 Junxian Chen
Junxian Chen Qingyu Liu
Qingyu Liu Hao Li3
Hao Li3 Dingguo Xu
Dingguo Xu- 1MOE Key Laboratory of Green Chemistry and Technology, College of Chemistry, Sichuan University, Chengdu, China
- 2College of Chemistry and Environment Protection Engineering, SouthWest University for Nationalities, Chengdu, China
- 3Department of Chemistry, University of Science and Technology of China, Hefei, China
Squaraine core based small molecules in bulk heterojunction organic solar cells have received extensive attentions due to their distinguished photochemical properties in far red and infrared domain. In this paper, combining theoretical simulations and experimental syntheses and characterizations, three major factors (fill factor, short circuit and open-cirvuit voltage) have been carried out together to achieve improvement of power conversion efficiencies of solar cells. As model material systems with D-A-D' framework, two asymmetric squaraines (CNSQ and CCSQ-Tol) as donor materials in bulk heterojunction organic solar cell were synthesized and characterized. Intensive density functional theory computations were applied to identify some direct connections between three factors and corresponding molecular structural properties. It then helps us to predict one new molecule of CCSQ'-Ox that matches all the requirements to improve the power conversion efficiency.
Introduction
Compared with polymers as donor materials in organic solar cells (OSCs), small organic molecules were developed even faster due to the several advantages, high purity, tunable electronic specialties and better device reproduction (Kan et al., 2014, 2015; Fan and Zhu, 2015; Collins et al., 2017). Moreover, solution-processed bulk heterojunction (BHJ) OSCs have great potential in realizing commercial solar cell devices due to the unique features like flexibility over a large area and low-cost fabrication (Huang et al., 2014). However, it is still challengeable to improve the power conversion efficiency (PCE) even higher to be commercialized although lots of efforts have been made to the synthesis (Li N. et al., 2013; Mulligan et al., 2014; Zhang et al., 2017).
Inspired by donor-acceptor (D-A) copolymers leading solar cells to 5~6% PCE (Peet et al., 2007; Park et al., 2009), where donor and acceptor unit are respectively conjugated electron-rich and electron- deficient moieties, the breakthroughs in PCE for small molecule donors were then achieved (Li et al., 2010). Moreover, such structural framework have been expanded to D-A-D, A-D-A, A-D-D-D-A, and A-π-D-π-A patterns, even their oligomers (Collins et al., 2017). It is notable that the asymmetric derivatives have a larger transition dipole moment so that they have higher performance than those centrosymmetric ones. Among those small molecules used in the BHJ OSCs, squaraine core based molecules have attracted extensive attentions due to their high extinction coefficients and relatively strong absorption in the far red and infrared domain (Chen et al., 2015; Gsänger et al., 2016), and thus have good overlap with solar spectra. In particular, squaraines have been demonstrated to have excellent thermal and (photo-) chemical stability as the dye and pigment chromophores. Squaraines can be generally assigned to a DL-A-DR system. The center four-ring part behaves as an electron-acceptor, which is covalently linked to two electron donators. They can be accessible by using straightforward synthetic protocols. Potentials for commercial applications are thus highly expected. Further, asymmetrical squaraines (ASQs) bearing D-A-D' molecular frameworks can afford more room for molecular tailoring than symmetrical squaraines (SSQs) (Pandey et al., 2011). Our group reported an breakthrough in BHJ OSCs by using ASQs as electron donors (PCEmax = 6.0%) (Yang et al., 2015), although it is lower than other small-molecule including D-A structural frameworks based devices (10%) (Ni et al., 2015; Collins et al., 2017). In short, it is valuable to explore how the structural details of D-A-D' framework molecules as electron donor affect the performance in BHJ OSCs.
Albeit the PCEs of organic photovoltaic (OPV) cells have been improved to some extent, it is still far from commercial application in comparison with 22.1% PCE of lead halide perovkite based thin-film solar cells (Xiao and Yan, 2017). Further improvement in OPV performance is necessary. Basically, the PCE of a photovoltaic cell can be expressed as
in which JSC means the short-circuit current density, Voc denotes the open-circuit photovoltage, FF is the fill factor, and IS represents the intensity of incident (Bérubé et al., 2013). Clearly, to get a better PCE, none of these four components can be ignored in the material design.
Generally, the Voc is related to the energy difference between the highest occupied molecular orbital (HOMO) of the donor material and the lowest unoccupied molecular orbital (LUMO) of the acceptor material. It is also affected by the rates of carrier generation and recombination, the presence of energetic tail states or trap states (Lange et al., 2013; Sweetnam et al., 2014). The JSC is defined as the integration of the external quantum efficiency (EQE) along the wavelength across the solar spectrum (Bérubé et al., 2013). The EQE can be further divided into five fundamental processes,
in which each term has the name of light absorption (ηA), exciton migration (ηED), exciton dissociation or charge separation (ηCD), charge transport (ηCT), and charge collection to the electrodes (ηCC) (Li, 2012). Therefore, to get better JSC, we should consider the combination of all five terms. Meanwhile, to achieve a high FF for BHJ OSCs, more efforts should be put on the device features, e.g., domain size or purity, gradated BHJ structures and π stacking distance or direction (Jao et al., 2016).
In contrast to experimental efforts in understanding fundamental mechanisms responsible for the photovoltaic properties of materials in OSCs, theoretical computational approaches represent an alternative way to establish the structure-property relationships. (Mennucci, 2015; Brückner and Engels, 2016; Volpi et al., 2016a,b; Alessandri et al., 2017; Brückner et al., 2017). Clearly, based on Equations (1, 2), various factors should work together to affect the PCE. However, rare work in material structural design includes all fundamental terms. Therefore, the objective in this work is to find the principle structural factors that regulate the fundamental photochemical properties. The asymmetrical squaraines (ASQs) with Fullerene based BHJ OSCs will be used as the example material systems.
In our previous work (Yang D. et al., 2014; Yang J. et al., 2014; Yang et al., 2015), a series of ASQs bearing benzindole-squarate-4-amino-2, 6-dihydroxyphenyl skeleton with different N-substitution like carbazole, indole, and indoline, were synthesized and characterized. All of them have good solubility, high film quality and achieved PCEs in range of 1.54–4.29% for solution-processable ASQ-based BHJ-OSC. Specifically, indoline group seems to be one of good candidates for further modification to get better solar cell performance. As shown in Figure 1, indoline subunit has two different linking positions (4 or 7), which can be attached by different groups. Particularly, the 4-position can be linked to a squaraine 4-member ring via dihydroxyphenyl group, which is denoted as C-N linkage. The 7-position can be linked to an electron-deficient core, namely C-C linkage. Indeed, thanks to different electronic structures and molecular skeleton of groups linked at 4 or 7 position, distinguished device performances in these two kinds of materials based dye-sensitized solar cell (DSSC) can be found like absorption, aggregation, or morphology (Li G. et al., 2013; Yang D. et al., 2014).
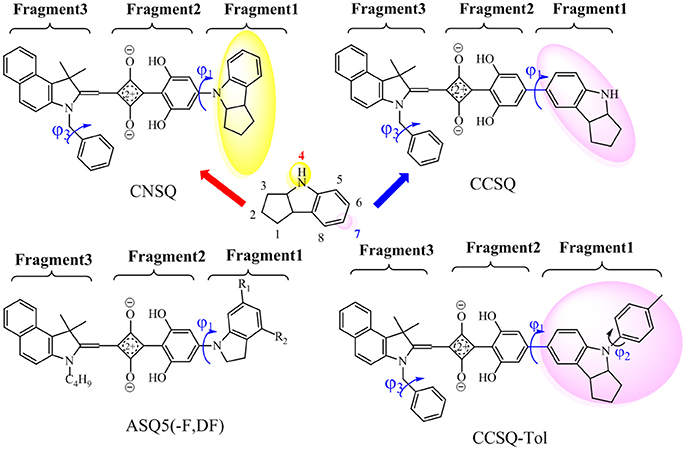
Figure 1. Investigated molecules [CNSQ, cCSQ, CCSQ-Tol and ASQ5(-F, DF); Yang et al., 2015] and definitions of three fragments used in this work.
In this work, two asymmetrical squaraines bearing indoline moiety were designed with different linkages, in which the 4- and 7-positions of indoline were linked to squarate core via dihydroxyphenyl group. They are defined as CNSQ and CCSQ, respectively. An additional molecule, namely CCSQ-Tol, was also synthesized by us with the H atom of CCSQ replaced by a toluene group as shown in Figure 1. In addition, three other well-characterized ASQ derivatives have been reported by us (Yang et al., 2015). Their good photophysical performances have been considered as the result of particular π-π stacking effects. Therefore, with all these different squaraine based donor molecules systematically theoretical computations can help us establish reliable connections between structural elements and the fundamental factors of PCE in BHJ OSCs. This can provide more opportunities to enhance photophysical properties of OSCs based on asymmetrical squaraines core.
Materials and Methods
First of all, only two target molecules CNSQ and CCSQ-Tol, due to instability of CCSQ's N-H bond in BHJ OSC, were synthesized and characterized. All experimental details can be found in Supplementary Material. As we can see in Table 1, the PCEs for two donor molecules, CNSQ and CCSQ-Tol, were not significantly improved as expected. In addition, the device based on CCSQ-Tol has higher JSC than that based on CNSQ. Of course, some apparently linking topological or N-H substitution effects can be identified. Theoretically, the molecule of CCSQ is also included in our simulation. In addition, in order to shed more lights to this interesting issue, more molecules based on asymmetric squaraines core, e.g., ASQ5, ASQ5-F, and ASQ5-DF, have also been evaluated in this work.
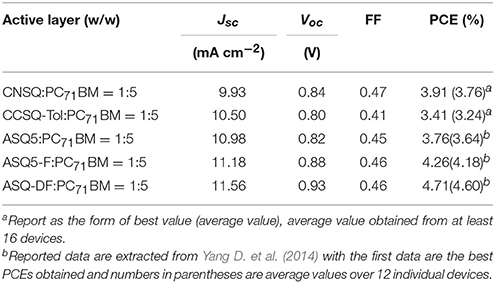
Table 1. Optimized OSC performance data with blended ratio of ASQs:PC71BM = 1:5 (w/w).
Previous studies (Yang D. et al., 2014; Yang J. et al., 2014; Yang L. et al., 2014) suggested that the hydroxyl groups of phenyl rings on the asymmetric squaraine system can form intramolecular hydrogen bonds with oxygen atoms on squarate core. A co-planar topology is displayed in Figure S9. Therefore, all targeted asymmetrical squaraines can be divided into three fragments with D-A-D' framework according to Figure 1. Specifically, Fragment 1 as the donor unit contains the indoline moiety with different linking topology or substitution; Fragment 2 as the accept unit includes square acid and dihydroxyl benzene; and Fragment 3 as the donor unit denotes the benzoindole derivative. Since the linking topological and substitution effects mainly occur in Fragment 1 of D-A-D' system, the torsion angle between Fragment 1 and Fragment 2 denoted as ϕ1 and the covalent bond distance between them denoted B12 need more attentions. Two additional torsion angles of ϕ2 and ϕ3 have also been defined as shown in Figure 1 for further discussion.
To fully understand the relationship between the molecular structures and corresponding photophysical properties of target materials, intensive density functional theory (DFT) calculations were then applied. The Becke3-Lee-Yang-Parr (B3LYP) exchange-correlation functional and a standard basis set of 6-31G(d) were first used in the full geometry optimization to locate the ground state (S0) configurations. Subsequently, the geometric optimization and property analyses of electronic excited states of S1 for all molecules were carried out at B3LYP/6-31G(d) and M06-2X/6-31++G(d, p) levels of theory within TD-DFT (Scalmani et al., 2006) framework. Meanwhile, to account for the solvent effects, the polarizable continuum model (PCM) (Tomasi et al., 2005) was applied using chloroform as the solvent. All DFT computations were performed using the Gaussian 09 suite of program (Frisch et al., 2009). The relationship between the structures and properties were based on electronic structure analyses, which were achieved using the package of Multiwfn (Lu and Chen, 2012). In addition, the ESP plots were rendered by Visual Molecular Dynamics (VMD) software (Humphrey et al., 1996).
Results and Discussion
Geometry and Spectra
For the systems investigated in this work, the method of B3LYP/6-31G(d) has good performance for the ground state of all target molecules, but it failed to correctly characterize the electronic excitation of the asymmetric squares. According to Table 2, the calculated stokes shifts of CNSQ and CCSQ-Tol at M06-2X/6-31++G(d, p) level are 0.09 and 0.20 eV, which is consistent with experimental 0.06 and 0.14 eV despite a little bit red-shift. However, such 2 values at B3LYP/6-31G(d) are 0.21 and 0.22 eV, which is too close and inconsistent with experimental observations. Basically, larger stokes shifts usually occurs on account of more conformational differences between ground and excited states (van Duren et al., 2004). According to Table S1, we can find that CCSQ-Tol exhibits much larger conformational change than CNSQ from states of S0 to S1, e.g., the torsion angle ϕ1 (8° vs. 2.7°) and the bond distance B12 (0.021 vs. 0.0 Å). Clearly, results based on B3LYP/6-31G(d) cannot reproduce this geometrical variation. Therefore, M06-2X/6-31++G(d,p) with solvent effect was then selected for all density functional theory calculations if not otherwise stated.
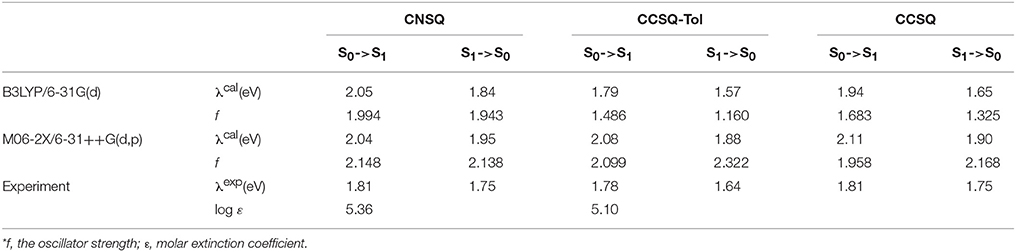
Table 2. The calculated absorption and emission spectra at B3LYP/6-31G(d) and M06-2X/6-31++G(d, p) level in chloroform in comparision with experimental data*.
As we have mentioned above that there two linking sites on indoline group, we noticed that this could cause large geometric variations for both S0 and S1 states for all three molecules of CCSQ, CCSQ-Tol, and CNSQ. According to Table S1, we can also find that the substitution of tolune group to CCSQ cannot change the overall geometry too much. Moreover, these different geometries cause distinct features in crystal structures. As shown in Figure S1, CNSQ shows stronger aggregation with π-π distance of 3.93 Å (2θ = 21.88°), while CCSQ-Tol exhibits weaker reflection peaks at 2θ = 23.78°, corresponding to π-π or π-edge distance of 3.68 Å. Unfortunately, efforts to cultivate single crystal for CCSQ-Tol finally failed. The experimentally monomer and superposition structures have been shown in Figure S2. In particular, the calculated torsion angles ϕ1 in chloroform for CNSQ is 13.1°, which is close to 4° in its X-ray structure. The nearly planar conformation of CNSQ in crystal structure might be due to the π-π stacking effects. Compared to CNSQ, much poorer π-π stacking effect for CCSQ-Tol can be seen based on the geometrical optimization. Indeed, ϕ1 for CCSQ-Tol is calculated to be 31.6°, and ϕ2 of 38.9°Can be also obtained.
Such poor π-π stacking effect can be further confirmed by the experimental absorption wavelength in chloroform and film (see in Table S4). In chloroform solution, the maximum absorption peaks of CNSQ and CCSQ-Tol are around 690 nm. The CNSQ shows higher molar extinction coefficient (log ε = 5.36) when compared to CCSQ-Tol (log ε = 5.10). However, CNSQ shows a typical sharp absorption band, which is much narrower than that of CCSQ-Tol, evidenced by full width at half maximum (FWHM) of 38 nm (CNSQ) vs. 108 nm (CCSQ-Tol). Relative to their absorption spectra in solution, CNSQ exhibits a more pronounced red-shift (43 nm) and broad absorption spectrum in thin film than that of CCSQ-Tol, which can be attributed to strong aggregation in solid state. Interestingly, the absorption spectrum of CCSQ-Tol in thin film is only slightly red-shift (16 nm), which indicates the weaker packing (Pommerehne et al., 1995; Wei et al., 2011). Nevertheless, the absorption spectrum of CCSQ-Tol still cover a wide range of 430–860 nm with a relatively broad FWHM of 195 nm, which is desirable for efficient light harvest.
Short-Circuit Current Density
Significantly different structural and π-π stacking features caused by linking topological and substitution effect of CNSQ, CCSQ and CCSQ-Tol can be observed based on experimental and theoretical characterizations. Therefore, it would be important to explore how they affect the fundamental factors of PCE, esp., for short-circuit current density (JSC). Basically, JSC is affected by five factors as shown in Equation (2). The property of ηCC to the electrodes is considered to be only related to the device fabrication, but not with the molecule itself. Thus, it was simply ignored here. We will then discuss other four terms individually below, and try to find out which term has dominant contribution to JSC.
Electronic Transition Analysis
The electronic transition properties for CNSQ and CCSQ-Tol are presented in Table 3. CNSQ has the largest transition dipole moment μtr (6.34 a.u.) and the smallest distance index Δr (1.90 Å) compared to CCSQ and CCSQ-Tol. Generally, norm of transition dipole moment μtr is related with the oscillator strength, which can explain the strength of light absorption (ηA). The distance index, Δr, between centroids of hole and electron usually represents a measure of intra-molecular charge-transfer (iCT). The smaller Δr is, the more likely the excitation is a local excitation mode (LE). To this point, the excitation for CCSQ-Tol is closer to an iCT mode. The CNSQ with more LE transition mode should have larger light absorption ηA. This is consistent with the fact that CNSQ in chloroform solution has higher molar extinction coefficient (log ε = 5.36) when compared to CCSQ-Tol (log ε = 5.10). All these data were summarized in Figure S4 and Table S4. In last part, we have already suggested the substitution has little effects on the overall geometry of molecular moieties. Furthermore, peak positions of adsorption and emission spectra are close for CCSQ and CCSQ-Tol, shown in Figure S4.
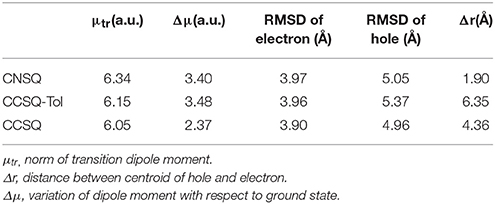
Table 3. Electron transition properties.
On the other hand, to characterize the carrier transport property ηCT, the hole mobility of the pristine and blend films (ASQs: PC71BM = 1: 5) were measured by the space charge limited current (SCLC) method, and the structures were ITO/MoO3 (8 nm)/active layer (80 nm)/Au (100 nm) (see Figure S6 in Supplementary Material). In pristine film, the hole mobility of the CCSQ-Tol (2.21 × 10−5 cm2 V−1 s−1) is slightly higher than that of CNSQ (1.68 × 10−5 cm2 V−1 s−1), which is consistent with the RMSD values of hole distribution according to Table 3. RMSD of hole or electron distribution denotes the region that an electron leaves or enters in the single-electron excitation process between S0 and S1. To get a better view of the electronic structures, we then plotted the hole, electron distributions and their corresponding overlaps for all three molecules in Figure 2. Obvious difference for the hole distributions (blue marked region) occurs in Fragment 3. From Table 3, CCSQ-Tol has broader hole distribution than CNSQ, while all three molecules have similar electron distributions. It could thus be expected that the development of a larger hole distribution for donor-materials could lead better ηCT. However, in blend film, the hole mobility of the CCSQ-Tol: PC71BM = 1:5 film (1.46 × 10−6 cm2 V−1 s−1) is much lower than that of CNSQ: PC71BM film (3.72 × 10−6 cm2 V−1 s−1).
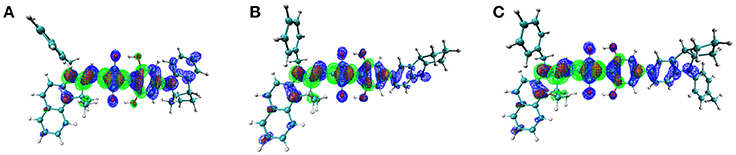
Figure 2. Electron (green), Hole (blue), and Overlap (red) Distribution for (A) CNSQ, (B) CCSQ, and (C) CCSQ-Tol.
Obviously, although larger values of ηA and ηCT for CNSQ than CCSQ-Tol have been identified via experimental measurements and theoretical computations, the JSC has a reverse performance between two molecules. This could tell us that we could not expect to enhance JSC (Table 1) via increasing ηA and ηCT. In other words, simple adjustment of the molecular topology might not work here, esp., for asymmetric squaraine based OSCs. We might focus on other two parameters of ηED and ηCD.
Electrostatic Analysis
There are no effective experimental methods that can be used to measure the ηCD and ηED. Theoretical simulation represents an alternative way to address this issue. Basically, there are two kinds of excitons in OSCs, molecular exciton and charge-transfer exciton (Zhugayevych and Tretiak, 2015; Vandewal, 2016). The exciton migration of ηED is related to the former case, while the exciton dissociation of ηCD is related to the latter case. According to the electrostatic interaction, monomers at S0 or S1 state tend to contact each other in complementary manner of electrostatic potential (ESP), i.e., the electron deficient part prefers interacting with the electron rich part. Therefore, the ESP information of both ground and excited states could give a hand to connect two factors of JSC (ηED and ηCD) with structural features of asymmetric squaraines.
The ESPs of CNSQ, CCSQ, and CCSQ-Tol were plotted for vertical excited states in Figure 3 at the 0.001 electron/bohr3 isosurfaces of electron density. The color ranges from −25 to 25 kcal/mol. From Figure 3, the ESP values are negative inside the aromatic group or on electron-withdrawn group, featuring electron-deficient region. ESP values are positive on hydrogen atoms of aromatic group or atoms on alkyl group, which means an electron-rich feature. The electro-deficient region covers not only the square acid but also the dihydroxyl benzene group in the asymmetric squaraine. The global positive maxima for three molecules can be located nearly at the benzoindole derivative, while much smaller local maxima with positive ESP values can be found at the right counterpart. It should be noted that the excited state discussed here is at Franck-Condon region via vertical excitation since the molecule of BHJ OSC in condensed phase might not have sufficient time to change its conformation after light absorption.
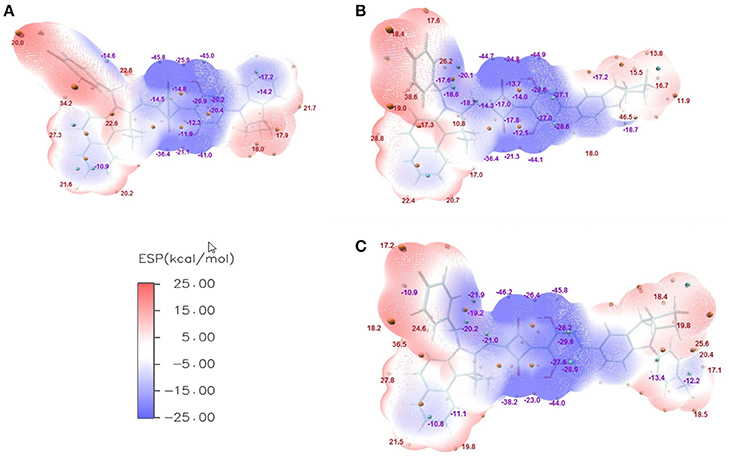
Figure 3. ESPs on the vdW surface for (A) CNSQ, (B) CCSQ, and (C) CCSQ-Tol at vertical excited states with blue denoting extremely electron-deficient region (corresponding to negative value), red denoting electron-rich region (corresponding to positive value) and white denoting neutral region. The cyan and orange spheres correspond to ESP surface minima and maxima, respectively. For simplification, only the most negative and positive values have been labeled.
For ηED, the exciton could transfer (or hop) from one molecule to the other until it arrives to the donor/acceptor (D/A) interface. The interaction type between two molecules is the key during this process. Generally, it tends to be achieved in the direction of electron transferring from the most electron-rich region of the excited state to the most electron deficient region of the ground state. Or in the reverse direction of hole transferring, it is from the most electron deficient region of the excited state to the most electron-rich region of the ground state. Hence, the electron transferring ability (denoted as Te) and the hole transferring ability (denoted as Th) can be used to elucidate exiton transfer (ηED). According to the calculation, CCSQ-Tol has larger Te and Th (83.2 and 90.3 kcal/mol) than CNSQ (79.8 and 83.2 kcal/mol), which is consistent with the values of observed JSC. That means the property of ηED shows similar trend with JSC for the squaraine based system.
On the other hand, formation of charge-transfer (CT) excitons also depends on electrostatic interaction between the most electron-rich region of donor molecules and the most electron deficient region of acceptor molecules at both excited state. The value of ηCD is mainly affected by formation of CT excitons. Therefore, a more favorable ηCD should be connected with the larger ESP extrema of electron-rich region on donor molecule. Here, the most positive extrema of the electron-rich region is defined as Xe. In donor materials investigated in this work, CCSQ-Tol has a larger Xe (36.6 kcal/mol) on electron rich region than CNSQ (34.2 kcal/mol). That might partially mean ηCD also has the same trend with JSC, although current samples are not sufficient.
According to above analyses, compared to ηA and ηCT, the improvement of both ηED and ηCD seems to be feasible to enhance JSC for the squaraine based materials. Now the question is which one is dominant. To address this issue, we then include three other ASQ based molecules (ASQ5, ASQ5-F, ASQ5-DF) in our computations, which have been synthesized and characterized by us (Yang et al., 2015). They all show some good performances of OSC devices. Since the ASQ5 holds nearly the same topology as CNSQ/CCSQ, it is then expected to provide us more information about how morphology or electronic structures affecting material properties.
Like CNSQ and CCSQ-Tol, ηA and ηCT of the devices based on these three donor molecules ASQ5, ASQ5-F, and ASQ5-DF were also shown to show opposite performance with JSC. Therefore, we can rule out the efforts to improve JSC with helping of either ηA or ηCT. As we have mentioned above, the variation tendency of Te and Th matches the trend of JSC of CCSQ-Tol and CNSQ. However, no such principle can be found for the fluorine substitution cases. According to Table S6, we could see that there is no connection between Te or Th with JSC. We then plot the possible correlation in Figure 4A. Clearly, we could not see any real correlation between Te (or Th) and JSC for all five systems investigated here. In other words, we also could not expect to improve JSC by regulating exiton transfer or ηED. Quite interestingly, similar with CNSQ and CCSQ-Tol, values of ηCD of ASQ5, ASQ5-F, and ASQ5-DF have the same trends with JSC (see more details in Table S6). In particular, with the fluorine substitution, JSC improves as well. Accordingly, the Xe also increases, i.e., 37.5 kcal/mol for ASQ5 and 38.3 kcal/mol for ASQ5-F, and 39.3 kcal/mol for ASQ5-DF. Moreover, a perfect linear correlation between JSC and Xe can be established according to Figure 4B. Since the Xe value is directly connected with the ηCD, we could safely conclude that a better ηCD can lead a higher JSC, esp., for asymmetric squaraine based OSC devices.
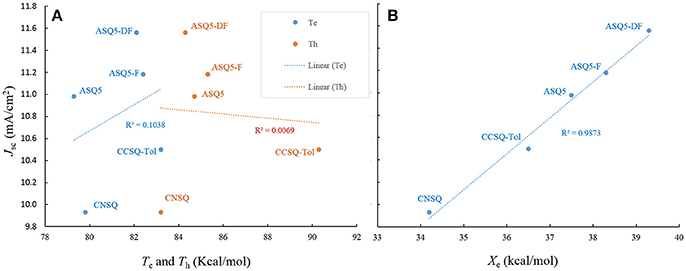
Figure 4. The Jsc as a function of electron/hole transferring ability (Te/Th) (A), and electron-rich extreme value on ESP surface Xe (B).
Analysis of VOC
It has been well accepted that a higher VOC in OSC usually correlates with higher HOMO level for donor material molecules or lower LUMO level for acceptor material molecules. Considering the same acceptor material molecule used in this work, it is not necessary to pay much attention to LUMO. First of all, the HOMO levels of CNSQ and CCSQ-Tol are calculated to be −6.14 and −6.12 eV, respectively. The calculated absolute values are larger than experimental data (see details in Table S4). From Table 1, we still can find their difference is consistent with VOC, i.e., a bit higher VOC (0.84 V) for the CNSQ-based OPV device than CCSQ-Tol (0.80 V) based one. On the other hand, considering much lower HOMO level of −6.30 eV for CCSQ and a little bit smaller value of VOC for CCSQ-Tol based device, it can be postulated that the backbone framework of CCSQ has positive contributions to VOC compared with CNSQ, while the toluene group contributes negatively to VOC.
To further understand the effects of electronic structures of all three molecules, total (TDOS), partial (PDOS) and overlap density of states between Fragment 1 and Fragment 2 (OPDOS) were depicted in Figure 5. In fact, suitable electronic structure of material is crucial to determine fundamental photophysical features of OSCs. The PDOS distribution clearly suggests that three fragments of CNSQ and CCSQ contribute similarly to their HOMOs of TDOS. However, Fragment 1 of CCSQ-Tol contributes much more to HOMO than Fragments 2 and 3. It further indicates that the toluene group has negative contribution to Voc. On the other hand, the overlap DOS between Fragments 1 and 2 show negative contributions to the frontier orbitals, which clearly indicates that those molecular orbitals are antibonding orbitals. To this point, we might say that the N-substitution on the position of the Fragment 1 of CCSQ could significantly affect the VOC.
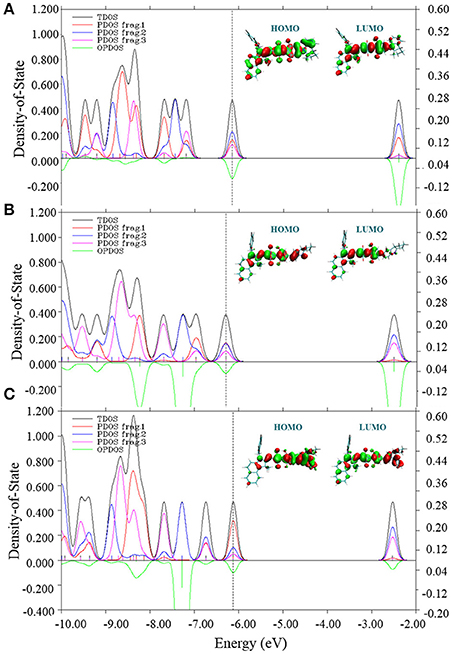
Figure 5. Total density of states (TDOS), partial DOS (PDOS) and overlap DOS (OPDOS) between fragment 1 and fragment 2 with HOMO and LUMO for (A) CNSQ, (B) CCSQ, and (C) CCSQ-Tol.
Analysis of Fill Factor
Gupta et al. (2010) have suggested that the FF depends on combination of some intrinsic device properties including the product of mobility and lifetime of the bulk material, thickness of the active-polymer layer and morphology of the cathode/polymer interface. Since all device parameters based on these donor materials are measured under the same conditions, the FF mainly relies on the charge carrier mobility of the electron (acceptor) and hole (donor) transport materials. It also improves with the presence of crystalline or relatively pure aggregate domains in the BHJ blend. In the system of ASQ5, we found that the FF value systematically increases with the fluorine substitution. Specifically, it is 0.51 for ASQ5, 0.52 for ASQ5-F, and 0.56 for ASQ5-D (Feng et al., 2015). These phenomena can be attributed to their special packing patterns between layers in crystals, e.g., Fragment 3 of ASQ5 interacts with Fragment 2 of the nearest neighbor layer, whereas Fragment 3 of ASQ5-DF has contacts with Fragment 1 of the nearest neighbor layer. To understand this issue, we then plotted their ESPs in Figure 6. The Fragment 1 is generally electron rich, except the position at the fluorine substitution, which appears to be electron deficient. It has been suggested by us (Yang et al., 2015) that this special electronic distribution should be the reason to cause different crystal packing patterns between ASQ5 and ASQ5-DF. To find out the direct relationship between molecular structure and the FF value, we summarized selected geometric parameters for three ASQ5 based molecules in Table 4. The biggest variation can be found is in ϕ1, which increases from 25.2 to 28.9° with fluorine substitution. Thus it could be deduced that the larger ϕ1 will benefit the complementary approaching between Fragments 1 and 3 of different molecules due to the substitution on N atom of Fragment 3. Meanwhile, if we recall the torsion angles of ϕ1 for CNSQ and CCSQ-Tol in Table 4, we can easily find that the latter molecule has a larger ϕ1 but smaller FF value. The reason is that much less contact between Fragment 3 and Fragment 2 of different molecules can take place after toluene substitution on CCSQ. Clearly, our results can strongly support that the morphology of material plays a key role in regulating FF value.
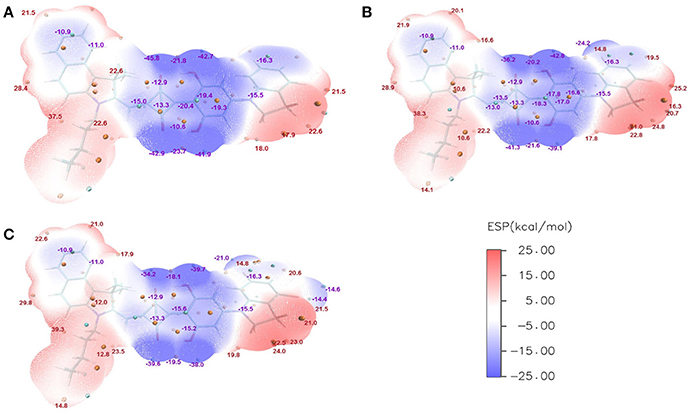
Figure 6. ESPs for (A) ASQ, (B) ASQ-F and (C) ASQ-DF at vertical excited state.
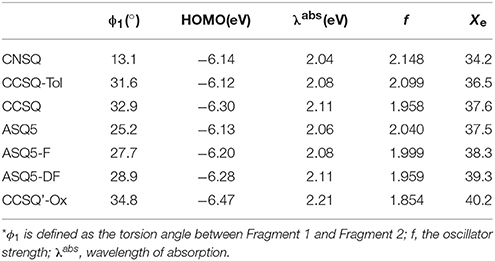
Table 4. Theoretical characterizations based on molecular design (the data about all the structures has been listed here to be compared)*.
Finally, we can summarize a basic principle to design a new donor molecule with D-A-D' electronic structure used in OSC devices here. The preferred molecules should have good intermolecular packing, bigger positive extremes on ESP at vertical excited state and lower HOMO energy level. With these basic rules, new molecules with better performances could be designed. Experimentally, CCSQ-Tol has better JSC but poorer VOC than CNSQ, which is consistent with our theoretical investigations presented above. However, the topological and substitution analyses do show that CCSQ has lower energy level of HOMO than CNSQ. We can simply deduce that the linking topology like CCSQ is beneficial to improve VOC, but the toluene substitution acts on the contrary.
Further, the design of new material molecule should consider how to increase FF, which can be inspired by the case of ASQ5-DF. The fluorine substitution on the electron-rich region of asymmetric squaraine makes the atom at the linking position more electron deficiently, thus a better packing style between Fragment 1 and Fragment 3 can thereby be achieved. To match the packing pattern like ASQ5-DF, the newly designed CCSQ based molecule was developed with O group replaced by N group on Fragment 1 on account of its strong electron-deficient property. In addition, the group of -CH2Ph was replaced with the group of -C4H9 on Fragment 3. This newly constructed molecule, namely CCSQ'-Ox, is depicted in Figure 7. According to Table 4, relatively large ϕ1 of 34.8°For CCSQ'-Ox can be found, which has been considered to be a sign for the good intermolecular stacking between Fragments 1 and 3. Meanwhile, the lowest HOMO at −6.47 eV for CCSQ'-Ox also means the best VOC among molecules we have consulted in this work. Moreover, the biggest positive extrema of 40.2 kcal/mol on ESP for CCSQ'-Ox can be seen from Table 4. It is thus expected a larger JSC. Theoretically, the CCSQ'-Ox seems to include all three aspects that affect the final PCE of BHJ OSC, and thus represents one possible candidate to develop a new BHJ OSC with better properties. Experimental characterizations currently are underway in our lab.
Figure 7. Newly designed molecule.
Conclusions
It has been well accepted that experimental and theoretical groups can work together to enhance the R&D speed of new materials. Our objective is to establish the reliable connection between intrinsic molecular structure and corresponding device performances of OSCs. For the donor molecules with D-A-D' framework, we have carried out systematically experimental and theoretical characterizations for a series of asymmetric squaraines.
Based on the previous work, we have designed three new molecules, namely CNSQ, CCSQ and CCSQ-Tol. Considering the stability of materials and devices, only features for CNSQ and CCSQ-Tol based devices were measured and characterized. The device based on CNSQ has higher PCE than that based on CCSQ-Tol. Combined experimental characterizations and theoretical calculations suggest that the improvement of the device performance cannot just depend on one single factor. Specifically, we can confirm that simple adjustment of the overall topology for the donor molecules might not work for improving JSC, we have to focus on regulating their intrinsic electronic structure properties. A linear correlation between JSC and the positive extrema (Xe) on ESP can be established. In summary, our simulations indicate that a good intermolecular stacking interaction is required for FF, the bigger positive extrema on ESP at vertical excited state is desired for improving JSC, and lower HOMO energy level is for better VOC.
Finally, based on our first principle computations, one new molecule, namely CCSQ'-Ox, was predicted with better values of FF, VOC, and JSC, which might have promising PCE. Of course, extensive experimental investigations should be required to confirm our prediction. It is also our hope to encourage improving device performance for new BHJ OSC with the help of theoretical simulations.
Author Contributions
JC, YH, and DX contributed conception and design of the study. QL, HL, ZZ, and ZL organized the database. JC performed the statistical analysis. JC wrote the first draft of the manuscript. JC, QL, YH, and DX wrote sections of the manuscript. All authors contributed to manuscript revision, read and approved the submitted version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was funded by the National Key R&D Program (No. 2016YFB0700801) and by the National Natural Science Foundation of China (nos. 21473117). This work was also supported by Post-doctoral Foundation of Sichuan University (No.0020307602031). Some of results described in this paper were obtained on the Supercomputing Center of Chinese Academy of Science.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2018.00200/full#supplementary-material
References
Alessandri, R., Uusitalo, J. J., de Vries, A. H., Havenith, R. W. A., and Marrink, S. J. (2017). Bulk heterojunction morphologies with atomistic resolution from coarse-grain solvent evaporation simulations. J. Am. Chem. Soc. 139, 3697–3705. doi: 10.1021/jacs.6b11717
Bérubé, N., Gosselin, V., Gaudreau, J., and Côté, M. (2013). Designing Polymers for photovoltaic applications using ab initio calculations. J. Phys. Chem. C 117, 7964–7972. doi: 10.1021/jp309800f
Brückner, C., and Engels, B. (2016). A theoretical description of charge reorganization energies in molecular organic P-type semiconductors. J. Comput. Chem. 37, 1335–1344. doi: 10.1002/jcc.24325
Brückner, C., Würthner, F., Meerholz, K., and Engels, B. (2017). Structure–property relationships from atomistic multiscale simulations of the relevant processes in organic solar cells. I. thermodynamic aspects. J. Phys. Chem. C 121, 4–25. doi: 10.1021/acs.jpcc.6b06755
Chen, G., Sasabe, H., Igarashi, T., Hong, Z., and Kido, J. (2015). Squaraine dyes for organic photovoltaic cells. J. Mater. Chem. A 3, 14517–14534. doi: 10.1039/C5TA01879J
Collins, S. D., Ran, N. A., Heiber, M. C., and Nguyen, T. Q. (2017). Small is powerful: recent progress in solution-processed small molecule solar cells. Adv. Energy Mater. 7:1602242. doi: 10.1002/aenm.201602242
Fan, H., and Zhu, X. (2015). Development of small-molecule materials for high-performance organic solar cells. Sci. China Chem. 58, 922–936. doi: 10.1007/s11426-015-5418-6
Feng, L., Liu, Z.-M., Hou, J., Lv, X., Ning, J., Ge, G.-B., et al. (2015). A highly selective fluorescent ESIPT probe for the detection of Human carboxylesterase 2 and its biological applications. Biosens. Bioelectron. 65, 9–15. doi: 10.1016/j.bios.2014.10.002
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2009). Gaussian 09. Wallingford, CT: Gaussian, Inc.
Gsänger, M., Bialas, D., Huang, L., Stolte, M., and Würthner, F. (2016). Organic semiconductors based on dyes and color pigments. Adv. Mater. Weinheim. 28, 3615–3645. doi: 10.1002/adma.201505440
Gupta, D., Mukhopadhyay, S., and Narayan, K. S. (2010). Fill factor in organic solar cells. Sol. Energ. Mat. Sol. C. 94, 1309–1313. doi: 10.1016/j.solmat.2008.06.001
Huang, Y., Kramer, E. J., Heeger, A. J., and Bazan, G. C. (2014). Bulk heterojunction solar cells: morphology and performance relationships. Chem. Rev. 114, 7006–7043. doi: 10.1021/cr400353v
Humphrey, W., Dalke, A., and Schulten, K. (1996). VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38. doi: 10.1016/0263-7855(96)00018-5
Jao, M.-H., Liao, H.-C., and Su, W.-F. (2016). Achieving a high fill factor for organic solar cells. J. Mater. Chem. A 4, 5784–5801. doi: 10.1039/C6TA00126B
Kan, B., Li, M., Zhang, Q., Liu, F., Wan, X., Wang, Y., et al. (2015). A series of simple oligomer-like small molecules based on oligothiophenes for solution-processed solar cells with high efficiency. J. Am. Chem. Soc. 137, 3886–3893. doi: 10.1021/jacs.5b00305
Kan, B., Zhang, Q., Li, M., Wan, X., Ni, W., Long, G., et al. (2014). Solution-processed organic solar cells based on dialkylthiol-substituted benzodithiophene unit with efficiency near 10%. J. Am. Chem. Soc. 136, 15529–15532. doi: 10.1021/ja509703k
Lange, I., Kniepert, J., Pingel, P., Dumsch, I., Allard, S., Janietz, S., et al. (2013). Correlation between the open circuit voltage and the energetics of organic bulk heterojunction solar cells. J. Phys. Chem. Lett. 4, 3865–3871. doi: 10.1021/jz401971e
Li, G., Liang, M., Wang, H., Sun, Z., Wang, L., Wang, Z., et al. (2013). Significant enhancement of open-circuit voltage in indoline-based dye-sensitized solar cells via retarding charge recombination. Chem. Mater. 25, 1713–1722. doi: 10.1021/cm400196w
Li, N., Baran, D., Forberich, K., Machui, F., Ameri, T., Turbiez, M., et al. (2013). Towards 15% energy conversion efficiency: a systematic study of the solution-processed organic tandem solar cells based on commercially available materials. Energ. Environ. Sci. 6, 3407–3413. doi: 10.1039/C3EE42307G
Li, Y. (2012). Molecular design of photovoltaic materials for polymer solar cells: toward suitable electronic energy levels and broad absorption. Accounts Chem. Res. 45, 723–733. doi: 10.1021/ar2002446
Li, Y., Guo, Q., Li, Z., Pei, J., and Tian, W. (2010). Solution processable D-A small molecules for bulk-heterojunction solar cells. Energ. Environ. Sci. 3, 1427–1436. doi: 10.1039/c003946b
Lu, T., and Chen, F. (2012). Quantitative analysis of molecular surface based on improved Marching Tetrahedra algorithm. J. Mol. Graph. Model. 38, 314–323. doi: 10.1016/j.jmgm.2012.07.004
Mennucci, B. (2015). Modeling absorption and fluorescence solvatochromism with QM/Classical approaches. Int. J. Quantum Chem. 115, 1202–1208. doi: 10.1002/qua.24889
Mulligan, C. J., Wilson, M., Bryant, G., Vaughan, B., Zhou, X., Belcher, W. J., et al. (2014). A projection of commercial-scale organic photovoltaic module costs. Sol. Energ. Mat. Sol. C. 120, 9–17. doi: 10.1016/j.solmat.2013.07.041
Ni, W., Wan, X., Li, M., Wang, Y., and Chen, Y. (2015). A-D-A small molecules for solution-processed organic photovoltaic cells. Chem. Commun. 51, 4936–4950. doi: 10.1039/C4CC09758K
Pandey, S. S., Watanabe, R., Fujikawa, N., Ogomi, Y., Yamaguchi, Y., and Hayase, S. (2011). “Fine tuning the structure of unsymmetrical squaraine dyes towards the development of efficient dye-sensitized solar cells”, in SPIE Solar Energy + Technology (San Diego, CA: SPIE, 10).
Park, S. H., Roy, A., Beaupre, S., Cho, S., Coates, N., Moon, J. S., et al. (2009). Bulk heterojunction solar cells with internal quantum efficiency approaching 100%. Nat Photon 3, 297–302. doi: 10.1038/nphoton.2009.69
Peet, J., Kim, J. Y., Coates, N. E., Ma, W. L., Moses, D., Heeger, A. J., et al. (2007). Efficiency enhancement in low-bandgap polymer solar cells by processing with alkane dithiols. Nat. Mater. 6, 497–500. doi: 10.1038/nmat1928
Pommerehne, J., Vestweber, H., Guss, W., Mahrt, R. F., Bassler, H., Porsch, M., et al. (1995). Effcient 2-layer LEDs on a polymer blend basis. Adv. Mater. Weinheim. 7, 551–554. doi: 10.1002/adma.19950070608
Scalmani, G., Frisch, M. J., Mennucci, B., Tomasi, J., Cammi, R., and Barone, V. (2006). Geometries and properties of excited states in the gas phase and in solution: theory and application of a time-dependent density functional theory polarizable continuum model. J. Chem. Phys. 124:094107. doi: 10.1063/1.2173258
Sweetnam, S., Graham, K. R., Ngongang Ndjawa, G. O., Heumüller, T., Bartelt, J. A., Burke, T. M., et al. (2014). Characterization of the polymer energy landscape in polymer:fullerene bulk heterojunctions with pure and mixed phases. J. Am. Chem. Soc. 136, 14078–14088. doi: 10.1021/ja505463r
Tomasi, J., Mennucci, B., and Cammi, R. (2005). Quantum mechanical continuum solvation models. Chem. Rev. 105, 2999–3094. doi: 10.1021/cr9904009
van Duren, J. K. J., Yang, X., Loos, J., Bulle-Lieuwma, C. W. T., Sieval, A. B., Hummelen, J.C., et al. (2004). Relating the morphology of poly(p-Phenylene vinylene)/methanofullerene blends to solar-cell performance. Adv. Funct. Mater. 14, 425–434. doi: 10.1002/adfm.200305049
Vandewal, K. (2016). Interfacial charge transfer states in condensed phase systems. Annu. Rev. Phys. Chem. 67, 113–133. doi: 10.1146/annurev-physchem-040215-112144
Volpi, R., Kottravel, S., Nørby, M. S., Stafström, S., and Linares, M. (2016a). Effect of polarization on the mobility of C 60: a kinetic monte carlo study. J. Chem. Theory Comput. 12, 812–824. doi: 10.1021/acs.jctc.5b00975
Volpi, R., Nassau, R., Nørby, M. S., and Linares, M. (2016b). Theoretical study of the charge-transfer state separation within marcus theory: the C 60 -anthracene case study. ACS Appl. Mater. Interfaces 8, 24722–24736. doi: 10.1021/acsami.6b06645
Wei, G., Xiao, X., Wang, S., Zimmerman, J. D., Sun, K., Diev, V. V., et al. (2011). Arylamine-based squaraine donors for use in organic solar cells. Nano Lett. 11, 4261–4264. doi: 10.1021/nl2022515
Xiao, Z., and Yan, Y. (2017). Progress in theoretical study of metal halide perovskite solar cell materials. Adv. Energy Mater. 7:1701136. doi: 10.1002/aenm.201701136
Yang, D., Yang, L., Huang, Y., Jiao, Y., Igarashi, T., Chen, Y., et al. (2015). Asymmetrical squaraines bearing fluorine-substituted indoline moieties for high-performance solution-processed small-molecule organic solar cells. ACS Appl. Mater. Interfaces 7, 13675–13684. doi: 10.1021/acsami.5b03558
Yang, D., Yang, Q., Yang, L., Luo, Q., Chen, Y., Zhu, Y., et al. (2014). A low bandgap asymmetrical squaraine for high-performance solution-processed small molecule organic solar cells. Chem. Commun. 50, 9346–9348. doi: 10.1039/C4CC03831B
Yang, J., Ganesan, P., Teuscher, J., Moehl, T., Kim, Y. J., Yi, C., et al. (2014). Influence of the donor size in D-π-A organic dyes for dye-sensitized solar cells. J. Am. Chem. Soc. 136, 5722–5730. doi: 10.1021/ja500280r
Yang, L., Yang, Q., Yang, D., Luo, Q., Zhu, Y., Huang, Y., et al. (2014). Marked effects of indolyl vs. indolinyl substituent on solid-state structure, carrier mobility and photovoltaic efficiency of asymmetrical squaraine dyes. J. Mater. Chem. A 2, 18313–18321. doi: 10.1039/C4TA03859B
Zhang, J., Zhu, L., and Wei, Z. (2017). Toward over 15% power conversion efficiency for organic solar cells: current status and perspectives. Small Methods 1:1700258-n/a. doi: 10.1002/smtd.201700258
Keywords: organic solar cell, squaraine, DFT, D-A-D' framework, open-circuit voltage, short-circuit current density, fill factor
Citation: Chen J, Liu Q, Li H, Zhao Z, Lu Z, Huang Y and Xu D (2018) Density Functional Theory Investigations of D-A-D' Structural Molecules as Donor Materials in Organic Solar Cell. Front. Chem. 6:200. doi: 10.3389/fchem.2018.00200
Received: 07 April 2018; Accepted: 15 May 2018;
Published: 04 June 2018.
Edited by:
Yong Wang, Lanzhou Institute of Chemical Physics (CAS), ChinaReviewed by:
Zhigang Sun, Dalian Institute of Chemical Physics (CAS), ChinaMinghui Yang, Wuhan Institute of Physics and Mathematics (CAS), China
Copyright © 2018 Chen, Liu, Li, Zhao, Lu, Huang and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dingguo Xu, ZGd4dUBzY3UuZWR1LmNu
Yan Huang, aHVhbmd5YW5Ac2N1LmVkdS5jbg==