 Akina Yoshizawa
Akina Yoshizawa Antonio Feula
Antonio Feula Andrew G. Leach
Andrew G. Leach Louise Male3
Louise Male3 John S. Fossey
John S. Fossey- 1School of Chemistry, University of Birmingham, Birmingham, United Kingdom
- 2School of Pharmacy and Biomolecular Sciences, Liverpool John Moores University, Liverpool, United Kingdom
- 3X-Ray Crystallography Facility, School of Chemistry, University of Birmingham, Birmingham, United Kingdom
Seven N,N'-palladium(II) chloride complexes, one N,N'-palladium(II) acetate complex of 2,4-cis-azetidines where prepared and analyzed by single crystal XRD. Two platinum(II) chloride N,N'-complexes of 2,4-cis-azetidines where prepared and analyzed by single crystal XRD. Computational analysis and determination of the %Vbur was examined conducted. A CNN' metallocyclic complex was prepared by oxidative addition of palladium(0) to an ortho bromo 2,4-cis-disubstituted azetidine and its crystal structure displays a slightly pyramidalized metal-ligand orientation.
Introduction
Complexes of group 10 metals coordinated by N,N'-ligands have found wide use from therapeutics (Graham et al., 2004) through to catalysis (Fossey et al., 2005; Saito and Fu, 2007; Binder et al., 2012; Martínez-Olid et al., 2014; Álvarez-Casao et al., 2015). In our research, through studying the synthesis and cyclisation of homoallylamine derivatives (Fossey et al.; Rixe et al., 1996; Jamieson and Lippard, 1999; Feula et al., 2010, 2013; Feula and Fossey, 2013; Hama Salih et al., 2015), the potential for use of the cis-aminoazetidine products as ligands emerged, and their use in asymmetric copper-catalyzed Henry reactions was developed and reported elsewhere (Yoshizawa et al., 2018a,b,c). Group 10 complexes of azetidine derivatives have been reported previously (Voureka et al., 1996; Keller et al., 2005; Lee et al., 2008, 2009a,b; Choi et al., 2011), and during our aforementioned studies various attempts to form and isolate racemic group 10 metal complexes have been made. Successful attempts to prepare crystals of metal complexes suitable for single crystal X-ray diffraction structure determination are reported herein, along with crystal structures of unexpected by-products, a complex formed from a pyrrolidine ligand (an isomer of the azetidines of primary interest) as well as an observation pertaining to diastereoisomer differentiation upon complexation and sample preparation under slightly different conditions. In most cases small amounts of (or even just single) crystals were obtained sometimes precluding full characterisation, however in some cases elemental analyses are reported. The structural observations facilitated by surveying the obtained crystal structures have the potential to influence catalysis design. Thus, whilst this report primarily serves as a crystal structure report the interpretation and understanding garnered is gathered here with a view to informing studies across organic and inorganic synthesis.
Results and Discussion
Our previously documented interest in the synthesis of 2,4-cis-disubstituted azetidines (Fossey et al.; Feula et al., 2010, 2013; Feula and Fossey, 2013; Hama Salih et al., 2015), and our recent use of the same family of azetidines as chiral ligands in asymmetric copper-catalyzed Henry reactions afforded us the opportunity to probe the complexation of these ligands and related compounds with various metal sources, in this report examples leading to samples suitable for analysis by X-ray single crystal diffraction studies are collected and compared. A previously revealed platinum complex (4a) is re-reported here since it is compared directly with a diastereomeric complex (4b) of the same ligand, and the racemic ligands used in this study have all been previously reported in the aforementioned publications arising from this programme of research1.
Treatment of a series of amino-substituted azetidines with various palladium(II) chloride derivatives afforded crystals suitable for analysis by single crystal XRD analysis in seven cases (Scheme 1). Racemic ligands 1a to g were converted to the corresponding square planar palladium chloride complexes 2a to g, by combination of various palladium(II) chloride sources and ligands in methanol and heating at reflux until ligand had been consumed by TLC analysis, see Figure 1 (and Supplementary Material) for representations of the palladium chloride complexes thus obtained. Treatment of ligand 1a with palladium(II) acetate in dichloromethane afforded mono-nuclear palladium complex 3, bearing a coordinated water molecule and displaying hydrogen bonding between the coordinated water molecule and the two acetate anions (coordinated to palladium and bridging ligand NH…H2O respectively), see Figure 2, for a representation of the crystal structure obtained for compound 3.
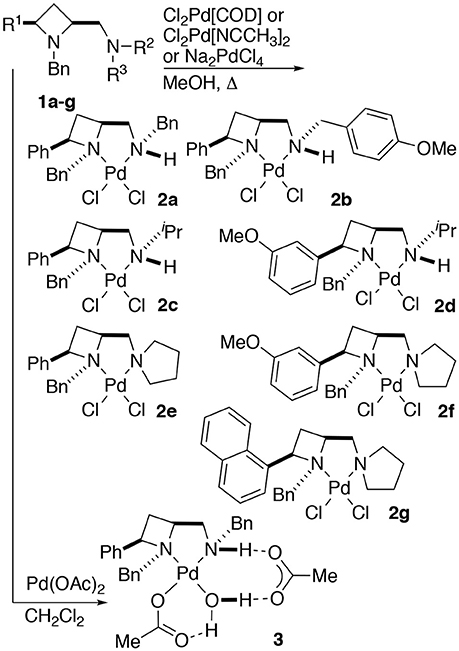
Scheme 1. General protocol for the preparation of identified palladium(II) chloride (2a–g) and acetate (3) complexes of 2,4-cis-disubstituted azetidine N,N'-ligands.
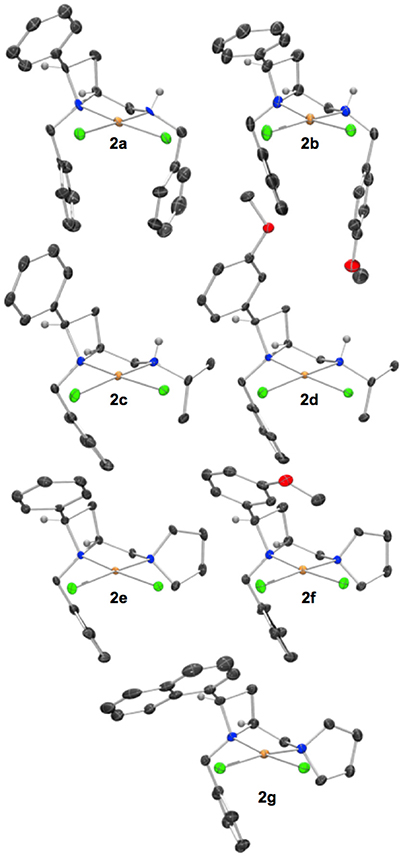
Figure 1. Representations of one molecule from the unit cells of the single crystal XRD structures of racemic complexes 2a-g. Elipsoids plot at 50% probability, and rendered using Ortep-III for Windows and PovRay v3.7. For selected bond lengths, angles and torsions see ESI.
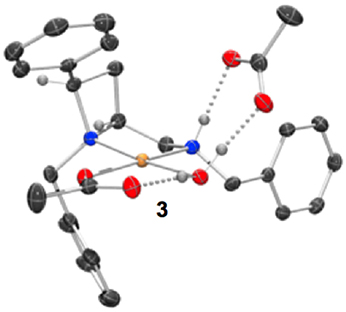
Figure 2. Representation of one molecule from the unit cell of the single crystal XRD structures of racemic complex 3. Elipsoids plot at 50% probability, and rendered using Ortep-III for Windows and PovRay v3.7.
Reactions were routinely conducted under anhydrous, oxygen-free conditions, but materials obtained from reactions were handled in air and no special precautions to exclude moisture or oxygen were taken. Crystals of palladium(II) chloride complexes (Figure 1) suitable for single crystal XRD analysis were typically obtained by crystallization from acetonitrile or acetonitrile/diethyl ether mixtures, pre-purification by flash chromatography or filtration through silica in dichloromethane/methanol mobile phase was sometimes advantageous. The XRD structures typically displayed single diastereoisomers of products as racemic mixtures. The figures of the main text show one molecule from the unit cell, displayed such that the stereochemistry of those drawn in the main text match permitting ready comparison of steric effects among the ligands compared.
Single crystals of complex 3 (Figure 2) were obtained by slow diffusion of hexane directly into the dichloromethane solvated reaction mixture.
The platinum(II) chloride complex 4a, formed by heating ligand 1a at reflux in methanol with potassium tetrachloroplatinate, was previously reported by us. We were intrigued when we later repeated the protocol, with ethanol as solvent, instead of methanol, and extended the reaction time somewhat (from 16 to 72 h), to find an alternative diastereoisomer, racemic 4b, was formed, differing only by the coordinational chirality of the amine-part's nitrogen, both complexes are discussed in this report (Scheme 2). Gagné and co-workers previous discussed persistent nitrogen chirality in square planar diamine complexes of palladium(II) in their seminal work on the topic (Pelz and Gagné, 2003; Pelz et al., 2004), and their work should be consulted for discussion about complexes displaying chirality only as a result of nitrogen's stereogenicity as a result of coordination to a metal.
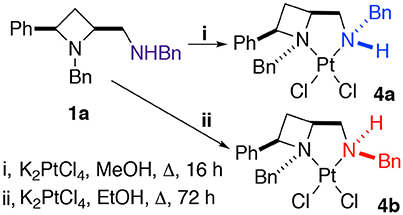
Scheme 2. The protocol used to obtain crystals suitable for single crystal XRD analysis of N-diastereomeric racemic platinum(II) chloride complexes 4a and 4b.
Crystallization from acetonitrile/diethyl ether or slow evaporation of an acetonitrile solution led to the isolation of crystals suitable for single crystal XRD analysis of 4a and 4b respectively, Figure 3.
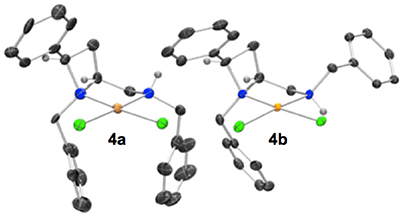
Figure 3. Representation of one molecule from each unit cell of racemic 4a (left) and 4b (right). The diastereoisomers drawn differ by inversion of the amine-centered nitrogen stereogenicity. The two diastereoisomers depicted are 4a N(azet)RN(amin)R (left) and 4b N(azet)RN(amin)R (right). Elipsoids plot at 50% probability, and rendered using Ortep-III for Windows and PovRay v3.7. For selected bond lengths, angles and torsions see ESI.
Noting that platinum complex 4a was formed under reflux in methanol (boiling point 64.7 °C) and the N-diastereomeric complex 4b at reflux in ethanol (boiling point 78.4 °C) and over a longer time, it was reasoned that 4a may be a kinetic product whilst 4b the thermodynamic. If this were the case, there may be implications for the future use of such complexes in catalysis. Computational analysis revealed this assumption to be incorrect, importantly demonstrating that XRD analysis, especially in the case of this report where, in some cases, very few crystals were isolated from the bulk. Nevertheless, the computational analysis does demonstrate the constrained nature of the cis-azetidine architecture to be important in determining the underlying steric parameters around the coordination environment. The square planar N-diastereomeric complexes 4a and 4b were studied with density functional theory in order to establish the intrinsic thermodynamic preference. Calculations employed the M06-2X functional with the 6-31G* basis set for all atoms apart from platinum atom which was treated with the LANL double zeta basis set and pseudopotential. Optimized geometries were subject to single point energy evaluations to evaluate the effects of solvation. Three conformations of the N-benzyl sidechain were studied, and energies of the lowest energy conformation reported. These calculations revealed that the lowest energy structure is that of 4a. In these calculations, the preferred conformation of the sidechain is an extended one, Figure 4 (left). The alternative conformation [Figure 4 (center)], resembling that observed in the crystal structures is close in energy (0.8 kcal/mol) and so is readily accessible. The preferred conformation for 4b is 1.6 kcal/mol higher in energy than the lowest energy conformation of 4a, and its geometry as shown in Figure 4 (right) resembles the crystal structure of 4b in Figure 3. The energy difference between 4a and 4b narrows slightly (by 0.01 kcal/mol) on changing from methanol solvation to ethanol. The origin of the preference for 4a is likely the close approach required by the N-benzyl group to the hydrogen at the 2-position of the azetidine; this rigid system does not permit this close contact to be avoided. A table comparing and contrasting both crystallographically and computationally determined (minima) selected bond lengths, angles and torsions of 4a and 4b is included in the Supplementary Material. Good agreement between these data confirms the computed coordination environment matches that experimentally observed.
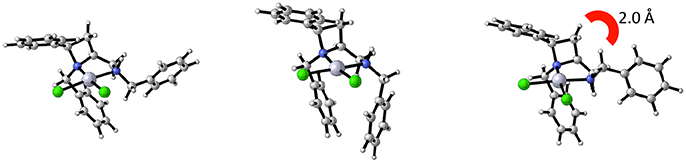
Figure 4. Calculated structural minima for 4a (left) and 4b (right), 4b being 1.6 kcal/mol higher in relative free energy then 4a. A crystal-structure-like accessible (0.8 kcal/mol higher in relative free energy) minima for 4a depicted in the center.
This computational analysis reveals that at equilibrium a mixture of 4a and 4b would consist mostly of 4a. Yet in our slightly divergent preparation and perhaps more importantly slow crystallization conditions is was possible to favor crystalline deposition of the minor component (diffusion of diethyl ether into, vs. slow evaporation of acetonitrile).
Closer inspection of the wider sphere about the metal atoms in the crystal structures shows that compound 4b places the azetidine ring N-benzyl group closer to the platinum, potentially due to the space created by placing the exocyclic N-benzyl group on the opposite face of the square plane. This may be judged by a shortest aryl-H…Pt distance of 2.8806 (3) Å for 4b (Figure 5) whereas aryl-H…Pt distances in 4a are ≥3.00 Å (Hambley, 1998).
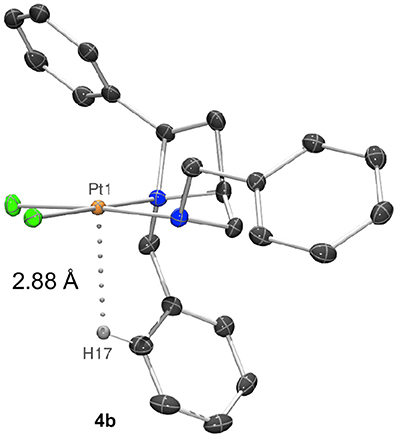
Figure 5. Representation of a molecule of 4b showing a 2.8806 (3) Å distance between and aryl-H (H17) and the platinum(II) center (Pt1).
Whilst computationally reasoned to be primarily due to solid state, crystallization condition-dependent, phenomena we were interested to probe the consequences of the nitrogen atom stereogenicity upon the asymmetric, or more precisely the differing topological, properties of the cleft described by ligand 1a in complexes 4a and 4b, as such the buried volume (%Vbur) described by the ligand geometries was calculated. Inspired by the work of Nolan (Clavier and Nolan, 2010), Cavallo (Poater et al., 2009) and their co-workers the SambVca 2.0 tool was employed (Falivene et al., 2016), topological maps generated and their features compared2, to describe the steric constraints imposed about the exchangeable (halide) sites. PDB files containing one molecule of the single crystal X-ray diffraction-determined unit cell were uploaded to the SambVca 2.0 web-portal. The x axis being described as parallel with the N-N orientation of the square plane about the metal; the y axis lies perpendicular to the square plane of the metals' coordination; and the z axis bisects the both the two nitrogens and the two chlorides. The chlorides were removed in the web-portal and the %Vbur calculation run under otherwise default settings.
Viewed from the front (down the z-axis as depicted in Figure 6) the diastereoisomeric complexes show similar steric constraints on the left upper and lower (NW and SW) quadrants. As expected the right side (Easterly) differs markedly between the diastereomeric complexes 4a and 4b, essentially leaving the NE and SE quadrants sterically unencumbered respectively. Thus, demonstrating the significance of nitrogen's coordinational chirality in defining the asymmetric environment a chiral ligand describes around a metal. In principle it may be reasoned that were the formed diastereomeric complexes stable and isolated as single enantiomers, that the same sense of carbon-centered stereogenicity could deliver opposite enantiomers of products in catalysis as a result of deviation in nitrogen-centered stereogenicity, a phenomenon warranting future further investigation. In order to permit direct comparison, steric maps of the percentage buried volume of all complexes of this report, generated by the SambVca 2.0 online tool, are included in the Supporting Information of this manuscript.
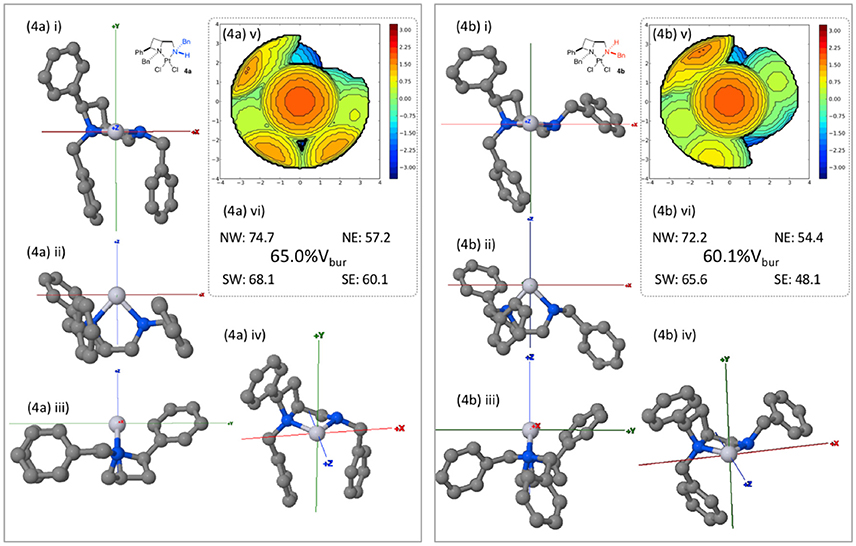
Figure 6. Burried volume analysis of ligand geometries in complexes 4a and 4b, conducted using online tool SambVca 2.0. (i-iv) Various orientations of the considered structures; (v) SambVca-generated %Vbur map ; and (vi) %Vbur and quadrantwise contributions.
Having previously accessed ortho bromo ligand 1i and having previously investigated oxidative addition of 10 metals to form NCN pincer complexes (Fossey and Richards, 2004; Fossey et al., 2007), it was reasoned that ligand 1i may form a metallocyclic complex upon reaction with palladium(0), Scheme 3. To our delight a small amount of a CNN' metallocyclic complex (5) was identified, see Figure 7 for a representation of the crystal structure.
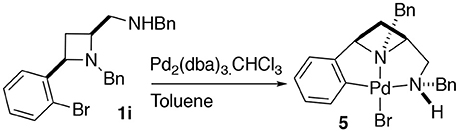
Scheme 3. Oxidative addition of palladium(0) to 1i to generate a palladium(II) CNN' metallocyclic complex.
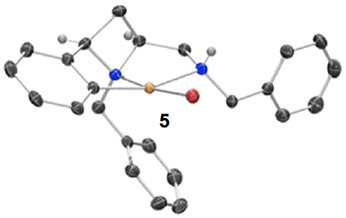
Figure 7. Representation of one molecule from the unit cell of the single crystal XRD structure of racemic complex 5. Elipsoids plot at 50% probability, and rendered using Ortep-III for Windows and PovRay v3.7.
A unique opportunity provided by the 2,4-cis-disubstituted azetidine geometry is that a tridentate complexation of palladium is possible and concave ligand architecture is able to asymmetrically envelop the metal, and distort the metal coordination geometry away from an ideal square plane. The trans bond angles C-Pd-N and N-Pd-Br deviate from 180°, being 160.8° and 173.8° respectively, leading to slight pyramidalisation of the palladium(II) center (Figure 7).
When a salt of an amino azetidine ligand (6) was exposed to sodium tetrachloropalladate and heated to reflux in methanol or ethanol ring-opened complexes (7a and b) were obtained, Scheme 4. Very few crystals were formed precluding analysis other than by single crystal XRD analysis alongside intractable material and it is not yet possible to say if all the ligand was consumed in this manner (Figure 8). But since prolonged heating of free-base ligands in methanol or ethanol with a metal source did not furnish any detectable ring-opened material it was reasoned that the small amount of crystals obtained resulted from acid promoted ring opening followed by coordination to palladium rather than the complex being unstable to the reaction conditions. This could be the subject of further future study, especially if the accessed ring opened complexes offer advantages in catalysis or other applications. The %Vbur of complexes 7a and b are also calculated, and details are provided in the Supporting Information to allow for comparison amongst the complexes detailed in this manuscript.
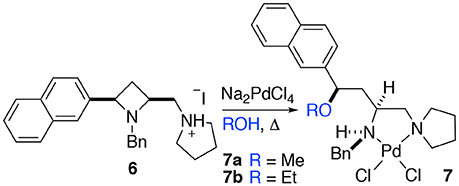
Scheme 4. Methyl and ethyl ether complexes 7a and 7b resulting from azetidine ring opening of salt 6 and complexation to palladium(II) identified by single crystal XRD investigation.
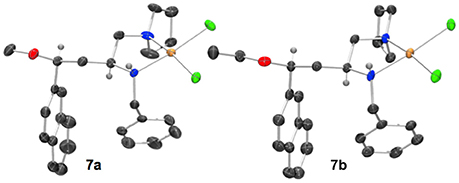
Figure 8. Representation of one molecule from the unit cells of the single crystal XRD structures of racemic complex 7a and 7b. Elipsoids plot at 50% probability, and rendered using Ortep-III for Windows and PovRay v3.7.
The crystal structures of racemic complexes 7a and 7b display typical NN' binding modes in these palladium(II) chloride complexes, the stereogenic center on the carbon backbone imposes a trans stereochemical relationship upon the coordinationally chiral neighboring nitrogen atom (Fossey et al., 2005, 2008). The bulky groups point away from each other, and there is no evidence, in the solid state, of ancillary ether oxygen coordination. The crystals analyzed are single diastereoisomer racemates, meaning the ring opening step appears to have proceeded with high stereochemical fidelity, i.e., with clean inversion upon O-attack on the aryl substitute azetidinium ring stereogenic carbon (Gaertner, 1968; Leonard and Durand, 1968).
The synthesis of 2,4-cis-disubstituted azetidines by an iodocyclisation protocol used in the synthesis of azetidines 1a-g can give rise to pyrrolidine by-products (see previous reports for details). In the case of the synthesis of complex 2d a minor pyrrolidine impurity led to the formation and initial isolation of a complex derived from 2,4-cis-disubstituted pyrrolidine 8.
Whilst only a single crystal of the formed complex 9 was isolated and analyzed Scheme 5, as the minor component of the mixture, the complex itself displays some noteworthy features (Figure 9). Firstly, that even as a minor component, complex 9 readily formed a prominent crystal bodes well for the future targeted synthesis and isolation of it and its analogs. Secondly the cis relative stereochemistry across the 2,4-positions confers a geometry similar to, but subtly different from, azetidine analogs (compare 2d with 9). Importantly, in our previous work we showed how we can access both diastereoisomers of 8-like pyrrolidines and a future planned programme of study will look in detail at the potential of 2,4-cis-pyrrolidines as scaffolds for catalyst construction (Feula et al., 2010, 2013).
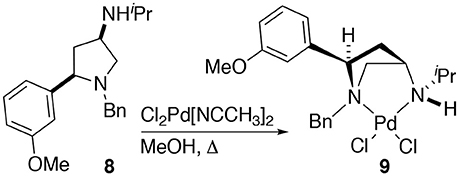
Scheme 5. The formation of complex 9. Namely through complexation of a minor impurity (8), leading to a crystalline material, one crystal, of a prominent crystal upon attempted crystallization, alter leading to 2d after removal of the obtain crystal of 9.
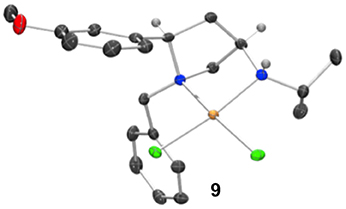
Figure 9. Representation of one molecule from the unit cell of the single crystal XRD structure of racemic complex 9. Elipsoids plot at 50% probability, and rendered using Ortep-III for Windows and PovRay v3.7.
Since ligands 1d and 8 are accessible form the same source, depending on divergent synthesis conditions (Feula et al., 2013), complexes 2d and 9 were compared using the SambVca 2.0 online tool, Figure 10.
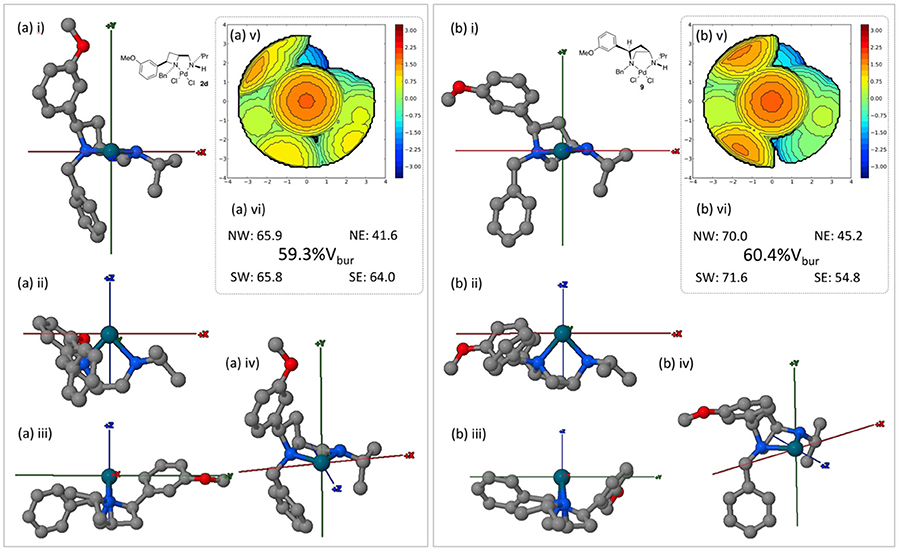
Figure 10. Burried volume analysis of ligand geometries in complexes 2d and 9, conducted using online tool SambVca 2.0. (i-iv) Various orientations of the considered structures; (v) SambVca-generated %Vbur map ; and (vi) %Vbur and quadrantwise contributions.
Under the same inspection protocols both the azetidine (2d) and pyrrolidine (9) complexes offer a similar level of steric constraint about their respective palladium(II) metal centers. However, the subtle impact of employing isomeric 4- and 5-membered heterocyclic ligands (1d and 8 respectively) can be witnessed in the buried volume differences between western and eastern hemispheres (as drawn). The average difference between the (western) left vs. (eastern) right increases from 13 to 21% across 2d and 9 respectively. Thus, were single enantiomer asymmetric catalyst to be developed it may be expected that 9 offers greater steric discrimination across the described x axis. Whilst beyond the scope of this crystal structure report the displayed steric divergence between these isomeric ligands warrants further attention in an onward programme of research.
Conclusions
Seven N,N'-palladium(II) chloride complexes, one N,N'-palladium(II) acetate complex of 2,4-cis-azetidines where prepared and analyzed by single crystal XRD. The racemic ligands adopted a single diastereoismer form upon coordination to palladium the same chirality at nitrogen. In the palladium(II) acetate complex a coordinated water molecule and H-bonding acetates formed the identified complex. Two platinum(II) chloride N,N'-complexes of 2,4-cis-azetidines where prepared and analyzed by single crystal XRD, and two diastereoisomers were generated upon amine coordination to platinum (under different preparation conditions). Computational analysis revealed which diastereoisomer was more stable and provided a rationale for why this is the case, and the %Vbur described by the diastereomeric coordination geometry was examined. A CNN' metallocyclic complex was prepared by oxidative addition of palladium(0) to an ortho bromo 2,4-cis-disubstituted azetidine and its crystal structure displays a slightly pyramidalized metal-ligand orientation. Ligand salts were not suitable for the synthesis of azetidine complexes, instead leading to N,N' complexation of stereospecifically ring-opened congeners. A minor, pyrrolidine, impurity in an azetidine ligand sample led, initially, to the formation of a highly crystalline complex, identified by single crystal XRD, as well as (later) from the same sample the expected azetidine complex. The isomeric azetidine and pyrrolidine complexes, characterized by single crystal XRD, were studied using the SambVca 2.0 online tool and their steric parameters contrasted revealing the potential for both azetidine and pyrrolidine ligands in future catalytic applications3.
Author Contributions
AF developed the ligand synthesis protocol (reported elsewhere) and synthesized some cis-aminoazetidines and prepared XRD-quality crystals of the first N,N'-platinum and -palladium complexes of them, the first organometallic CNN'-palladium azetidine complex was also prepared, AF co-wrote sections of the manuscript; JF is the lead and corresponding author who conceived and led the project, JF conceived the project and co-wrote the manuscript; AL: calculated the free energies of the diastereomeric platinum complexes and wrote the aspects of the manuscript pertaining to that aspect; LM recorded, analyzed and interpreted some of the X-ray crystallography data of this report and has oversight of all of the X-ray crystallography data herein. LM conducted training of AY, in order for AY to carry out XRD analysis, LM co-wrote aspects of the report pertaining to XRD data collection and analysis; AY synthesized some cis-aminoazetidines and prepared XRD-quality crystals of N,N'-platinum and -palladium complexes of them. AY obtained the crystal of the cis-pyrrolidine palladium complex and probed the solvent-mediated ring opening of an azetidine salt during a palladium complexation protocol. AY prepared ligands and complexes, analyzed and interpreted some of the XRD data in this report and co-wrote aspects of the report. All authors made critical contributions to the report, gave significant input into the report writing process and analysis of data herein.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The University of Birmingham is thanked for support, including travel funds permitting AY and AF to undertake overseas training placements within the CASE network (Fossey and Brittain, 2015; Payne et al., 2016), and for AY to attend a Crystallography Summer School in Durham. JF would like to thank the Royal Society for an Industrial Fellowship and the EPSRC for funding (EP/J003220/1). AL thanks Liverpool John Moores University for support. Dr Chi Tsang and Dr Peter Ashton are thanked for helpful discussions about mass spectrometry. Dr Cécile S. Le Duff, and Dr Neil Spencer gave advice on aspects of NMR spectroscopy underpinning the preliminary findings. We thank the EPSRC UK National Crystallography Service at the University of Southampton for the collection of the crystallographic data pertaining to 2b, 4a, and 5 (Coles and Gale, 2012).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2018.00211/full#supplementary-material
Footnotes
1. ^The strucutre of 4a was published previously as compound see (rac)-7 in reference 4a. With a UK National Crystallography Service Code: 2010src0915.
2. ^The crystal structure and SambVca analysis of complex 4a was already reported by us in a paper pertaining to copper-cataylzed Henry reactions. Data for compound 4a is included here to permit facile comparison amongst the platinum and palladium complexes of this report.
3. ^A preprint version of this manuscript was submitted to ChemRxiv prior to the peer revewing process.
References
Álvarez-Casao, Y., Monge, D., Álvarez, E., Fernández, R., and Lassaletta, J. M. (2015). Pyridine–hydrazones as N,N′-Ligands in asymmetric catalysis: Pd(II)-catalyzed addition of boronic acids to cyclic sulfonylketimines. Org. Lett. 17, 5104–5107. doi: 10.1021/acs.orglett.5b02613
Binder, J. T., Cordier, C. J., and Fu, G. C. (2012). Catalytic enantioselective cross-couplings of secondary alkyl electrophiles with secondary alkylmetal nucleophiles: negishi reactions of racemic benzylic bromides with achiral alkylzinc reagents. J. Am. Chem. Soc. 134, 17003–17006. doi: 10.1021/ja308460z
Choi, J. Y., Byun, J. C., Harrowfield, J. M., Lim, W. T., Thuéry, P., Kim, Y., et al. (2011). Small-ring heterocycles as components of multidentate ligands: some structural studies relating to ring opening. J. Incl. Phenom. Macro 71, 395–407. doi: 10.1007/s10847-011-9931-8
Clavier, H., and Nolan, S. P. (2010). Percent buried volume for phosphine and N-heterocyclic carbene ligands: steric properties in organometallic chemistry. Chem. Commun. 46, 841–861. doi: 10.1039/B922984A
Coles, S. J., and Gale, P. A. (2012). Changing and challenging times for service crystallography. Chem. Sci. 3, 683–689. doi: 10.1039/C2SC00955B
Falivene, L., Credendino, R., Poater, A., Petta, A., Serra, L., Oliva, R., et al. (2016). SambVca 2. a web tool for analyzing catalytic pockets with topographic steric maps. Organometallics 35, 2286–2293. doi: 10.1021/acs.organomet.6b00371
Feula, A., and Fossey, J. S. (2013). Base and solvent dependency of an oxidative retro-alkylation of secondary and tertiary benzylamines. RSC Adv. 3, 5370–5373. doi: 10.1039/c3ra40228b
Feula, A., Dhillon, S. S., Byravan, R., Sangha, M., Ebanks, R., Salih, M. A., et al. (2013). Synthesis of azetidines and pyrrolidines via iodocyclisation of homoallyl amines and exploration of activity in a zebrafish embryo assay. Org. Biomol. Chem. 11, 5083–5093. doi: 10.1039/c3ob41007b
Feula, A., Male, L., and Fossey, J. S. (2010). Diastereoselective preparation of azetidines and pyrrolidines. Org. Lett. 12, 5044–5047. doi: 10.1021/ol102215e
Fossey, J. S., and Brittain, W. D. G. (2015). The CASE 2014 symposium: catalysis and sensing for our environment, Xiamen 7th-9th November 2014. Org. Chem. Front. 2, 101–105. doi: 10.1039/C4QO90045F
Fossey, J. S., and Richards, C. J. (2004). Synthesis and X-ray crystal structure analysis of the first nickel bisoxazoline pincer complex. J. Organomet. Chem. 689, 3056–3059. doi: 10.1016/j.jorganchem.2004.06.055
Fossey, J. S., Matsubara, R., Kiyohara, H., and Kobayashi, S. (2008). Heterochiral triangulo nickel complex as evidence of a large positive nonlinear effect in catalysis. Inorg. Chem. 47, 781–783. doi: 10.1021/Ic7017727
Fossey, J. S., Matsubara, R., Vital, P., and Kobayashi, S. (2005). A C-2-symmetric nickel diamine complex as an asymmetric catalyst for enecarbamate additions to butane-2,3-dione. Org. Biomol. Chem. 3, 2910–2913. doi: 10.1039/b505404d
Fossey, J. S., Russell, M. L., Malik, K. M. A., and Richards, C. J. (2007). Synthesis and crystal structures of the first C-2-symmetric bis-aldimine NCN-pincer complexes of platinum and palladium. J. Organomet. Chem. 692, 4843–4848. doi: 10.1016/j.jorganchem.2007.06.066
Fossey, J. S., Yoshizawa, A., and Feula, A. (2015). “ORGN-408 American Chemical Society,” in Abstracts of Papers, 250th ACS National Meeting & Exposition (Boston, MA).
Gaertner, V. R. (1968). Ring-opening alkylations of 1,5-dialkyl-3-substituted azetidinium cations. Substituent entropy-controlled strained ring-chain equilibria. J. Org. Chem. 33, 523–530. doi: 10.1021/jo01266a010
Graham, J., Muhsin, M., and Kirkpatrick, P. (2004). Oxaliplatin. Nat. Rev. Drug Disc. 3, 11–12. doi: 10.1038/nrd1287
Hama Salih, M. A., Male, L., Spencer, N., and Fossey, J. S. (2015). [gamma]-Lactams and furan bispyrrolidines via iodine mediated cyclisation of homoallylamines. Org. Chem. Front. 2, 1445–1449. doi: 10.1039/C5QO00183H
Hambley, T. W. (1998). van der Waals Radii of Pt(II) and Pd(II) in Molecular Mechanics Models and an Analysis of Their Relevance to the Description of Axial M···H(–C), M···H(–N), M···S, and M···M (M = Pd(II) or Pt(II)) Interactions. Inorg. Chem. 37, 3767–3774. doi: 10.1021/ic971392m
Jamieson, E. R., and Lippard, S. J. (1999). Structure, Recognition, and Processing of Cisplatin–DNA Adducts. Chem. Rev. 99, 2467–2498. doi: 10.1021/cr980421n
Keller, L., Keller, L., Sanchez, M. V., Prim, D., Couty, F., Evano, G., et al. (2005). Azetidines as ligands in the Pd(II) complexes series. J. Organomet. Chem. 690, 2306–2311. doi: 10.1016/j.jorganchem.2005.02.041
Lee, D.-H., Lee, D.-H., Lee, Y. H., Kim, D. I., Kim, Y., Lim, W. T., Harrowfield, J. M., et al. (2008). A highly effective azetidine–Pd(II) catalyst for Suzuki–Miyaura coupling reactions in water. Tetrahedron 64, 7178–7182. doi: 10.1016/j.tet.2008.05.093
Lee, Y. H., Harrowfield, J., Kim, Y., Lim, W. T., Park, Y. C., and Thuéry, P. (2009a). Functionalised azetidines as ligands: pyridyl-complemented coordination. Dalton Trans. 434–442. doi: 10.1039/B812295D
Lee, Y. H., Kim, H. H., Thuéry, P., Harrowfield, J. M., Lim, W. T., Kim, B. J., et al. (2009b). Functionalised azetidines as ligands: some basic coordination chemistry. J. Incl. Phenom. Macro 65, 65–71. doi: 10.1007/s10847-009-9635-5
Leonard, N. J., and Durand, D. A. (1968). Small charged rings. XI. Synthesis and reactions of 1,1,2,2-tetrasubstituted azetidinium salts. J. Org. Chem. 33, 1322–1333. doi: 10.1021/jo01268a003
Martínez-Olid, F., Jesús, E., and d. Flores, J. C. (2014). Monometallic nickel(II) complexes containing N,N′-iminopyridine chelating ligands with dendritic substituents: the influence of dendrimer topology on the catalytic oligomerization and polymerization of ethylene. Inorgan. Chim. Acta 409, 156–162. doi: 10.1016/j.ica.2013.07.025
Payne, D. T., Fossey, J. S., and Elmes, R. B. P. (2016). Catalysis and sensing for our environment (CASE2015) and the supramolecular chemistry ireland meeting (SCI 2015): Dublin and Maynooth, Ireland. 8th−11th July. Supramol. Chem. 28, 921–931. doi: 10.1080/10610278.2016.1150595
Pelz, K. A., and Gagné, M. R. (2003). Synthesis and reactivity of N-chiral diamine palladium(II) complexes for Lewis acid catalysis. Abstr. Pap. Am. Chem. S. 226, U754–U754.
Pelz, K. A., White, P. S., and Gagné, M. R. (2004). Persistent N-chirality as the only source of asymmetry in nonracemic N2PdCl2 complexes. Organometallics 23, 3210–3217. doi: 10.1021/om0498495
Poater, A., Cosenza, B., Correa, A., Giudice, S., Ragone, F., Scarano, V., et al. (2009). SambVca: a Web Application for the Calculation of the Buried Volume of N-Heterocyclic Carbene Ligands. Eur. J. Inorg. Chem. 2009, 1759–1766. doi: 10.1002/ejic.200801160
Rixe, O., Ortuzar, W., Alvarez, M., Parker, R., Reed, E., Paull, K., et al. (1996). Oxaliplatin, tetraplatin, cisplatin, and carboplatin: spectrum of activity in drug-resistant cell lines and in the cell lines of the national cancer institute's anticancer drug screen panel. Biochem. Pharmacol. 52, 1855–1865. doi: 10.1016/S0006-2952(97)81490-6
Saito, B., and Fu, G. C. (2007). Alkyl–alkyl suzuki cross-couplings of unactivated secondary alkyl halides at room temperature. J. Am. Chem. Soc. 129, 9602–9603. doi: 10.1021/ja074008l
Voureka, E., Tsangaris, J. M., Terzis, A., and Raptopoulou, C. P. (1996). Coordination complexes of l-azetidine-2-carboxylic acid with platinum(II) and palladium(II). Trans. Met. Chem. 21, 244–249. doi: 10.1007/BF00165976
Yoshizawa, A., Feula, A., Leach, A. G., Male, L., and Fossey, J. S. (2018a). Palladium and platinum 2,4-cis-amino azetidine and related complexes. ChemRxiv. doi: 10.26434/chemrxiv.6104054.v1
Yoshizawa, A., Feula, A., Male, L., Leach, A. G., and Fossey, J. S. (2018c). Rigid and concave, 2,4-cis-substituted azetidine derivatives: a platform for asymmetric catalysis. Sci. Reports 8:6541. doi: 10.1038/s41598-018-24784-3
Keywords: azetidine, ligand, platinum, palladium, complex, pyrrolidine
Citation: Yoshizawa A, Feula A, Leach AG, Male L and Fossey JS (2018) Palladium and Platinum 2,4-cis-amino Azetidine and Related Complexes. Front. Chem. 6:211. doi: 10.3389/fchem.2018.00211
Received: 05 April 2018; Accepted: 23 May 2018;
Published: 21 June 2018.
Edited by:
Christian Hartinger, University of Auckland, New ZealandReviewed by:
Vladimir Boris Arion, Universität Wien, AustriaSanja Grguric-Sipka, University of Belgrade, Serbia
Fernando Novio, Instituto Catalán de Nanociencia y Nanotecnología (CIN2), Spain
Copyright © 2018 Yoshizawa, Feula, Leach, Male and Fossey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John S. Fossey, ai5zLmZvc3NleUBiaGFtLmFjLnVr