR inhibitor, the ribose-modified anilinopyrimidine derivative 1a might be used as a promising lead compound for further development of selective EGFR inhibitors to overcome EGFR L858R/T790M resistance mutation.
Author Contributions
YY and YX designed and guided this study. XH conducted the chemical synthesis. YT, LT, LZ, and HX performed the kinase activity assays. DW, XW, and SL performed the molecular docking studies. XH, YY, and YX analyzed the data and wrote the manuscript with input from all authors.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Financial support from the National Thousand Young Talents Program (YC0130518, YC0140103), the Shanghai Committee of Science and Technology (14431902100), and the Shanghai Pujiang Program (15PJ1401500), is gratefully acknowledged.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2017.00101/full#supplementary-material
References
Balak, M. N., Gong, Y., Riely, G. J., Somwar, R., Li, A. R., Zakowski, M. F., et al. (2006). Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor–mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin. Cancer Res. 12, 6494–6501. doi: 10.1158/1078-0432.CCR-06-1570
Bonache, M. A., Nuti, F., Le Chevalier Isaad, A., Real-Fernandez, F., Chelli, M., Rovero, P., et al. (2009). Synthesis of new ribosylated Asn building blocks as useful tools for glycopeptide and glycoprotein synthesis. Tetrahedron Lett. 50, 4151–4153. doi: 10.1016/j.tetlet.2009.04.124
Boschelli, D. H., Powell, D., Sharky, V., and Semmelhack, M. F. (1989). An improved synthesis of glycinamide ribonucleotide. Tetrahedron Lett. 30, 1599–1600. doi: 10.1016/S0040-4039(00)99530-3
Castellanos, E. H., and Horn, L. (2015). Generations of epidermal growth factor receptor tyrosine kinase inhibitors: perils and progress. Curr. Treat. Options Oncol. 16, 51. doi: 10.1007/s11864-015-0365-1
Cheng, H., Nair, S. K., and Murray, B. W. (2016). Recent progress on third generation covalent EGFR inhibitors. Bioorg. Med. Chem. Lett. 26, 1861–1868. doi: 10.1016/j.bmcl.2016.02.067
Cohen, M. H., Johnson, J. R., Chen, Y. F., Sridhara, R., and Pazdur, R. (2005). FDA drug approval summary: erlotinib (Tarceva®) tablets. Oncologist 10, 461–466. doi: 10.1634/theoncologist.10-7-461
Cross, D. A., Ashton, S. E., Ghiorghiu, S., Eberlein, C., Nebhan, C. A., Spitzler, P. J., et al. (2014). AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 4, 1046–1061. doi: 10.1158/2159-8290.CD-14-0337
De Luca, A., Carotenuto, A., Rachiglio, A., Gallo, M., Maiello, M. R., Aldinucci, D., et al. (2008). The role of the EGFR signaling in tumor microenvironment. J. Cell. Physiol. 214, 559–567. doi: 10.1002/jcp.21260
Dungo, R. T., and Keating, G. M. (2013). Afatinib: first global approval. Drugs 73, 1503–1515. doi: 10.1007/s40265-013-0111-6
Eberlein, C. A., Stetson, D., Markovets, A. A., Al-Kadhimi, K. J., Lai, Z., Fisher, P. R., et al. (2015). Acquired resistance to the mutant-selective EGFR inhibitor AZD9291 is associated with increased dependence on RAS signaling in preclinical models. Cancer Res. 75, 2489–2500. doi: 10.1158/0008-5472.CAN-14-3167
Finlay, M. R., Anderton, M., Ashton, S., Ballard, P., Bethel, P. A., Box, M. R., et al. (2014). Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J. Med. Chem. 57, 8249–8267. doi: 10.1021/jm500973a
Gray, J., and Haura, E. (2014). Update on third-generation EGFR tyrosine kinase inhibitors. Transl. Lung Cancer Res. 3, 360–362. doi: 10.3978/j.issn.2218-6751.2014.09.08
Grünwald, V., and Hidalgo, M. (2003). Developing inhibitors of the epidermal growth factor receptor for cancer treatment. J. Natl. Cancer Inst. 95, 851–867. doi: 10.1093/jnci/95.12.851
Gschwind, A., Fischer, O. M., and Ullrich, A. (2004). The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat. Rev. Cancer 4, 361–370. doi: 10.1038/nrc1360
Günther, M., Juchum, M., Kelter, G., Fiebig, H., and Laufer, S. (2016). Lung cancer: EGFR inhibitors with low nanomolar activity against a therapy-resistant L858R/T790M/C797S mutant. Angew. Chem. Int. Ed. 55, 10890–10894. doi: 10.1002/anie.201603736
Günther, M., Lategahn, J., Juchum, M., Döring, E., Keul, M., Tumbrink, H. L., et al. (2017). Trisubstituted pyridinylimidazoles as potent inhibitors of the clinically resistant L858R/T790M/C797S EGFR mutant: targeting of both hydrophobic regions and the phosphate binding site. J. Med. Chem. 60, 5613–5637. doi: 10.1021/acs.jmedchem.7b00316
Han, C., Wan, L., Ji, H., Ding, K., Huang, Z., Lai, Y., et al. (2014). Synthesis and evaluation of 2-anilinopyrimidines bearing 3-aminopropamides as potential epidermal growth factor receptor inhibitors. Eur. J. Med. Chem. 77, 75–83. doi: 10.1016/j.ejmech.2014.02.032
Harrington, K. J., El-Hariry, I. A., Holford, C. S., Lusinchi, A., Nutting, C. M., Rosine, D., et al. (2009). Phase I study of lapatinib in combination with chemoradiation in patients with locally advanced squamous cell carcinoma of the head and neck. J. Clin. Oncol. 27, 1100–1107. doi: 10.1200/JCO.2008.17.5349
Huang, S. M., and Harari, P. M. (1999). Epidermal growth factor receptor inhibition in cancer therapy: biology, rationale and preliminary clinical results. Invest. New Drugs 17, 259–269. doi: 10.1023/A:1006384521198
Jia, Y., Yun, C. H., Park, E., Ercan, D., Manuia, M., Juarez, J., et al. (2016). Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 534, 129–132. doi: 10.1038/nature17960
Juchum, M., Günther, M., Döring, E., Sievers-Engler, A., Lämmerhofer, M., and Laufer, S. (2017). Trisubstituted imidazoles with a rigidized hinge binding motif act as single digit nM inhibitors of clinically relevant EGFR L858R/T790M and L858R/T790M/C797S mutants: an example of target hopping. J. Med. Chem. 60, 4636–4656. doi: 10.1021/acs.jmedchem.7b00178
Kris, M. G., Natale, R. B., Herbst, R. S., Lynch, T. J., Prager, D., Belani, C. P., et al. (2003). Efficacy of gefitinib, an inhibitor of the epidermal growth factor receptor tyrosine kinase, in symptomatic patients with non-small cell lung cancer. JAMA 290, 2149–2158. doi: 10.1001/jama.290.16.2149
Moore, M. J., Goldstein, D., Hamm, J., Figer, A., Hecht, J. R., Gallinger, S., et al. (2007). Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the national cancer institute of Canada clinical trials group. J. Clin. Oncol. 25, 1960–1966. doi: 10.1200/JCO.2006.07.9525
Niederst, M. J., Hu, H., Mulvey, H. E., Lockerman, E. L., Garcia, A. R., Piotrowska, Z., et al. (2015). The allelic context of the C797S mutation acquired upon treatment with third generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin. Cancer Res. 21, 3924–3933. doi: 10.1158/1078-0432.CCR-15-0560
Normanno, N., Bianco, C., De Luca, A., Maiello, M. R., and Salomon, D. S. (2003). Target-based agents against ErbB receptors and their ligands: a novel approach to cancer treatment. Endocr. Relat. Cancer 10, 1–21. doi: 10.1677/erc.0.0100001
Numao, N., Hemmi, H., Naujokaitis, S. A., Rabinovitz, M., and Beisler, J. A. (1981). Showdomycin analogues: synthesis and antitumor evaluation. J. Med. Chem. 24, 515–520. doi: 10.1021/jm00137a008
Ozvegy-Laczka, C., Cserepes, J., Elkind, N. B., and Sarkadi, B. (2005). Tyrosine kinase inhibitor resistance in cancer: role of ABC multidrug transporters. Drug Resist. Updates 8, 15–26. doi: 10.1016/j.drup.2005.02.002
Pao, W., and Chmielecki, J. (2010). Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat. Rev. Cancer 10, 760–774. doi: 10.1038/nrc2947
Park, H., Jung, H. Y., Mah, S., and Hong, S. (2017). Discovery of EGF receptor inhibitors that are selective for the d746-750/T790M/C797S mutant through structure-based de novo design. Angew. Chem. 56, 7634–7638. doi: 10.1002/anie.201703389
Piotrowska, Z., Niederst, M. J., Karlovich, C. A., Wakelee, H. A., Neal, J. W., Mino-Kenudson, M., et al. (2015). Heterogeneity underlies the emergence of EGFRT790 wild-type clones following treatment of T790M-positive cancers with a third-generation EGFR inhibitor. Cancer Discov. 5, 713–722. doi: 10.1158/2159-8290.CD-15-0399
Shie, J. J., Fang, J. M., Wang, S. Y., Tsai, K. C., Cheng, Y. S. E., Yang, A. S., et al. (2007). Synthesis of tamiflu and its phosphonate congeners possessing potent anti-influenza activity. J. Am. Chem. Soc. 129, 11892–11893. doi: 10.1021/ja073992i
Steuer, C. E., Khuri, F. R., and Ramalingam, S. S. (2015). The next generation of epidermal growth factor receptor tyrosine kinase inhibitors in the treatment of lung cancer. Cancer 121, E1–E6. doi: 10.1002/cncr.29139
Thress, K. S., Paweletz, C. P., Felip, E., Cho, B. C., Stetson, D., Dougherty, B., et al. (2015). Acquired EGFR C797S mutation mediates resistance to AZD9291 in non–small cell lung cancer harboring EGFR T790M. Nat. Med. 21, 560–562. doi: 10.1038/nm.3854
Traxler, P., and Furet, P. (1999). Strategies toward the design of novel and selective protein tyrosine kinase inhibitors. Pharmacol. Ther. 82, 195–206. doi: 10.1016/s0163-7258(98)00044-8
Walter, A. O., Sjin, R. T., Haringsma, H. J., Ohashi, K., Sun, J., Lee, K., et al. (2013). Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov. 3, 1404–1415. doi: 10.1158/2159-8290.CD-13-0314
Keywords: EGFR, tyrosine kinase inhibitors, anilinopyrimidine, glycosides, carbohydrate-based drugs
Citation: Hu X, Wang D, Tong Y, Tong L, Wang X, Zhu L, Xie H, Li S, Yang Y and Xu Y (2017) Design, Synthesis, and Evaluation of Ribose-Modified Anilinopyrimidine Derivatives as EGFR Tyrosine Kinase Inhibitors. Front. Chem. 5:101. doi: 10.3389/fchem.2017.00101
Received: 02 September 2017; Accepted: 30 October 2017;
Published: 15 November 2017.
Edited by:
Daniela Schuster, University of Innsbruck, AustriaReviewed by:
Johannes Kirchmair, University of Hamburg, GermanyDharmendra Kumar Yadav, All India Institute of Medical Sciences Jodhpur, India
Copyright © 2017 Hu, Wang, Tong, Tong, Wang, Zhu, Xie, Li, Yang and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: You Yang, eWFuZ3lvdUBlY3VzdC5lZHUuY24=
Yufang Xu, eWZ4dUBlY3VzdC5lZHUuY24=