 Alexander Wurzer1†Adrienn Vágner2†‡Dávid Horváth2
Alexander Wurzer1†Adrienn Vágner2†‡Dávid Horváth2 Flóra Fellegi2Hans-Jürgen Wester1
Flóra Fellegi2Hans-Jürgen Wester1 Ferenc K. Kálmán2
Ferenc K. Kálmán2 Johannes Notni1*
Johannes Notni1*- 1Pharmaceutical Radiochemistry, Technische Universität München, Munich, Germany
- 2Department of Inorganic and Analytical Chemistry, University of Debrecen, Debrecen, Hungary
Due to its 4 carbonic acid groups being available for bioconjugation, the cyclen tetraphosphinate chelator DOTPI, 1,4,7,10-tetraazacyclododecane-1,4,7, 10-tetrakis[methylene(2-carboxyethylphosphinic acid)], represents an ideal scaffold for synthesis of tetrameric bioconjugates for labeling with radiolanthanides, to be applied as endoradiotherapeuticals. We optimized a protocol for bio-orthogonal DOTPI conjugation via Cu(I)-catalyzed Huisgen-cycloaddition of terminal azides and alkynes (CuAAC), based on the building block DOTPI(azide)4. A detailed investigation of kinetic properties of Cu(II)-DOTPI complexes aimed at optimization of removal of DOTPI-bound copper by transchelation. Protonation and equilibrium properties of Ca(II)-, Zn(II), and Cu(II)-complexes of DOTPI and its tetra-cyclohexylamide DOTPI(Chx)4 (a model for DOTPI conjugates) as well as kinetic inertness (transchelation challenge in the presence of 20 to 40-fold excess of EDTA) were investigated by pH-potentiometry and spectrophotometry. Similar stability constants of CaII-, ZnII, and CuII-complexes of DOTPI (logK(CaL) = 8.65, logK(ZnL = 15.40, logK(CuL) = 20.30) and DOTPI(Chx)4 (logK(CaL) = 8.99, logK(ZnL) = 15.13, logK(CuL) = 20.42) were found. Transchelation of Cu(II)-complexes occurs via proton-assisted dissociation, whereafter released Cu(II) is scavenged by EDTA. The corresponding dissociation rates [kd = 25 × 10−7 and 5 × 10−7 s−1 for Cu(DOTPI) and Cu(DOTPI(Chx)4), respectively, at pH 4 and 298 K] indicate that conjugation increases the kinetic inertness by a factor of 5. However, demetallation is completed within 4.5 and 7.2 h at pH 2 and 25°C, respectively, indicating that Cu(II) removal after formation of CuAAC can be achieved in an uncomplicated manner by addition of excess H4EDTA. For proof-of-principle, tetrameric DOTPI conjugates of the prostate-specific membrane antigen (PSMA) targeting motif Lys-urea-Glu (KuE) were synthesized via CuAAC as well as dibenzo-azacyclooctine (DBCO) based, strain-promoted click chemistry (SPAAC), which were labeled with Lu-177 and subsequently evaluated in vitro and in SCID mice bearing subcutaneous LNCaP tumor (PSMA+ human prostate carcinoma) xenografts. High affinities (3.4 and 1.4 nM, respectively) and persistent tumor uptakes (approx. 3.5% 24 h after injection) confirm suitability of DOTPI-based tetramers for application in targeted radionuclide therapy.
Introduction
Endoradiotherapy (also termed molecular radiotherapy, radioligand therapy or, if addressing peptide receptors, peptide receptor radionuclide therapy (PRRT) or, if involving alpha-emitting nuclides, targeted alpha therapy) (Oyen et al., 2007) refers to the internal application of radionuclides for therapeutic purposes, above all, for curing cancer. In the respective disease management schemes, radiotherapeutics represent the natural complements to imaging tracers. In a tandem application of both types of agents, referred to as “theranostics,” one targeting mechanism is exploited for delivery of different sorts of radionuclides to tissues, either for diagnostic purposes, that is, with the intention to localize lesions by means of external detection of emitted gamma photons, or to achieve a therapeutic effect via local absorption of particle (i.e., alpha- or beta) radiation. The corresponding radiopharmaceuticals are frequently based on peptides, peptidomimetics, enzyme inhibitors, or similar molecules capable of recognizing a specific cell surface receptor, membrane-bound enzyme, ion channel, or comparable target. The bioactive structures are often decorated with a chelate ligand for kinetically inert complexation of a metal ion radionuclide (Wadas et al., 2010). At present, radiotherapeutics most frequently rely on lanthanide(III)- or chemically related ions, such as 177Lu, 90Y, 225Ac, or 213Bi (Notni and Wester, 2018). Recognizing malignant cells by overexpression of above-mentioned surface markers, the radiolabeled bioconjugates deliver these beta- or alpha-emitting isotopes in the lesion, resulting in a local irradiation which kills malignant cells.
In this context, the chelator DOTA (Figure 1) plays an important role (Stasiuk and Long, 2013) since it forms stable and sufficiently inert complexes with virtually all relevant metal ions. DOTA is usually attached to e.g., peptides via amide formation on one side arm, resulting in derivatives of DOTA-monoamide which is the actual chelator structure in such conjugates (commonly dubbed “DOTA peptides”). However, a likewise functionalization of more than one acetic acid side arm of DOTA yields conjugates whose metal complexes either lack kinetic inertness (e.g., in case of the positron emitter 68GaIII) or show very slow formation kinetics (namely, with lanthanide(III) ions) (Baranyai et al., 2007; Pasha et al., 2007), limiting practical applicability in radiopharmacy. Conjugates comprising more than one biomolecule (multimers) are nevertheless desirable because a multitude of similar bioactive structure elements in a single framework usually results in enhanced target affinities, and sometimes increases uptake of the respective radiolabeled compounds in target tissues (Maschauer et al., 2017).
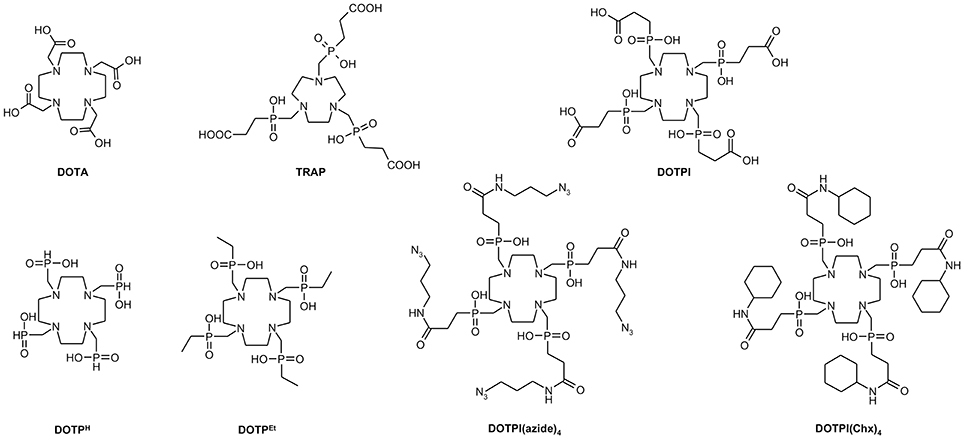
Figure 1. Structures of chelate ligands discussed in the text.
For this purpose, we recently designed the DOTA-analog tetraphosphinate chelator DOTPI (Figure 1) featuring four terminal carboxylic acid moieties, which are not required for metal ion complexation and, therefore, are available for bioconjugation (Šimeček et al., 2013). However, similar to the observations made for its smaller congener TRAP (1,4,7-triazacyclononane-1,4,7-tris[methylene(2-carboxyethylphosphinic acid)]) (Notni et al., 2014), amide functionalization of these carboxylates using standard peptide coupling reagents is sometimes complicated by instability of the active ester intermediates (Baranyai et al., 2015). Similar to TRAP, functionalization of DOTPI via “click chemistry,” (CuI-catalyzed alkyne-azide cycloaddition, CuAAC) (Meldal and Tornøe, 2008) employing respective DOTPI-derivatives decorated with “clickable” functional groups, such as DOTPI(azide)4 (Figure 1) (Wurzer et al., 2018), appeared to be a valuable alternative. Since the presence of ionic Cu inevitably results in copper complexes of the desired conjugates wherein the Cu ion blocks the coordination site intended for accommodation of the radiometal, removal of chelator-bound Cu, ultimately being transformed to oxidation state +2 during workup, is mandatory to restore the radiolabeling properties. As discussed previously, demetalation with sulfide or cyanide frequently causes problems, while proton-assisted dissociation, promoted by excess of a competing chelator, should be widely applicable and compatible with many synthetic tasks (Notni and Wester, 2016). Against this background, we investigated the metal coordination properties of DOTPI and a model conjugate thereof, DOTPI(Chx)4 (Figure 1), with a particular emphasis on CuII complexes, in order to facilitate optimization of respective demetallation protocols and to support application of CuAAC for elaboration of DOTPI-based multimers.
Results
In order to enhance legibility, charge, and protonation state of ligands and complexes is indicated only where necessary for comprehension and where possible without ambiguity, e.g., in case it is referred to a single, well defined species and not to a mixture, as is mostly the case due to protonation equilibria. The animal experiments were conducted in accordance with the German Animal Welfare Act (Tierschutzgesetz), and an ethics approval was obtained from the responsible authority (Regierung von Oberbayern).
Solution Thermodynamics
Protonation schemes of cyclens bearing four N-substitutents with additional donors (often referred to as DOTA-like ligands) have been thoroughly investigated in the past (Desreux et al., 1981; Bianchi et al., 2000; Takács et al., 2014). Table 1 displays the respective data for DOTPI in comparison to DOTA (Baranyai et al., 2010) and the most closely related tetraphosphinate analogs, DOTPH (Bazakas and Lukeš, 1995; Kotková et al., 2009) and DOTPEt (Lázár et al., 1991) (Figure 1). According to previous literature, it is assumed that the first and second protonation of all chelators under investigation occur at two opposite ring nitrogen atoms. The subsequent protonation steps most likely take place at protonation sites located at the side arms (if available, at the carboxylate groups), due to greater charge separation and lower electrostatic repulsion between the distant protonated donor atoms. Protonations of the phosphinate moieties, characterized by the log and log for DOTPI and log for DOTPI(Chx)4, occur only in the acidic pH region (Rohovec et al., 2000), which is why these phosphinate ligands are generally capable of metal complexation and rapid radiolabeling at much lower pH values than chelators with acetic acid side arms, such as DOTA.
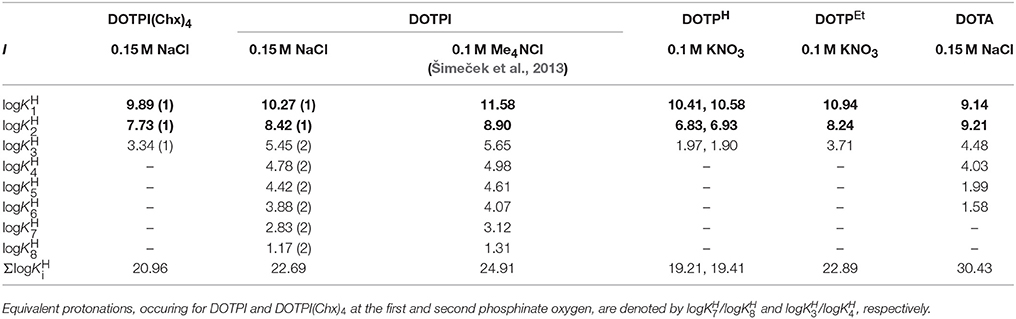
Table 1. Protonation constants of DOTPI(Chx)4, DOTPI, DOTPH, DOTPEt, and DOTA at 25°C (for structures see Figure 1).
The equilibrium properties of chelates based on the 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetrakis(methylenephosphinic acid) scaffold depend on the electronic properties of the substituents on the phosphorus atoms (Kotková et al., 2009). log and log values for DOTPI(Chx)4 are somewhat lower than those of DOTPI, while this lower basicity of the ring nitrogens might be explained by the presence of the more electronegative amide substituents on the pendant arms. However, the lower log value measured for DOTA in 0.15 M NaCl has another reason; it is explained by the formation of relatively stable [Na(DOTA)]3− complex (logKNaL = 4.38) (Chaves et al., 1992). Our data indicate that DOTPI forms a similar NaL complex, entailing a lower log value in presence of 0.15 M NaCl than in 0.1 M Me4NCl (Šimeček et al., 2013).
Total basicity of ligands (Σlog, Table 1) generally correlates with thermodynamic stability constants (KML) of their metal complexes, while in order to obtain chemically meaningful values, the log values of the distant carboxylate groups were not considered for calculation of Σlog value of DOTPI because they are not involved in metal ion coordination. That being said, it is not surprising that the logKML value of [Ca(DOTA)], [Zn(DOTA)], and [Cu(DOTA)] complexes is about 2–5 logK unit higher than phosphinic acid analogs, because DOTA shows a significantly higher total basicity (Table 2).
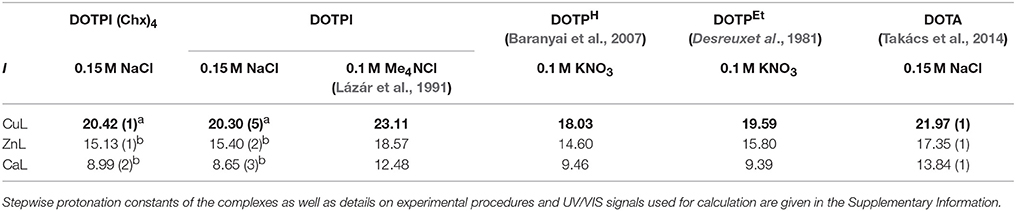
Table 2. Stability constants (logKML) for non-protonated DOTPI(Chx)4, DOTPI, DOTPH, DOTPEt, and DOTA complexes formed with divalent metals at 25°C, determined by UV/Vis spectroscopy (a) or potentiometry (b).
Among the tetraphosphinates, total basicities Σlog of DOTPI(Chx)4, DOTPI, and DOTPEt ligands are similar but about 1–3 orders of magnitude higher than that of DOTPH which, as expected, is reflected by similar logKCuL and logKZnL values for CuII and ZnII complexes of the P-substitued ligands but somewhat lower stabilities for those of DOTPH. Thermodynamic stabilities of the CaII complexes are, however, similar for all phosphinate ligands. This might be explained by differences in the preferred coordination number of these divalent metal ions (CuII and ZnII: 6, CaII: 6–8) owing to their different size (CuII: 73 p.m., ZnII: 74 p.m., CaII: 100 p.m.), entailing different structures for CuII and ZnII complexes as observed for CaII complexes. Based on available structural data (Riesen et al., 1986), it is assumed that the smaller cations are bound in an N4O2 coordination environment involving only two side arm oxygens, whereas CaII is surrounded by a N4O4 donor set involving all side arms. Hence, side arm basicity and protonation presumably affects the complexation equilibria in a different manner.
Formation of several protonated Cu(HxL) species at pH < 7 has been confirmed by pH-potentiometric and spectrophotometric studies of solutions containing CuII and DOTPI(Chx)4 or DOTPI, respectively, while both ligands form CuII complexes already in acidic solution (pH 1.5 and lower). Since molar absorptivities of the protonated Cu(HxDOTPI) and non-protonated Cu(DOTPI) species were found to be essentially equal, it can be assumed that stepwise protonation of Cu(DOTPI) starts with the non-coordinating carboxylates, resulting in Cu(HL), Cu(H2L), Cu(H3L), and Cu(H4L). Two further protonations at comparable pH are observed for CuII complexes of both DOTPI(Chx)4 and DOTPI, while it appears most plausible that they occur on phosphinate oxygens because two of these moieties do not coordinate to CuII and can thus be protonated.
Kinetic Inertness and Transchelation
In order to provide the necessary information for optimization of demetallation protocols, we investigated the pH-dependent kinetics of the transchelation of CuII from CuDOTPI and CuDOTPI(Chx)4 complexes to ethylenediamine-tetraacetic acid (EDTA). Reactions were monitored by UV spectrophotometry at the absorption bands of Cu(DOTPI) and Cu(DOTPI(Chx)4) over a pH range of 1.5–4.5, using a 20- and 40-fold excess of EDTA (see Supplementary information).
Figure 2 shows that the obtained pseudo-first-order rate constants do not depend on the excess of EDTA, and increase with decreasing pH. Hence, it can be assumed that the transchelation of Cu(DOTPI) and Cu(DOTPI(Chx)4) occurs by initial protonation, followed by spontaneous dissociation of the protonated CuII complexes as rate-determining step, while irreversibility is granted by scavenging the released CuII with EDTA. Similar data and mechanistic implications have been found for CuII complexes of the smaller triphosphinate macrocycle TRAP (Baranyai et al., 2015), underscoring wide applicability of the transchelation approach for demetallation of chelator conjugates after CuAAC coupling (Notni and Wester, 2016).
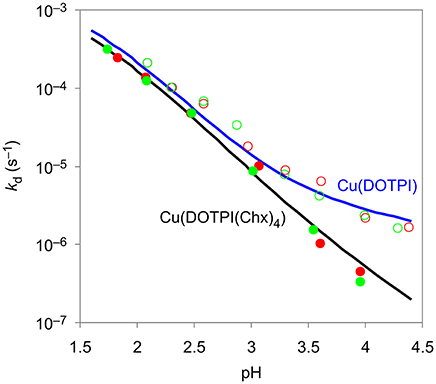
Figure 2. Pseudo-first-order rate constants at 25°C in 0.15 M NaCl for the reaction of CuII complexes of DOTPI (empty symbols) and DOTPI(Chx)4 (filled symbols) with a 20- (red) and 40-fold (green) excess of EDTA at different pH values. Respective kd functions were calculated from kinetic and equilibrium data.
Table 3 summarizes rate (k) and equilibrium constants (K) of all possible pathways for dissociation of the protonated species Cu(HxDOTPI) and Cu(HyDOTPI(Chx)4) (x = 3,4,5,6; y = 1,2; for details see Supplementary Information). According to the proposed mechanism, that is, a fast protonation equilibrium followed by a first-order dissociation reaction rendered irreversible by competitor excess, cleavage of the protonated Cu(HxL) complex is promoted by transfer of the proton, which is initially located on the pendant arm, to a ring nitrogen, resulting in the most labile intermediate, an off-cage complex (Baranyai et al., 2015). Overall, the proton displaces the CuII ion from the coordination cage, causing dissociation of the Cu(HxL) complex.
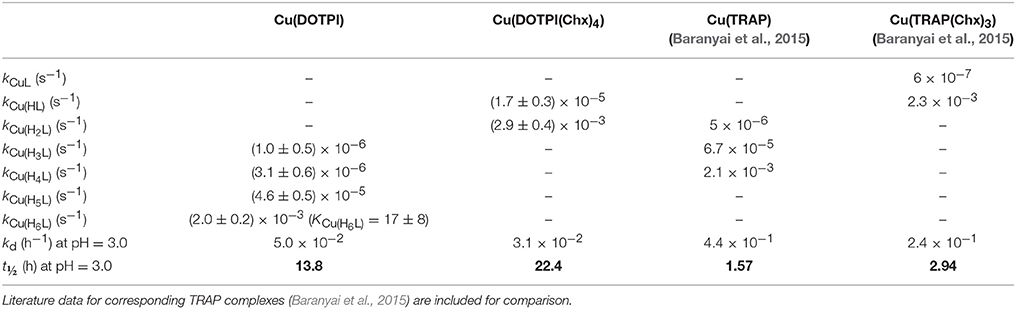
Table 3. Rate constants (kCu(HiL)) and half-lives (t½ = ln2/kd) characterizing the dissociation reactions of Cu(DOTPI) and Cu(DOTPI(Chx)4) complexes (0.15 M NaCl, 25°C).
A comparison of the rate constants (kCu(HiL)) obtained for protonated species Cu(HxDOTPI) and Cu(HyDOTPI(Chx)4) confirms that dissociation of the uncharged species, [Cu(H2DOTPI(Chx)4)] and [Cu(H6DOTPI)], which likely contains two non-coordinated and protonated phosphinic oxygen atoms, occurs with a very similar rate. Moreover, the dissociation rates of [Cu(H6DOTPI)] and [Cu(H2DOTPI(Chx)4)] are very similar to those of [Cu(H4TRAP)] and [Cu(HTRAP(Chx)3)] (Baranyai et al., 2015), which might be explained by similar activation parameters characterizing the proton transfer process from the phosphinate oxygen to the ring nitrogen, resulting in comparable dissociation rate constants at pH < 2.
Figure 3 shows the calculated contributions of individual dissociation rates for all protonated Cu(HxDOTPI) and Cu(HyDOTPI(Chx)4) species to the overall demetallation rates (see also Figures S3, S6), which decrease with increasing pH. In the pH range 1.5–4.5, dissociation of Cu(DOTPI) and Cu(DOTPI(Chx)4) occurs by the decomplexation of the various protonated species, respectively. In this respect, [Cu(H6DOTPI)] and [Cu(H2DOTPI(Chx)4)] are equivalent as they both carry two phosphinate-bound protons and hence, show similar dissociation rates. Since these species are prevailing at pH < 2, they govern overall dissociation rates which are, therefore, nearly identical (see also Figure 2). On the other hand, at pH > 4, a substantial amount of protonated (and thus, labile) species is observed only for the Cu(DOTPI) system, explaining the higher inertness of Cu(DOTPI(Chx)4) complexes.
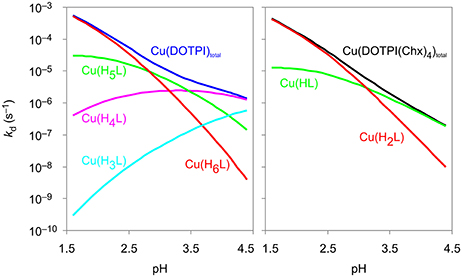
Figure 3. Contributions of Cu(DOTPI) and Cu(DOTPI(Chx)4) complexes in different protonation states to the overall rates of the dissociation reaction.
Figure 4 displays the quintessence of the kinetic studies for practical consideration, namely, the dissociation half-lives (t½) of Cu(DOTPI) and Cu(DOTPI(Chx)4) as functions of pH (see Supplemental information, Equation 18). It shows that loss of protonable carboxylates by amide functionalization results in higher t½ of the respective Cu(DOTPI(Chx)4) complexes at higher pH; at pH > 4, the increase exceeds one order of magnitude. On the other hand, CuII removal from both neat and decorated DOTPI occurs with similar efficiency at lower pH. In view of the calculated t½, it appears recommendable to carry out such reactions at pH values below 3, preferably at pH = 2, while slightly elevated temperature (e.g., 50°C) will also substantially accelerate the reaction (Baranyai et al., 2015). Apart from that, a t½ of > 4,000 h for dissociation of Cu(DOTPI(Chx)4) at pH > 5 suggests compatibility with the positron emitter 64Cu (T½ = 12.7 h) for application in 64Cu-PET imaging.
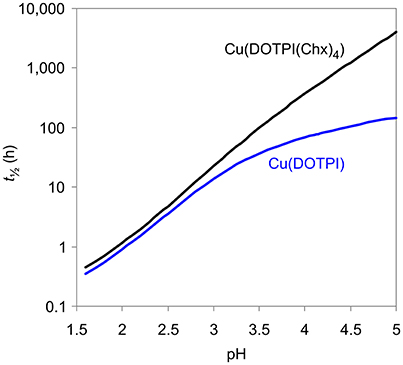
Figure 4. Dissociation half lives (t½ = ln2/kd) of Cu(DOTPI) and Cu(DOTPI(Chx)4) as functions of pH (25°C, 0.15 M NaCl).
Application
In order to demonstrate utility of the CuAAC/demetallation tandem reaction for assembly of large multimeric bioconjugates and application in the design of therapeutic radiopharmaceuticals, the 4-fold azide-decorated building block DOTPI(azide)4 (Wurzer et al., 2018) was reacted with alkyne-functionalized KuE (lysine-urea-glutamic acid, an inhibitor motif for prostate-specific membrane antigen, PSMA, EC 3.4.17.21; synonyms: glutamate carboxypeptidase II, NAALADase (Mesters et al., 2006); a membrane-bound zinc hydrolase which is overexpressed by malignant human prostate cancers). As an alternative, the inhibitor was equipped with dibenzo-azacyclooctyne (DBCO) (Agard et al., 2004) for conjugation via “copper-free click chemistry” (strain-promoted alkyne-azide cycloaddition, SPAAC) according to path B in Figure 5. Although this approach circumvents the entire demetallation problem, potentially obviating the above study, the disadvantages of SPAAC are limiting its practical value in the present context. Firstly, reaction rates for SPAAC are several orders of magnitude lower than those of CuAAC, requiring adjustment of reaction conditions, for example, a higher excess of reactants, in order to achieve reasonable yields. Secondly, the isomerism of the formed linker moiety gives rise to a total of 6 stereoisomers of DOTPI(DBCO-KuE)4 which are hard to separate, if at all. While the isomers are not likely to exhibit noticeable differences regarding their pharmacodynamics, justifying use of the mixture for in-vivo application, such action might nonetheless lead to regulatory issues upon clinical translation. Third, introduction of several large, non-polar linker groups usually affects pharmacokinetics, above all, due to increased plasma protein binding. The overall simplicity of the CuAAC/demetallation approach (Figure 5, path A) thus appears preferable for this type of synthetic task, not least because of the known robustness and pharmacokinetic inertness of the 1,3-triazole linkage (Horne et al., 2004, 2009; Bock et al., 2007; Liu et al., 2008; Pedersen and Abell, 2011; Davis et al., 2012; Tischler et al., 2012).
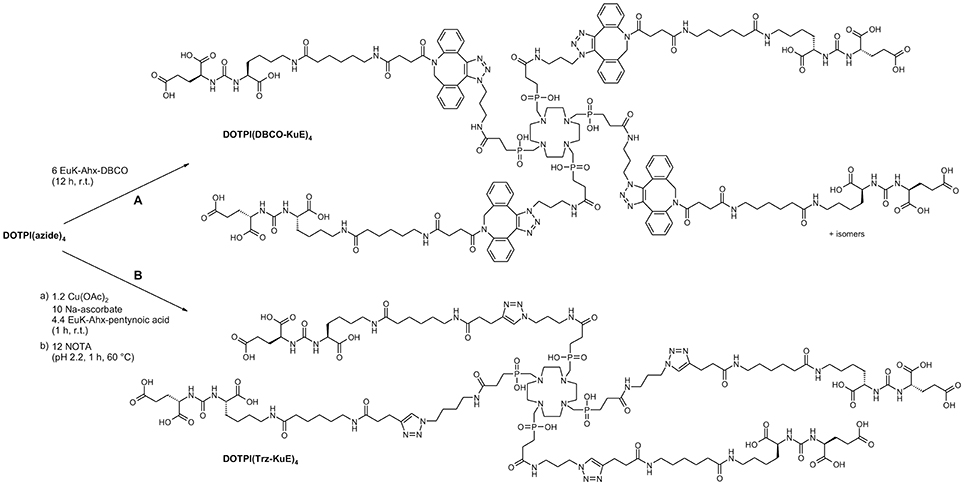
Figure 5. Synthesis of symmetrical tetrameric prostate-specific membrane antigen (PSMA) inhibitor conjugates via DBCO-driven, strain-promoted (SPAAC, path A), or CuI-catalyzed (CuAAC, path B) alkyne-azide cycloaddition (Huisgen-reaction).
Because the 177Lu-labeled tetramers feature nearly identical linker lengths and differ only in the type of linkage, the effect of the dibenzo-azacyclooctane system annulated to the triazole moiety becomes clearly apparent. While PSMA affinities of both tetramers are, as expected (Maschauer et al., 2017), higher than that of clinically applied monomers (Weineisen et al., 2014; Benešová et al., 2016), it is conspicuous that the natLu-DOTPI(DBCO-KuE)4 exhibits a higher affinity than natLu-DOTPI(Trz-KuE)4 which is featuring simple 1,2,3-triazole linkages (IC50 = 1.4 ± 0.5 vs. 3.4 ± 0.1, respectively). This is because apart from the primary KuE binding site, namely, the catalytic center which contains two hydroxo-bridged ZnII ions, the enzyme PSMA features another hydrophobic site in close proximity (Zhang et al., 2010) which can be addressed by lipophilic groups in the linker (Kularatne et al., 2009; Banerjee et al., 2010). With regard to the DBCO moieties, a virtue is hence made of necessity, because these hydrophobic structure elements contribute to a divalent binding mode. Since the observed increase of affinity is linked to the particular combination of binding sites of the target PSMA, this favorable effect of DBCO cannot be generalized.
Apart from that, Figure 6 shows that the higher degree of hydrophilicity of 177Lu-DOTPI(Trz-KuE)4 (logD = −5.0 ± 0.1) results in a remarkably low kidney uptake and a much faster washout from non-target tissues as compared to 177Lu-DOTPI(DBCO-KuE)4 (logD = −4.0 ± 0.1). A virtually similar tumor uptake of both compounds after 24 h gives rise to substantially better tumor-to-organ ratios for 177Lu-DOTPI(Trz-KuE)4. However, although the acquired data are sufficient for proof-of-principle in the context of this study, a more detailed investigation of the compounds is necessary in order to draw a reliable conclusion regarding their clinical potential.
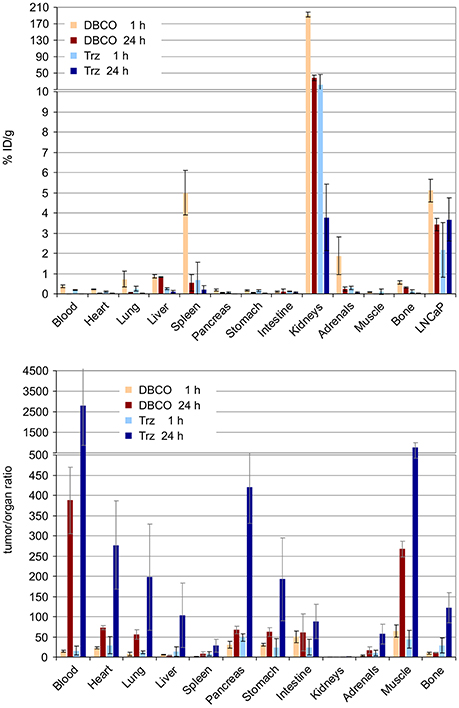
Figure 6. Biodistribution (Top) and tumor-to-organ ratios (Bottom) for 177Lu-DOTPI(DBCO-KuE)4 and 177Lu-DOTPI(Trz-KuE)4 (1.1–4.5 MBq, 0.11–0.15 nmol, n = 3), determined 1 and 24 h post injection in SCID mice bearing subcutaneous LNCaP (PSMA-expressing human prostate carcinoma) xenografts.
Discussion
Replacement of the acetate with amide groups in the pendant arms of DOTPI does not affect the thermodynamic stability (logKML values) of the CaII-, ZnII-, and CuII-complexes, while dissociation half-lives of the CuII chelates at pH > 3 show a clear trend toward higher kinetic inertness. This is another example for the notion that thermodynamic stability constants of complexes do not necessarily correlate with their kinetic inertness, requiring more than just equilibrium data for prediction or confirmation of the suitability of (radio-)metal chelates for in-vivo applications. This applies even more because any data acquired under ideal conditions, e.g., in neat solvents or well-defined buffers, should be treated with care in view of a limited transferability to more complex systems, such as living organisms. For example, the aforementioned demetallation half-life of more than 4,000 h for Cu(DOTPI(Chx)4) at pH > 5 is no ultimate proof that such conjugates are indeed applicable for 64Cu-PET imaging, because other mechanisms than proton-assisted dissociation may contribute to in-vivo loss of 64Cu, even from highly stable and inert chelates (Bass et al., 2000; Zarschler et al., 2014). A final conclusion on this matter cannot be drawn without further evidence and an in-vivo proof of concept, which however is beyond the focus of this study.
Conclusion
A sufficiently low kinetic inertness of CuII complexes of amide-functionalized DOTPI derivatives at low pH warrants the practical applicability of the CuAAC/demetallation tandem protocol, which can be conveniently conducted as a one-pot reaction, for the synthesis of symmetrical tetrameric DOTPI conjugates. In view of the inherent limitations of strain-promoted AAC, such as slower reaction kinetics and isomerism/lipophilicity of linkages, CuAAC appears to be the preferable method for this purpose. A pilot in-vivo study showed that the DOTPI-based PSMA-targeted radiotherapeutics combine high affinity with excellent clearance from non-target tissues, thus demonstrating the potential of DOTPI as a scaffold for the elaboration of therapeutic radiopharmaceuticals.
Materials and Methods
Chemical Synthesis
General
The protected amino acid analogs were purchased from Bachem (Bubendorf, Switzerland) or Iris Biotech (Marktredwitz, Germany). All necessary solvents and other organic reagents were purchased from either, Alfa Aesar (Karlsruhe, Germany), Sigma-Aldrich (Munich, Germany), or VWR (Darmstadt, Germany). The DOTPI chelator (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetrakis[methylene(2-carboxyethylphosphinic acid)]) (Šimeček et al., 2013) and its azide-functionalized derivative DOTPI(azide)4 (Wurzer et al., 2018) were synthesized, as described previously. CheMatech (Dijon, France) delivered the NOTA chelator (1,4,7-triazacyclononane-1,4,7-triacetic acid). The PSMA-addressing binding motifs (DBCO-KuE and Trz-KuE) were prepared according to previously published procedures (Wurzer et al., 2018). Analytical and preparative HPLC were performed on Shimadzu gradient systems with a SPD-20A dual wavelength UV/Vis detector (220, 254 nm) with mobile phase gradients combined of purified water (component A; from Millipore system) and acetonitrile (component B; J.T.Baker®Ultra Gradient HPLC grade, supplemented with 5% H2O), both containing 0.1% trifluoroacetic acid. A Nucleosil 100-5 C18 column (125 × 4.6 mm) was used for analytical measurements at a flow rate of 1 mL/min. Preparative HPLC purification was done using a Multospher 100 RP 18-5 μ column (250 × 10 mm) at a flow rate of 5 mL/min. Electrospray ionization (ESI) mass spectra were acquired on a Varian 500-MS Ion Trap spectrometer (Varian, by Agilent Technologies).
DOTPI(Chx)4
DOTPI·2 H2O (85.0 mg, 105 μmol, 1.0 eq) was dissolved in a mixture of anhydrous DMSO (420 μL) and DIPEA (320 μL, 1.89 mmol, 20 eq). Then cyclohexylamine (72.9 mg, 736 μmol, 7.0 eq) and HATU (439 mg, 1.16 mmol, 11 eq) were added in one portion with stirring. After 1 h at room temperature, the red reaction mixture was quenched with water (900 μL). The crude product was purified by size exclusion chromatography (Sephadex G-10 medium, column size: 40 × 3 cm, mobile phase: water, adjusted to pH 3 with HCl), yielding DOTPI(Chx)4, as a yellow-green solid (57.7 mg, 53 μmol, 51%). MW (calcd. for C48H92N8O12P4): 1097.20. HPLC (30–90% B in 15 min): tR = 13.2 min. 1H-NMR (300 MHz, D2O, 300 K) δ = 1.13–1.36 (m, 20H), 1.57–1.60 (m, 4H), 1.69–1.74 (m, 8H, C(O)-CH2), 1.78–1.87 (m, 16H), 2.37–2.45 (m, 8H, P-CH2-C), 3.13–3.29 (m, 8H, P-CH2-N), 3.42 (bs, 16H, ring-CH2), 3.52–3.59 (m, 4H, C(O)-NH-CH) ppm. MS (ESI, positive): m/z = 1098.2 [M+H+], 1119.9 [M+Na+], 549.7 [M+2 H+].
DOTPI(DBCO-KuE)4
DOTPI(azide)4·TFA (2.00 mg, 1.65 μmol, 1.0 eq) and DBCO-KuE (7.13 mg, 9.90 μmol, 6.0 eq) were dissolved in a 1:1 mixture (v/v, 2.2 mL) of H2O and tert-butanol and stirred for 12 h at room temperature. Subsequent purification by preparative HPLC yielded DOTPI(DBCO-KuE)4·TFA as a colorless solid (6.02 mg, 1.47 μmol, 89%). MW (calcd. for C184H252N40O52P4): 3980.10. HPLC (10–90% B in 15 min): tR = 9.2 min. MS (ESI, positive): m/z = 1327.9 [M+3H+], 1335.6 [M+2H++Na+], 1991.4 [M+2H+].
DOTPI(Trz-KuE)4
DOTPI(azide)4 (6.0 mg, 4.94 μmol, 1.0 eq), sodium ascorbate (97.9 mg, 494 μmol, 100 eq), and Trz-KuE (11.1 mg, 21.7 μmol, 4.4 eq) were dissolved in a 1:1 mixture (v/v, 2 mL) of H2O and tert-butanol. After adding copper(II) acetate monohydrate (1.18 mg, 5.93 μmol, 1.2 eq) dissolved in H2O (250 μL) a deep blue reaction mixture was formed, which was stirred for 1 h at room temperature. For demetallation, the mixture was diluted to 6 mL with H2O, and NOTA (37.6 mg, 124 μmol, 25 eq) was added. After adjusting the pH to 2.2 by adding 1 M HCl, the solution was heated to 60°C for 2 h. Subsequent preparative HPLC-purification afforded DOTPI(Trz-KuE)4·TFA as a colorless solid (1.6 mg, 0.49 μmol, 10%). MW (calcd. for C128H216N36O48P4): 3151.24. HPLC (10–60% B in 15 min): tR = 6.9 min. MS (ESI, positive): m/z = 1576.6 [M+2H+], 1051.2 [M+3H+], 788.7 [M+4H+].
Equilibrium Studies (Protonation and Stability Constants)
The chemicals used for the experiments were of the highest analytical grade. CaCl2, ZnCl2, and CuCl2 solutions were prepared from solid MCl2 (Aldrich; 99.9%). Concentration of CaCl2, ZnCl2, and CuCl2 solutions were determined by complexometric titration with standardized Na2H2EDTA and xylenol orange (ZnCl2), murexid (CuCl2), and Patton & Reeder (Ca2+) as indicators. The concentration of the DOTPI(Chx)4, DOTPI, and EDTA was determined by pH-potentiometric titration in the presence and absence of a large (40-fold) excess of CaCl2. All the equilibrium measurements were made at constant ionic strength maintained by 0.15 M NaCl at 25°C.
For determining the protonation constants of DOTPI(Chx)4 and DOTPI three parallel pH-potentiometric titration were made with 0.2 M NaOH in 0.002 M ligand solutions. The stability and protonation constants of the CaII and ZnII complexes formed with DOTPI(Chx)4 and DOTPI ligands have been determined by direct pH-potentiometric titration made at 1:1 and 2:1 metal to ligand concentration ratios. (the concentration of ligands was 0.002 M). For the calculation of the logKML and logKMLHi values, the mL base—pH data used were obtained in the pH range 1.7–12.0.
Stability constant of Cu(DOTPI) complex was determined by spectrophotometry, studying the CuII-DOTPI systems at the absorption band of CuII-complex at [H+] = 0.01– 0.2 M over the wavelength range of 400–800 nm. Concentrations of CuII and DOTPI were 1 mM. The H+ concentration in the samples was adjusted by addition of calculated amounts of 2.0 M HCl, while ionic strength was not constant in these samples. Samples were kept at 25°C for a week. Absorbance values were determined at 9 wavelengths (550, 575, 600, 625, 650, 675, 700, 725, and 750 nm). For calculation of stability and protonation constants of Cu(DOTPI), molar absorptivities of CuCl2, and Cu(HxL) species (wherein x = 0, 1, 2 …5) were determined by recording the spectra of 1.0 × 10−3, 2.0 × 10−3 and 3.0 × 10−3 M solutions of CuCl2 and Cu(DOTPI) in the pH range of 1.7–11.7. The protonation constants of the complexes Cu(DOTPI) and the stability and protonation constants of the dinuclear Cu2(DOTPI) complexes were determined by pH-potentiometric titrations, made at 1:1 and 2:1 metal to ligand concentration ratios.
The stability constant of the Cu(DOTPI(Chx)4) complex has been determined by spectrophotometry with the use of competition reactions between DOTPI(Chx)4 and EDTA for the Cu2+ at pH = 5.0. The concentration of Cu(EDTA) was 0.2 mM, while that of the DOTPI(Chx)4 was varied between 0.1 and 1.0 mM (5 samples). The samples were kept at 25°C for 2 weeks. The absorbance values and the molar absorptivities of CuCl2, Cu(DOTPI(Chx)4), and Cu(EDTA) have been determined at 11 wavelength (300, 304, 308, 312, 316, 320, 324, 328, 332, 336, and 340 nm) values between 300 and 340 nm. The molar absorptivities of CuCl2, Cu(DOTPI(Chx)4), and Cu(EDTA) were deteremined in 0.05, 0.1, 0.2, 0.3, and 0.4 mM solutions. The absorbance and pH values were determined in the samples after the equilibrium was reached (the time needed to reach the equilibria was determined by spectrophotometry). The spectrophotometric measurements were made with the use of 1.0 cm cells with a Cary 1E spectrophotometer at 25°C. The protonation constants of Cu(DOTPI(Chx)4) complex were determined by pH-potentiometric titrations at 1:1 metal to ligand molar ratio.
For pH measurements and titrations, a Metrohm 785 DMP Titrino titration workstation and a Metrohm-6.0233.100 combined electrode were used. Equilibrium measurements were carried out at a constant ionic strength (0.15 M NaCl) in 6 mL samples at 25°C. The solutions were stirred, and constantly purged with N2. The titrations were performed in a pH range of 1.7–11.7. KH-phthalate (pH = 4.005) and borax (pH = 9.177) buffers were used to calibrate the pH meter. For calculation of [H+] from measured pH values, the method proposed by Irving et al. was used (Irving et al., 1967). A 0.01 M HCl solution was titrated with the standardized NaOH solution in the presence of 0.1 M NaCl. Differences between the measured (pHread) and calculated pH (–log[H+]) values were used to obtain the equilibrium H+ concentration from the pH values, measured in the titration experiments. For the equilibrium calculations, the stoichiometric water ionic product (pKw) is also needed to calculate [H+] values in basic conditions. The VNaOH – pHread data pairs of the HCl – NaOH titration obtained in the pH range 10.5–12 have been used to calculate the pKw value (pKw = 13.85). For the calculation of the equilibrium constants the program PSEQUAD (Zekany and Nagypal, 1985) was used.
Kinetic Studies
The rates of the ligand exchange reactions of Cu(DOTPI) and Cu(DOTPI(Chx)4) with EDTA ligand were studied by following the dissociation of Cu(DOTPI) and Cu(DOTPI(Chx)4) complexes with spectrophotometry at 340 nm, in the pH range 1.7–4.5, in the presence of the 20- and 40-fold excess of EDTA in order to maintain pseudo-first order kinetic conditions. Concentrations of Cu(DOTPI) and Cu(DOTPI(Chx)4) were 0.1 mM. Kinetic studies were performed with Cary 1E and Cary 100 Bio spectrophotometers, using cell holders thermostated to 25°C. The pre-thermostated solutions were mixed in tandem cells (l = 0.874 cm). The ionic strength of the solutions was kept constant at 0.15 M with NaCl. In order to keep the pH values constant, dichloro-acetic acid (DCA) (pH range 1.5–2.5), chloro-acetic acid (MCA) (pH range 2.5–3.5) and 1,4-dimethylpiperazine (DMP) (pH = 3.1–4.5) buffers (0.01 M) were used. Pseudo-first-order rate constants (kd) were calculated by fitting the absorbance values to the equation
wherein A0, Ae, and At are the absorbance values at the start, at equilibrium and at the time t of the reaction, respectively. The calculation of the kinetic parameters were performed by the fitting of the absorbance–time data pairs with the Micromath Scientist computer program (version 2.0, Salt Lake City, UT, USA).
Radiochemistry
For 177Lu-labeling, 10 μL aqueous NH4OAc buffer (1 M, pH = 5.9) were added to 1.0 nmol of the labeling precursor (1 mM in DMSO), 10–40 MBq 177LuCl3 (Specific Activity > 3,000 GBq/mg, 740 MBq/mL, 0.04 M HCl, ITG, Garching, Germany) and finally filled up to 100 μL with H2O. The reaction mixture was heated for 30 min at 95°C and the radiochemical purity was determined using radio-TLC (Silica gel 60, mobile phase: 1:1 mixture of 1 M aqueous ammonium acetate and DMF).
In Vitro and in Vivo Evaluation
Determination of PSMA Affinities
PSMA-expressing LNCaP (human prostate carcinoma) cells were grown in Dublecco modified Eagle medium/Nutrition Mixture F-12 with Glutamax-I (1:1) (Invitrogen), supplemented with 10% fetal calf serum and maintained at 37°C in a humidified 5% CO2 atmosphere. For determination of the PSMA affinity (IC50), cells were harvested 24 ± 2 h before the experiment and seeded in 24-well plates (1.5 × 105 cells per 1 mL well). The competitive binding assay was carried out as described previously, using the radioiodinated PSMA ligand (125I-BA)KuE (Weineisen et al., 2014).
Octanol-Water Distribution Coefficients
Approximately 1 MBq of the 177Lu-labeled tracer was added to a mixture of 0.5 mL phosphate buffered saline (PBS, pH 7.4) and 0.5 mL n-octanol in an Eppendorf tube (n = 6). After vigorous mixing of the suspension for 3 min, the vial was centrifuged at 11.500 g for 3 min for phase separation. 200 μL aliquots of each phase were withdrawn and measured in a gamma counter.
Biodistribution
The experiments were carried out in accordance with the German Animal Welfare Act (Tierschutzgesetz), and were previously approved by the responsible authority (Regierung von Oberbayern). The animal model, male CB-17 SCID mice bearing subcutaneous LNCaP tumor xenografts, were generated as described before (Weineisen et al., 2014). Approximately 1–4 MBq (varying molar activities; absolute molar amounts of active compound ranging from 0.11 to 0.15 nmol) of the 177Lu-labeled PSMA inhibitors were injected into the tail vein of the animals, which were sacrificed 1 and 24 h post injection (n = 3 per tracer per time point). Selected organs were removed, weighted and the activities contained were measured in a γ-counter.
Author Contributions
AW: Performed chemical synthesis, radiochemistry, and in-vivo studies; AV and DH: Performed the equilibrium and kinetic measurements; FF, FK, and H-JW: Performed interpretation of data and critically reviewed the manuscript; JN: Conceived the study, interpreted the data, and wrote the manuscript. All authors approved the final version of the manuscript.
Conflict of Interest Statement
The handling editor declared a past co-authorship with several of the authors, AV, H-JW, JN.
The other authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Support by the Deutsche Forschungsgemeinschaft (grant #NO822/4-1 and SFB 824, project A10), the EU and the European Regional Development Fund (projects GINOP-2.3.2-15-2016-00008 and GINOP-2.3.3-15-2016-00004), and a János Bolyai Research Scholarship of the Hungarian Academy of Science awarded to FK is gratefully acknowledged. We furthermore thank Prof. Imre Tóth and particularly Dr. Zsolt Baranyai (University of Debrecen & Bracco Imaging) for helpful discussions and proofreading.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2018.00107/full#supplementary-material
References
Agard, N. J., Prescher, J. A., and Bertozzi, C. A. (2004). A strain-promoted [3 + 2] azide–alkyne cycloaddition for covalent modification of biomolecules in living systems. J. Am. Chem. Soc. 126, 15046–15047. doi: 10.1021/ja044996f
Banerjee, S. R., Pullambhatla, M., Byun, Y., Nimmagadda, S., Green, G., Fox, J. J., et al. (2010). 68Ga-labeled inhibitors of prostate-specific membrane antigen (PSMA) for Imaging prostate cancer. J. Med. Chem. 53, 5333–5341. doi: 10.1021/jm100623e
Baranyai, Z., Bányai, I., Brücher, E., Király, R., and Terreno, E. (2007). Kinetics of the formation of [Ln(DOTAM)]3+ complexes. Eur. J. Inorg. Chem. 49, 3639–3645. doi: 10.1002/ejic.200700178
Baranyai, Z., Palinkas, Z., Uggeri, F., and Brücher, E. (2010). Equilibrium studies on the Gd3+, Cu2+ and Zn2+ complexes of BOPTA, DTPA and DTPA-BMA ligands: kinetics of metal-exchange reactions of [Gd(BOPTA)]2−. Eur. J. Inorg. Chem. 1948–1956. doi: 10.1002/ejic.200901261
Baranyai, Z., Reich, D., Vágner, A., Weineisen, M., Tóth, I., Wester, H. J., et al. (2015). A shortcut to high-affinity Ga-68 and Cu-64 radiopharmaceuticals: one-pot click chemistry trimerisation on the TRAP platform. Dalton Trans. 44, 11137–11146. doi: 10.1039/C5DT00576K
Bass, L. A., Wang, M., Welch, M. J., and Anderson, C. J. (2000). In vivo transchelation of copper-64 from TETA-octreotide to superoxide dismutase in rat liver. Bioconjug. Chem. 11, 527–532. doi: 10.1021/bc990167l
Bazakas, K., and Lukeš, I. (1995). Synthesis and complexing properties of polyazamacrocycles with pendant N-methylenephosphinic acid. J. Chem. Soc. Dalton Trans. 1133–1137. doi: 10.1039/dt9950001133
Benešová, M., Bauder-Wüst, U., Schäfer, M., Klika, K. D., Mier, W., Haberkorn, U., et al. (2016). Linker modification strategies to control the prostate-specific membrane antigen (PSMA)-targeting and pharmacokinetic properties of DOTA-conjugated PSMA inhibitors. J. Med. Chem. 59, 1761–1775. doi: 10.1021/acs.jmedchem.5b01210
Bianchi, A., Calabi, L., Giorgi, C., Losi, P., Mariani, P., Paoli, P., et al. (2000). Thermodynamic and structural properties of Gd3+ complexes with functionalized macrocyclic ligands based upon 1,4,7,10-tetraazacyclododecane. J. Chem. Soc. Dalton Trans. 697–705. doi: 10.1039/a909098c
Bock, V. D., Speijer, D., Hiemstra, H,., and van Maarseveen, J. H. (2007). 1,2,3-Triazoles as peptide bond isosteres: synthesis and biological evaluation of cyclotetrapeptide mimics. Org. Biomol. Chem. 5, 971–975. doi: 10.1039/b616751a
Chaves, S., Delgado, R., and Da Silva, J. J. (1992). The stability of the metal complexes of cyclic tetra-aza tetra-acetic acids. Talanta 39, 249–254. doi: 10.1016/0039-9140(92)80028-C
Davis, M. R., Singh, E. K., Wahyudi, H., Alexander, L. D., Kunicki, J. B., Nazarova, L. A., et al. (2012). Synthesis of sansalvamide A peptidomimetics: triazole, oxazole, thiazole, and pseudoproline containing compounds. Tetrahedron 68, 1029–1051. doi: 10.1016/j.tet.2011.11.089
Desreux, J. F., Merciny, E., and Loncin, M. F. (1981). Nuclear magnetic resonance and potentiometric studies of the protonation scheme of two tetraaza tetraacetic macrocycles. Inorg. Chem. 20, 987–991. doi: 10.1021/ic50218a008
Horne, S. W., Olsen, C. A., Beierle, J. M., Montero, A., and Ghadiri, R. M. (2009). Probing the bioactive conformation of an archetypal natural product HDAC inhibitor using conformationally homogeneous triazole-modified cyclic tetrapeptides. Angew. Chem. Int. Ed. Engl. 48, 4718–4724. doi: 10.1002/anie.200805900
Horne, W. S., Yadav, M. K., Stout, C. D., and Ghadiri, M. R. (2004). Heterocyclic peptide backbone modifications in an alpha-helical coiled coil. J. Am. Chem. Soc. 126, 15366–15367. doi: 10.1021/ja0450408
Irving, H. M., Miles, M. G., and Pettit, L. D. (1967). A study of some problems in determining the stoicheiometric proton dissociation constants of complexes by potentiometric titrations using a glass electrode. Anal. Chim. Acta 38, 475–488. doi: 10.1016/S0003-2670(01)80616-4
Šimeček, J., Hermann, P., Havlíčková, J., Herdtweck, E., Kapp, T. G., Engelbogen, N., et al. (2013). A cyclen-based tetraphosphinate chelator for preparation of radiolabeled tetrameric bioconjugates. Chem. Eur. J. 19, 7748–7757. doi: 10.1002/chem.201300338
Kotková, Z., Pereira, G. A., Djanashvili, K., Kotek, J., Rudovsky, J., Hermann, P., et al. (2009). Lanthanide(III) complexes of phosphorus acid analogues of H4DOTA as model compounds for the evaluation of the second-sphere hydration. Eur. J. Inorg. Chem. 119–136. doi: 10.1002/ejic.200800859
Kularatne, S. A., Zhou, Z., Yang, J., Post, C. B., and Low, P. S. (2009). Design, synthesis, and preclinical evaluation of prostate-specific membrane antigen targeted 99mTc-radioimaging agents. Mol. Pharm. 6, 790–800. doi: 10.1021/mp9000712
Lázár, I., Sherry, A. D., Ramasamy, R., and Brücher, E. (1991). Synthesis and complexation properties of a new macrocyclic polyaza polyphosphinate ligand, DOTEP (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetrakis(methyleneethylphosphinate)). Inorg. Chem. 30, 5016–5019. doi: 10.1021/ic00026a030
Liu, Y. Q., Zhang, L. H., Wan, J. P., Li, Y. S., Xu, Y. H., and Pan, Y. J. (2008). Design and synthesis of cyclo[-Arg-Gly-Asp-Ψ(triazole)-Gly-Xaa-] peptide analogues by click chemistry. Tetrahedron 64, 10728–10734. doi: 10.1016/j.tet.2008.08.090
Maschauer, S., Einsiedel, J., Reich, D., Hübner, H., Gmeiner, P., Wester, H.-J., et al. (2017). Theranostic value of multimers: lessons learned from trimerization of neurotensin receptor ligands and other targeting vectors. Pharmaceuticals 10:29. doi: 10.3390/ph10010029
Meldal, M., and Tornøe, C. W. (2008). Cu-catalyzed azide–alkyne cycloaddition. Chem. Rev. 108, 2952–3015. doi: 10.1021/cr0783479
Mesters, J. R., Barinka, C., Li, W., Tsukamoto, T., Majer, P., Slusher, B. S., et al. (2006). Structure of glutamate carboxypeptidase II, a drug target in neuronal damage and prostate cancer. EMBO J. 25, 1375–1384. doi: 10.1038/sj.emboj.7600969
Notni, J., Šimeček, J., and Wester, H. J. (2014). Phosphinic acid functionalized polyazacycloalkane chelators for radiodiagnostics and radiotherapeutics: unique characteristics and applications. Chem. Med. Chem. 9, 1107–1115. doi: 10.1002/cmdc.201400055
Notni, J., and Wester, H. J. (2016). A practical guide on the synthesis of metal chelates for molecular imaging and therapy by means of click chemistry. Chem. Eur. J. 22, 11500–11508. doi: 10.1002/chem.201600928
Notni, J., and Wester, H. J. (2018). Re-thinking the role of radiometal isotopes: towards a future concept for theranostic radiopharmaceuticals. J. Label. Compd. Radiopharm. 61, 141–153. doi: 10.1002/jlcr.3582
Oyen, W. J., Bodei, L., Giammarile, F., Maecke, H. R., Tennvall, J., Luster, M., et al. (2007). Targeted therapy in nuclear medicine—current status and future prospects. Ann. Onc. 18, 1782–1792. doi: 10.1093/annonc/mdm111
Pasha, A., Tircsó, G., Benyó, E. T., Brücher, E., and Sherry, A. D. (2007). Synthesis and Characterization of DOTA-(amide)4 derivatives: equilibrium and kinetic behavior of their lanthanide(III) complexes. Eur. J. Inorg. Chem. 49, 4340–4349. doi: 10.1002/ejic.200700354
Pedersen, D. S., and Abell, A. (2011). 1,2,3-Triazoles in peptidomimetic chemistry. Eur. J. Org. Chem. 2399–2411. doi: 10.1002/ejoc.201100157
Riesen, A., Zehnder, M., and Kaden, T. A. (1986). Metal complexes of macrocyclic ligands. Part XXIII. Synthesis properties, and structures of mononuclear complexes with 12- and 14-membered tetraazamacrocycle-N,N′,N″,N‴-tetraacetic Acids. Helv. Chim. Acta 69, 2067–2073. doi: 10.1002/hlca.19860690830
Rohovec, J., Kyvala, M., Vojtíšek, P., Hermann, P., and Lukeš, I. (2000). Synthesis and coordination behavior of symmetrical tetraamine phosphinic acids. Eur. J. Inorg. Chem. 195–203. doi: 10.1002/(SICI)1099-0682(200001)2000:1<195::AID-EJIC195>3.0.CO;2-6
Stasiuk, G. J., and Long, N. J. (2013). The ubiquitous DOTA and its derivatives: the impact of 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid on biomedical imaging. Chem. Commun. 49, 2732–2746. doi: 10.1039/c3cc38507h
Takács, A., Napolitano, R., Purgel, M., Benyei, A. C., Zekany, L., Brücher, E., et al. (2014). Solution structures, stabilities, kinetics, and dynamics of DO3A and DO3A–sulphonamide complexes. Inorg. Chem. 53, 2858–2872. doi: 10.1021/ic4025958
Tischler, M., Nasu, D., Empting, M., Schmelz, S., Heinz, D. W., Rottmann, P., et al. (2012). Braces for the peptide backbone: insights into structure-activity relationships of protease inhibitor mimics with locked amide conformations. Angew. Chem. Int. Ed. Engl. 51, 3708–3712. doi: 10.1002/anie.201108983
Wadas, T. J., Wong, E. H., Weisman, G. R., and Anderson, C. J. (2010). Coordinating radiometals of copper, gallium, indium, yttrium, and zirconium for PET and SPECT imaging of disease. Chem. Rev. 110, 2858–2902. doi: 10.1021/cr900325h
Weineisen, M., Šimeček, J., Schottelius, M., Schwaiger, M., and Wester, H. J. (2014). Synthesis and preclinical evaluation of DOTAGA-conjugated PSMA ligands for functional imaging and endoradiotherapy of prostate cancer. EJNMMI Res. 4:63. doi: 10.1186/s13550-014-0063-1
Wurzer, A., Seidl, C., Morgenstern, A., Bruchertseifer, F., Schwaiger, M., Wester, H.-J., et al. (2018). Dual-nuclide radiopharmaceuticals for positron emission tomography based dosimetry in radiotherapy. Chem. Eur. J. 24, 547–550. doi: 10.1002/chem.201702335
Zarschler, K., Kubeil, M., and Stephan, H. (2014). Establishment of two complementary in vitro assays for radiocopper complexes achieving reliable and comparable evaluation of in vivo stability. RSC Adv. 4, 10157–10164. doi: 10.1039/c3ra47302c
Zekany, L., and Nagypal, I. (1985). “PSEQUAD,” in Computational Methods for Determination of Formation Constants, ed D. J. Leget (New York, NY: Plenum Press), 291–353.
Keywords: Huisgen-reaction, potentiometry, spectrophotometry, phosphinate, radiopharmaceuticals, endoradiotherapy, prostate-specific membrane antigen, theranostics
Citation: Wurzer A, Vágner A, Horváth D, Fellegi F, Wester H-J, Kálmán FK and Notni J (2018) Synthesis of Symmetrical Tetrameric Conjugates of the Radiolanthanide Chelator DOTPI for Application in Endoradiotherapy by Means of Click Chemistry. Front. Chem. 6:107. doi: 10.3389/fchem.2018.00107
Received: 24 January 2018; Accepted: 22 March 2018;
Published: 10 April 2018.
Edited by:
Lorenzo Tei, Università degli Studi del Piemonte Orientale, ItalyReviewed by:
Steve J. Archibald, University of Hull, United KingdomKristina Djanashvili, Delft University of Technology, Netherlands
Copyright © 2018 Wurzer, Vágner, Horváth, Fellegi, Wester, Kálmán and Notni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Johannes Notni, am9oYW5uZXMubm90bmlAdHVtLmRl
†These authors have contributed equally to this work.
‡Present Address: Adrienn Vágner, Scanomed Ltd., Debrecen, Hungary