 Yuri Alves de Oliveira Só1
Yuri Alves de Oliveira Só1 Pedro Henrique de Oliveira Neto1
Pedro Henrique de Oliveira Neto1 Luiz Guilherme Machado de Macedo2
Luiz Guilherme Machado de Macedo2 Ricardo Gargano1*
Ricardo Gargano1*- 1Institute of Physics, University of Brasília, Brasília, Brazil
- 2Institute of Biological Sciences, Faculty of Biotechnology, Federal University of Pará, Belém, Brazil
Although molecular collisions of noble gases (Ng) can be theoretically used to distinguish between the enantiomers of hydrogen peroxide - H2O2 (HP), little is known about the effects of HP-Ng interactions on the chiral rate. In this work, the chiral rate as a function of temperature (CRT) between enantiomeric conformations of HP and Ng (Ng=He, Ne, Ar, Kr, Xe, and Rn) are presented at MP2(full)/aug-cc-pVTZ level of theory through a fully basis set superposition error (BSSE) corrected potential energy surface. The results show that: (a) the CRT is highly affected even at a small decrease in the height of trans-barrier; (b) its smallest values occur with Ne for all temperatures between 100 and 4,000 K; (c) that the decrease of CRT shows an inverse correlation with respect to the average valence electron energy of the Ng and (d) Ne and He may be the noble gases more suitable for study the oriented collision dynamics of HP. In addition to binding energies, the electron density ρ and its Laplacian ∇2ρ topological analyses were also performed within the atoms in molecules (AIM) theory in order to determine the nature of the HP-Ng interactions. The results of this work provide a more complete foundation on experiments to study HP's chirality using Ng in crossed molecular beams without a light source.
1. Introduction
Hydrogen peroxide - H2O2 (HP) is a molecule of interest in a large and diverse number of fields in addition to its industrial uses. For example, it has emerged as a major metabolite in redox signaling and regulation (Antunes and Brito, 2017; Sies, 2017), and its presence was observed in Martian atmosphere (Encrenaz et al., 2004) and also on the surface of Jupiter's moon Europa (Carlson et al., 1999). The HP is interesting since it is simplest molecule that exhibits internal (torsional) rotation and chirality. Furthermore, this molecule can form dimers (Dobado and Molina, 1993; González et al., 1997), clusters (Yu and Yang, 2011), complexes with water (Mo et al., 1994; González et al., 1997) and with biologically important molecules such as adenine (Dobado and Molina, 1999), DNA (Piatnytskyi et al., 2016), glycine (Shi and Zhou, 2004) or nitrosamines (Roohi et al., 2010). These features indicate that HP should be a better proton donor for hydrogen bonding than water. Thus, the understanding of how the relative orientation of the O-H can lead to a weakly complex or a chemical reaction has also been paid considerable attention due to its implication in atmospheric chemistry and oxidation reactions (Lundell et al., 1998, 2001; Daza et al., 2000; Goebel et al., 2000, 2001a,b, 2002; Molina et al., 2002; Pehkonen et al., 2004; Mucha and Mielke, 2009; Grzechnik et al., 2013). Moreover, HP's properties have been investigated, such as its isolated chirality (Roncaratti and Aquilanti, 2010), stereomutation (Fehrensen et al., 2007; Bitencourt et al., 2008), size-dimensional wave packets (Wang et al., 2012), spectroscopy (Hunt et al., 1965; Małyszek and Koput, 2013; Al-Refaie et al., 2015) and rotation barriers (Song et al., 2005).
On the other hand, the hydrogen peroxide seems to be a prototypical model to be used into experiments to observe chirality in crossed molecular beam without a light source (Palazzetti et al., 2013), a frontier in research of stereodynamics which is still at early stages (Su et al., 2013; Lombardi and Palazzetti, 2018). In these kind of experiments, the molecular orientation control on the intense continuous beam is mandatory to the phenomena of chiral selectivity to be demonstrated (Aquilanti et al., 2005). For this reason, the interaction between HP and atoms, molecules and ions is so relevant to sterodynamics studies (Barreto et al., 2007, 2010; Lombardi et al., 2011; Roncaratti et al., 2014; Leal et al., 2016).
In the present paper we investigated the dynamics of the chiral molecule HP interacting by van der Waals forces with noble gases Ng (Ng=He, Ne, Ar, Kr, Xe and Rn) in order to obtain the chiral rate as a function of temperature (CRT) for these complexes, as well as its consequences for the chiral kinetic interconversion when tunneling effect is included. The aim of this work was to understand how the kinetic interconversion of the two HP's isomers is affected along collisional events through a basis set superposition error (Boys and Bernardi, 1970) (BSSE) corrected potential energy surface (PES). In addition, the electron density ρ(r) and its Laplacian ∇2ρ(r) topological analyses were also performed within the atoms in molecules (AIM) theory in order to determine the nature of the intermolecular interactions.
2. Methodology
2.1. Computational Details
All calculations were performed using the Gaussian09 package (Frisch et al., 2009). The structures were optimized without constraints at MP2(full) method in conjunction with aug-cc-pVTZ-PP for Xe and Rn (Peterson et al., 2003) and aug-cc-pVTZ for the remaining atoms (Dunning, 1989; Woon and Dunning Jr, 1993; Wilson et al., 1999). Vibrational frequencies at the same level of theory were also performed in order to ensure that each minimum has only positive frequencies and that each transition state has only a single imaginary frequency, as well as to obtain the zero point vibrational energy (ZPE). The counterpoise method of Boys and Bernardi (1970) was used to correct the BSSE for binding energy.
AIM analysis (Matta and Boyd, 2007) and graphic representations were performed with the AIMALL program (Keith, 2017) using the MP2(full) density (wavefunction) as input as described in the AIM theory (Dobado et al., 1998; Cortés-Guzmán and Bader, 2005).
2.2. Overview of the Transition State Theory
The transition state theory (TST)(Truhlar et al., 1996) was developed primarily by Henry Eyring (Eyring, 1935) and Michael Polanyi (Polanyi and Wigner, 1928) between 1928 and 1935. The TST is an improvement over the so-called theory of collisions (Lewis, 1967), and it is widely used to calculate the rate constants of chemical reactions.
The start point of TST is the existence of a transition state (TS) between the reagents and products. Located at the top of the potential energy barrier and it assumes a quasi-equilibrium between reactants and activated transition state complexes. For a bimolecular reaction given by
The TS is characterized by a single imaginary frequency along the reaction coordinate of the molecular system which is represented here by . In its turn, the reaction coordinate can be represented by angular changes in bond distances during the chemical reaction (Henkelman et al., 2002).
The equation that determines the reaction rate is known as the Eyring equation, given by
where 0 < κ(T) ≤ 1 is the so-called transmission coefficient, kB is the Boltzmann constant, h is the Planck constant, is the standard molar partition function, NA is the Avogadro constant, R is the gas constant and is the barrier energy with zero-point energy correction. In addition, the TS, R1 and R2 subscripts stand for the transition state and reagents, respectively. Thus, the rate constant is determined by the parameters that characterize both reagents and the TS.
The general partition function is formed by the product of translational qtrans., rotational qrot., vibrational qvib. and electronic qele. partition functions. The translational partition function for a free particle with mass m moving along the length dimension lx can be evaluated by considering that the separation of energy levels is small and that a large number of states are accessible at room temperatures. Therefore, the energy levels should be continuous and the sum contribution of the translational partition function becomes an integral. Which the solution for the three-dimensional case is (Atkins et al., 2013)
Although the system can be excited at normal modes, the energy levels are discrete for the rotational mode. The three degrees of freedom of spatial rotation and the three moments of inertia IA, IB and IC must be taken into account for a non-linear molecule (Atkins et al., 2013), thus
where σ is the so-called number of symmetry. The vibrational mode has reasonably spaced energy which must be taken into account since they are partially occupied. As a consequence, the vibrational partition function is strictly calculated as a sum over the occupied states. In the case of n vibrational degrees of freedom, the vibrational partition function is given by the product of n partition functions,
where νi is each of the fundamental vibrational frequencies. In most cases, only the lowest energy state is occupied and the electronic energies should not contribute considerably to the total partition function (Atkins et al., 2013). A good approximation is to disregard the contributions of the nuclear and electronic spins and to vanish the fundamental energy level for the electronic partition function. Under these considerations the electronic partition function should be equal to unity (Atkins et al., 2013)
On the other hand, the coefficient κ(T) represents the tunneling effect of the reaction coordinate of the chemical system and it is usually important for light atoms or molecules at low temperatures. Thus, tunneling estimates were made using both Wigner (Polanyi and Wigner, 1928) and Eckart (Eckart, 1930) methods.
The Wigner tunneling correction proposes a parabolic potential,
where Eb corresponds the energy potential barrier of MEP, is the imaginary frequency of transition state and s is the coordinate reaction. This implies in a transmission coefficient given by Bell (1959)
For very low temperatures, the Wigner tunneling effect is not very effective, and for this reason, it was also employed Eckart tunneling correction (Truhlar et al., 1985).
The Eckart tunneling correction uses a potential of the type
where α is a parameter described by
and μ is the reduced mass of the system. These parameters determine the barrier width. Here it is important to note that the A and B can be positive, negative or zero. The A parameter corresponds to the energy difference V(x → −∞) and V(x → +∞), and B is a parameter that measures the height of the barrier given by
So the most usual form for the Eckart's potential in the study of reaction rates is (Truhlar et al., 1985)
where s is the coordinate of the reaction and s0 is the reaction coordinate corresponding to the maximum of the barrier, which is given by
Finally, the transmission probability (Bell, 1980), obtained through the solution of the Schrödinger equation with Eckart's potential, is expressed by the following equation
where k, β and δ depend on , A, B and energy (E).
The quantum tunneling correction κ(T) can thus be calculated from the ratio between the quantum rate kquan.(T) and the classical rate kclass.(T) in which the particles cross the barrier. Thus, the Eckart tunneling correction with transmission coefficient is given by
where integration is performed over all possible energies.
3. Results and Discussion
3.0.1. Geometric Parameters, Interactions and AIM Analysis
The details about the generation of the potential energy surface are described in another work of our group (Roncaratti et al., 2014), so it will be commented briefly here. First, all HP geometry parameters were kept frozen at their equilibrium values of DOO = 1.45Å, DOH = 0.966Å and the angle HOO = 100.8°. The Ng's position is expressed in terms of the polar coordinates as represented in Figure 1, where R is the distance of Ng relative to the middle of O-O bond and α is the polar angle with respect to an axis perpendicular to the O-O bond (z axis). The two planes defined by O-O-H atoms are then rotated around the O-O axis, with steps of 1°. In addition, α was equal to 0°, 45°, 90° and R distance was varied from 2 to 5Å with steps 0.1Å.
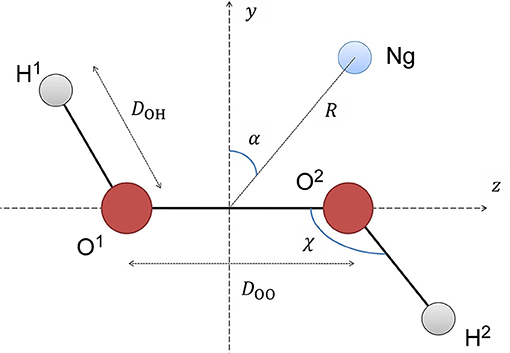
Figure 1. Definitions of the coordinate system used to represent the H2O2-Ng PES (see text for more details).
Topological studies performed on this adjusted potential energy surface (PES) showed that the HP and HP-Ng complexes have two overall minimum configurations, termed cis (labeled as θ−) and trans (labeled as θ+), separated by two potential barriers, denoted here as cis-barrier and trans-barrier. The potential energy curves (PEC) obtained from the PES are then presented in Figure 2.
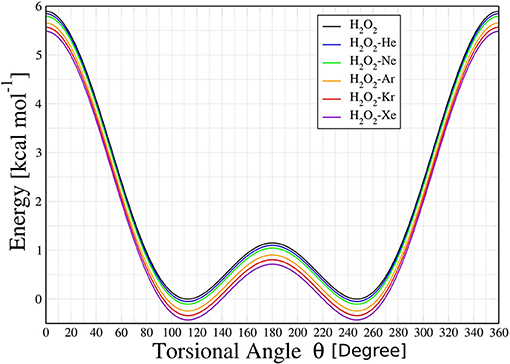
Figure 2. Potential energy curves for H2O2 and H2O2-Ng (Ng = He, Ne, Ar, Kr, and Xe) complexes as a function of torsional angle θ (in degrees).
The PECs obtained for HP-Ng complexes are similar in shape and depth. The cis-barriers for the HP-Ng complexes are all smaller than the respective value for the free HP. The free HP has a cis-barrier of 7.5594 kcal/mol whereas the values for the complexes increase monotonically from 6.9828 kcal/mol for HP-Rn up to 7.5107 kcal/mol for HP-He. In addition the trans-barrier values are also lower than the respective value for the free HP, which is 1.0427 kcal/mol, and their values are 1.0928 kcal/mol for HP-He, 1.0817 kcal/mol for HP-Ne, 1.0651 kcal/mol for HP-Ar, 1.0676 kcal/mol for HP-Kr, 1.0736 kcal/mol for HP-Rn and 1.0749 kcal/mol for HP-Xe complexes. For the free HP, the cis-barrier and trans-barrier experimental energies (Hunt et al., 1965) are 7.0334 ± 0.0715 kcal/mol and 1.1036 ± 0.0114 kcal/mol, respectively. These values are in a good agreement with our results. However, for the HP-Ng complexes we did not find experimental data for comparison.
The results concerning geometric parameters, interactions and their characterization are summarized in Tables 1–3. The geometrical parameters obtained at MP2(full)/aug-cc-pVTZ, optimized without any constraints for two minimum structures and transition states (presented as cis and trans barriers) are given in Table 1 together with the graphical representation in Figure 3. Table 2 lists the binding energies corrected and uncorrected for the BSSE. Table 3 shows the numerical results for AIM analysis and Figure 4 depicts the ∇2ρ(r) contour plots for cis and trans barrier configurations.
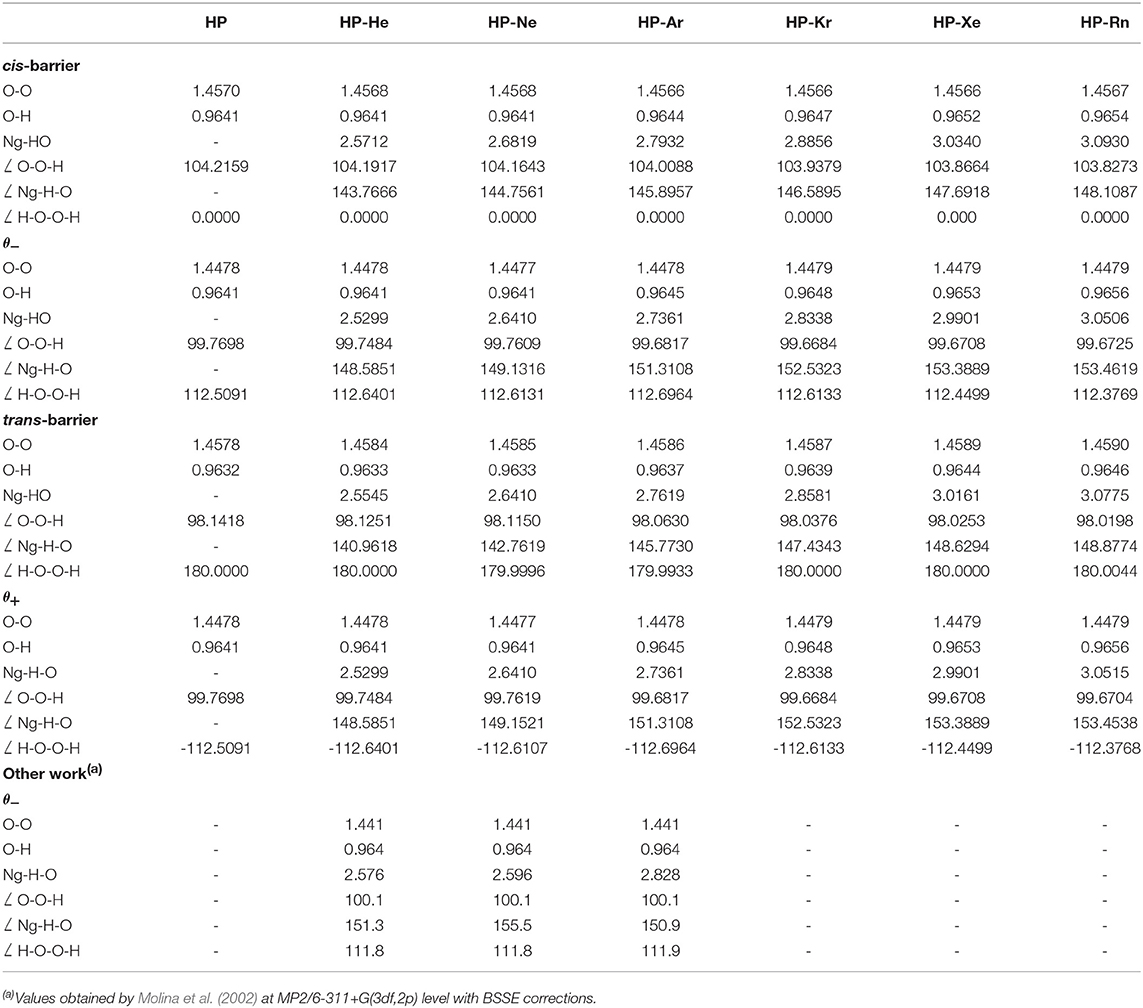
Table 1. Geometrical parameters (in Å and degree) obtained at MP2(full)/aug-cc-pVTZ level for isolated HP and HP-Ng (Ng=He, Ne, Ar, and Kr) complexes and MP2(full)/aug-cc-pVTZ-PP level for HP-Ng (Ng=Xe and Rn) complexes.
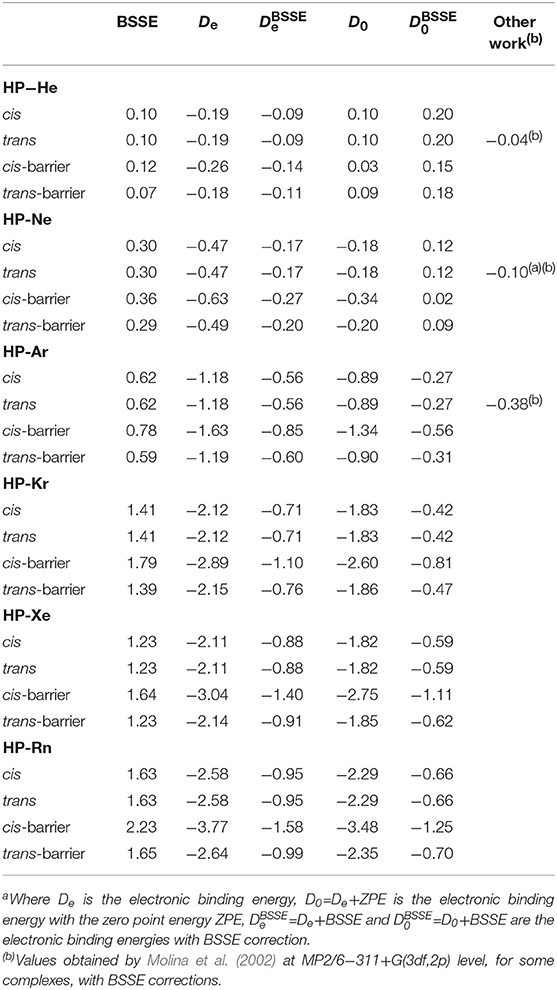
Table 2. Binding energies (in kcal/mol) of HP−Ng complexes obtained at MP2(full)/aug−cc−pVTZ level for HP−Ng (Ng=He, Ne, Ar, and Kr) and MP2(full)/aug−cc−pVTZ−PP level for HP−Ng (Ng=Xe and Rn)a.
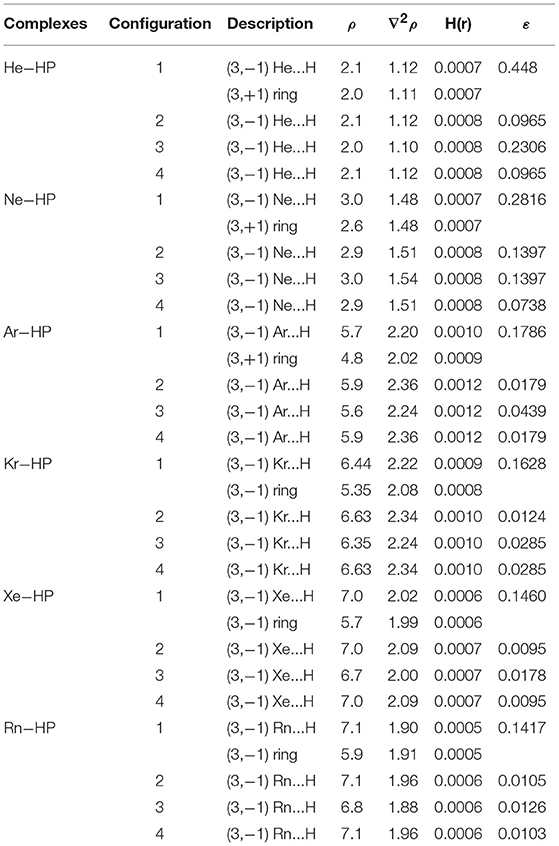
Table 3. Bond critical point (BCP) data for charge density ρ (in ), Laplacian of the charge density ∇2ρ (in ), electronic energy density H(r) and ellipticity ε for configurations 1(cis), 2(cis-barrier), 3(trans) and 4(trans-barrier) of the HP-Ng complexes.
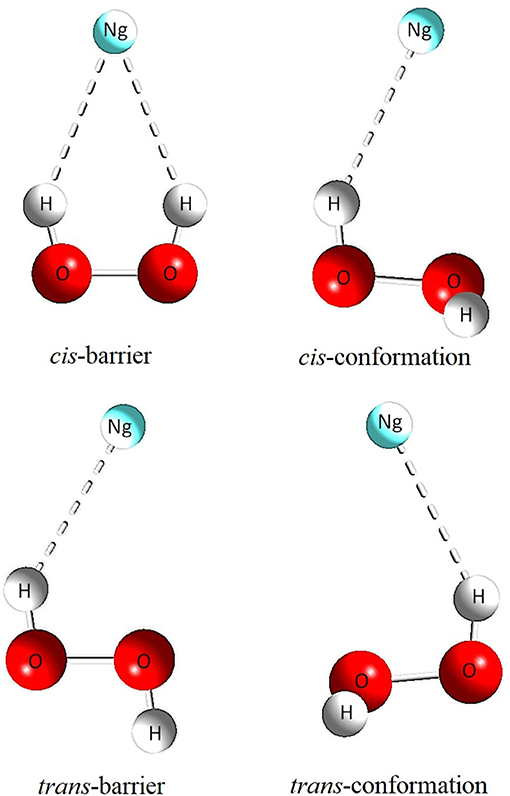
Figure 3. Geometric representation of the cis-barrier, θ−, trans-barrier and θ+ structures.
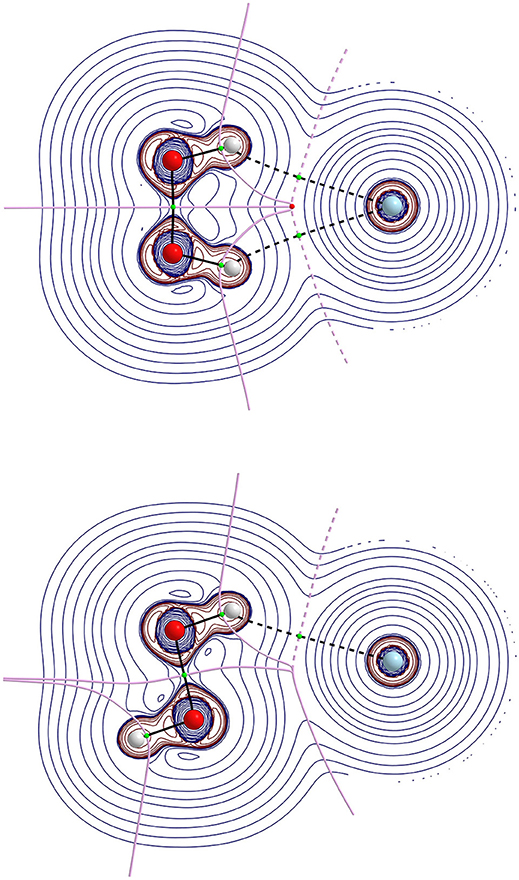
Figure 4. Laplacian of the electron density, ∇2ρ, contours map in the molecular plane for cis-barrier (top) and trans-barrier (bottom) conformations of HP-Ne at MP2(full)/aug-cc-pVTZ level. The thick solid lines represent the molecular graph that joints the nuclei, the bond critical point and ring critical point, and also represent the zero flux surface.
The PESs yield θ− and θ+ as true minima, i.e. without any imaginary frequencies in accordance with results from literature (Maciel et al., 2006; Roncaratti et al., 2014). In addition, all transition state structures displayed a well characterized imaginary frequency around 600 cm−1 for cis and 400 cm−1 for trans barriers (see Supplementary Information for further details). Figure 5 describes a schematic representation of vibrational modes of the isolated HP in the transition state with the actual frequencies and an imaginary frequency, which represents the frequency along the reaction coordinate.
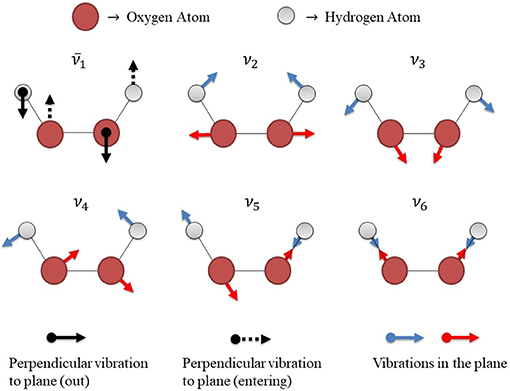
Figure 5. Schematic representation of vibrational modes of the H2O2 isolated in the transition state. There are 6 vibrational modes, 5 of them correspond to the actual frequencies (ν2, ν3, ν4, ν5, and ν6) and one of them corresponds to the imaginary frequency , which represents the frequency along the reaction coordinate.
For the HP-Ng complexes, the geometrical parameters are almost the same when compared with isolated HP in agreement with the weak interaction of these systems. The HP-Ng distances increase from He up Rn. On average, they are close to 2.55Å(He), 2.65Å(Ne), 2.75Å(Ar), 2.85Å(Kr), 3.00Å(Xe), and 3.06Å(Rn).
Regarding the binding energies, HP-He and HP-Ne are all repulsive, being less repulsive for the cis barrier configuration. This can be understood as a consequence of the fact that the noble gases turn out to be the hardest elements (Furtado et al., 2015) and this hardness decreases when the Ng atomic number is increased (the hardness in this context is a resistance to changes in its electronic population Furtado et al., 2015 coupled to Ng's high electronegativity Allen and Huheey, 1980). Although the BSSE increases monotonically from He to Rn, the binding energies also become more attractive as the atomic number increases.
For the four structures of each HP-Ng, the higher binding values are always observed for the cis-barrier configuration. As it will pointed latter, the decrease of the rate through the two barriers are not correlated with the binding energy, suggesting the hyperconjugation effects on HP may be important for the decrease of the interaction rate.
Regarding the AIM analysis, the existence of (3,−1) bond critical point (BCP) and its associated atomic interaction line indicates that electronic charge density is accumulated between the linked nuclei (Bader, 1991). In its turn, the values of the charge density ρ(r) in BCP are small while their corresponding ∇2ρ(r) are positive in accordance with a closed shell type of interaction. As a consequence, all configurations of all complexes show an interaction of a van der Waals type. Since higher ellipticity suggests conjugation and hyperconjugation effects of electron delocalization, these effects seem more pronounced in the HP-He and HP-Ne complexes. Another interesting feature is that all cis-barrier configurations of all complexes show a (3,+1) BCP indicating a cyclic nature.
3.0.2. Thermal Chiral Rate Analysis
The temperature dependence of the rate constant for cis to trans (i.e., through trans-barrier) and trans to cis (i.e., through cis-barrier) conformations for HP and HP-Ng complexes are presented in Figure 6. These results, in addition to conventional rate, are also exhibited with Eckart's or Wigner's tunneling corrections.
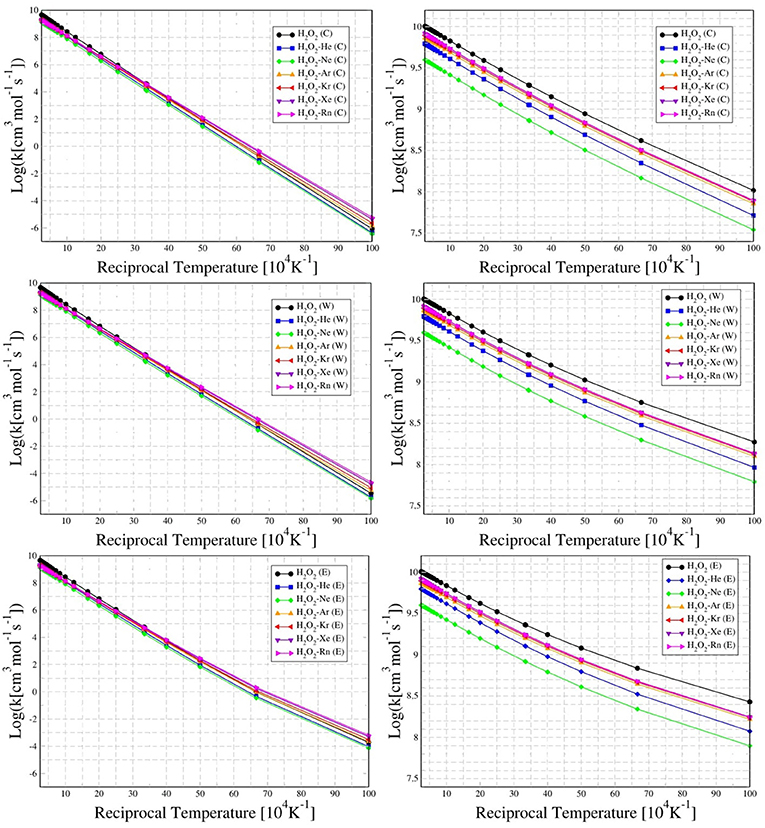
Figure 6. Temperature dependence (from 100 K up to 4,000 K) of the rate constant for conventional (C), Wigner (W), and Eckart (E) tunneling corrections, for cis to trans (left column) and trans to cis (right column) chiral conformations of H2O2 and H2O2-Ng complexes.
It was found that for the entire 100 K up to 4,000 K range the HP-Ne has the lowest rates for both barriers among all noble gas complexes, followed by HP-He. This result suggests that Ne and He are the noble gases more suitable for study the oriented collision dynamics with HP. In fact, the decrease of CRT shows an inverse correlation with respect the average valence electron energy (Allen, 1989), which follows the sequence (from higher to lower values): Ne, He, Ar, Kr; with Xe and Rn having very close values.
Nevertheless, there is a trend of rate increase as are move from Ar up to Rn. It is interesting to note that although this behavior is very similar regarding the cis-barrier for all rates (conventional, Wigner e Eckart), it seems that the tunneling is more important to describe the trans barrier's rate, where there is a significant difference for Eckart's values specially in the 100–200 K range when compared to respective Wigner and conventional results.
The final thermal rate constant can be expressed in the two familiar Arrhenius forms. In this work, the first is the Arrhenius modified form given by
where A is the pre-exponential factor, T a temperature, n is a real number, R is the universal gas constant and Ea is the activation energy. The second is the d-Arrhenius form (Aquilanti et al., 2010; Silva et al., 2013; Carvalho-Silva et al., 2017) expressed by
where d is a parameter that yield the degree of deformation of the exponential function.
The curve obtained by the reaction rate constant vs. the temperature can be fitted (Ramalho et al., 2011) to obtain the parameters A, n and Ea for the Arrhenius modified form, as presented in Table 4, and the parameters A, d, and Ea for d-Arrhenius form, as presented in Table 5. This feature confirms the trend of lower k(T) observed for HP-Ng complexes for both barriers when compared to isolated HP.
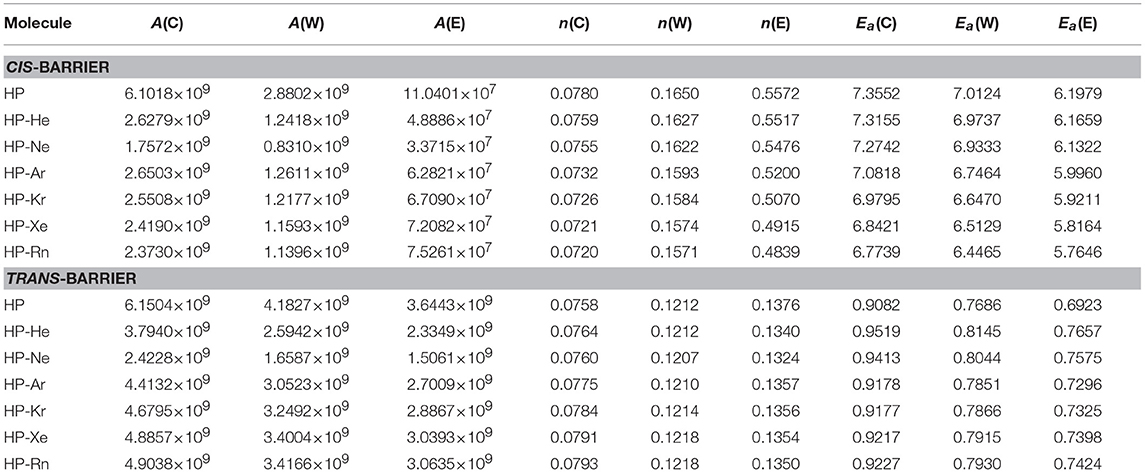
Table 4. Adjusted parameters for the modified Arrhenius equation for conventional (C), Wigner (W) and Eckart (E) models with Ea in kcal/mol.
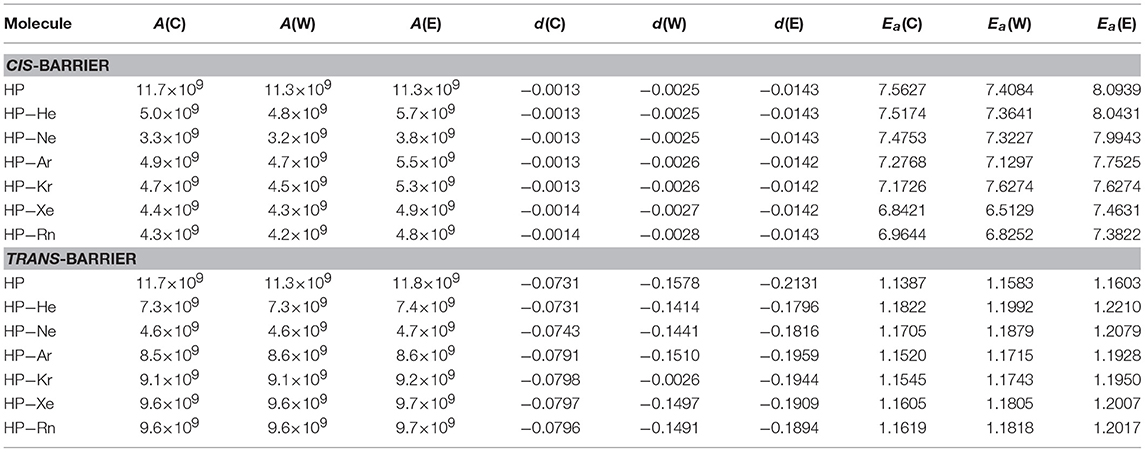
Table 5. Adjusted parameters for the d-Arrhenius equation for conventional (C), Wigner (W) and Eckart (E) models with Ea in kcal/mol.
It can be also observed in Figure 6 that the trans to cis conformation rate of HP is lower (in the range 100–200 K) than the corresponding ones for HP-Ar, HP-Kr, HP-Xe, and HP-Rn. In the case of the chiral transition from cis to trans, the rates of all HP-Ng complexes are lower than that of the isolated HP. These results showed that the transition rate from cis to trans is greater than the corresponding trans to cis for both the isolated HP molecule and for all HP-Ng complexes. This suggests that the most important barrier that separates the chiral configurations of the isolated HP and the HP-Ng complexes is the trans-barrier, since it is the smallest. The energy of the HP's trans-barrier is relatively small (1.0427 kcal/mol) compared to its cis-barrier (7.5595 kcal/mol) as already seen in Figure 2.
An interesting result is presented in the Table 6. Although the increase in the trans-barrier of the HP-Ng complexes relative to HP is considerably small (see Figure 7), the change in the transition rate from cis to trans is relatively high. This is verified for high (4,000 K), room (298.15 K) and also for low temperatures (100 K). The most pronounced decrease in the rate corresponds to the HP-Ne complex, in which the decrease of the trans-barrier of just 0.0389 kcal/mol (see Figure 6) corresponds to a decrease of over 60% for the cis-trans transition rate, followed by HP-He. It is also interesting to note that this small change in energy barrier but with a substantial change in rate was also observed for other HP-Ng complexes. For example, HP-Ar complex showed a decrease of just 0.0224 kcal/mol but a 28.68% decrease of rate at 100 K.
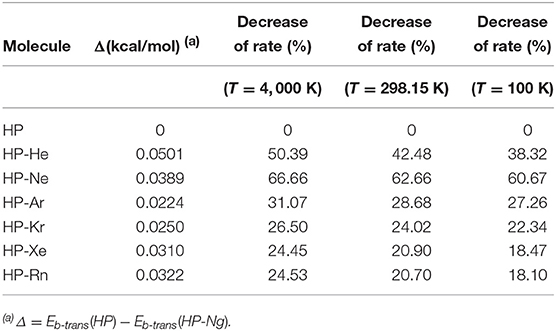
Table 6. Difference between heights of trans-barrier of HP and HP-Ng complexes and relative decrease of the transition rate of cis to trans configuration for representative temperatures (4, 000, 298.15, and 100 K).
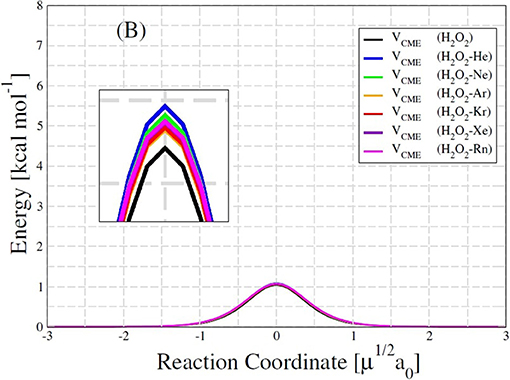
Figure 7. Trans-barrier (B) energies of HP and H2O2-Ng complexes.
Finally, at a temperature close to 300K, the Boltzmann distribution shows that about 16% of HP's population has higher energy than the trans-barrier with thermal fluctuations of approximately 1.7686 kcal/mol (Ball and Brindley, 2016). It has also been found that at low temperatures the chiral interconversion quantum encapsulation time of HP is very small. At a temperature of 100 K this time is <1 pico-second (Bitencourt et al., 2008), and at temperatures close to 0 K which can reach 3 pico-seconds.
4. Conclusions
The obtained results indicate that the chiral transition rate of trans to cis configuration of hydrogen peroxide in the presence of the noble gases He and Ne were the lowest over the entire temperature range of 4,000–100 K.
The AIM analysis shows that the interaction between H2O2 and the noble gases should be a van der Waals type. Although the H2O2 acts as an acid in the context of this investigation, the high hardness and high electronegativity of the nobles gases hold their electrons very tight to permit a covalence bond between H2O2 and Ng. On the other hand, it seems that both He and Ne are better able to affect the hyperconjugation effect and destabilizing repulsion among the lone pairs that are responsible for rotational barriers (Song et al., 2005). This may explain why the chiral transition rate decreases more for the complexes composed by Ne and He atoms, the hardest and more electronegative noble gases (Furtado et al., 2015).
Finally, the trans-barrier plays an important role because it is much smaller than the cis-barrier. The results showed that a small increase in the trans-barrier height in the complexes is responsible for a significant decrease in the rate of transition from cis to trans. Thus, these effects may contribute to the feasibility of separating one or the other enantiomer of the H2O2 molecule.
Author Contributions
RG conceived and supervised the study. RG also helped write the paper. YS performed the H2O2-Ng electronic and thermal chiral rate calculations. PN determined the H2O2-Ng minimum and transition state configurations and LdM used the AIM theory to perform the H2O2-Ng topological analyses and wrote the manuscript, which was reviewed by all authors.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We gratefully acknowledge the financial support from the Brazilian Research Councils CNPq and FAPDF.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2018.00671/full#supplementary-material
References
Allen, L. C. (1989). Electronegativity is the average one-electron energy of the valence-shell electrons in ground-state free atoms. J. Am. Chem. Soc. 111, 9003–9014. doi: 10.1021/ja00207a003
Allen, L. C., and Huheey, J. E. (1980). The definition of electronegativity and the chemistry of the noble gases. J. Inorg. Nuclear Chem. 42, 1523–1524. doi: 10.1016/0022-1902(80)80132-1
Al-Refaie, A. F., Ovsyannikov, R. I., Polyansky, O. L., Yurchenko, S. N., and Tennyson, J. (2015). A variationally calculated room temperature line-list for H2O2. J. Mol. Spectrosc. 318, 84–90. doi: 10.1016/j.jms.2015.10.004
Antunes, F., and Brito, P. M. (2017). Quantitative biology of hydrogen peroxide signaling. Redox Biol. 13, 1–7. doi: 10.1016/j.redox.2017.04.039
Aquilanti, V., Bartolomei, M., Pirani, F., Cappelletti, D., Vecchiocattivi, F., Shimizu, Y., et al. (2005). Orienting and aligning molecules for stereochemistry and photodynamics. Phys. Chem. Chem. Phys. 7, 291–300. doi: 10.1039/B415212C
Aquilanti, V., Mundim, K. C., Elango, M., Kleijn, S., and Kasai, T. (2010). Temperature dependence of chemical and biophysical rate processes: Phenomenological approach to deviations from arrhenius law. Chem. Phys. Lett. 498, 209–213. doi: 10.1016/j.cplett.2010.08.035
Atkins, P., De Paula, J., and Friedman, R. (2013). Physical Chemistry: Quanta, Matter, and Change. New York, NY: Oxford University Press.
Bader, R. F. (1991). A quantum theory of molecular structure and its applications. Chem. Rev. 91, 893–928. doi: 10.1021/cr00005a013
Ball, R., and Brindley, J. (2016). The life story of hydrogen peroxide iii: chirality and physical effects at the dawn of life. Orig. Life Evol. Biospheres 46, 81–93. doi: 10.1007/s11084-015-9465-y
Barreto, P. R., Palazzetti, F., Grossi, G., Lombardi, A., Maciel, G., and Vilela, A. (2010). Range and strength of intermolecular forces for van der waals complexes of the type h2xn-rg, with x= o, s and n= 1, 2. Int. J. Quantum Chem. 110, 777–786. doi: 10.1002/qua.22127
Barreto, P. R., Vilela, A. F., Lombardi, A., Maciel, G. S., Palazzetti, F., and Aquilanti, V. (2007). The hydrogen peroxide- rare gas systems: Quantum chemical calculations and hyperspherical harmonic representation of the potential energy surface for atom- floppy molecule interactions. J. Phys. Chem. A 111, 12754–12762. doi: 10.1021/jp076268v
Bell, R. P. (1959). The tunnel effect correction for parabolic potential barriers. Trans. Faraday Soc. 55, 1–4. doi: 10.1039/tf9595500001
Bitencourt, A. C., Ragni, M., Maciel, G. S., Aquilanti, V., and Prudente, F. V. (2008). Level distributions, partition functions, and rates of chirality changing processes for the torsional mode around o–o bonds. J. Chem. Phys. 129:154316. doi: 10.1063/1.2992554
Boys, S. F., and Bernardi, F. d. (1970). The calculation of small molecular interactions by the differences of separate total energies. some procedures with reduced errors. Mol. Phys. 19, 553–566. doi: 10.1080/00268977000101561
Carlson, R., Anderson, M., Johnson, R., Smythe, W., Hendrix, A., Barth, C., et al. (1999). Hydrogen peroxide on the surface of europa. Science 283, 2062–2064. doi: 10.1126/science.283.5410.2062
Carvalho-Silva, V. H., Aquilanti, V., de Oliveira, H. C., and Mundim, K. C. (2017). Deformed transition-state theory: Deviation from a rrhenius behavior and application to bimolecular hydrogen transfer reaction rates in the tunneling regime. J. Comput. Chem. 38, 178–188. doi: 10.1002/jcc.24529
Cortés-Guzmán, F., and Bader, R. F. (2005). Complementarity of qtaim and mo theory in the study of bonding in donor–acceptor complexes. Coord. Chem. Rev. 249, 633–662. doi: 10.1016/j.ccr.2004.08.022
Daza, M. C., Dobado, J., Molina, J. M., and Villaveces, J. L. (2000). Structure and bonding of h 2 o 2··· x complexes with (x= no+, cn-, hcn, hnc, co). Phys. Chem. Chem. Phys. 2, 4089–4094. doi: 10.1039/b001885f
Dobado, J., Martínez-García, H., Molina, J. M., and Sundberg, M. R. (1998). Chemical bonding in hypervalent molecules revised. application of the atoms in molecules theory to y3 x and y3 xz (y= h or ch3; x= n, p or as; z= o or s) compounds. J. Am. Chem. Soc. 120, 8461–8471. doi: 10.1021/ja980141p
Dobado, J., and Molina, J. (1999). Adenine- hydrogen peroxide system: Dft and mp2 investigation. J. Phys. Chem. A 103, 4755–4761. doi: 10.1021/jp990671n
Dobado, J., and Molina, J. M. (1993). Ab initio molecular orbital calculation of the hydrogen peroxide dimer: study of basis set superposition error. J. Phys. Chem. 97, 7499–7504. doi: 10.1021/j100131a018
Dunning, T. H. Jr. (1989). Gaussian basis sets for use in correlated molecular calculations. i. the atoms boron through neon and hydrogen. J. Chem. Phys. 90, 1007–1023. doi: 10.1063/1.456153
Encrenaz, T., Bézard, B., Greathouse, T., Richter, M., Lacy, J., Atreya, S., et al. (2004). Hydrogen peroxide on mars: evidence for spatial and seasonal variations. Icarus 170, 424–429. doi: 10.1016/j.icarus.2004.05.008
Eyring, H. (1935). The activated complex in chemical reactions. J. Chem. Phys. 3, 107–115. doi: 10.1063/1.1749604
Fehrensen, B., Luckhaus, D., and Quack, M. (2007). Stereomutation dynamics in hydrogen peroxide. Chem. Physics, 338, 90–105. doi: 10.1016/j.chemphys.2007.06.012
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2009) Gaussian 09 Revision e.01. Wallingford, CT: Gaussian Inc.
Furtado, J., De Proft, F., and Geerlings, P. (2015). The noble gases: how their electronegativity and hardness determines their chemistry. J. Phys. Chem. 119, 1339–1346. doi: 10.1021/jp5098876
Goebel, J., Ault, B. S., and Del Bene, J. E. (2000). Matrix isolation and ab initio study of the hydrogen-bonded complex between h2o2 and (ch3) 2o. J. Phys. Chem. 104, 2033–2037. doi: 10.1021/jp9941716
Goebel, J. R., Antle, K. A., Ault, B. S., and Del Bene, J. E. (2002). Matrix isolation and ab initio study of 1: 1 hydrogen-bonded complexes of h2o2 with hf, hcl, and hbr. J. Phys. Chem. 106, 6406–6414. doi: 10.1021/jp020520b
Goebel, J. R., Ault, B. S., and Del Bene, J. E. (2001a). Matrix isolation and ab initio study of 1: 1 hydrogen-bonded complexes of h2o2 with nh3 and n (ch3) 3. J. Phys. Chem. 105, 6430–6435. doi: 10.1021/jp010813p
Goebel, J. R., Ault, B. S., and Del Bene, J. E. (2001b). Matrix isolation and ab initio study of 1: 1 hydrogen-bonded complexes of h2o2 with phosphorus and sulfur bases. J. Phys. Chem. 105, 11365–11370. doi: 10.1021/jp013262
González, L., Mó, O., and Yáñez, M. (1997). High-level ab initio versus dft calculations on (h2o2) 2 and h2o2–h2o complexes as prototypes of multiple hydrogen bond systems. J. Comput. Chem. 18, 1124–1135.
Grzechnik, K., Mierzwicki, K., and Mielke, Z. (2013). Matrix-isolated hydrogen-bonded and van der waals complexes of hydrogen peroxide with ocs and cs2. ChemPhysChem 14, 777–787. doi: 10.1002/cphc.201200696
Henkelman, G., Jóhannesson, G., and Jónsson, H. (2002). “Methods for finding saddle points and minimum energy paths,” in Theoretical Methods in Condensed Phase Chemistry, ed S. D. Schwartz (Dordrecht: Springer), 269–302.
Hunt, R. H., Leacock, R. A., Peters, C. W., and Hecht, K. T. (1965). Internal-rotation in hydrogen peroxide: the far-infrared spectrum and the determination of the hindering potential. J. Chem. Phys. 42, 1931–1946. doi: 10.1063/1.1696228
Keith, T. A. (2017). Aimall (version 17.11.04). TK gristmill software: Overland Park, KS. Available online at: aim.tkgristmill.com
Leal, L. A., da Cunha, W. F., Roncaratti, L. F., Silva, G. M. E., and Gargano, R. (2016). H2O2–ng dynamics predictions using an accurate potential energy surface. Mol. Phys. 114, 440–445. doi: 10.1080/00268976.2015.1078507
Lewis, W. M. (1967). “Studies in catalysis. Part ix. the calculation in absolute measure of velocity constants and equilibrium constants in gaseous systems,” in Selected Readings in Chemical Kinetics, eds M. H. Back and K. J. Laidler (Pergamon: Elsevier), 36–40.
Lombardi, A., and Palazzetti, F. (2018). Chirality in molecular collision dynamics. J. Phys. Condensed Matter 30:063003. doi: 10.1088/1361-648X/aaa1c8
Lombardi, A., Palazzetti, F., Maciel, G., Aquilanti, V., and Sevryuk, M. (2011). Simulation of oriented collision dynamics of simple chiral molecules. Int. J. Quantum Chem. 111, 1651–1658. doi: 10.1002/qua.22816
Lundell, J., Jolkkonen, S., Khriachtchev, L., Pettersson, M., and Räsänen, M. (2001). Matrix isolation and ab initio study of the hydrogen-bonded h2o2–co complex. Chem. Eur. J. 7, 1670–1678. doi: 10.1002/1521-3765(20010417)7:8<1670::AID-CHEM16700>3.0.CO;2-N
Lundell, J., Pehkonen, S., Pettersson, M., and Räsänen, M. (1998). Interaction between hydrogen peroxide and molecular nitrogen. Chem. Phys. Lett. 286, 382–388. doi: 10.1016/S0009-2614(98)00003-7
Maciel, G. S., Bitencourt, A. C. P., Ragni, M., and Aquilanti, V. (2006). Studies of the dynamics around the o–o bond: orthogonal local modes of hydrogen peroxide. Chem. Phys. Lett. 432, 383–390. doi: 10.1016/j.cplett.2006.10.073
Małyszek, P., and Koput, J. (2013). Accurate ab initio potential energy surface and vibration-rotation energy levels of hydrogen peroxide. J. Comput. Chem. 34, 337–345. doi: 10.1002/jcc.23137
Matta, C., and Boyd, R. (2007). Quantum Theory of Atoms in Molecules: Recent Progress in Theory and Application. New York, NY: Wiley-VCH.
Mo, O., Yanez, M., Rozas, I., and Elguero, J. (1994). Structure, vibrational frequencies, and thermodynamic properties of hydrogen peroxide dimers: an ab initio molecular orbital study. J. Chem. Phys. 100, 2871–2877. doi: 10.1063/1.466429
Molina, J. M., Dobado, J. A., Daza, M. C., and Villaveces, J. L. (2002). Structure and bonding of weak hydrogen peroxide complexes. J. Mol. Struct. 580, 117–126. doi: 10.1016/S0166-1280(01)00602-9
Mucha, M., and Mielke, Z. (2009). Photochemistry of the glyoxal–hydrogen peroxide complexes in solid argon: formation of 2-hydroxy-2-hydroperoxyethanal. Chem. Phys. Lett. 482, 87–92. doi: 10.1016/j.cplett.2009.09.082
Palazzetti, F., Tsai, P.-Y., Lombardi, A., Nakamura, M., Che, D.-C., Kasai, T., et al. (2013). Aligned molecules: chirality discrimination in photodissociation and in molecular dynamics. Rendiconti Lincei 24, 299–308. doi: 10.1007/s12210-013-0248-y
Pehkonen, S., Lundell, J., Khriachtchev, L., Pettersson, M., and Räsänen, M. (2004). Matrix isolation and quantum chemical studies on the h 2 o 2–so 2 complex. Phys. Chem. Chem. Phys. 6, 4607–4613. doi: 10.1039/B410223A
Peterson, K. A., Figgen, D., Goll, E., Stoll, H., and Dolg, M. (2003). Systematically convergent basis sets with relativistic pseudopotentials. ii. small-core pseudopotentials and correlation consistent basis sets for the post-d group 16–18 elements. J. Chem. Phys. 119, 11113–11123. doi: 10.1063/1.1622924
Piatnytskyi, D., Zdorevsky, O., Perepelytsya, S., and Volkov, S. (2016). Formation of complexes of hydrogen peroxide molecules with dna. Ukr. J. Phys. 61, 219–225. doi: 10.15407/ujpe61.03.0219
Polanyi, M., and Wigner, E. (1928). Über die interferenz von eigenschwingungen als ursache von energieschwankungen und chemischer umsetzungen. Zeitschrift für Phys. Chem. 139, 439–452. doi: 10.1515/zpch-1928-13930
Ramalho, S. S., da Cunha, W. F., Barreto, P. R., Neto, P. H., Roncaratti, L. F., e Silva, G. M., and Gargano, R. (2011). Thermal rate constant calculation of the nf+ f reactive system multiple arrangements. J. Phys. Chem. 115, 8248–8254. doi: 10.1021/jp204229d
Roncaratti, L., Leal, L., Pirani, F., Aquilanti, V., e Silva, G., and Gargano, R. (2014). Chirality of weakly bound complexes: the potential energy surfaces for the hydrogen-peroxide- noble-gas interactions. J. Chem. Phys. 141:134309. doi: 10.1063/1.4897136
Roncaratti, L. F., and Aquilanti, V. (2010). Whittaker–hill equation, ince polynomials, and molecular torsional modes. Int. J. Quantum Chem. 110, 716–730. doi: 10.1002/qua.22255
Roohi, H., Nowroozi, A., Bavafa, S., Akbary, F., and Eshghi, F. (2010). Interaction between nh2no and h2o2: a quantum chemistry study. Int. J. Quantum Chem. 110, 1972–1981. doi: 10.1002/qua.22364
Shi, Y., and Zhou, Z.-Y. (2004). Density functional theory study of the hydrogen bonding interaction complexes of hydrogen peroxide with glycine. J. Mol. Struct. 674, 113–119. doi: 10.1016/j.theochem.2003.12.018
Sies, H. (2017). Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: oxidative eustress. Redox Biol. 11, 613–619. doi: 10.1016/j.redox.2016.12.035
Silva, V. H., Aquilanti, V., de Oliveira, H. C., and Mundim, K. C. (2013). Uniform description of non-arrhenius temperature dependence of reaction rates, and a heuristic criterion for quantum tunneling vs classical non-extensive distribution. Chem. Phys. Lett. 590, 201–207. doi: 10.1016/j.cplett.2013.10.051
Song, L., Liu, M., Wu, W., Zhang, Q., and Mo, Y. (2005). Origins of rotational barriers in hydrogen peroxide and hydrazine. J. Chem. Theory Comput. 1, 394–402. doi: 10.1021/ct049843x
Su, T.-M., Palazzetti, F., Lombardi, A., Grossi, G., and Aquilanti, V. (2013). Molecular alignment and chirality in gaseous streams and vortices. Rendiconti Lincei 24, 291–297. doi: 10.1007/s12210-013-0249-x
Truhlar, D. G., Garrett, B. C., and Klippenstein, S. J. (1996). Current status of transition-state theory. J. Phys. Chem. 100, 12771–12800. doi: 10.1021/jp953748q
Truhlar, D. G., Isaacson, A. D., and Garrett, B. C. (1985). Generalized Transition State Sheory, Volume 4. Boca Raton, FL: CRC Press.
Wang, C., Zhang, D. H., and Skodje, R. T. (2012). A six-dimensional wave packet study of the vibrational overtone induced decomposition of hydrogen peroxide. J. Chem. Phys. 136:164314. doi: 10.1063/1.4705755
Wilson, A. K., Woon, D. E., Peterson, K. A., and Dunning, T. H. Jr. (1999). Gaussian basis sets for use in correlated molecular calculations. ix. The atoms gallium through krypton. J. Chem. Phys. 110, 7667–7676. doi: 10.1063/1.478678
Woon, D. E., and Dunning Jr, T. H. (1993). Gaussian basis sets for use in correlated molecular calculations. iii. The atoms aluminum through argon. J. Chem. Phys. 98, 1358–1371. doi: 10.1063/1.464303
Yu, C.-Y., and Yang, Z.-Z. (2011). A systemic investigation of hydrogen peroxide clusters (h2o2) n (n= 1- 6) and liquid-state hydrogen peroxide: based on atom-bond electronegativity equalization method fused into molecular mechanics and molecular dynamics. J. Phys. Chem. 115, 2615–2626. doi: 10.1021/jp111284t
Keywords: hydrogen peroxide, noble gases, stereodynamics, chirality, thermal chiral rate, enantiomers, TST method, AIM theory
Citation: Só YAdO, Neto PHdO, de Macedo LGM and Gargano R (2019) Theoretical Investigation on H2O2-Ng (He, Ne, Ar, Kr, Xe, and Rn) Complexes Suitable for Stereodynamics: Interactions and Thermal Chiral Rate Consequences. Front. Chem. 6:671. doi: 10.3389/fchem.2018.00671
Received: 08 November 2018; Accepted: 24 December 2018;
Published: 18 January 2019.
Edited by:
Antonio Aguilar, University of Barcelona, SpainReviewed by:
Vincenzo Aquilanti, University of Perugia, ItalyLuca Evangelisti, University of Bologna, Italy
Copyright © 2019 Só, Neto, de Macedo and Gargano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ricardo Gargano, Z2FyZ2Fub0B1bmIuYnI=