 Yongqi Tian
Yongqi Tian Xiuping Lin2
Xiuping Lin2 Xuefeng Zhou
Xuefeng Zhou Yonghong Liu
Yonghong Liu- 1College of Biological Science and Technology, Fuzhou University, Fuzhou, China
- 2CAS Key Laboratory of Tropical Marine Bio-resources and Ecology, Guangdong Key Laboratory of Marine Materia Medica, South China Sea Institute of Oceanology, Chinese Academy of Sciences, Guangzhou, China
- 3University of Chinese Academy of Sciences, Beijing, China
Seven new phenol derivatives named coleophomones E and F (1, 2), diorcinols L and M (3, 4), 1-hydroxy-6-methyl-11-methoxy-8-hydroxymethylxanthone (5), porric acid E (6), and 7-(2-hydroxyphenyl) butane-7,8,9-triol (7), were isolated from the EtOAc extract of the marine sponge-derived fungus Didymellaceae sp. SCSIO F46, together with 10 known compounds. Their structures were determined by spectroscopic analyses, including NMR, MS, X-ray diffraction, and theoretical calculations. Each of 1 and 2 contains an unusual spiro [cyclohexane-1,2′-inden] moiety, which is relatively seldom in nature products. Cytotoxic and COX-2 inhibitory activities of all purified compounds were tested and evaluated. Compound 3 displayed obvious cytotoxicities against Huh-7, HeLa, DU145 and HL60 cells (IC50 values 5.7–9.6 μM) and weak activities against other five cell lines, while 8 showed weak cytotoxicities against HeLa and HL7702 cells. Compound 6 displayed COX-2 inhibitory activity with IC50 value of 3.3 μM.
Introduction
Natural products are still irreplaceable and continuing sources of novel drug leads, especially in the anti-infective area (Newman and Cragg, 2016). The marine ecosystem is one of the most complex and largest aquatic systems on earth, and host a huge microbial biodiversity (Agrawal et al., 2017; Corinaldesi et al., 2017). The unique and extreme characteristics of marine systems have driven a variety of biological adaptations, leading to the production of a large number of novel molecules for the treatment of many diseases (Gerwick and Fenner, 2013; Blunt et al., 2017). Marine sponges, a kind of precious marine organisms for new drug discovery, are hosts for a large community of microbes (up to 50–60% of the biomass of the sponge host) (Bergmann and Burke, 1955; Wang, 2006; Zhang et al., 2017). It was indicated that the symbiotic microbes of marine sponges might be the true producers of bioactive chemical defense substance of the sponge ecosystem (Richelle-Maurer et al., 2003; Thomas et al., 2010).
Sponge-derived fungi have been proven to be a treasure trove of novel biomolecules (Indraningrat et al., 2016; Blunt et al., 2017). During an ongoing search for new bioactive metabolites from the sponge-derived fungi (Tian et al., 2015a, 2018a,b), a strain of Didymellaceae sp. (SCSIO F46) isolated from a sponge Callyspongia sp. was subjected to chemical study. The EtOAc extract of rice fermentation of F46 showed toxicity against brine shrimp. Further isolation yielded seven new phenol derivatives, coleophomones E, F (1, 2), diorcinols L, M (3,4), 1-hydroxy-6-methyl-11-methoxy-8-hydroxymethylxanthone (5), porric acid E (6), and 7-(2-hydroxyphenyl) butane-7,8,9-triol (7), together with ten known compounds (Figure 1). The cytotoxic and COX-2 inhibitory activities of all compounds were evaluated. Details of the isolation structure elucidation, and bioactivity screening of these metabolites are reported herein.
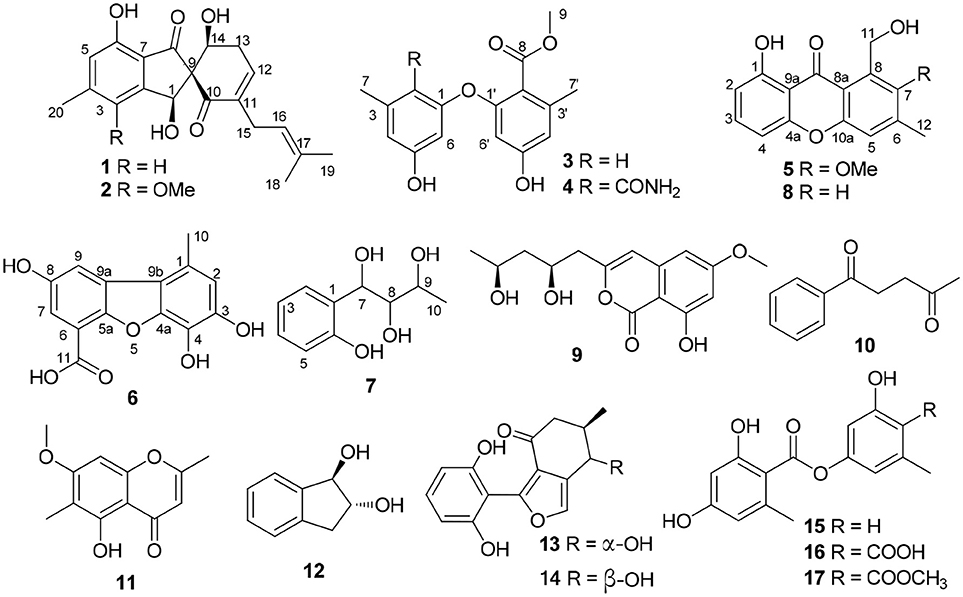
Figure 1. Structures of 1–17.
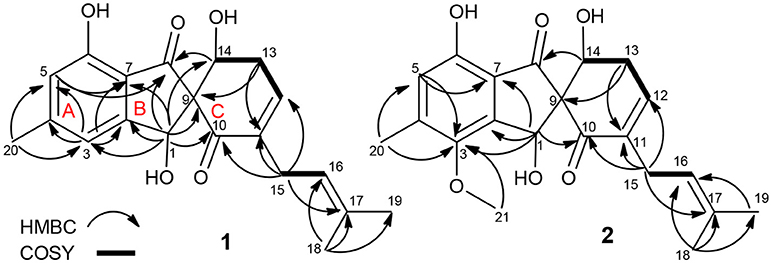
Figure 2. Key COSY and HMBC correlations of 1 and 2.
Materials and Methods
General Experimental Procedures
The NMR spectra were recorded on a Bruker AC 500 NMR (Bruker, Fällanden, Switzerland) spectrometer with TMS as an internal standard. HRESIMS data were measured on a Bruker micro TOF-QII mass spectrometer (Bruker, Fällanden, Switzerland). UV spectra were recorded on a Shimadzu UV-2600 UV-Vis spectrophotometer (Shimadzu, Kyoto, Japan). ECD spectra were performed on a Chirascan circular dichroism spectrometer (Applied Photophysics). X-ray diffraction intensity data were collected on a CrysAlis PRO charge-coupled device (CCD) area detector diffractometer with graphite monochromated Cu Kα radiation (λ = 1.54178 Å). Semi-preparative reversed-phase HPLC (RP-HPLC) was performed on a YMC-Pack Pro C18 RS column (5 μm, 250 × 10 mm id; YMC, Kyoto, Japan) with a Agilent 1260 separation module equipped with a Photodiode Array (PDA) detector. Silica gel GF254 used for TLC were supplied by the Qingdao Marine Chemical Factory, Qingdao, China. Sephadex LH-20 gel (GE Healthcare, Uppsala, Sweden) was used. Spots were detected on TLC under UV light or by heating by spraying with 12% H2SO4 in H2O.
Fungal Material
The fungal strain SCSIO F46 was isolated from a sponge Callyspongia sp., collected from the sea area near Xuwen County, Guangdong Province, China, during August 2013. The isolate was stored on MB agar (malt extract 15 g, sea salt 10 g, agar 15 g) slants at 4°C and then deposited at CAS Key Laboratory of Tropical Marine Bio-resources and Ecology. The fungus was identified using a molecular biological protocol by DNA amplification and sequencing of the ITS region. The nucleotide sequence of the ITS region reported in this article was assigned the GenBank accession number KU361223.
Extraction and Isolation
Didymellaceae sp. SCSIO F46 was cultured on MB-agar plates at 25°C for 7 days. The seed medium (malt extract 15 g, sea salt 10 g, distilled water 1,000 mL, pH 7.4–7.8) was inoculated with strain F46 and incubated at 25°C for 72 h on a rotating shaker (170 rpm). Mass scale fermentation of F46 was carried out using solid rice medium in 1,000 mL flasks (rice 200 g, sea salt 2.5 g, distilled water 200 mL), and inoculated with 10 mL of seed solution. Flasks were incubated at 25°C under normal day night cycle. After 30 days, cultures from 30 flasks were harvested. The culture of solid rice medium was soaked in acetone and cut into small pieces and kept for 1 day. The content was filtered and evaporated under vacuum and extracted with EtOAc thrice. The extract was partitioned between petroleum ether, and 90% aqueous MeOH to obtain the crude extract (43.0 g). The crude extract was subjected to silica gel column chromatography (CC) eluted with petroleum ether/EtOAc in a gradient eluent (v/v, 50:1, 30:1, 20:1, 10:1, 5:1, 1:1, 0:1) to obtain 8 fractions (fractions 1–8). Fr. 2 was subjected to ODS CC eluted with MeOH/H2O in a gradient eluent (1:9, 2:3, 3:2, 4;1, 9:1), to give 3 sub fractions (fr. 2.1–2.3). Fr. 2.2 was further purified by HPLC eluting with MeOH/H2O (60:40) to afford 9 (5.5 mg), 10 (4.7 mg), and 11 (3.8 mg). Fr. 3 was purified by Sephadex LH-20 (CH3Cl/MeOH, 1:1) to give 3 sub fractions (fr. 3.1–3.3). Fr. 3.2 was further purified by silica gel CC eluted with petroleum ether/EtOAc in a gradient eluent (v/v, 10:1) to obtain 5 (20.1 mg) and 8 (10.3 mg). Fr. 4 was subjected to an ODS column (MeOH/H2O: 10–100%) to give 4 sub fractions (fr. 4.1–4.4). Fr. 4.2 was further purified by HPLC eluting with MeOH/H2O (50:50, 1%0TFA) to afford 3 (20.8 mg). Compounds 16 (20.3 mg) and 17 (43.2 mg) were purified from Fr. 4.4 by HPLC (35% MeCN, 1%0 TFA). Fr.5 was purified by ODS CC (MeOH/H2O: 10–100%) and HPLC (35% MeCN, 1%0 TFA) to afford 1 (7.4 mg), 2 (4.3 mg) and 6 (5.5 mg). Compound 12 (2.3 mg) was isolated from Fr.6 by an ODS CC and followed by HPLC using 10% MeCN. Fr.7 was subjected to Sephadex LH-20 CC (CH3Cl/MeOH, 1:1) to provide 4 subfractions (Frs.7.1–7.4). Fr. 7.1 was further purified on HPLC by 25% MeCN (2.5 mL/min) to give 13 (32.3 mg), 14 (43.7 mg). Fr. 7.2 was purified on HPLC by 10% MeCN to give 7 (15 mg). Finally, compounds 15 (4.4 mg) and 4 (3.5 mg) were isolated from Fr.7.3 and Fr. 7.4 by HPLC using 25% MeCN and 30% MeCN, respectively.
Coleophomone E (1): Yellow amorphous solid; [α]25 D+14.1 (c 0.31, MeOH); UV (MeOH) λmax (log ε) 266 (3.16), 322 (2.25) nm, HRESIMS m/z 343.1541 [M + H]+ (calcd for C20H23O5, 343.1541); 1H and 13C NMR data, Table 1.
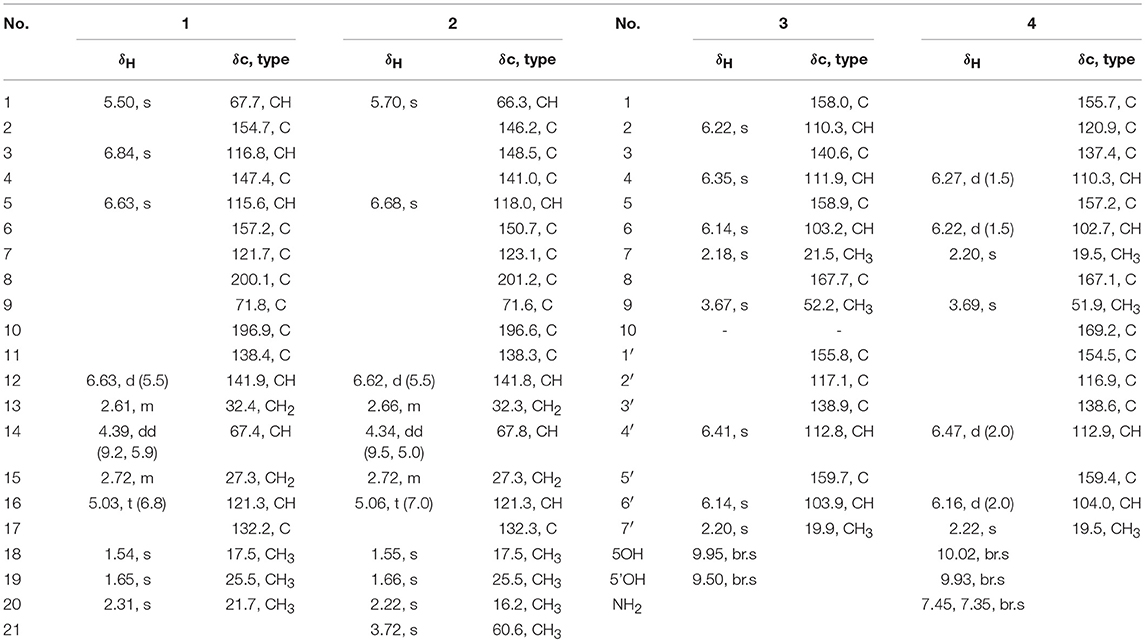
Table 1. 1H, 13C NMR data of 1–4 (500/125 MHz, in DMSO-d6, δ ppm, and J in Hz).
Coleophomone F (2): Yellow amorphous solid; [α]25 D+13.2 (c 0.46, MeOH); UV (MeOH) λmax 220 (3.85), 266 (3.20), 313 (2.43) nm, HRESIMS m/z 373.1638 [M + H]+ (calcd for C21H25O6, 373.1646); 1H and 13C NMR data, Table 1.
Diorcinol L (3): Yellow amorphous solid; HRESIMS m/z 289.1070 [M + H]+ (calcd for C16H17O5, 289.1071); 1H and 13C NMR data, Table 1; The structure of 3 have been deposited in the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 1502322.
Diorcinol M (4): Yellow amorphous solid; HRESIMS m/z 332.1127 [M + H]+ (calcd for C17H18NO6, 332.1129). 1H and 13C NMR data, Table 1.
1-Hydroxy-6-methyl-11-methoxy-8-hydroxymethylxanthone (5): Yellow needle crystals; HRESIMS m/z 287.0913 [M + H]+ (calcd for C16H15O5, 287.0919); 1H and 13C NMR data, Table 2.
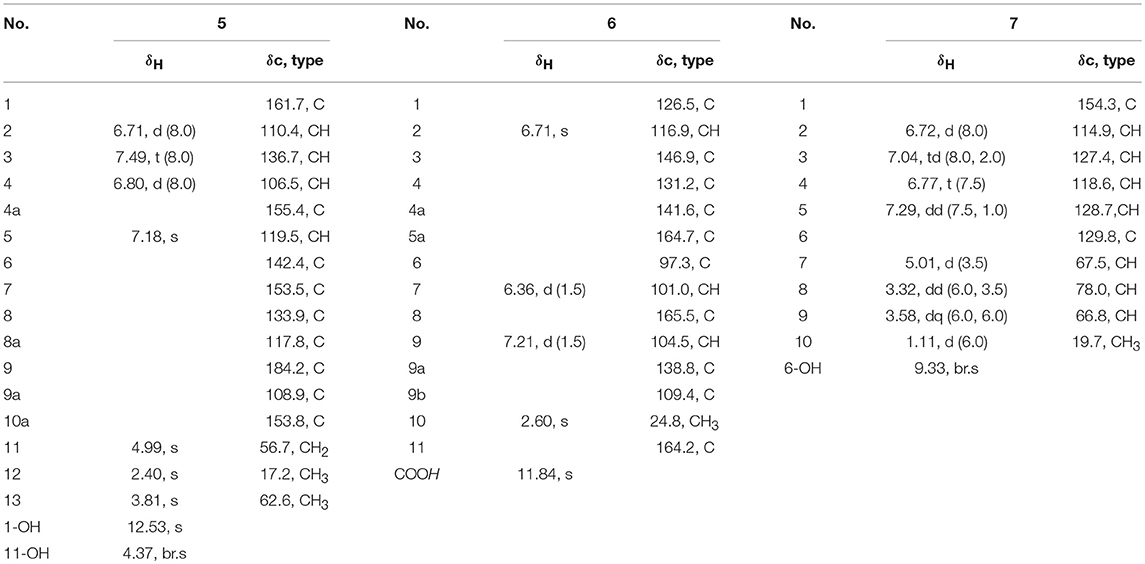
Table 2. 1H, 13C NMR data of 5–7 (500/125 MHz, in CDCl3, δ ppm, J in Hz).
Porric acid E (6): Colorless needle crystals; HRESIMS m/z 275.0544 [M + H]+ (calcd for C14H11O6, 275.0550); 1H and 13C NMR data, Table 2.
7-(2-Hydroxyphenyl) butane-7,8,9-triol (7): Yellow oil; HRESIMS m/z 221.0781 [M + Na]+ (calcd for C10H14NaO4, 221.0784); 1H and 13C NMR data, Table 2.
ECD Calculation
The eight possible stereoisomers (a-h) of 1 were initially performed using Confab (O'Boyle et al., 2011) with systematic search at MMFF94 force field. Room-temperature equilibrium populations were calculated according to Boltzmann distribution law. The conformers with Boltzmann-population of over 1% were chosen were chosen for ECD calculations using Gaussian 09 (Frisch et al., 2009) software, and the stable conformers were initially optimized at the B3LYP/6-311G(d,p) in methanol using the IEFPCM model. Vibrational frequency analysis confirmed the stable structures. Under the same condition, the ECD calculation was conducted using Time-dependent Density functional theory (TD-DFT). Rotatory strengths for a total of 30 excited states were calculated. The ECD spectrum was simulated in SpecDis (University of Würzburg) with a half-bandwidth of 0.3–0.4 eV, according to the Boltzmann-calculated contribution of each conformer after UV correction.
NMR Calculation
The two stereoisomers 1e and 1f were delivered to geometry optimization at B3LYP/6-31+G(d,p) in gas phase. The theoretical calculation of NMR was conducted using the Gauge-Independent Atomic Orbitals (GIAO) method at mPW1PW91/6-311+G(2d,p) in methanol by the IEFPCM model. Finally, the TMS-corrected NMR chemical shift values were averaged according to Boltzmann distribution for each conformer and fitting to the experimental values by linear regression. The calculated 13C-NMR chemical shift values of TMS in methanol were 0 ppm.
Cytotoxicity Assay
The cytotoxic activities of 1-17 were screened against the growth panel of 10 tumor cell lines (K562, MCF-7, A549, Huh-7, H1975, HeLa, HL7702, HL60, MOLT-4, and DU145) (Bergeron et al., 1994) (Supplementary Material).
COX-2 Inhibitory Activity Assay
According to the manufacturer's instructions. The test compounds were dissolved in DMSO and the final concentration was set as 10 μM. The percentage inhibition has been calculated by comparison with control incubations (Tian et al., 2015b) (Supplementary Material).
Results and Discussion
Compound 1 was assigned a molecular formula of C20H22O5 (10 degrees of unsaturation) by HRESIMS (m/z 343.1541 [M + H]+). Its NMR spectra showed resonances for three methyls, two methylenes, two oxygenated sp3 methine, one sp3 quaternary carbon, four sp2 methine, six sp2 quaternary carbon, and two ketocarbonyl carbons (δC 200.1 and 196.9) (Table 1). These data suggested a tricyclic skeleton of 1. The 1H NMR spectrum exhibited aromatic signals at δH 6.84 (s, H-3), 6.63 (s, H-5) indicating the presence of a tetrasubstituted aromatic ring. The HMBC correlations of H-3/C-2, C-4, C-5, C-7, and C-20, H3-20 (δH 2.31, s)/C-4, and C-5, and H-5/C-6, and C-7 suggested a methyl (C-20) and a hydroxy at C-4 and C-6, respectively. The HMBC correlations of H-1 (δH 5.50, s)/C-2, C-3, C-4, C-6, C-7, C-8 (δC 200.1) and C-9 (δC 71.8), and H-5/C-8 indicated linkage of ring B and the connection of C-1 to C-2 and C-7 to C-8. The COSY cross-peaks of H-13 (δH 2.61, m) /H-14 (δH 4.39, dd, J = 9.2, 5.9 Hz), and H-12 (δH 6.33, d, J = 5.9 Hz) delineated the spin system C12-C13-C14. Moreover, the HMBC correlations of H-12/C-10 and C-14, H-13/C-9, C-11, and C-12, and H-14/C-1, C-8 and C-9 indicated the presence of a α,β-unsaturated ketone (ring C) and rings B/C are connected by C-9. Finally, a 2-methyl-2 butene group was attached to C-11 (δC 138.4) by the evidence of the HMBC correlations of H3-18 (δH 1.54, s)/C-16 (δC 121.3), C-17 (δC 132.2), and C-19 and H2-15 (δH 2.72, s)/C-10, C-11, C-12, C-13, C-16, and C-17.
NOESY correlations and Mosher method were failure to determine the configurations of 1, so theoretical calculations were used to solve it. There are eight possible stereoisomers (a-h) of 1, as shown in Figure 3A. Computational studies of electron circular dichroism (ECD) were carried out. All stereoisomers (a–h) were selected for theoretical calculations using time dependent density functional theory (TDDFT) B3LYP/6-311G (d,p) level with the IEFPCM model in MeOH (Tables S1, 2, 4). A comparison of the experimental spectrum of 1 with the calculated ECD spectra of eight possible stereoisomers (a-h) were presented in Figure 3B. The measured ECD curve exhibits two negative Cotton effects (CEs) at 219 and 315 nm, and two positive cotton effects at 238 and 267 nm, matching well with the calculated ECD curve of 1e (1S, 9R, 14S) and 1f (1S, 9R, 14R). Then, computed 13C-NMR chemical shifts was carried out to define the stereochemistry of C-14. Computed 13C-NMR chemical shifts of each conformer were first Boltzmann-weighted averaged, and then fitted to experimental values by Ordinary Least Squares (OLS) Linear Regression method in order to remove systematic error that results from the conformational search and random error from experimental conditions (Tables S3, 5). As a result, the computed chemical shift for C-14 of 1e is δC = 68.7 ppm, with only a deviation of 1.3 ppm from the experimental value (δC = 67.4 ppm) (Table S8). All in all, the computed chemical shifts of 1e showed good agreement with the experimental values and has the higher R2 and R values than that of 1f, which suggesting that 1e (1S, 9R, 14S) be the true isomer of 1 (Figure 3C, Table S7).
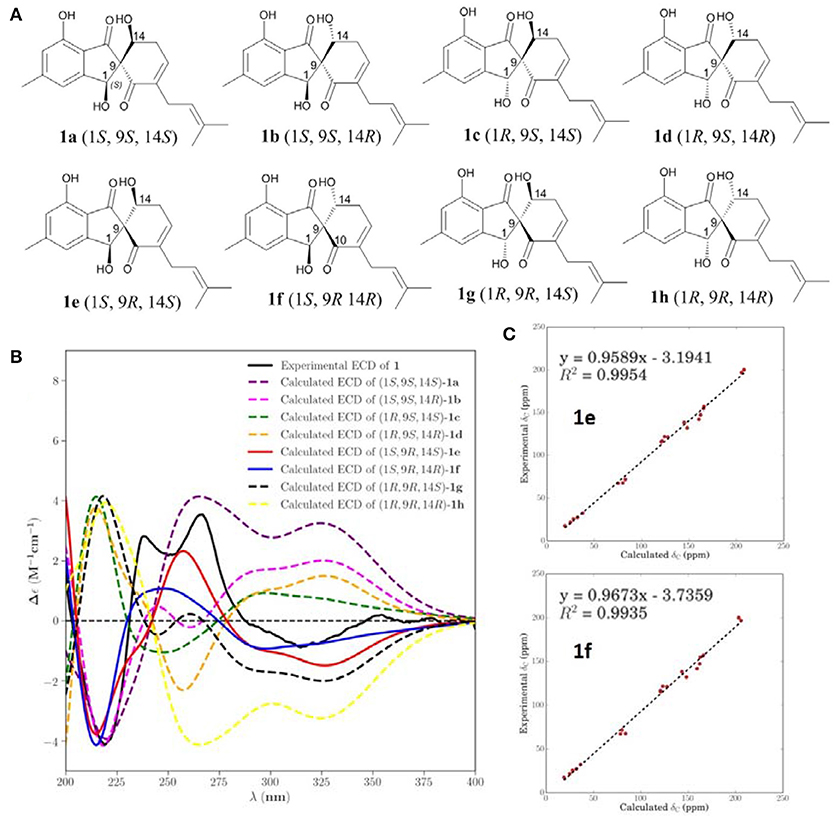
Figure 3. Calculated ECD and NMR studies of 1. (A) Eight possible stereoisomers (a-h) of 1. (B) Calculated ECD spectra of configurations a-h were compared with the experimental ECD spectra. (C) Linear regression fitting of calculated 13C-NMR chemical shifts of 1e and 1f with experimental values.
The molecular formula of 2 was determined as C21H24O6 by its HRESIMS (m/z 373.1638 [M + H]+), corresponding to 10 units of unsaturation. Its UV and NMR date were similar to those of 1, except for the presence of a methoxy (δH 3.72, δC 60.6) in 2 (Table 1). The extra methoxy (C-21) was located at C-3 by HMBC correlations from H3-21 to C-3 (δC 148.5) (Figure 2). The absolute configuration of 2 was suggested as (1S,9R,14S), as its chemical shift, coupling constant, optical rotation and CD effect (Table 1, Figure S20) almost the same as those of 1.
The molecular formula of 3 was assigned as C16H16O5 by its HRESIMS ion peak at m/z 289.1070 [M + H]+ and NMR date. The 1H NMR spectrum of 3 exhibited two hydroxyl proton at δH 9.95 and 9.50, five aromatic signals at δH 6.41, 6.35, 6.22, and 6.14×xs2, one O-methyl at δH 3.67, and two single methyls at δH 2.18 and 2.20 (Table 2). Analysis of the 13C and DEPT-135 NMR spectra of 3 indicated the presence of 16 carbons, including one carbonyl carbon (δC 167.7), 12 aromatic carbons (four oxygenated ones at δC 159.7, 158.9, 158.0 and 155.8), one methoxy and two methyls. These spectra of 3 were similar to those of diorcinol (Tian et al., 2015b), except for the presence of a metheyl formate group (one carbonyl carbon at δC 167.7 and one methoxy at δH 3.67/δC 52.2). However, relying solely on the NMR date, the location of metheyl formate was more difficult to determine. In order to determine location of metheyl formate of 3, a single-crystal X-ray diffraction pattern was obtained using the anomalous scattering of Cu Kα radiation shows an ORTEP drawing (Figure 4, Table S9) and unambiguously determined metheyl formate at C-2′. Thus, the structure of 3 was determined, and named as diorcinol L.
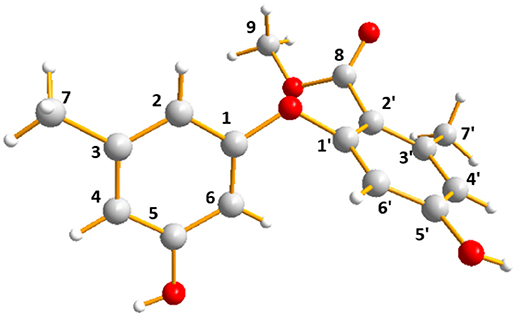
Figure 4. X-ray crystallographic structure of 3.
Compound 4 was obtained as brown powder. The molecular formula of was determined as C17H17NO6 by its HRESIMS (m/z 332.1131 [M + H]+), which corresponded to ten units of unsaturation. The 1H and 13C NMR data of 4 were similar to those of 3, except for the presence of one amide [δC 169.2 (CONH2)/ δH 7.45, 7.35 (CONH2)] (Table 1). The extra amide was determined at C-2 by the HMBC correlations of H-4, H-6/C-2, as well as H3-7/C-2, C-3, C-4, and C-10 (Figure 5). Hence, the structure of was elucidated and the trivial name diorcinol M was assigned.
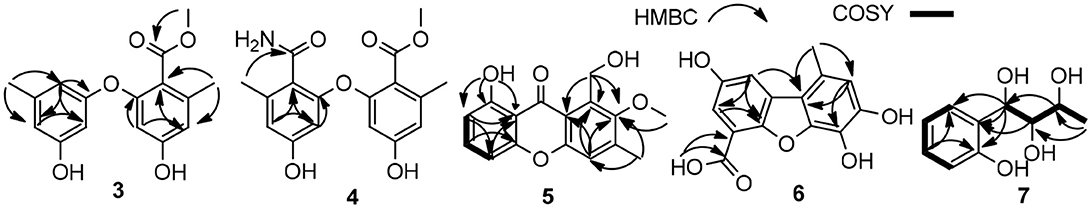
Figure 5. Key COSY and HMBC correlations of 3–7.
Compound 5 was isolated as pale yellow needle-like crystals. Its molecular formula was determined to be C16H14O5, by HR-ESI-MS, indicating 10 degrees of unsaturation. The 1D NMR data (Table 3) of 5 contained resonances for one carbonyl carbon, eight sp2 quarternary carbons, four sp2 methines, one sp3 methylene, two sp3 methys. Comparison of UV-vis and NMR data with those of 1-Hydroxy-6-methyl-8-hydroxy- methylxanthone (8) revealed a high degree of similarity skeleton, where the only obvious differences is in the presence of one methoxy (δH 3.81; δC 62.6) and low-field chemical shift of C-7 (from δC 127.2 in 8 to 153.5 in 1). Meanwhile, this difference can also be ascribed by the HMBC correlations (Figure 5) of δH (3.81, s, H-13) with δC (153.5, C-7) and requirements of HRESIMS spectrum. Thus, the structure of 5 was determined and named as 1-hydroxy-6-methyl-11-methoxy-8-hydroxymethylxanthone.
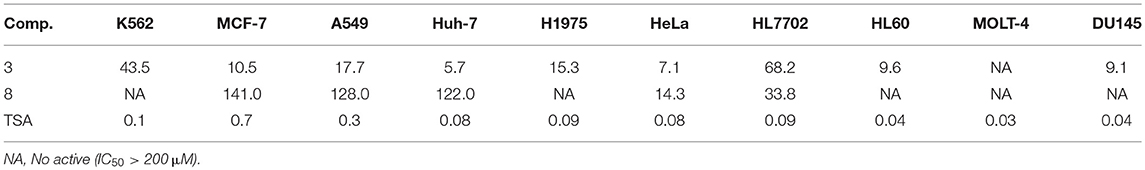
Table 3. Cytotoxic results of the compounds (IC50, μM).
Compound 6 showed a molecular ion peak at m/z 275.0544 [M + H]+ in the HR-ESI-MS, in accordance with the molecular formula C14H10O6, which was also supported by NMR data. The 1H NMR data displayed one sharp hydroxy proton singlet (11.84, s, COOH), two aromatic protons at δH 7.21 (1H, d, J = 1.5 Hz), 6.71 (1H, s), 6.36 (1H, d, J = 1.5 Hz), and a singlet methyl at δH 2.60 (3H, s). The 13C and DEPT-135 NMR spectra (Table 2) showed 14 carbons, including one carboxyl group (δC 164.2), three aromatic methine carbons (δC 116.9, 104.5, 101.0), 11 aromatic quaternary carbons (δC 165.5, 164.7, 146.9, 141.6, 138.8, 131.2, 126.5, 109.4, 97.3), and one methyl (δC 24.8). The aforementioned NMR data showed 1 was closely related structurally to the porric acid C (Carotenuto et al., 1998). The only difference was the substituent of C-4, the chemical shift to low field of C-4 (δC 131.2 in 4; δC 101.0 in porric acid C), the HMBC correlation (Figure 5, Table 2) from H-2 (δH 6.71, s) to C-3 (δC 146.9) and C-4 (δC 131.2) requirements of HRESIMS spectrum suggested that a hydroxy proton group was connected to C-4. Thus, the structure of 6 was determined and named porric acid E.
Compound 7 was obtained as a yellow oil. Its HRESIMS gave the molecular formula C10H14O4, requiring four degrees of unsaturation. The 1H NMR spectrum (Table 2) exhibited aromatic signals at δH 7.29 (dd, J = 7.5, 1.0 Hz, H-5), 7.04 (td, J = 8.0, 2.0 Hz, H-3), 6.77 (t, J = 8.0, Hz, H-4), and 6.72 (d, J = 8.0 Hz, H-2), indicating the presence of a disubstituted aromatic ring. The substitutions of a trihydroxybutyl group (C-7—C-10) and a hydroxy (δH 9.33, br.s, OH-6) at C-1 (δC 154.3) and C-6 (δC 129.8), were assigned by the COSY and HMBC interactions (Figure 5, Table 2).
Biological Activity
Cytotoxic activities of the isolated compounds 1–17 and trichostatin A (TSA, positive control) against 10 tumor cell lines (K562, MCF-7, A549, Huh-7, H1975, HeLa, HL7702, HL60, MOLT-4, and DU145) were tested. Among all of them, 3 displayed a wide range of cytotoxicities with IC50 values in the range of 5.7–68.2 μM, and 8 showed weak selective cytotoxicities against HeLa and HL7702 cells (Table 3). Furthermore, compound 6 displayed COX-2 inhibitory activity with the prominent IC50 values of 3.3 μM. Celecoxib was used as the positive control with IC50 values of 0.01 μM.
Conclusion
In conclusion, 17 phenol derivatives were isolated from the EtOAc extract of a marine sponge-derived fungus Didymellaceae sp. SCSIO F46. Coleophomones E and F (1 and 2) possess unprecedented spiro [cyclohexane-1,2′-inden] moiety, which is relatively seldom in natural products. Other new compounds 3-7 represent common types of phenol derivatives, which are widely found in fungal metabolites. Amongst, compounds 3 and 8 displayed a wide range of cytotoxicities against several tumor cell lines. In addition, 6 displayed significant COX-2 inhibitory activity with the prominent IC50 value of 3.3 μM.
Author Contributions
YT designed the experiments and performed the isolation and characterization of all the compounds and wrote the manuscript. XL performed the isolation and purification of the fungal strain. XZ designed the research work and revised the manuscript. YL contributed in project design and manuscript preparation. All authors reviewed the manuscript and approved for submission.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. 21502204, 31270402, 21172230, 81741154 and 41476135), the Foundation for Scholars of Fuzhou University (No. XRC-1672, 2017T022), the Scientific and Technological Project of Fuzhou City of Fujian Province (No. 2017-G-39), the Natural Science Foundation of Fujian Provincial Department of Education (No. JAT170077), Project from Department of Ocean and Fishery of Guangdong Province (GDME-2018C010), and the Open Project of CAS Key Laboratory of Tropical Marine Bio-resources and Ecology (2018011009) and Guangdong Key Laboratory of Marine Materia Medica (LMM2018-3).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2018.00536/full#supplementary-material
References
Agrawal, S., Acharya, D., Adholeya, A., Barrow, C. J., and Deshmukh, S. K. (2017). Nonribosomal peptides from marine microbes and their antimicrobial and anticancer potential. Front. Pharmacol. 8:828. doi: 10.3389/fphar.2017.00828
Bergeron, J. J., Brenner, M. B., Thomas, D. Y., and Williams, D. B. (1994). Calnexin: a membrane bound chaperone of the endoplasmic reticulum. Trends Biochem. Sci. 19, 124–128.
Bergmann, W. B., and Burke, D. C. (1955). Contributions to the study of marineproducts. XXXIX. The nucleosides of sponges. III. Spongothymidine and spongouridine. J. Org. Chem. 20, 1501–1507.
Blunt, J. W., Carroll, A. R., Copp, B. R., Davis, R. A., Keyzers, R. A., and Prinsep, M. R. (2017). Marine natural products. Nat. Pro. Rep. 34, 235–294. doi: 10.1039/c7np00052a
Carotenuto, A., Fattorusso, E., Lanzotti, V., and Magno, S. (1998). Porric acids A-C—new antifungal dibenzofurans from the Bulbs of Allium porrum L. Eur. J. Org. Chem. 29, 661–663.
Corinaldesi, C., Barone, G., Marcellini, F., Dell'Anno, A., and Danovaro, R. (2017). Marine microbial-derived molecules and their potential use in cosmeceutical and cosmetic products. Mar Drugs 15:118. doi: 10.3390/md15040118
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2009). Gaussian 09. (Wallingford, CT: Gaussian. Inc).
Gerwick, W. H., and Fenner, A. M. (2013). Drug discovery from marine microbes. Microb Ecol. 65, 800–806. doi: 10.1007/s00248-012-0169-9
Indraningrat, A. A., Smidt, H., and Sipkema, D. (2016). Bioprospecting sponge-associated microbes for antimicrobial compounds. Mar Drugs. 14:87. doi: 10.3390/md14050087
Newman, D. J., and Cragg, G. M (2016). Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 79, 629–661. doi: 10.1021/acs.jnatprod.5b01055
O'Boyle, N. M., Vandermeersch, T., Flynn, C. J., Maguire, A. R., and Hutchison, G. R. (2011). Confab-systematic generation of diverse low-energy conformers. J. Cheminformatics 3, 1–9. doi: 10.1186/1758-2946-3-8
Richelle-Maurer, E. G., Gomez, R., Braekman, J. C., van de Vyver, G., van Soest, R. W., and Devijver, C. (2003). Primary cultures from the marine sponge Xestospongia muta (petrosiidae, haplosclerida). J. Biotechnol. 100, 169–176. doi: 10.1016/S0168-1656(02)00251-1
Thomas, T., Rusch, D., DeMaere, M. Z., Yung, P. Y., Lewis, M., Halpern, A., et al. (2010). Functional genomic signatures of sponge bacteria reveal unique and shared features of symbiosis. ISME J. 4, 1557–1567. doi: 10.1038/ismej.2010.74
Tian, Y., Qin, X., Lin, X., Kaliyaperumal, K., Zhou, X., Liu, J., et al. (2015b). Sydoxanthone C and acremolin B produced by deep-sea-derived fungus Aspergillus sp. SCSIO Ind09F01. J. Antibiot. 68, 703–706. doi: 10.1038/ja.2015.55
Tian, Y. Q., Lin, S. N., Kaliyaperumal, K., Zhou, H., Wang, S. Y., and Liu, Y. H. (2018b). Polyketide-derived metabolites from the sponge-derived fungus Aspergillus sp. F40. Phytochem Lett. 27, 74–77. doi: 10.1016/j.phytol.2018.06.009
Tian, Y. Q., Lin, S. N., Zhou, H., Lin, S. T., Wang, S. Y., and Liu, Y. H. (2018a). Protuboxepin C and protuboxepin D from the sponge-derived fungus Aspergillus sp SCSIO XWS02F40. Nat. Prod. Res. 32, 2510–2515. doi: 10.1080/14786419.2017.1423303
Tian, Y. Q., Lin, X. P., Wang, Z., Zhou, X. F., Qin, X. C., Kaliyaperumal, K., et al. (2015a). Asteltoxins with antiviral activities from the marine sponge-derived fungus Aspergillus sp. SCSIOXWS02F40. Molecules 21:E34. doi: 10.3390/molecules21010034
Wang, G. (2006). Diversity and biotechnological potential of the sponge-associated microbial consortia. J. Ind. Microbiol. Biotechnol. 33, 545–551. doi: 10.1007/s10295-006-0123-2
Keywords: sponge-derived fungus, Didymellaceae sp., Phenol derivatives, cytotoxic, COX-2 inhibitory
Citation: Tian Y, Lin X, Zhou X and Liu Y (2018) Phenol Derivatives From the Sponge-Derived Fungus Didymellaceae sp. SCSIO F46. Front. Chem. 6:536. doi: 10.3389/fchem.2018.00536
Received: 09 August 2018; Accepted: 16 October 2018;
Published: 01 November 2018.
Edited by:
Xian-Wen Yang, Third Institute of Oceanography, State Oceanic Administration, ChinaReviewed by:
José Carlos Menéndez, Complutense University of Madrid, SpainShan He, Ningbo University, China
Copyright © 2018 Tian, Lin, Zhou and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuefeng Zhou, eGZ6aG91QHNjc2lvLmFjLmNu
Yonghong Liu, eW9uZ2hvbmdsaXVAc2NzaW8uYWMuY24=