 Elena Lenci
Elena Lenci Riccardo Innocenti
Riccardo Innocenti Gloria Menchi
Gloria Menchi Andrea Trabocchi
Andrea Trabocchi- Department of Chemistry “Ugo Schiff”, University of Florence, Florence, Italy
Diversity-Oriented Synthesis (DOS) consists of generating structurally diverse compounds from a complexity-generating reaction followed by cyclization steps and appendage diversity. DOS has gathered interest to systematically explore the chemical space by generating high-quality small-molecule collections as probes to investigate biological pathways. The generation of heterocycles using amino acid and sugar derivatives as building blocks is a powerful approach to access chemical and geometrical diversity thanks to the high number of stereocenters and the polyfunctionality of such compounds. Our efforts in this field are focused on the generation of diversity-oriented molecules of peptidomimetic nature as a tool addressing protein-protein interactions, taking advantage of amino acid- and sugar-derived polyfunctional building blocks to be applied in couple-pair synthetic approaches. In this paper, the combination of diversity-oriented synthesis and chemoinformatics analysis of chemical space and molecular diversity of heterocyclic peptidomimetics are reported, with particular interest toward carbohydrate- and amino acid-derived morpholine scaffolds with a higher fraction of sp3 carbon atoms. Also, the chemoinformatic analysis of chemical space and molecular diversity of 186 morpholine peptidomimetics is outlined.
Introduction
When the molecular targets behind a disease are poorly characterized or difficult to identify, the screening of small-molecule libraries is a powerful starting point for drug discovery programmes (Gerry and Schreiber, 2018). This is especially true considering that many biological mechanisms, such as signal transduction or gene expression, are regulated by protein-protein interaction (PPI), “undruggable” targets that cannot be addressed with existing chemical tools (Wells and McClendon, 2007). Even though many synthetic efforts have given a great advance in improving peptide druggability, this class of compounds covers only 2% of the worldwide drug market (Sun, 2013) and the development of new peptidomimetic scaffolds is still a growing field of medicinal chemistry and chemical biology (Kaminker et al., 2018; Ramaswamy et al., 2018). In this context, Diversity-Oriented Synthesis (DOS) (Trabocchi, 2013; Chauhan et al., 2017; Zeng et al., 2017), where many different molecular scaffolds possessing a high structural complexity are developed using short synthetic strategies, is a convenient approach for the generation of large sets of small molecule peptidomimetics. In particular, in view of creating sp3-rich molecular entities, with polyfunctional and stereochemically dense characteristics, building blocks from the chiral pool are increasingly used in DOS, as showed by the relevance recently gained by the biosynthetically inspired divergent approach (Yang et al., 2014; Bender et al., 2018), or the diversity-oriented synthesis of natural-product inspired libraries (Huigens et al., 2013; McLeod et al., 2014; Annamalai et al., 2017; Saleeb et al., 2018). Our efforts in this field are focused on the exploitation of amino acid and sugar derivatives for the generation of peptidomimetic libraries around the morpholine skeleton, as this key nucleus is contained in many natural products and drugs (Figure 1) (Wijtmans et al., 2004; Pal'chikov, 2013).
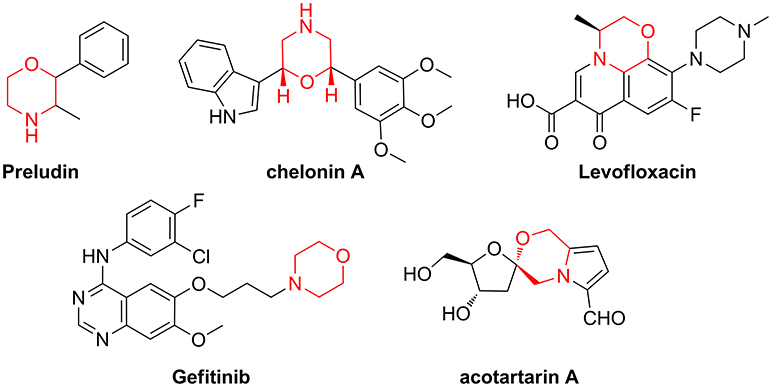
Figure 1. Representative examples of natural products and drugs containing a morpholine ring.
Over the years, we reported the synthesis of many different bicyclic compounds 3 based on the 6,8-dioxa-3-azabicyclo[3.2.1]octane core, an atom-by-atom dipeptide isostere. They proved to be active as aspartyl protease inhibitors (SAP2: Trabocchi et al., 2010; Calugi et al., 2012; HIV: Calugi et al., 2014; BACE1: Innocenti et al., 2017) and as RGD integrin ligands (Cini et al., 2009; Bianchini et al., 2012). The short and convenient synthetic strategies consist of combining two key components from the chiral pool, an amino carbonyl derivative 1 and a diol species 2, followed by the acid-catalyzed acetalization of the resulting coupling intermediate (Scheme 1A) (Trabocchi et al., 2006). Representative follow-up chemistry was achieved generating spirocylic scaffolds 4 (Trabocchi et al., 2007). This couple/pair approach proved to be even more interesting in a diversity-oriented point of view when α-amino acid derivatives 6 were combined with dimethoxyacetaldehyde 5, as morpholine acetal scaffold 7 was a good starting point for the generation of many bi- and tricyclic compounds, such as diketopiperazines 8 and 2-oxa-5-azabicyclo[4.1.0]heptanes 9 (Sladojevich et al., 2008; Lalli et al., 2009; Lenci et al., 2015a) (Scheme 1B). Also, bicyclic morpholine lactone 12, coming from aminoacetaldehyde dimethylacetal 10 and protected methyl threonate derivative 11, gave structures 13, taking advantage of lactone aminolysis, and structures 14, when the aminolysis was combined with diketopiperazine synthesis (Lalli et al., 2009; Ciofi et al., 2010) (Scheme 1C).
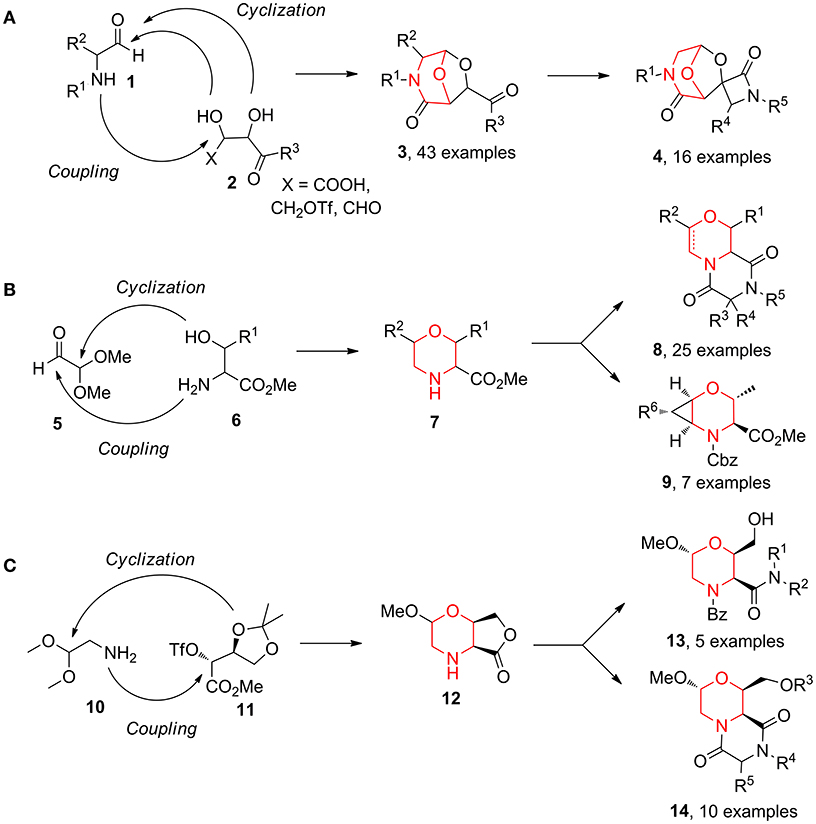
Scheme 1. Representative diversity-oriented synthesis of morpholines, starting from amino carbonyl derivative 1 and diol 2 (A); from dimethoxyacetaldehyde 5 and amino acid derivative 6 (B); and from aminoacetaldehyde 10 and threonate derivative 11 (C).
Also, the combination of mannose 15 with aminoacetaldehyde 10 allowed to obtain morpholine-derived compounds enriched with polyhydroxylated chains (compounds 16–19, Scheme 2a) exploiting the reactivity of sugar hydroxyl groups toward the acetal moiety (Lenci et al., 2015b, 2016). Similarly, the application of lactone formation and trans-acetalization pairing reactions were used in the synthesis of 21–22 starting from the Petasis coupling intermediate obtained by glycolaldehyde 20 (Scheme 2b) (Lenci et al., 2017).
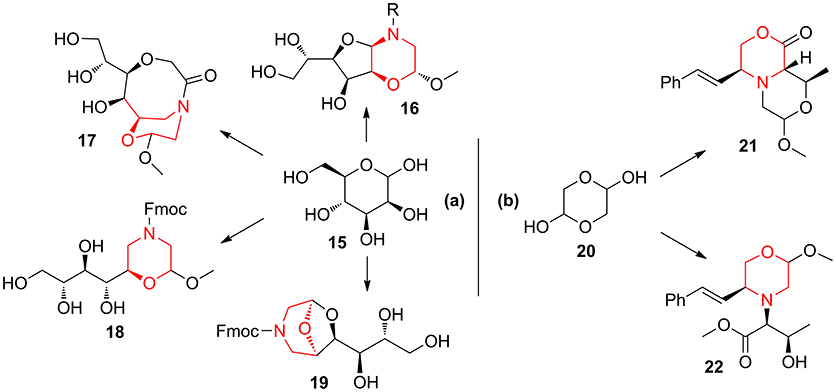
Scheme 2. Representative syntheses of morpholine-derived compounds starting from mannose 15 (a) and from glycolaldehyde 20 (b).
Considering that a higher scaffold complexity is generally associated with a more successful outcome in drug discovery and development (Clemons et al., 2010; Galloway et al., 2011; Flagstad et al., 2016; Stotani et al., 2016), we recently turned our attention on exploiting the chemistry useful to develop skeletally complex sp3-rich morpholines, for example by using multicomponent reactions. In this work, as a further improvement in this direction, we envisioned to install quaternary stereocenters on this nucleus, as they are often present in the structure of many biologically active compounds and pharmaceutical agents (Christoffers and Baro, 2006; Hawner and Alexakis, 2010). This was envisaged by transforming the sp3 carbon atom in α-position of the carbomethoxy group of different morpholin-3-one starting materials, by means of the Staudinger reaction, to generate morpholinone-derived spiro-β-lactams (Scheme 3A), and by different alkylation strategies (Scheme 3B).
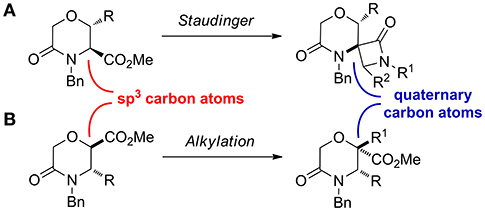
Scheme 3. Synthetic approaches exploited to install quaternary carbon atoms on morpholine-3-one. (A) the Staudinger ketene-imine reaction on methyl 5-oxomorpholine-3-carboxylate and (B) alkylation reaction on methyl 5-oxomorpholine-2-carboxylate.
Finally, the exploration of the chemical space accessed by these new compounds was analyzed using PCA (Principal Component Analysis) and PMI (Principal Moment of Inertia) graphical representation in relation to our in-house library of more than 170 morpholine compounds developed over the years in our laboratory. The entire collection of morpholines was also studied using different chemoinformatic approaches (Colomer et al., 2016) by characterizing the degree of complexity of each library member, by using the Fsp3 definition (Lovering et al., 2009), and through the relationship between different drug- and lead-like properties.
Materials and Methods
Chemistry
Experimental procedures, compound characterization data for newly synthesized compounds 35–38 and 42 and NOESY 1D spectra for compounds 37 and 42, are reported in the Supplementary Material. NMR spectra were collected on a Varian INOVA 400 spectrometer operating at 400 MHz for 1 H. The spectra were obtained in CDCl3 solutions. Proton signals were assigned via TOCSY spectra, and NOESY spectra provided the data used in the conformational analyses. TOCSY spectra were recorded with 2,048 points in t1, 200 points in t2, and 8 scans per t2 increment, and 80 ms as mixing time. NOESY spectra were recorded with a similar number of t1 and t2 points unless otherwise noted, 32 per t2 increment, and 500 ms as mixing time. 1D NOESY experiments were carried out using 64 increments and 500 ms as mixing time.
Molecular Modeling Methods
Molecular modeling calculations were carried out on compounds 35–38 and compound 42 so as to assess the global minimum conformer and to gain insight into the detailed structure of the molecular scaffolds. Energy-minimized conformations of 35–38 and compound 42 were achieved using SPARTAN Version 5.1 (Wavefunction, Inc., Irvine, C). Conformational searches were carried out using Monte Carlo method within MMFF94 force field (Halgren, 1996) and the AM1 semiempirical method (Dewar et al., 1985) was used to optimize the global minimum conformer.
Chemoinformatics Analysis
PCA Analysis
The web-based public tool ChemGPS-NP (http://chemgps.bmc.uu.se/) was used for the PCA analysis of compounds 35–38 and compound 42, to compare their chemical properties with those of an in-house library of morpholine-derived compounds. ChemGPS-NP can be applied for comprehensive chemical space navigation and exploration in terms of global mapping on to a consistent 8-dimensional map of structural characteristics. The first four dimensions of the ChemGPS-NP map capture 77% of data variance. Chemical compounds were positioned onto this map using interpolation in terms of PCA score prediction. SMILES codes for all compounds were retrieved using ChemBioDraw Ultra 12.0 and submitted to ChemGPS-NP for achieving the corresponding PC scores. The PCA data were then used for the construction of PC1 (representing size, shape, and polarizability) vs. PC2 (representing aromatic and conjugation related properties).
PMI Analysis
Principal moments of inertia analysis was carried out by calculating the lowest energy conformation of compounds 35–38 and compound 42, and each compound from an in-house library of morpholine-derived compounds. The conformational search was performed using the built-in AMMP molecular mechanics algorithm with default parameters of the VEGA ZZ molecular modeling software package v.3.0.1 (Pedretti et al., 2002). Once the lowest energy conformer was calculated, the three principal moments of inertia (Ixx, Iyy, Izz) and the normalized principal moments of inertia were determined. Specifically, the three calculated principal moments of inertia were sorted by ascending magnitude I1, I2, and I3. Subsequently, in order to eliminate completely the dependency of the chosen representation on the size of the molecules, normalization was performed by dividing the two lower PMI-values (I1 and I2) by the highest value (I3), generating two characteristic values of normalized PMI ratios (NPRs) for each compound (I1/I3 and I2/I3). Then, NPR1 (I1/I3) and NPR2 (I2/I3) were plotted on a triangular graph with the vertices (0,1), (0.5,0.5), and (1,1) representing a perfect rod, disc and sphere, respectively.
Calculation of Medicinally-Relevant Molecular Properties
Molecular weight, cLogP, and the number of sp3 carbon atoms, stereogenic centers, rotatable bonds, hydrogen bond acceptors and donors were calculated using the web-based public tool FAFDrugs (Free ADME-Tox Filtering Tool), developed at the Paris Diderot University (Lagorce et al., 2015). LogP values are computed by using the xLogP3 program (Cheng et al., 2007), enhanced by employing an in-house library of experimental logP-values from the PHYSPROP database (Lobell et al., 2006) as several models showed that xLogP3 and cLogP methods give similar results (Mannhold et al., 2009). Fsp3 was calculated as the number of sp3 hybridized carbon atoms vs. the total carbon count. FC* was calculated as the number of stereocenters vs. the total carbon count. Rotatable bonds were defined as any single bond, not in a ring, bound to a non-terminal heavy (i.e., non-hydrogen) atom, excluding amide C-N bonds. Hydrogen bond donors were taken as the sum of all OHs and NHs, and hydrogen bond acceptors were taken as the sum of all oxygen and nitrogen atoms without a formal positive charge, excluding pyrrole nitrogen, heteroaromatic oxygen and higher oxidation states of nitrogen, in agreement with the Lipinski definition (Lipinski, 1997).
Results and Discussion
Synthesis
As case study to install quaternary stereocenters on the morpholine nucleus, we explored simple synthetic methodologies capable of transforming the sp3 carbon atom in the α-position of the carbomethoxy group of different morpholin-3-one compounds. In particular, we selected methyl 5-oxomorpholine-2-carboxylate 25 derived by the application of the Castagnoli-Cushman reaction (Dar'in et al., 2015) between imine 23 and 1,4-dioxane-2,6-dione (24), and methyl 5-oxomorpholine-3-carboxylates 28 and 29, obtained respectively from serine and threonine derivatives 26-27 after the acylation with α-bromoacetylbromide and subsequent NaH-mediated intramolecular cyclization reaction (Scheme 4). To improve the scaffold complexity and to install quaternary stereocenters on these compounds, we firstly studied the Staudinger reaction (Alcaide et al., 2007; Cossío et al., 2008; Omidvari and Zarei, 2018) with different aromatic imines to generate polycyclic spiro-β-lactams, in agreement with previous studies on 3-aza-6,8-dioxabicyclo[3.2.1]octane bicycles giving compounds 4 (Trabocchi et al., 2007). In particular, compounds 28 and 29 were transformed into the more reactive acyl chloride derivatives 30–31 in order to generate the intermediate ketene more easily and to avoid the formation of amide by-products (Scheme 4).
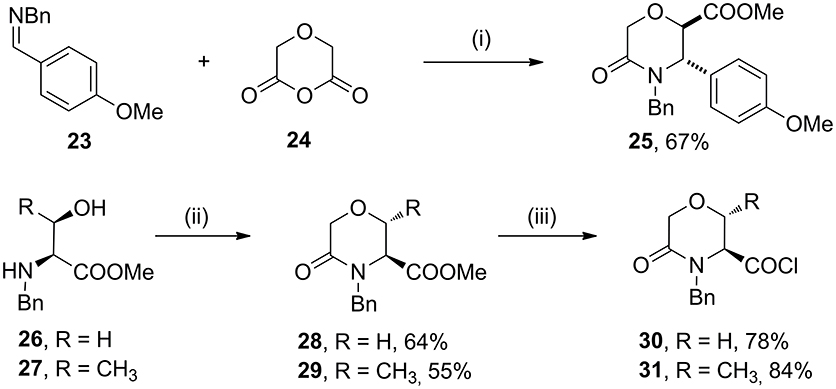
Scheme 4. Synthesis of methyl 5-oxomorpholine-2-carboxylate 25 and methyl 5-oxomorpholine-3-carboxylates 28 and 29 and preparation of acyl chloride derivatives 30 and 31. Reagents and conditions: (i) dry toluene, 80°C, 4 h; then SOCl2, MeOH, reflux, 2 h; (ii) BrCOCH2Br, Et3N, dry CH2Cl2, −15°C, 1 h; then NaH, THF dry, 0 °C - r. t., 1 h; (iii) LiOH, H2O/THF, r. t., 16 h; then SOCl2, reflux, 2 h.
However, after refluxing the acyl chloride 30-31 in the presence of triethylamine as a base and aromatic imines 32–34 in toluene for 16 h, the spiro-β-lactams 35–38, characterized by the 8-oxa-2,5-diazaspiro[3.5]nonane-1,6-dione molecular framework, were obtained in moderate yields, as a consequence of the formation of amide by-products 39–41 (Table 1).
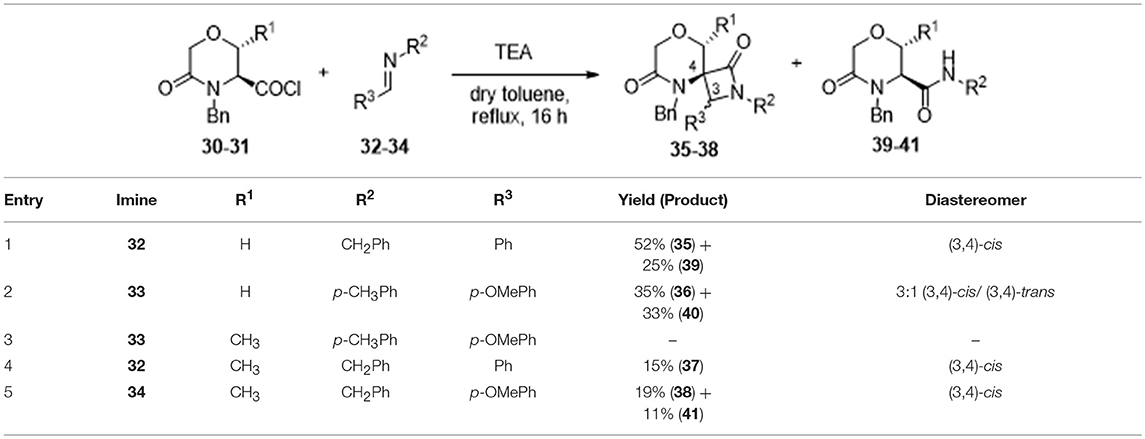
Table 1. Synthesis of spiro-β-lactams 35–38 from serine and threonine-derived morpholine derivatives 30 and 31.
Considering that the nucleophilicity of the amine derivatives comprising the imine proved to affect the yields, only aromatic imines were taken into account. Also, as shown in Table 1, the steric hindrance of both imine and morpholine counterparts resulted in reducing drastically the yield. In particular, best results were obtained starting from serine-derived morpholine 30 using N-benzylidene-1-phenylmethanamine 32 and N-(4-methoxybenzylidene)-4-methylaniline 33, even though the higher steric hindrance of this second imine resulted in the achievement of compound 36 in lower yield (35% instead of 52%, Table 1, entry 1 and 2). On the other hand, threonine-derived morpholine 31 was found to be less reactive and unstable, as a consequence of the presence of the methyl group adjacent to the ketene functionality. In fact, no reaction was observed with imine 33 (Table 1, entry 3), whereas the use of N-benzylidene-1-phenylmethanamine 32 and N-(4-methoxybenzylidene)-4-phenylmethanamine 34 yielded the spiro compounds 37 and 38 in low yields (Table 1, entry 4 and 5, respectively) and with many degradation products, confirming the difficulty in achieving highly substituted spiro-β-lactams, as also reported (Bari and Bhalla, 2010).
Nevertheless, interesting results were obtained as regarding the diastereoselectivity. In fact, despite the four theoretically possible diastereomers, in all cases the cis-products were obtained as a major or single stereoisomer, as shown by 1D and 2D NOESY experiments carried out on spiro compound 37 and 35 (see Figures S13, S14). In particular, the existence of a NOESY peak between H-3 and the methyl group at C-9 for compound 37 proved the relative configuration as reported in Figure 2. The absence of any correlation between the methyl group and H-7 suggested that the methyl group is oriented in equatorial position. Although purely indicative, this observation was found to be reasonable for such a constrained structure and was in agreement with the global minimum conformer resulting from molecular modeling calculations (Figure 2, right). Specifically, the calculated distance between H-3 and the CH3 atoms was 2.1 Å, whereas for the other possible diastereomer at the spiro position this distance was found being more than 4 Å. Similar structural arrangement was ascertained for compound 35, with the C-1 carbonyl group pointing toward C-9 and the H-3 showing a strong NOESY correlation with H-9 protons, whereas the same cis-configuration was evinced for the other compounds by comparing the diagnostic signal of the H-3 proton, which appeared as a singlet in an unambiguous region of 1H-NMR spectrum between 4.74 and 4.84 ppm. This diastereoselectivity is in agreement with what observed for similar spiro-β-lactams obtained starting from proline-derived ketenes (Khasanov et al., 2004) and 6,8-dioxabicyclo[3.2.1]octane-derived ketenes (Trabocchi et al., 2007), as the widely accepted mechanism of the reaction involves the nucleophilic attack of the imine on the ketene species to give a zwitterionic intermediate, which preferentially undergoes an outward conrotatory ring closure, due to stabilizing stereoelectronic effects.
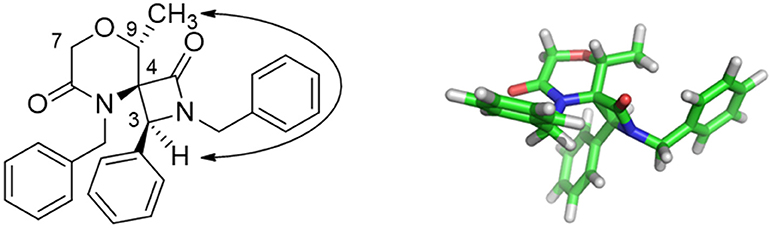
Figure 2. Key NOESY peak (Left), and lowest energy conformer (Right) of compound 37 showing the major stereoisomer resulting from the two newly-generated stereocenters at C-3 and C-4.
Unfortunately, when the Staudinger reaction was performed between the acid chloride of methyl 5-oxomorpholine-2-carboxylate 25 and aromatic imines 32–34, only degradation products were observed. Thus, in order to install a quaternary stereocenter on this morpholin-3-one, we explored a complementary approach based on an alkylation strategy, and in particular, as a case study, we performed the methylation of the α-carbon of the carbomethoxy group of 25 using NaHDMS as a strong base to generate the intermediate carbanion (Scheme 5).
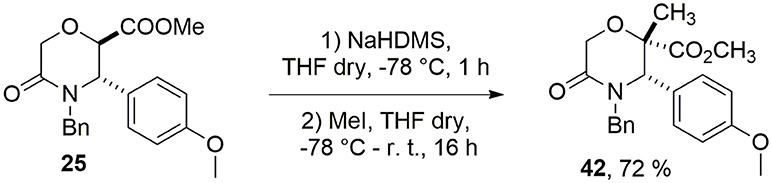
Scheme 5. Methylation of 5-oxomorpholine-2-carboxylate 25.
Compound 42 was obtained with 72% yield as a single stereoisomer, showing inversion of the configuration at the α-carbon. Structure analysis performed by NMR and molecular modeling calculations showed a half-chair conformation for the morpholinone scaffold possessing both the methyl and aryl groups in axial position and with a trans geometry. Specifically, the trans arrangement was ascertained by key NOESY peaks between H-3 and CH3 at C-2, and a strong NOESY interaction between H-6 and the methyl group at C-2, suggesting the methyl group being positioned in axial orientation (Figure 3).
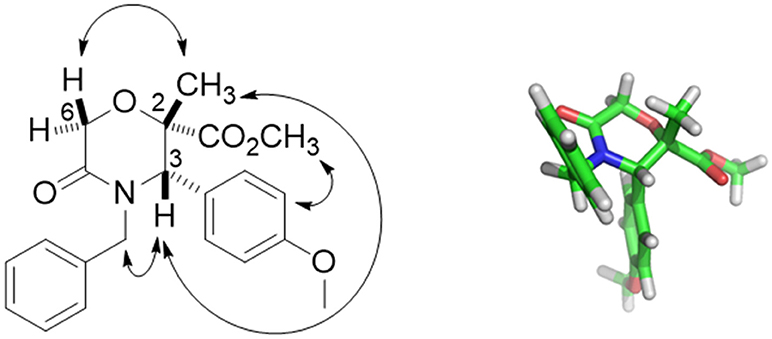
Figure 3. Key NOESY peaks (Left), and lowest energy conformer (Right) of compound 42 showing the aryl and methyl groups in axial positions.
Chemoinformatic Analysis
The exploration of the chemical space accessed by newly synthesized compounds 35–42, in relation to the pool of 176 morpholine-derived small molecules previously synthesized in our laboratories, was then studied by using different chemoinformatic approaches (see Figure 4 for a scaffold tree composed by all the 16 different molecular frameworks present in this library).
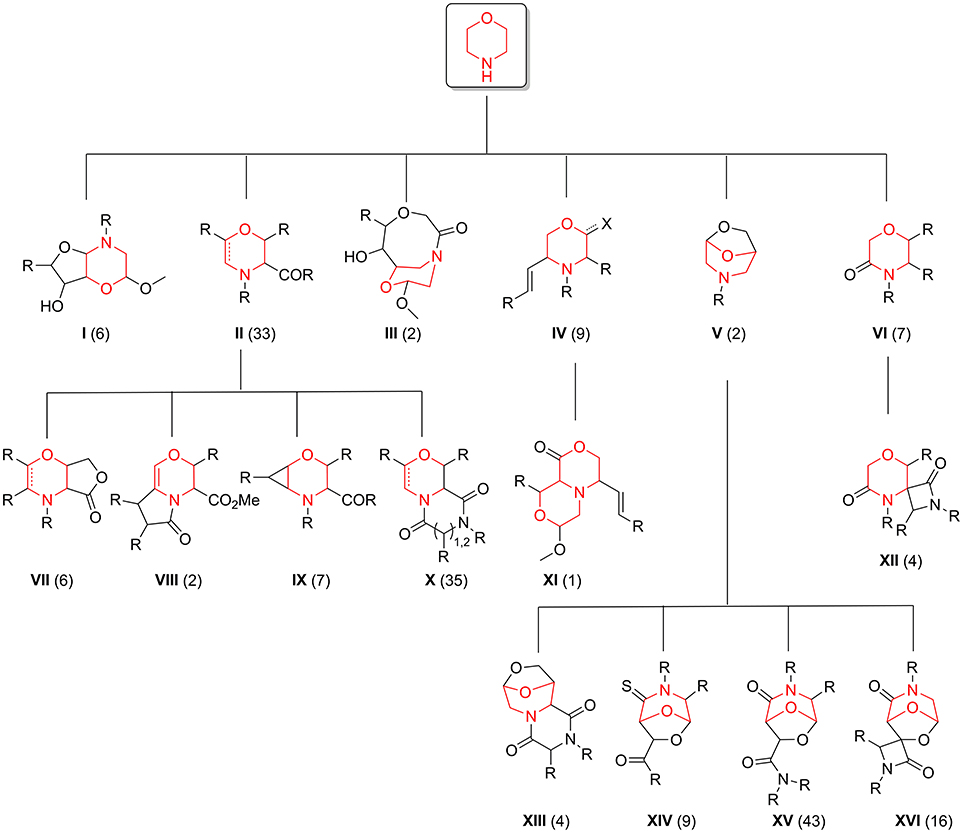
Figure 4. Morpholine-containing molecular scaffolds of our in-house library. The number of compounds in the library representing each scaffold is displayed in brackets.
Firstly, Principal Component Analysis (PCA), performed using the web-based public tool ChemGPS-NP, was used to simplify the comparison of all these molecules on the basis of different chemical properties (Xue et al., 2004; Tan, 2005). A pool of 186 compounds was analyzed, focusing in particular on principal component one (PC1), representing size, shape and polarizability, and the principal component two (PC2), that is a direct expression of aromatic and conjugation related properties, and plotted in a graph (Figure 5), where compounds 35–42 are shown as red diamonds, their parent analogs 25, 28, and 29 as blue diamonds, and the previously synthesized morpholines as black squares. All the library members were found being grouped in four different clusters (Figure 5, I–IV), depending on both the structure of the skeletons and side chain properties. As regarding to the introduction of quaternary stereocenters in the morpholine nucleus, a peculiar effect was found for the Staudinger reaction products. In fact, although the methylation did not induce any movement within the chemical space, as both compounds 25 and 42 reside in the second cluster, the Staudinger chemistry proved to shift the serine and threonine-derived morpholinone compounds 28–29 from the third cluster to the first one (Figure 5, red arrow), being populated also by spiro-β-lactams derived from the bicycle 3-aza-6,8-dioxabicyclo[3.2.1]octane, possibly due to the contribution to aromaticity given by the Staudinger reaction with aromatic imines.
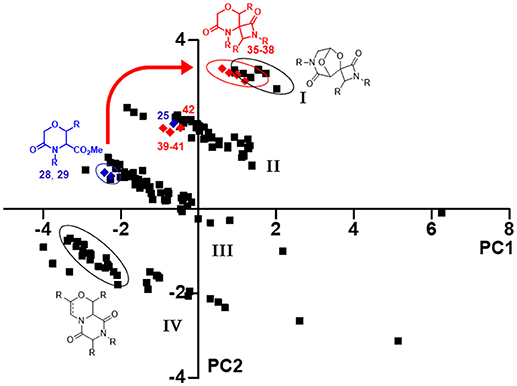
Figure 5. PCA plot resulting from the correlation between PC1 vs. PC2, showing the positioning in the chemical space of compounds 35–42 (red diamonds) and their parent analog 25, 28, and 29 (blue diamonds), in relation to an in-house library of 176 morpholine compounds (black squares). The thick red arrow indicates the shift in the chemical space induced by Staudinger chemistry from compounds 28–29 to spiro-β-lactams 35–38. The ellipses highlight the various compounds subclasses; the compound clusters are numbered as I–IV.
This significant movement in the chemical space achieved by the Staudinger chemistry was also observed in the Principal Moment of Inertia (PMI) analysis graph (Figure 6), obtained by calculating the three principal moments of inertia (Ixx, Iyy, Izz) and plotting their corresponding normalized values (I1/I3 and I2/I3) on a triangular graph, where the vertices (0,1), (0.5,0.5), and (1,1) represent a perfect rod (acetylene), disc (benzene) and sphere (adamantane), respectively (Sauer and Schwarz, 2003). As evinced from this graph, morpholine-derived compounds were found to lie along the center-left side of the triangle, as usually observed in the PMI analysis of small molecules. However, while the Staudinger chemistry performed on bicyclic 3-aza-6,8-dioxabicyclo[3.2.1]octanes did not result in a relevant shift in the PMI graph (Figure 6, green arrow), the installation of spiro-β-lactams on the morpholin-3-ones 28–29 proved to modulate significantly the three-dimensional complexity of these molecular frameworks. Compounds 28–29 were found to move from the center of the graph toward the rod-sphere axis (as for spiro-β-lactams 35, 37, 38) or the disc corner (as for spiro-β-lactam 36 that contains a N-p-tolyl group instead of a N-benzyl group) (Figure 6, red arrows). Also, amide by-products 39–41 were found lying closer to the rod-disc axis, as a result of the less three-dimensional character possessed by these structures, when compared to 28–29. On the contrary, the effect of the extra methyl group in compound 42 did not prove to change significantly the shape of the morpholine nucleus, as this compound was found to be close to its parent 25 in the PMI plot. Interestingly, the bicyclic compounds based on the 6,8-dioxa-3-azabicyclo[3.2.1]octane core were found to be not close to the sphere region, as expected, possibly due to the major contribution in exploring the space toward the sphere-disc axis given by the side chains, as in the case of some dihydro-1,4-oxazine compounds with peculiar functional groups like the myristoyl chain.
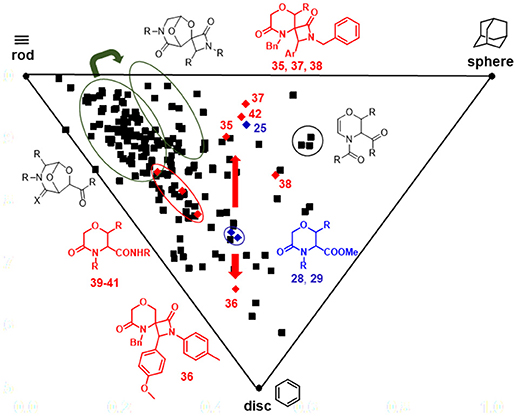
Figure 6. PMI plot showing the skeletal diversity of compounds 35–42 and their parent analogs 25, 28, and 29 (red diamonds), in relation to an in-house library of 176 morpholine compounds (black squares). The thick red arrows indicate the shift in the chemical space induced by Staudinger chemistry from compounds 28, 29 to spiro-β-lactams 35–38, whereas the green arrow indicates the shift induced by the same chemistry performed on 6,8-dioxa-3-azabicyclo[3.2.1]octane core. The ellipses highlight the various compounds subclasses.
To gain insight into a chemoinformatic evaluation of our in-house morpholine library, we calculated the saturation index (Fsp3) of each compound collection, as a measure of the molecular complexity (Lovering et al., 2009). This value was calculated as the ratio between the number of sp3 hybridized carbons in the molecule vs. the total carbon count and compared with those of a reference set of 40 brand-name blockbuster (BB) drugs as reported by Tan (Bauer et al., 2010; Kopp et al., 2012) (Figure 7, left). A similar approach was applied also to quantify the presence of stereocenters (Figure 7, right), by defining FC* as the ratio of stereogenic center vs. the total carbon count. These two parameters (Fsp3 and FC*) allow to evaluate the quality of small molecule collections as regarding to the ability of both accessing new areas of the chemical space and giving successful results in drug discovery programmes. Sp3-rich DOS-derived small molecule collections proved to be more selective and more effective in binding to specific targets, as compared to analog small molecule libraries with lower Fsp3 ratio (Clemons et al., 2010), although the hit rate trend was found to be opposite in fragment-based screening (Hall et al., 2014). The analysis of the Fsp3 and FC* parameters revealed that our library possesses higher frequency of molecules with a Fsp3 in the range between 0.4 and 0.6, as compared to the drugs, and also higher mean value of Fsp3 (Fsp3 morpholines = 0.52, Fsp3 BB drugs = 0.40) and FC* ratio (FC* morpholines = 0.19, FC* BB drugs = 0.05). However, the Staudinger ketene-imine reaction, despite the possibility to introduce a quaternary stereocenter in the molecule, proved not to be a good strategy in terms of improving the Fsp3 ratio of the overall molecule, since it introduced a high number of sp2 carbon atoms due to the presence of aromatic appendages. In fact, the Fsp3 of starting compounds 28 and 29 (respectively 0.38 and 0.43) were reduced dramatically after the reaction to a mean value of 0.26 for the spiro-β-lactams 35–38.
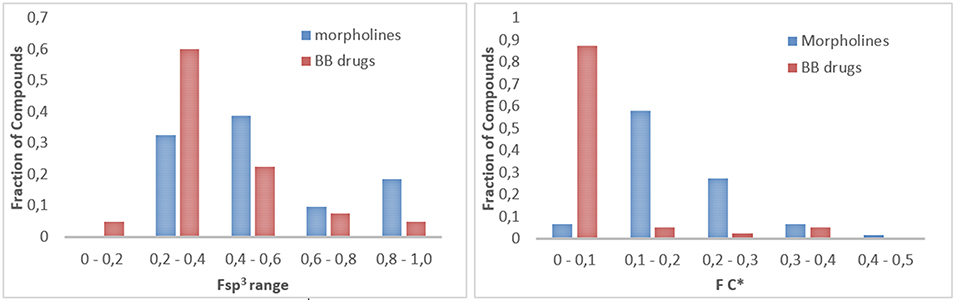
Figure 7. (Left) Fraction of compounds (morpholines in blue, blockbuster drugs in red) with different Fsp3 value, subdivided into 5 different ranges. (Right) Fraction of compounds (morpholines in blue, blockbuster drugs in red) with different FC* value, subdivided into 5 different ranges.
Finally, the investigation of small molecule physicochemical properties was carried out in order to establish the “druggability” and “lead-likeness” of our library, according to Lipinski's “rule of five” (Lipinski, 1997, 2004; Lipinski and Hopkins, 2004) and Congreve's “rule of three” (Congreve et al., 2003), respectively. In particular, we evaluated the lipophilicity and the molecular weight as key parameters to achieve good solubility, membrane permeability and subsequent oral bioavailability, by plotting clogP values (calculated as the logarithm of the partition coefficient between n-octanol and water) and the molecular weight of each library member in a graph (Figure 8, left). Only 12 out of 186 compounds were not compliant with Lipinski's “rule of five,” as they showed cLogP values higher than 5 and molecular weight higher than 500. This was evinced for compounds where morpholine was installed in a pentapeptide, or in the case of few bicyclic or morpholines characterized by a large number of aromatic substituents. Forty-five of these compounds were found following the restricted “lead-likeness” filters as proposed by the Congreve's “rule of three,” too, proving to be good starting points for potential drug optimization (Teague et al., 1999). Similarly, Veber et al. (2002) have proposed that the number of rotatable bonds (RB), together with the number of hydrogen bond donors (HBD) and hydrogen bond acceptors (HBA), can give another good criteria for predicting oral bioavailability. According to such structural parameters, only 8 compounds of our library were found not following the Veber's rule (RB ≤ 10 and (HBA + HBD) ≤ 12) for a good bioavailability. The graph reported in Figure 8, right can easily show that most of the morpholine compounds are within the cut-off values of drug-like Lipinski's “rule of five” (HBA ≤ 10, HBD ≤ 5, RB ≤ 5), whereas only for the number of HBD (blue line) the Congreve lead-like “rule of three” is satisfied (HBD ≤ 3, HBA ≤ 3, RB ≤ 3) (Figure 8, right). As expected, no particular changing in the Lipinski drug-like properties were observed for the spiro-β-lactams 35–38 derived from the Staudinger reaction, since the molecular weight and the cLogP values increased significantly, but still remained under the cut-off values of Lipinski's “rule of five,” as well as the number of HBA, HBD and rotatable bonds. In particular, the introduction of the nitrogen atom brought another hydrogen bond acceptor to the molecule (moving from 5 to 6) and the number of rotatable bonds increased from 4 to 5 or 6, depending on the imine counterpart.
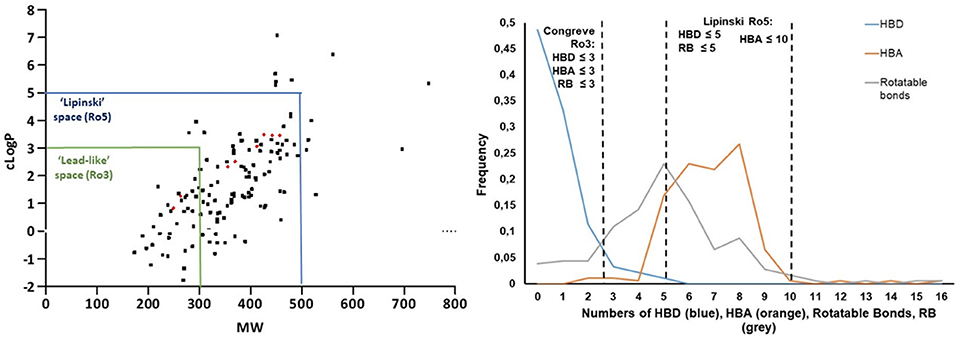
Figure 8. (Left) cLogP vs. molecular weight plot highlights all the compounds that follow Lipinski's “rule of five” (Lipiski space, blue box) and lead-likeness “rule of three” (Lead-like space, green box). (Right) Fraction of compounds with different number of hydrogen bond donors (HBD, blue line), hydrogen bond acceptors (HBA, orange line) and rotatable bonds (RB, gray line).
Conclusions
The development of new peptidomimetic scaffolds useful to address protein-protein interactions is still a growing field of medicinal chemistry and chemical biology. This approach requires efficient synthetic processes able to produce high-quality small molecule collections, as in the case of the use of Diversity Oriented Synthesis (DOS) strategies, especially starting from amino acid and sugar derivatives, to produce polyfunctional and sp3-rich building blocks. Our efforts in this field are focused on the generation of different peptidomimetic compounds around the morpholine nucleus, as this heterocycle is contained in many different bioactive molecules.
In order to increase the complexity and the sp3 character of this important nucleus, we studied different build/couple/pair strategies that exploit complexity-generating reactions. In this work, as a further improvement in this direction, we envisioned to transform the sp3 carbon atom in α-position of the carbomethoxy group of selected morpholin-3-one starting materials, by means of the Staudinger reaction, to generate morpholinone-derived spiro-β-lactams and of different alkylation strategies. This approach proved to be valuable, especially when assessing the structural diversity and complexity of these new compounds in comparison with 176 morpholine-derived small molecules previously synthesized in our laboratories, by analyzing the populated chemical space. In fact, both PCA (Principal Component Analysis) and PMI (Principal Moment of Inertia) analysis revealed that the Staudinger ketene-imine reaction proved to shift the serine and threonine-derived morpholine-3-one compounds in new areas of the chemical space, assessing a relevant change of positions, hardly achieved by using other synthetic approaches. Finally, we also investigated different small-molecule physicochemical parameters (cLogP, molecular weight, number of rotatable bonds, hydrogen bond acceptors, hydrogen bond donators, Fsp3, FC*) of all the 186 morpholines of the library in comparison with a reference set of 40 brand-name blockbuster (BB) drugs. These analyses revealed that only few compounds did not show “drug-like” values, as defined by the Lipinski rule of five, whereas most of the compounds showed higher Fsp3 and FC* values as compared to the drugs. Indeed, several applications in medicinal chemistry projects demonstrated over the years the value of morpholine as a scaffold for peptidomimetic design and drug discovery.
Author Contributions
AT and EL conceived the research. EL and RI carried out the synthesis. EL carried out the chemoinformatics analyses. AT carried out the molecular modeling calculations. AT and GM supervised the work. EL and AT wrote the paper. All the authors revised the manuscript.
Funding
Financial support from MIUR PRIN2015 (cod. 20157WW5EH), Fondazione CR Firenze and University of Florence are acknowledged.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2018.00522/full#supplementary-material
References
Alcaide, B., Almendros, P., and Aragoncillo, C. (2007). Chem. Rev. 107, 4437–4492. doi: 10.1021/cr0307300
Annamalai, M., Kumar, K., Hristeva, S., Bielska, M., and Ortega, R. (2017). Highly stereoselective synthesis of a compound collection based on the bicyclic scaffolds of natural products. Molecules 22:827. doi: 10.3390/molecules22050827
Bari, S. S., and Bhalla, A. (2010). Spirocyclic β-Lactams: synthesis and biological evaluation of novel heterocycles. Top. Heterocycl. Chem. 22, 49–99. doi: 10.1007/7081_2009_8
Bauer, R. A., Wurst, J. M., and Tan, D. S. (2010). Expanding the range of ‘druggable’ targets with natural product-based libraries: an academic perspective. Curr. Opin. Chem. Biol. 14, 308–314. doi: 10.1016/j.cbpa.2010.02.001
Bender, C. F., Paradise, C. L., Lynch, V. M., Yoshimoto, F. K., and De Brabander, J. K. (2018). A biosynthetically inspired synthesis of (-)-berkelic acid and analogs, Tetrahedron 74, 909–919. doi: 10.1016/j.tet.2018.01.021
Bianchini, F., Cini, N., Trabocchi, A., Bottoncetti, A., Raspanti, S., Vanzi, E., et al. (2012). 125I-Radiolabelled morpholine-containing Arg-Gly-Asp-ligand of αvβ3 integrin as a molecular imaging probe for angiogenesis. J. Med. Chem. 55, 5024–5033. doi: 10.1021/jm2016232
Calugi, C., Guarna, A., and Trabocchi, A. (2014). Identification of constrained peptidomimetic chemotypes as HIV protease inhibitors. Eur. J. Med. Chem. 84, 444–453. doi: 10.1016/j.ejmech.2014.07.049
Calugi, C., Trabocchi, A., De Bernardis, F., Arancia, S., Navarra, P., Cauda, R., et al. (2012). Bicyclic Peptidomimetics targeting secreted aspartic Protease 2 (SAP2) from Candida albicans reveal a constrained inhibitory chemotype. Bioorg. Med. Chem. 20, 7206–7213. doi: 10.1016/j.bmc.2012.09.031
Chauhan, J., Luthra, T., Gundla, R., Ferraro, A., Holzgrabe, U., and Sen, S. (2017). A Diversity oriented synthesis of natural product inspired molecular libraries. Org. Biomol. Chem. 15, 9108–9120. doi: 10.1039/C7OB02230A
Cheng, T., Zhao, Y., Li, X., Lin, F., Xu, Y., Zhang, X., et al. (2007). Computation of octanol-water partition coefficients by guiding an additive model with knowledge. J. Chem. Inf. Model. 47, 2140–2148. doi: 10.1021/ci700257y
Christoffers, J., and Baro, A. (2006). Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis. Hoboken, NJ: John Wiley & Sons.
Cini, N., Trabocchi, A., Menchi, G., Bottoncetti, A., Raspanti, S., Pupi, A., et al. (2009). Morpholine-based RGD-cyclopentapeptides as αvβ3/αvβ5 integrin ligands: role of configuration towards receptor binding affinity. Bioorg. Med. Chem. 17, 1542–1549. doi: 10.1016/j.bmc.2009.01.006
Ciofi, L., Morvillo, M., Sladojevich, F., Guarna, A., and Trabocchi, A. (2010). Skeletal diversity by sequential one-pot and stepwise routes using morpholine ester scaffolds. Tetrahedron Lett. 51, 6282–6285. doi: 10.1016/j.tetlet.2010.09.103
Clemons, P. A., Bodycombe, N. E., Carrinski, H. A., Wilson, J. A., Shamji, A. F., Wagner, B. K., et al. (2010). Small molecules of different origins have distinct distributions of structural complexity that correlate with protein-binding profiles. Proc. Natl. Acad. Sci. U.S.A. 107, 18787–18792. doi: 10.1073/pnas.1012741107
Colomer, I., Empson, C. J., Craven, P., Owen, Z., Doveston, R. G., Churcher, I., et al. (2016). A divergent synthetic approach to diverse molecular scaffolds: assessment of lead-likeness using LLAMA, an open-access computational tool. Chem. Commun. 52, 7209–7212. doi: 10.1039/C6CC03244C
Congreve, M., Carr, R., Murray, C., and Jhoti, H. (2003). A 'rule of three' for fragment-based lead discovery? Drug Discov. Today 8, 876–877. doi: 10.1016/S1359-6446(03)02831-9
Cossío, F. P., Arrieta, A., and Sierra, M. A. (2008). The mechanism of the Ketene–Imine (Staudinger) reaction in its centennial: still an unsolved problem? Acc. Chem. Res. 41, 925–936. doi: 10.1021/ar800033j
Dar'in, D., Bakulina, O., Chizhova, M., and Krasavin, M. (2015). New heterocyclic product space for the Castagnoli–Cushman three-component reaction. Org. Lett. 17, 3930–3933. doi: 10.1021/acs.orglett.5b02014
Dewar, M. J. S., Zoebisch, E. G., Healy, E. F., and Stewart, J. J. P. (1985). Development and use of quantum mechanical molecular models. 76. AM1: a new general purpose quantum mechanical molecular model. J. Am. Chem. Soc. 107, 3902 – 3909. doi: 10.1021/ja00299a024
Flagstad, T., Min, G., Bonnet, K., Morgentin, R., Roche, D., Clausen, M. H., et al. (2016). Synthesis of sp3-rich scaffolds for molecular libraries through complexity-generating cascade reactions. Org. Biomol. Chem. 14, 4943–4946. doi: 10.1039/C6OB00961A
Galloway, W. R. J. D., Isidro-Llobet, A., and Spring, D. R. (2011). Diversity-oriented synthesis as a tool for the discovery of novel biologically active small molecules. Nat. Commun. 1:80. doi: 10.1038/ncomms1081
Gerry, C. J., and Schreiber, S. L. (2018). Chemical probes and drug leads from advances in synthetic planning and methodology. Nat. Rev. Drug Discov. 17, 333–352. doi: 10.1038/nrd.2018.53
Halgren, T. A. (1996). Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 17, 490–519. doi: 10.1002/(SICI)1096-987X(199604)17:5/6<490::AID-JCC1>3.0.CO;2-P
Hall, R. J., Mortenson, P. N., and Murray, C. W. (2014). Efficient exploration of chemical space by fragment-based screening. Prog. Biophys. Mol. Biol. 116, 82–91. doi: 10.1016/j.pbiomolbio.2014.09.007
Hawner, C., and Alexakis, A. (2010). Metal-catalyzed asymmetric conjugate addition reaction: formation of quaternary stereocenters. Chem. Commun. 46, 7295–7306. doi: 10.1039/c0cc02309d
Huigens, R. W., Morrison, K. C., Hicklin, R. W., Flood, T. A., Richter, M. F., and Hergenrother, P. J. (2013). A ring-distortion strategy to construct stereochemically complex and structurally diverse compounds from natural products. Nat. Chem. 5, 195–202. doi: 10.1038/nchem.1549
Innocenti, R., Lenci, E., Menchi, G., Pupi, A., and Trabocchi, A. (2017). Design and synthesis of bicyclic acetals as Beta Secretase (BACE1) inhibitors. Bioorg. Med. Chem. 25, 5077–5083. doi: 10.1016/j.bmc.2017.03.030
Kaminker, R., Kaminker, I., Gutekunst, W. R., Luo, Y., Lee, S., Niu, J., et al. (2018). Tuning conformation and properties of peptidomimetic backbones through dual N/Cα-substitution, Chem. Commun. 54, 5237–5240. doi: 10.1039/C8CC01356J
Khasanov, A. B., Ramirez-Weinhouse, M. M., Webb, T. R., and Thiruvazhi, M. (2004). Novel asymmetric approach to proline-derived Spiro-β-lactams. J. Org. Chem. 69, 5766–5769. doi: 10.1021/jo049430o
Kopp, F., Stratton, C. F., Akella, L. B., and Tan, D. S. (2012). A diversity-oriented synthesis approach to macrocycles via oxidative ring expansion. Nat. Chem. Biol. 2012, 358–365. doi: 10.1038/nchembio.911.
Lagorce, D., Sperandio, O., Baell, J. B., Miteva, M. A., and Villoutreix, B. O. (2015). FAF-Drugs3: a web server for compound property calculation and chemical library design. Nucleic Acids Res. 1, 200–207. doi: 10.1093/nar/gkv353
Lalli, C., Trabocchi, A., Sladojevich, F., Menchi, G., and Guarna, A. (2009). Diversity-oriented synthesis of morpholine-containing molecular scaffolds. Chem. Eur. J. 5, 7871–7875. doi: 10.1002/chem.200900744
Lenci, E., Innocenti, R., Biagioni, A., Menchi, G., Bianchini, F., and Trabocchi, A. (2016). Identification of novel human breast Carcinoma (MDA-MB-231) cell growth modulators from a carbohydrate-based diversity oriented synthesis library. Molecules 21:1405. doi: 10.3390/molecules21101405
Lenci, E., Innocenti, R., Menchi, G., Faggi, C., and Trabocchi, A. (2015a). Two-step one-pot synthesis of dihydropyrazinones as Xaa-Ser dipeptide isosteres through morpholine acetal rearrangement. Org. Biomol. Chem. 13, 7013–7019. doi: 10.1039/C5OB00783F
Lenci, E., Menchi, G., Guarna, A., and Trabocchi, A. (2015b). Skeletal diversity from Carbohydrates: use of mannose for the diversity-oriented synthesis of Polyhydroxylated compounds. J. Org. Chem. 80, 2182–2191. doi: 10.1021/jo502701c
Lenci, E., Rossi, A., Menchi, G., and Trabocchi, A. (2017). Short synthesis of polyfunctional sp3-rich threonine-derived morpholine scaffolds. Org. Biomol. Chem. 15, 9710–9717. doi: 10.1039/C7OB02454A
Lipinski, C., and Hopkins, A. (2004). Navigating chemical space for biology and medicine. Nature 432, 855–861. doi: 10.1038/nature03193
Lipinski, C. A. (1997). Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 46, 3–26. doi: 10.1016/S0169-409X(96)00423-1
Lipinski, C. A. (2004). Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov. Today 1, 337–341. doi: 10.1016/j.ddtec.2004.11.007
Lobell, M., Hendrix, M., Hinzen, B., Keldenich, J., Meier, H., Schmeck, C., et al. (2006). In silico ADMET traffic lights as a tool for the prioritization of HTS hits. ChemMedChem 1, 1229–1236. doi: 10.1002/cmdc.200600168
Lovering, F., Bikker, J., and Humblet, C. (2009). Escape from Flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem. 2009, 6752–6756. doi: 10.1021/jm901241e
Mannhold, R., Poda, G. I., Ostermann, C., and Tetko, I. V. (2009). Calculation of molecular lipophilicity: state-of-the-art and comparison of logP methods on more than 96,000 compounds. J. Pharm. Sci. 98, 861–893. doi: 10.1002/jps.21494
McLeod, M. C., Singh, G., Plampin, J. N., Rane, D., Wang, J. L., Day, V. W., et al. (2014). Probing chemical space with alkaloid-inspired libraries. Nat. Chem. 6, 133–140. doi: 10.1038/nchem.1844
Omidvari, Z., and Zarei, M. (2018). Synthesis of novel β-Lactams from Phenothiazin-10-ylacetic acid. J. Heterocyclic Chem. 55, 1085–1091. doi: 10.1002/jhet.3138
Pal'chikov, V. A. (2013). Morpholines. Synthesis and biological activity. Russ. J. Org. Chem. 49, 787–814. doi: 10.1134/S1070428013060018
Pedretti, A., Villa, L., and Vistoli, G. (2002). VEGA: a versatile program to convert, handle and visualize molecular structure on Windows-based PCs. J. Mol. Graph. 21, 47–49. doi: 10.1016/S1093-3263(02)00123-7
Ramaswamy, K., Forbes, L., Minuesa, G., Gindin, T., Brown, F., Kharas, M. G., et al. (2018). Peptidomimetic blockade of MYB in acute myeloid leukemia. Nat. Commun. 9, 1–13. doi: 10.1038/s41467-017-02618-6
Saleeb, M., Mojica, S., Eriksson, A. U., Andersson, C. D., Gylfe, A., and Elofsson, M. (2018). Natural product inspired library synthesis - Identification of 2,3-diarylbenzofuran and 2,3-dihydrobenzofuran based inhibitors of Chlamydia trachomatis. Eur. J. Med. Chem. 143, 1077–1089. doi: 10.1016/j.ejmech.2017.11.099
Sauer, W. H. B., and Schwarz, M. K. (2003). Molecular shape diversity of combinatorial libraries: a prerequisite for broad bioactivity. J. Chem. Inf. Comput. Sci. 43, 987–1003. doi: 10.1021/ci025599w
Sladojevich, F., Trabocchi, A., and Guarna, A. (2008). Stereoselective cyclopropanation of serine- and threonine-derived oxazines to access new morpholine-based scaffolds. Org. Biomol. Chem. 6, 3328–3336. doi: 10.1039/b808895k
Stotani, S., Lorenz, C., Winkler, M., Medda, F., Picazo, E., Martinez, R. O., et al. (2016). Design and synthesis of Fsp3-Rich, Bis-Spirocyclic-based compound libraries for biological screening. ACS Comb. Sci. 18, 330–336. doi: 10.1021/acscombsci.6b00005
Sun, L. (2013). Peptide-based drug development. Mod. Chem. Appl. 1:e103. doi: 10.4172/2329-6798.1000e103
Tan, D. S. (2005). Diversity-oriented synthesis: exploring the intersections between chemistry and biology. Nat. Chem. Biol. 1, 74–84. doi: 10.1038/nchembio0705-74
Teague, S. J., Davis, A. M., Leeson, P. D., and Oprea, T. (1999). The design of leadlike combinatorial libraries. Angew. Chem. Int. Ed. 38, 3743–3748. doi: 10.1002/(SICI)15213773(19991216)38:24<3743::AID-ANIE3743>3.0.CO;2-U
Trabocchi, A. (2013). Diversity-Oriented Synthesis: Basics and Applications in Organic Synthesis, Drug Discovery, and Chemical Biology. Hoboken, NJ: John Wiley & Sons.
Trabocchi, A., Lalli, C., Guarna, F., and Guarna, A. (2007). Diastereoselective synthesis of highly constrained Spiro-β-Lactams by the Staudinger reaction using an unsymmetrical bicyclic ketene. Eur. J. Org. Chem. 16, 4594–4599. doi: 10.1002/ejoc.200700260
Trabocchi, A., Mannino, C., Machetti, F., De Bernardis, F., Arancia, S., Cauda, R., et al. (2010). Identification of inhibitors of drug-resistant Candida albicans strains from a library of bicyclic peptidomimetic compounds. J. Med. Chem. 53, 2502–2509. doi: 10.1021/jm901734u
Trabocchi, A., Menchi, G., Guarna, F., Machetti, F., Scarpi, D., and Guarna, A. (2006). Design, synthesis and applications of aza 6,8-dioxa-bicyclo[3.2.1]octane-based scaffolds for peptidominetic chemistry. Synlett 3, 331–353. doi: 10.1055/s-2006-926249
Veber, D. F., Johnson, S. R., Cheng, H. Y., Smith, B. R., Ward, K. W., and Kopple, K. D. (2002). Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 6, 2615–2623. doi: 10.1021/jm020017n
Wells, J. A., and McClendon, C. L. (2007). Reaching for high-hanging fruit in drug discovery at protein–protein interfaces. Nature 450, 1001–1009. doi: 10.1038/nature06526
Wijtmans, R., Vink, M. K. S., Schoemaker, H. E., van Delft, F. L., Blaauw, R. H., and Rutjes, F. P. J. T. (2004). Biological relevance and synthesis of C-substituted morpholine derivatives. Synthesis 5, 641–662. doi: 10.1055/s-2004-816003
Xue, L., Stahura, F., and Bajorath, J. (2004). “Cell-based partitioning,” in Chemoinformatics, ed J. Bajorath (Totowa, NJ: Humana Press), 279–289
Yang, Y., Bai, Y., Sun, S., and Dai, M. (2014). Biosynthetically inspired divergent approach to Monoterpene Indole alkaloids: total synthesis of Mersicarpine, Leuconodines, B., and D, Leuconoxine, Melodinine, E., Leuconolam, and Rhazinilam. Org. Lett. 16, 6216–6219. doi: 10.1021/ol503150c
Keywords: chemical diversity, heterocycles, amino acids, carbohydrates, small molecules, building blocks, spiro-lactam
Citation: Lenci E, Innocenti R, Menchi G and Trabocchi A (2018) Diversity-Oriented Synthesis and Chemoinformatic Analysis of the Molecular Diversity of sp3-Rich Morpholine Peptidomimetics. Front. Chem. 6:522. doi: 10.3389/fchem.2018.00522
Received: 29 June 2018; Accepted: 10 October 2018;
Published: 30 October 2018.
Edited by:
Andrea Basso, Università di Genova, ItalyReviewed by:
Mads Hartvig Clausen, Technical University of Denmark, DenmarkBruno Linclau, University of Southampton, United Kingdom
Copyright © 2018 Lenci, Innocenti, Menchi and Trabocchi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elena Lenci, ZWxlbmEubGVuY2lAdW5pZmkuaXQ=
Andrea Trabocchi, YW5kcmVhLnRyYWJvY2NoaUB1bmlmaS5pdA==