 Jing-fan Xin1,2
Jing-fan Xin1,2 Yi-hong Ding
Yi-hong Ding- 1Laboratory of Theoretical and Computational Chemistry, Institute of Theoretical Chemistry, Jilin University, Changchun, China
- 2Inner Mongolia Key Laboratory of Photoelectric Functional Materials, College of Chemistry and Chemical Engineering, Chifeng University, Chifeng, China
Despite the great interest in energy storage application, stable neutral CnOn (n > 1) structures either in thermodynamics or kinetics have yet been largely limited due to the rather high tendency to release the very stable CO molecule. The neutral cyclopropanetrione (C3O3) cluster has long remained elusive since no isomer with sufficient kinetic stability has been found either experimentally or theoretically. In this work, we constructed the first global potential energy surface of singlet C3O3 at the CCSD(T)/aug-cc-pVTZ//B3LYP/aug-cc-pVTZ level, from which the kinetic stability of a wide range of C3O3 isomers can be determined by investigating their isomerization and fragmentation pathways. Amongst, a three-membered ring structure 01 is the global C3O3 isomer with a barrier of 10.6 kcal/mol at the sophisticated W1BD level. In particular, two carbene-type isomers 02 and 04 possess appreciable destruction barriers of 20.3 and 24.7 kcal/mol at W1BD, respectively. Thus, 02 and 04 can be useful building blocks for constructing larger high-energy density carbon-oxygen clusters. Moreover, with the carbene center, both might effectively functionalize various nano-materials while retaining the electrochemical active carbonyl and epoxyl moieties that are very desirable in alkali metal-ion batteries.
Introduction
Carbon (C) and oxygen (O) are key elements on earth and in space. Clusters constituted simply by them form a special class of oxides of carbon, namely oxocarbons(Rubin and Gleiter, 2000; Horiuchi et al., 2010; Kikuchi et al., 2014; Davis and Sajeev, 2017; Wang et al., 2018a). The huge energy release from CnOn to nCO provides great promise that a kinetically stabilized CnOn might find applications in the so-called high-energy density materials (HEDMs) (Schmidt et al., 2000; Gambi et al., 2001; Corkran and Ball, 2004; Xia et al., 2017). In fact, the polymeric CO networks as potential HEDMs have been computationally predicted to exist under the high-pressure environments (Lipp et al., 2005; Ryu et al., 2016, 2017). Yet finite-sized CnOn clusters with both high-energy and appreciable kinetic stability against destruction (i.e., isomerization/fragmentation) still remain unknown. Moreover, there have been growing evidences that the rich oxygen density in form of carbonyl and epoxyl groups are key in development of alkali metal-ion electrodes for sustainable ion batteries (Chen et al., 2009; Seo et al., 2011; Kim et al., 2014; Zhao et al., 2016; Larm et al., 2018; Wang et al., 2018b). Kinetically stable CnOn that natively bear rich oxygens would surely find interest in such applications.
The structure, bonding and stability of chemically bound CnOn have been explored in a large number of publications (Frenking, 1990; Schröder et al., 1998, 1999; Talbi and Chandler, 2000; Sabzyan and Noorbala, 2003; Zhou et al., 2010; Bao et al., 2012; Guo et al., 2012; Hu et al., 2012; Dixon et al., 2015; Liu et al., 2015; Hansen et al., 2016). To our surprise, larger CO oligomers in poly-cyclic form with n = 8, 9, 10, 12 were reported early (in 1967 and 1984; Verter and Dominic, 1967; Nallaiah, 1984). Their easy accessibility is in accordance with their good thermodynamic stability with respect to nCO (Schröder et al., 1999), and these low-lying clusters surely cannot be used for HEDMs. However, the detection and characterization of CnOn with intermediate size has been found quite difficult, partly due to their worse thermodynamic stability (Schleyer et al., 2000). The possible existence of C2O2 was suggested more than 200 years ago (Staudinger and Anthes, 1913). Yet the combined spectroscopic and theoretical study has shown that the most feasible isomer, triplet linear OCCO, could only be transient or fleeting due to the low intersystem crossing barrier 3.0 kcal/mol (Schröder et al., 1998). Up to now, a conclusive spectroscopic characterization of OCCO still remains missing (Dixon et al., 2015; Lunny et al., 2018). For the monocyclic structures (n = 3–6), their generation and characterization were reported recently in the negative ion photoelectron (NIPE) mass spectroscopic studies (Hsu and Lin, 1978; Guo et al., 2012; Bao et al., 2013; Chen et al., 2014). We must note that the observed monocyclic C3O3 was just a hilltop structure with two imaginary frequencies (Chen et al., 2014). In spite of the available structural and thermodynamic information (Sabzyan and Noorbala, 2003; Sahu and Lee, 2005; Zhou et al., 2010; Bao et al., 2012; Liu et al., 2015; Hansen et al., 2016), to our best knowledge, very little study has been made to address the kinetic stability of CnOn (n > 2) isomers, i.e., their lowest barriers against isomerization/fragmentation.
In this work, we focus on the cyclopropanetrione cluster (C3O3) that could present the smallest mono C3-ring. Three distinct types of singlet C3O3 isomers (A–C in Scheme 1) have been reported in literatures (Hsu and Lin, 1978; Hu et al., 2012; Chen et al., 2014). The long expected and hotly studied monocyclic singlet isomer A is actually a second-order saddle point, and its observation can only be realized under very extreme experimental conditions (e.g., NIPE; Chen et al., 2014). The isomers B and C of C3O3 were computationally reported in 2012 (Hu et al., 2012), yet their kinetic stability is still uncertain. We can say that at present, no C3O3 with reasonable kinetic stability has been shown either experimentally or computationally. Thus, C3O3 represents a quite “elusive” oxocarbon system.
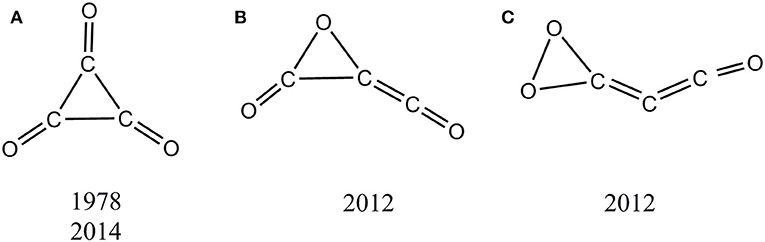
Scheme 1. Proposed singlet C3O3 isomers in literatures with its first proposed year and characterized year. (A) C3-ring isomer. (B) C2O- ring isomer. (C) CO2- ring isomer.
Surely, the key to resolve this problem is to build a global potential energy surface (PES) picture of C3O3, which involves both the C3O3 isomers and the isomerization/fragmentation transition states as many possible. With the PES, the kinetic stability of each C3O3 isomer can then be determined. Unfortunately, building the global PES for such a high-energy and hexatomic molecule should be time-consuming, tedious and even exhaustive. In the present work, applying an effective global PES search strategy, we computationally constructed the first global potential energy surface (PES) of singlet C3O3, which helped us determine the kinetic stability of a wide range of isomers. Amongst them we for the first time identified the global isomer of C3O3, i.e., 01. In particular, two carbene-like isomers 02 and 04 have the potential use in energy storage applications (e.g., HEDM and ion batteries). Such a thorough PES would provide a good base for future theoretical and laboratory studies of these C3O3 isomers.
Computational Details
The search of isomers and transition states of singlet C3O3 was carried out on a locally developed platform “global potential energy surface survey (GPESS)” (Shao and Ding, 2010; Ding, 2015; Bo and Ding, 2018). The flow chart of our strategies for constructing C3O3 PES is shown in the Supporting Information (SI_1). The isomeric search was based on the grid search program “grid-based isomeric search strategy” at the B3LYP/6-31G(d) level (Parr and Yang, 1989; Becke, 1992; Perdew et al., 1992) for both geometries and frequencies. The transition state (TS) search was divided into two types, i.e., the isomeric conversion and the isomeric decomposition. For the interconversion TS search, the “QST2” algorithm (Foresman and Frisch, 1996; Hrarchian and Schlegel, 2005), was applied, which yet has a difficulty in placing atoms in the same atomic order between reactant and product (especially for molecules with many homo-atomic elements). This was treated in GPESS by automatic enumeration of all possible combinations. Besides, the decomposition TS search was considered directing to some relatively stable molecular fragments like CO and CO2. The connection of each transition state was determined by the intrinsic reaction coordinate (IRC) (Fukui, 1981) calculations. The effectiveness of such isomeric and TS search strategies has been confirmed in study of various small to medium-sized systems (Cui et al., 2011; Gao and Ding, 2012; Tang et al., 2012; Guo et al., 2013a,b, 2014; Zhang and Ding, 2014, 2015; He and Ding, 2016; Xu et al., 2017; Bo et al., 2019).
Further, the geometry and frequencies of each isomeric and transition state structure were refined at the B3LYP/aug-cc-pVTZ optimization level followed by the CCSD(T)/aug-cc-pVTZ single-point energy calculations. The eventual energy includes the Gibbs free energy corrections (GFEC). The overall were included in CCSD(T)/aug-cc-pVTZ single-point energy calculation. For all the obtained isomers, we carried out the “stability” analysis of the wave-function at the B3LYP/aug-cc-pVTZ level, applying the broken-symmetry strategy of Noodleman (Noodleman, 1981; Noodleman and Davidson, 1986). For key species, the composite CBS-QB3 (Montgomery et al., 1999, 2000) and W1BD (Barnes et al., 2009) methods were applied to get more reliable energy. All the calculations were carried out with the GAUSSIAN03 and GAUSSIAN09 program packages (Frisch et al., 2004, 2013).
Results and Discussions
Potential Energy Surface of Singlet C3O3
By means of the extensive “grid” isomeric search and the transition state search strategies, we eventually located a total of 22 singlet chemically bound isomers and 46 transition states (Supporting Information, SI_2, SI_3). Note that our study found many other transition states whose imaginary frequency is only associated with the evolution of one separate part, while the other part is just a spectator. These transition states are not related to the evaluation of the kinetic stability of C3O3 isomers and thus not discussed. The relative energies (RE) with respect to P1 3CO (0.0) and the corresponding destruction barriers of each singlet isomer are listed in Table 1. For simplicity, “CCSD(T)//B3LYP+GFEC” represents the CCSD(T)/aug-cc-pVTZ//B3LYP/aug-cc-pVTZ values with GFEC. The structures of singlet isomers are listed in Figure 1. The schematic potential energy surface of singlet C3O3 isomers is given in Figure 2. Energies for singlet C3O3 isomers, transition states and products are shown in SI_4, SI_5, SI_6. The natural molecular orbitals for 01, 02, 04 are shown in SI_8.
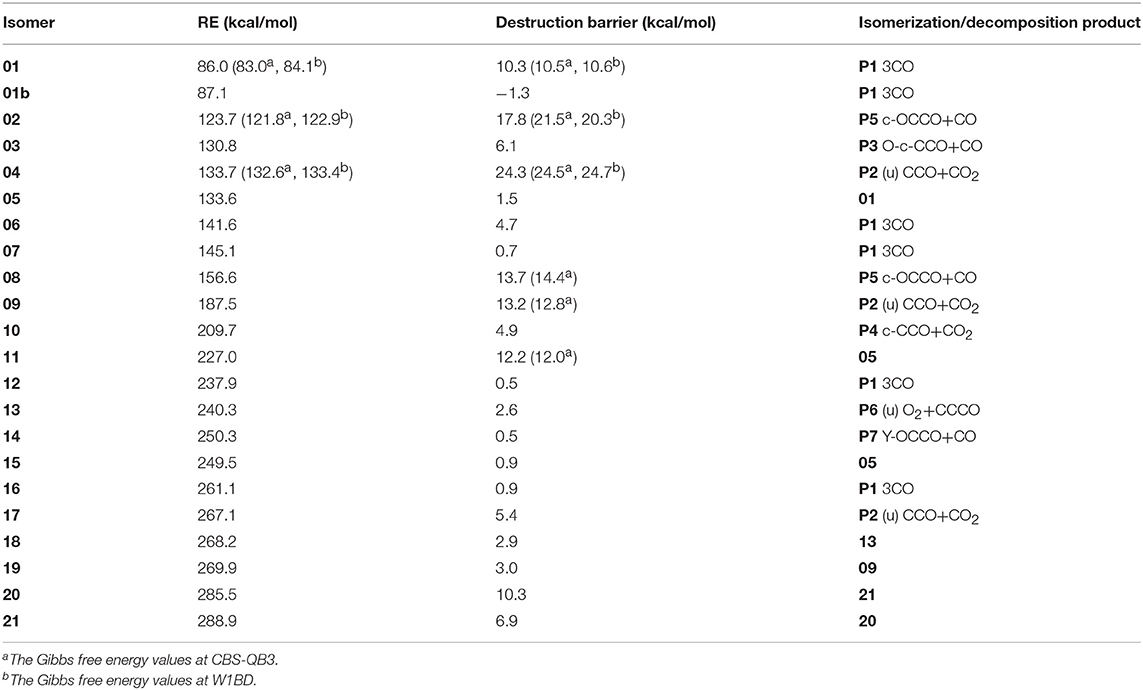
Table 1. RE values for singlet C3O3 isomers with respect to 3CO as well as the destruction barriers for each isomer at the CCSD(T)//B3LYP+GFEC level.
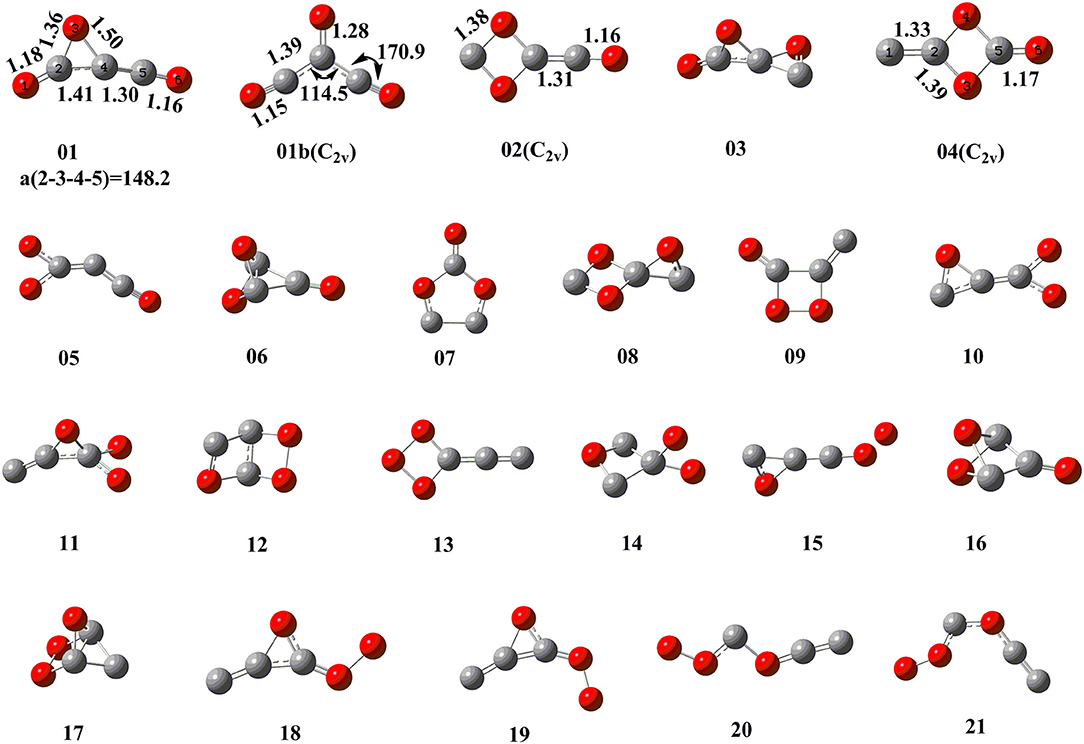
Figure 1. Structural information of singlet C3O3 isomers at the B3LYP/aug-cc-pVTZ level.
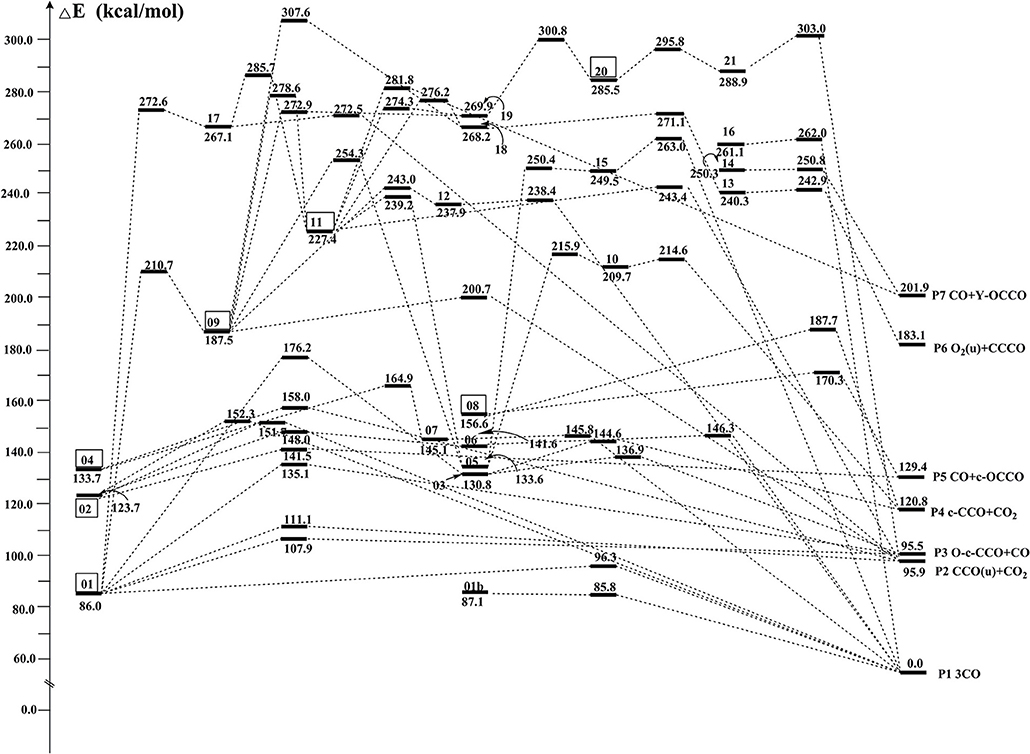
Figure 2. Schematic potential energy surface of singlet C3O3 at the CCSD(T)//B3LYP+GFEC level.
Global Isomer
The global C3O3 isomer is 01, which lies 86.0 kcal/mol higher than the fragments 3CO (0.0). This indicates that C3O3 is truly an energized system. As shown in Figure 1, the exocyclic C(4)-C(5) bond distance of 01 is 1.30 Å, indicative of the ketene-like >C = C = O bonding. To our surprise, C(4) is somewhat pyramidal in contrast to the usual sp2-C, for which three connected bonds are in a plane. So, 01 should have contribution from two resonant structures (A) >C = C = O and (B) >C(:)← CO (see Scheme 2).
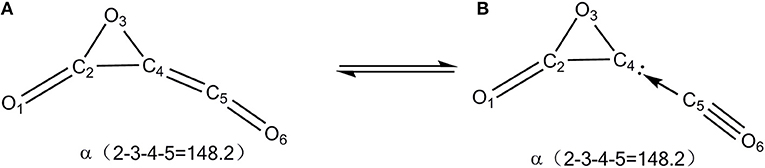
Scheme 2. Two resonant structures of 01. (A) Cumulenic type. (B) Donor-acceptor type.
To test the bonding picture B in isomer 01, we computed the three-membered ring (3MR) C2O2 when the exocyclic CO is formally removed from 01. The 3MR C2O2 is truly a singlet local minimum structure and is electronically stable during the “stability” analysis. Its triplet structure does not exist, which upon optimization would collapse to the ground structure, i.e., triplet OCCO. The electronic stability of the unsaturated carbon in 3MR C2O2 must originate from the lone-pair electronic stabilization of the neighboring O-atom. So, C(4) in 01 has the significant closed-shell divalent carbene character with one electron lone pair (:) and one vacant orbital that is occupied by the Lewis base CO. This bonding picture is supported by two facts. First, the leaving barrier of CO (22.5 kcal/mol) is roughly quarter of the typical C-C single bond energy (85.4 kcal/mol; Sanderson, 1983) and the reverse association barrier between CO and 3MR-C is negligibly tiny. Note that the barrier becomes negative as −1.3 kcal/mol at the CCSD(T)//B3LYP+GFEC level, which is just a result of the single-point energy calculation at a lower-level geometry. This phenomenon usually occurs for low-barrier processes. This is well-indicative of the donor-acceptor interaction as shown in B). Second, there is an appreciable binding energy between the 3MR-C atom and the Lewis acid AlCl3 (12.6 kcal/mol at the B3LYP/aug-cc-pVTZ+ZPVE level), suggestive of the existence of an electron lone pair on C(4).
Viable and Fleeting Isomers
According to Hoffmann et al. (2008), a viable molecule should be resistant to fragmentation, isomerization, and dimerization or higher chemical aggregation. For a gas-phase molecule like C3O3, the aggregation can usually be omitted (the chance of bimolecular association is very little). So, the kinetic stability against both fragmentation and isomerization is the key to determine the lifetime of a C3O3 structure. Species with several kcal/mol should better be viewed as fleeting or transient. For safety, in this work, the value 10 kcal/mol is artificially taken as the bar of “fleeting” or “transient.”
Among the 22 located singlet C3O3 isomers, a total of seven isomers (01, 02, 04, 08, 09, 11, 20) were identified to have the destruction barriers of ≥10 kcal/mol. The easiest conversion pathway for the global isomer 01 is to decompose into P1 3CO with a marginal barrier of 10.3 kcal/mol. Two four-membered ring (4MR) isomers 02 and 04 (lie at 123.7 and 133.7 kcal/mol, respectively) both contain a C(μ-O)2C = C core (μ for “bridge”). In particular, both 02 and 04 feature the unsaturated carbenes. Their most feasible primary pathways are different, i.e., P5 CO + c-OCCO for 02 and P2 (u)CCO + CO2 for 04 with the corresponding barriers of 17.8 and 24.3 kcal/mol. Isomers 08 and 11 (at 156.6 and 227.0 kcal/mol, respectively) are bicyclic and spiral. Their most feasible product is P5 CO+c-OCCO via the direct and indirect pathways with the corresponding barriers 13.7 and 12.2 kcal/mol. For isomer 09 with bridge-OO, the most favorable product is P2 (u)CCO + CO2 with the barriers 13.2 kcal/mol. The very high-energy cis/trans chainlike isomers 20 (285.5 kcal/mol) and 21 (288.9 kcal/mol) both have a terminal-OO and can be interconverted to each other. As shown in SI_9, 21 might undergo the intersystem crossing during fragmentation to CCO (triplet)+COO, which greatly decrease its kinetic stability (<10 kcal/mol). By contrast, the interconversion governs the kinetic stability of 20 and its the destruction barrier is 10.3 kcal/mol.
The remaining 15 singlet C3O3 isomers have smaller destructions barriers of ≤ 10 kcal/mol, i.e., 01b (-1.3), 03 (6.1), 05 (1.5), 06 (4.7), 07 (0.7), 10 (4.9), 12 (0.5), 13 (2.6), 14 (0.5), 15 (0.9), 16 (0.9), 17 (5.4), 18 (2.9), 19 (3.0), 20 (6.9). The values in () are the respective destruction barriers in kcal/mol. Amongst them isomer 20 has the largest destruction barrier 6.9 kcal/mol. Clearly, the 15 isomers should be considered as “fleeting” or “transient.” Note that isomer 01b has a physically troublesome negative barrier height value −1.3 kcal/mol according to the transition state theory, although the barrier height is a reasonable one (0.2 kcal/mol) at B3LYP/aug-cc-pVTZ+GFEC level. This is an indication that 01b is either not a minimum or faces a negligibly small barrier at the CCSD(T) level.
To determine whether a single-reference-based electron correlation procedure (here is CCSD(T)/aug-cc-pVTZ) is appropriate or not, the T1 diagnostic values (T1Diag) were computed. A large T1 (i.e., >0.02) probably indicates that a multireference electron correlation procedure is needed (Lee and Taylor, 1989). In our work, the T1Diag values lie below the threshold 0.02 for key isomers and transition states (see SI_7). Understandably, for the –OO isomers 20 and 21, the T1Diag values are greater, i.e., around 0.04 for isomers and around 0.03 for transition states. Other species have acceptable T1Diag values of around 0.02 except ts04/07, ts08/P1, uts09/P2, and ts11/P2, whose T1Diag values are close to 0.03.
In the present study on the C3O3 PES construction as well as the composite CBS and W1BD calculations were all based on the B3LYP method. We thus further performed the comparative study of B3LYP with B3LYP-D3BJ (Grimme et al., 2011) and B2PLYP-D3(Grimme, 2006; Grimme et al., 2010) using the same aug-cc-pVTZ basis set followed by the CCSD(T)/aug-cc-pVTZ single-point energy calculations for both the isomeric and destruction transition state strucutures for key structures, i.e., 01, 02, 04, 01-TS, 02-TS, and 04-TS (see SI_10, SI_11). For the geometries (bond length, bond angle, dihedral angle), B3LYP agrees excellently with B3LYP-D3BJ and B2PLYP-D3 for 02, 04, 02-TS, and 04-TS. The deviation is relatively larger for 01 and 01-TS with largest differences value of bond length, bond angle and dihedral angle are 0.11 Å, 4.2°, and 10.5°, respectively. This must be due to the unique electronic structure of 01 as bearing two resonant structures >C = C = O and >C(:)← CO (as discussed above). Yet the relative energies and destruction barriers at CCSD(T)//B3LYP agree quite well with those at CCSD(T)//B3LYP-D3BJ and CCSD(T)//B2PLYP-D3 within 1.0 kcal/mol for all the three isomers.
Implications
Let us compare our extensive potential energy surface study with literatures. Only three singlet C3O3 structures (A, B, C) have been proposed (see Scheme 1). The second-order saddle point nature of A was reproduced in our work. The structures B and C predicted as local minima in 2012 at B3LYP/6-31G(d) level correspond to 01 and 05, respectively in our work. To our great surprise, 01 (B) was reported to lie by 34.9 kcal/mol higher in energy than 05 (C) in the 2012 work, in sharp contrast to the present study that 01 (B) is the global minimum and 47.6 kcal/mol more stable than 05 (C). After careful repetition and analysis, the reason for such dramatic discrepancy was found to be that the 2012 study actually used the results at two different levels for comparison, i.e., B3LYP/6-31G (with no d function) for 01 (B) and B3LYP/6-31G(d) 05 (C). Thus, the present work for the first time predicted 01 as the global minimum of C3O3.
The two carbene-like isomers 02 and 04 with good kinetic stability deserve special attention. First, they provide much promise to design larger oxocarbon clusters with similar structural backbones. Second, the intrinsic carbene-reactivity allows them to be used in functionalizing various nano-materials such as graphenes (Bruce, 1991; Rit et al., 2016; Meyer, 2018). In particular, both isomers have one carbonyl (C = O) and two epoxyl (O<) groups, which could make them promising in the alkali metal-ion batteries (Wang et al., 2013; Zan, 2014; Deng et al., 2017; Zhao et al., 2017).
In particular, the destruction barrier 24.3 kcal/mol of the isomer 04 is quite close to those of the already synthesized species, e.g., N2CO (25.8 kcal/mol; Korkin et al., 1996) and pentazole anion N (25.2 kcal/mol; Rahm and Brinck, 2010). With very huge energy release to 3CO (133.7 kcal/mol), 04 deserves to be taken as a HEDM candidate.
In short, we constructed the first global potential energy surface of singlet C3O3 through the thorough isomeric and transition state search strategies. The detailed isomerization/fragmentation and stability data presented in this work should provide an important base for future laboratory study of C3O3 isomers.
Conclusions
The key contribution of our work can be summarized as follow:
1) We built up an extensive singlet potential energy surface of the ever elusive molecule, C3O3, covering 22 isomers and 46 transition states, among which seven isomers have the destruction barriers of 10–25 kcal/mol.
2) We for the first time identified the global isomer of C3O3, i.e., a three-membered ring structure 01 that has the destruction barrier of 10.3 kcal/mol against decomposition to three CO. Careful examination of its electronic structure indicates that 01 could be partly viewed as a CO-stabilized cyclic carbene.
3) With good kinetic stability, rich oxygen density and the active carbene center, the four-membered ring isomers 02 and 04 can functionalize nano-materials in alkali metal-ion batteries. In particular, with comparable destruction barrier to known HEDM molecules like N2CO and N, 04 can act as a suitable HEDM candidate.
Data Availability
All datasets generated for this study are included in the manuscript and/or the Supplementary Files.
Author Contributions
JX formulated the problem, did ab initio calculations, searched for literatures, wrote, and finalized the manuscript. XH did composite CBS-QB3 and W1BD calculations, participated in data collection, and analysis. FH formulated the problem, did ab initio calculations, and wrote the first manuscript draft. YD formulated the problem, generated key concepts, guided the whole research, and finalized the manuscript.
Funding
This work was funded by the National Natural Science Foundation of China (No. 21773082, 21473069).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The reviewers' helpful comments and suggestions are greatly acknowledged.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00193/full#supplementary-material
References
Bao, X., Hrovat, D. A., Borden, W. T., and Wang, X. B. (2013). Negative ion photoelectron spectroscopy confirms the prediction that (CO)5 and (CO)6 each has a singlet ground state. J. Am. Chem. Soc. 135, 4291–4298. doi: 10.1039/c5sc03542b
Bao, X., Zhou, X., Lovitt, C. F., Venkatraman, A., Hrovat, D. A., Gleiter, R., et al. (2012). Molecular orbitals of the oxocarbons (CO)n, n = 2-6. why does (CO)4 have a triplet ground state? J. Am. Chem. Soc. 134, 10259–10270. doi: 10.1021/ja3034087
Barnes, E. C., Petersson, G. A., Montgomery, J. J. A., Frisch, M. J., and Martin, J. M. (2009). Unrestricted coupled cluster and brueckner doubles variations of W1 Theory. J. Chem. Theory Comput. 5, 2687–2693. doi: 10.1021/ct900260g
Becke, A. D. (1992). Density-functional thermochemistry. I. The effect of the exchange-only gradient correction. J. Chem. Phys. 96, 2155–2160. doi: 10.1063/1.462066
Bo, X., Zheng, H., Xin, J., and Ding, Y. (2019). A kinetically persistent isomer found for pentazole: a global potential energy surface survey. Chem. Commun. 55, 2597–2600. doi: 10.1039/C8CC09626K
Bruce, M. I. (1991). Organometallic chemistry of vinylidene and related unsaturated carbenes. Chem. Rev. 91, 197–257. doi: 10.1021/cr00002a005
Chen, B., Hrovat, D. A., West, R., Deng, S. H., and Wang, X. (2014). The negative ion photoelectron spectrum of cyclopropane-1,2,3-trione radical anion, (CO) a joint experimental and computational study. J. Am. Chem. Soc. 136, 12345–12354. doi: 10.1021/ja505582k
Chen, H., Armand, M., Courty, M., Jiang, M., Grey, C. P., Dolhem, F., et al. (2009). Lithium salt of tetrahydroxybenzoquinone: toward the development of a sustainable Li-ion battery. J. Am. Chem. Soc. 131, 8984–8988. doi: 10.1021/ja9024897
Corkran, G., and Ball, D. W. (2004). The relative energies of cyclopropanone, cyclopropanedione, and cyclopropanetrione. Hartree-fock, density-functional,G2, and CBS calculations. J. Mol. Struct-Theochem. 668, 171–178. doi: 10.1016/j.theochem.2003.10.026
Cui, Z., Contreras, M., Ding, Y., and Merino, G. (2011). Planar tetracoordinate carbon versus planar tetracoordinate boron: the case of CB4 and its cation. J. Am. Chem. Soc. 133, 13228–13231. doi: 10.1021/ja203682a
Davis, D., and Sajeev, Y. (2017). Communication: low-energy free-electron driven molecular engineering: in situ preparation of intrinsically short-lived carbon-carbon covalent dimer of CO. J. Chem. Phys. 146:081101. doi: 10.1063/1.4976969
Deng, Q., Pei, J., Fan, C., Ma, J., Cao, B., Li, C., et al. (2017). Potassium salts of para-aromatic dicarboxylates as the highly efficient organic anodes for low-cost K-ion batteries. Nano Energy. 33, 350–355. doi: 10.1016/j.nanoen.2017.01.016
Dixon, A. R., Xue, T., and Sanov, A. (2015). Spectroscopy of ethylenedione. Chem. Int. Ed. 54, 8764–8767. doi: 10.1002/anie.201503423
Foresman, B., and Frisch, Æ. (1996). Exploring Chemistry with Electronic Structure Methods, 2nd Edn. Pittsburgh, PA: Gaussian Inc.
Frenking, G. (1990). The structure of cyclic C6S6 and C6O6. Angew. Chem. Int. Ed. Engl. 29, 1410–1412. doi: 10.1002/anieback.199014101
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2004). Gaussian 03, Revision C.02. Wallingford, CT: Gaussian, Inc.
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2013). Gaussian 09, Revision D.01. Wallingford, CT: Gaussian, Inc.
Fukui, K. (1981). The path of chemical reactions -the IRC approach. Acc. Chem. Res. 14, 363–367. doi: 10.1021/ar00072a001
Gambi, A., Giumanini, A. G., and Strazzolini, P. (2001). Theoretical investigations on (CO)n(CO2)m cyclic cooligomers. J. Mol. Struct. Theochem. 536, 9–16. doi: 10.1016/s0166-1280(00)00601-1
Gao, S., and Ding, Y. (2012). Theoretical study of nitrogen-rich CN anion and related salts M+[CN3]− (M = Li, Na, K). RSC Adv. 2, 11764–11776. doi: 10.1039/c2ra21250a
Grimme, S. (2006). Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 124:034108. doi: 10.1063/1.2148954
Grimme, S., Antony, J., Ehrlich, S., and Krieg, H. (2010). A consistent and accurate ab initio parametrization of density functional dispersion correction DFT-D… for the 94 elements H-Pu. J. Chem. Phys. 132:154104. doi: 10.1063/1.3382344
Grimme, S., Ehrlich, S., and Goerigk, L. (2011). Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32,1456–1465. doi: 10.1002/jcc.21759
Guo, C., Cui, Z., and Ding, Y. (2013a). Computational identification of a global carbon-sulfur triply bonded isomer SCBO. Struct. Chem. 24, 263–270. doi: 10.1007/s11224-012-0077-0
Guo, C., Cui, Z., and Ding, Y. (2013b). Structures, energetics, and isomerism of [Be, C, O, S]: stability of triply bonded sulfur. Int. J. Quant. Chem. 113, 2213–2219. doi: 10.1002/qua.24433
Guo, C., Wang, C., and Ding, Y. (2014). Structures and stability of SCBO+/− and SBCO+/−: prediction of very short yet classical triple bonding of sulfur. Struct. Chem. 25, 1023–1031. doi: 10.1007/s11224-013-0372-4
Guo, J., Hou, G., Li, S., and Wang, X. (2012). Probing the low-lying electronic states of cyclobutanetetraone (C4O4) and its radical anion: a low-temperature anion photoelectron spectroscopic approach. J. Phys. Chem. Lett. 3, 304–308. doi: 10.1021/jz201593z
Hansen, J. A., Bauman, N. P., Shen, J., Borden, W. T., and Piecuch, P. (2016). Ab initio coupled-cluster and multi-reference configuration interaction studies of the low-lying electronic states of 1,2,3,4-cyclobutanetetraone. Mol. Phys. 114, 695–708. doi: 10.1080/00268976.2015.1112926
He, F, and Ding, Y. (2016). Global picture of isomerization and dissociation of CN2O2: new metastable isomers. RSC Adv. 6, 26441–26450. doi: 10.1039/c5ra27576h
Hoffmann, R., Schleyer, P. V. R., and Schaefer, H. F. III. (2008). Predicting molecules—more realism, please! Angew. Chem. Int. Ed. 47, 7164–7167. doi: 10.1002/anie.200801206
Horiuchi, S., Tokunaga, Y., Giovannetti, G., Picozzi, S., Itoh, H., Shimano, R., et al. (2010). Above-room-temperature ferroelectricity in a single-component molecular crystal. Nature. 463, 789–792. doi: 10.1038/nature08731
Hrarchian, H. P., and Schlegel, H. B. (2005). Chapter 10: Finding minima, transition states, and following reaction pathways onab initio potential energy surfaces. Theory Appl. Comput. Chem. 2005, 195–249. doi: 10.1016/B978-044451719-7/50053-6
Hsu, D. S. Y., and Lin, M. C. (1978). Dynamics of the reaction of oxygen atoms with carbon suboxide. J. Chem. Phys. 68, 4347–4351. doi: 10.1063/1.435511
Hu, F., Zhang, T., Liu, X., Zhou, L., Ma, W., et al. (2012). A theoretical study on structures and dissociation mechanism of C3O3−5 cluster. J. Shan Dong Polytech. Univ. 26, 50–51.
Kikuchi, T., Nakajima, D., Kawashima, J., Natsui, S., and Suzuki, R. O. (2014). Fabrication of anodic porous alumina via anodizing in cyclic oxocarbon acids. Appl. Surf. Sci. 313, 276–285. doi: 10.1016/j.apsusc.2014.05.204
Kim, H., Seo, D. H., Goddard, W. A. III., Lee, Y. S., Yoon, W. S., Kang, K., et al. (2014). The reaction mechanism and capacity degradation model in lithium insertion organic cathodes, Li2C6O6, using combined experimental and first principle studies. J. Phys. Chem. Lett. 5, 3086–3092. doi: 10.1021/jz501557n
Korkin, A. A., Balkova, A., Bartlett, R. J., Boyd, R. J., and Schleyer, P. V. R. (1996). The 28-electron tetraatomic molecules: N4, CN2O, BFN2, C2O2, B2F2, CBFO, C2FN, and BNO2. Challenges for computational and experimental chemistry. J. Phys. Chem. 100, 5702–5714. doi: 10.1021/jp960125j
Larm, N. E., Essner, J. B., Pokpas, K., Canon, J. A., Jahed, N., Iwuoha, E. I., et al. (2018). Room-temperature turkevich method: formation of gold nanoparticles at the speed of mixing using cyclic oxocarbon reducing agents. J. Phys. Chem. C 122, 5105–5118. doi: 10.1021/acs.jpcc.7b10536
Lee, T. J., and Taylor, P. R. (1989). A diagnostic for determining the quality of single-reference electron correlation methods. Int. J. Quantum Chem. 36, 199–207. doi: 10.1002/qua.560360824
Lipp, M. J., Evans, W. J., Baer, B. J., and Yoo, C. (2005). High-energy-density extended CO solid. Nat. Mater. 4, 211–215. doi: 10.1038/nmat1321
Liu, J., Bao, X., Hrovat, D. A., and Borden, W. T. (2015). Theoretical analysis of the fragmentation of (CO)5: a symmetryallowed highly exothermic reaction that follows a stepwise pathway. J. Org. Chem. 80, 11788–11793. doi: 10.1021/acs.joc.5b01546
Lunny, K. G., Benitez, Y., Albeck, Y., Strasser, D., Stanton, J. F., and Continetti, R. E. (2018). Spectroscopy of ethylenedione and ethynediolide: a reinvestigation. Angew. Chem. 130, 5492–5495. doi: 10.1002/ange.201801848
Meyer, K. (2018). Pushing electrons-which carbene ligand for which application? Organometallics. 37, 273–274. doi: 10.1021/acs.organomet.8b00014
Montgomery, J. A., Frisch, M. J., Ochterski, J. W., and Petersson, G. A. (1999). A complete basis set model chemistry. VI. Use of density functional geometries and frequencies. J. Chem. Phys. 110, 2822–2827. doi: 10.1063/1.477924
Montgomery, J. A., Frisch, M. J., Ochterski, J. W., and Petersson, G. A. (2000). A complete basis set model chemistry. VII. Use of the minimum population localization method. J. Chem. Phys. 112, 6532–6542. doi: 10.1063/1.481224
Nallaiah, C. (1984). Synthesis of tetrahydroxy-1,4-benzoquinone biscarbonate and hexahydroxybenzene triscarbonate-new organic carbon oxides. Tetrahedron. 40, 4897–4900. doi: 10.1016/S0040-4020(01)91324-9
Noodleman, L. (1981). Valence bond description of antiferromagnetic coupling in transition metal dimers. J. Chem. Phys. 74, 5737–5743. doi: 10.1063/1.440939
Noodleman, L., and Davidson, E. R. (1986). Ligand spin polarization and antiferromagnetic coupling in transition metal dimers. Chem. Phys. 109, 131–143. doi: 10.1016/0301-0104(86)80192-6
Parr, R. G., and Yang, W. (1989). Density-Functional Theory of Atoms and Molecules. Oxford: Oxford University Press.
Perdew, J. P., Chevary, J. A., Vosko, S. H., Jackson, K. A., Pederson, M. R., and Fiolhais, C. (1992). Atoms, molecules, solids, and surfaces: applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 46, 6671–6687. doi: 10.1103/PhysRevB.46.6671
Rahm, M., and Brinck, T. (2010). Kinetic stability and propellant performance of green energetic materials. Chem. Eur. J. 16, 6590–6600. doi: 10.1002/chem.201000413
Rit, A., Campos, J., Niu, H, and Aldridge, S. (2016). A stable heavier group 14 analogue of vinylidene. Nat. Chem. 8, 1022–1026. doi: 10.1038/nchem.2597
Rubin, M. B., and Gleiter, R. (2000). The chemistry of vicinal polycarbonyl compounds. Chem. Rev. 100, 1121–1164. doi: 10.1021/cr960079j
Ryu, Y., Kim, M., Lim, J., Dias, R., Klug, D., and Yoo, C. (2016). Dense carbon monoxide to 160 GPa: stepwise polymerization to two-dimensional layered solid. J. Phys. Chem. C 120, 27548–27554. doi: 10.1021/acs.jpcc.6b09434
Ryu, Y., Yoo, C., Kim, M., Yong, X., Tse, J., Lee, S. K., et al. (2017). Hydrogen-doped polymeric carbon monoxide at high pressure. J. Phys. Chem. C 121, 10078–10086. doi: 10.1021/acs.jpcc.7b01506
Sabzyan, H., and Noorbala, M. R. (2003). Ab initio and DFT study of carbon monoxide cyclic oligomers, (CO)2 to (CO)6. J. Mol. Struct-Theochem. 626, 143–158. doi: 10.1016/s0166-1280(03)00029-0
Sahu, P. K., and Lee, S. L. (2005). Many-body interactions of carbon monoxide cyclic oligomers:a computational study. Int. J. Quant. Chem. 103, 314–321. doi: 10.1002/qua.20474
Sanderson, R. T. (1983). Electronegativity and bond energy, J. Am. Chem. Soc. 105, 2259–2261. doi: 10.1021/ja00346a026
Schleyer, P. v. R, Najafian, K., Kiran, B., and Jiao, H. (2000). Are oxocarbon dianions aromatic? J. Org. Chem. 65, 426–431. doi: 10.1021/jo991267n
Schmidt, M. W., Gordon, M. S., and Boatz, J. A. (2000). Cubic fuels? Int. J. Quantum Chem. 76, 434–446. doi: 10.1002/(SICI)1097-461X(2000)76:3<434::AID-QUA12>3.0.CO;2-W
Schröder, D., Heinemann, C., Schwarz, H., Harvey, J. N., Dua, S., Blanksby, S. J., et al. (1998). Ethylenedione: an intrinsically short-lived molecule. Chem. Eur. J. 4, 2550–2557. doi: 10.1002/(SICI)1521-3765(19981204)4:12<2550::AID-CHEM2550>3.0.CO;2-E
Schröder, D., Schwarz, H., Dua, S., Blanksby, S. J., and Bowie, J. H. (1999). Mass spectrometric studies of the oxocarbons CnOn (n = 3-6). Int. J. Mass. Spectrom. 188, 17–25. doi: 10.1016/s1387-3806(98)14208-2
Seo, D. H., Kim, H., Kim, H., Goddard, W. A. III., and Kang, K. (2011). The predicted crystal structure of Li4C6O6, an organic cathode material for Li-ion batteries, from first-principles multi-level computational methods. Energy Environ. Sci. 4, 4938–4941. doi: 10.1039/c1ee02410h
Shao, C., and Ding, Y. (2010). Grid-Based Comprehensive Isomeric Search Algorithm. Changchun: Jilin University.
Talbi, D., and Chandler, G. S. (2000). Extensive ab initio study of the C2O2, C2S2, and C2OS systems: stabilities and singlet-triplet energy gaps. J. Phys. Chem. A 104, 5872–5881. doi: 10.1021/jp000732d
Tang, X., Cui, Z., Shao, C., and Ding, Y. (2012). Structural and energetic exploration of a boron-rich sulfide cluster B6S. Int. J. Quant. Chem. 112, 1299–1306. doi: 10.1002/qua.23128
Verter, H. S., and Dominic, R. (1967). A new carbon oxide synthesis of hexahydroxybenzene tris oxalate. Tetrahedron. 23, 3863–3864. doi: 10.1016/S0040-4020(01)97894-9
Wang, D., Sun, C., Zhou, G., Li, F., Wen, L., et al. (2013). The examination of graphene oxide for rechargeable lithium storage as a novel cathode material. J. Mater. Chem. A 1, 3607–3612. doi: 10.1039/c3ta01658g
Wang, Z., Li, J., Zhang, Y., and Xu, H. (2018a). A study of the most condensed configuration of oxocarbon molecule adsorption on graphene surface: a first-principle investigation. Appl. Surf. Sci. 455, 216–220. doi: 10.1016/j.apsusc.2018.05.115
Wang, Z., Li, S., Zhang, Y., and Xu, H. (2018b). Oxocarbon-functionalized graphene as a lithium-ion battery cathode: a first-principles investigation. Phys. Chem. Chem. Phys. 20, 7447–7456. doi: 10.1039/c7cp07960e
Xia, K., Sun, J., Pickard, C. J., Klug, D. D., and Needs, R. J. (2017). Ground state structure of high-energy-density polymeric carbon monoxide. Phys. Rev. B 95:144102. doi: 10.1103/PhysRevB.95.144102
Xu, J., Zhang, X., Yu, S., Ding, Y., and Bowen, K. H. (2017). Identifying the hydrogenated planar tetracoordinate carbon: a combined experimental and theoretical study of CAl4H and CAl4H−. J. Phys. Chem. Lett. 8, 2263–2267. doi: 10.1021/acs.jpclett.7b00732
Zan, W. (2014). Chemical functionalization of graphene by carbene cycloaddition: a density functional theory study. Appl. Surf. Sci. 311, 377–383. doi: 10.1016/j.apsusc.2014.05.071
Zhang, X., and Ding, Y. (2014). Computational prediction of a global planar penta-coordinate carbon structure CAl4Ga+. Comput. Theor. Chem. 1048, 18–24. doi: 10.1016/j.comptc.2014.08.019
Zhang, X., and Ding, Y. (2015). Global structure of C2B4H4: hypercloso or not. RSC Adv. 5, 27134–27139. doi: 10.1039/c5ra02178b
Zhao, Q., Lu, Y., and Chen, J. (2017). Advanced organic electrode materials for rechargeable Sodium-ion batteries. Adv. Energy Mater. 7:1601792. doi: 10.1002/aenm.201601792
Zhao, Q., Wang, J., Lu, Y., Li, Y., and Liang, G. (2016). Oxocarbon salts for fast rechargeable batteries. Angew. Chem. Int. Ed. 55, 12528–12532. doi: 10.1002/ange.201607194
Zhou, X., Hrovat, D. A., and Borden, W. T. (2010). Calculations of the relative energies of the 2B1g and 2A2u states of cyclobutanetetraone radical cation and radical anion provide further evidence of a 3B2u ground state for the neutral molecule: a proposed experimental test of the prediction of a triplet ground state for (CO)4. J. Phys. Chem. A 114, 1304–1308. doi: 10.1021/jp905462b
Keywords: neutral cyclopropanetrione, C3O3, global isomeric survey, kinetic stability, computational study, high-energy density materials, alkali metal-ion battery
Citation: Xin J, Han X, He F and Ding Y (2019) Global Isomeric Survey of Elusive Cyclopropanetrione: Unknown but Viable Isomers. Front. Chem. 7:193. doi: 10.3389/fchem.2019.00193
Received: 09 January 2019; Accepted: 14 March 2019;
Published: 03 April 2019.
Edited by:
Marzio Rosi, University of Perugia, ItalyReviewed by:
Cecilia Coletti, Università degli Studi G. d'Annunzio Chieti e Pescara, ItalyFanny Vazart, Université Grenoble Alpes, France
Copyright © 2019 Xin, Han, He and Ding. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fei-fei He, aGZmMTZAdHNpbmdodWEub3JnLmNu
Yi-hong Ding, eWhkZEBqbHUuZWR1LmNu