 Yusuke Tamaki
Yusuke Tamaki Kazuma Tokuda
Kazuma Tokuda Yasuomi Yamazaki
Yasuomi Yamazaki- Department of Chemistry, Tokyo Institute of Technology, Tokyo, Japan
Ruthenium(II) picolinate complex, [Ru(dmb)2(pic)]+ (Ru(pic); dmb = 4,4′-dimethyl-2,2′-bipyridine; Hpic = picolinic acid) was newly synthesized as a potential redox photosensitizer with a wider wavelength range of visible-light absorption compared with [Ru(N∧N)3]2+ (N∧N = diimine ligand), which is the most widely used redox photosensitizer. Based on our investigation of its photophysical and electrochemical properties, Ru(pic) was found to display certain advantageous characteristics of wide-band absorption of visible light (λabs < 670 nm) and stronger reduction ability in a one-electron reduced state ( = −1.86 V vs. Ag/AgNO3), which should function favorably in photon-absorption and electron transfer to the catalyst, respectively. Performing photocatalysis using Ru(pic) as a redox photosensitizer combined with a Re(I) catalyst reduced CO2 to CO under red-light irradiation (λex > 600 nm). TONCO reached 235 and ΦCO was 8.0%. Under these conditions, [Ru(dmb)3]2+ (Ru(dmb)) is not capable of working as a redox photosensitizer because it does not absorb light at λ > 560 nm. Even in irradiation conditions where both Ru(pic) and Ru(dmb) absorb light (λex > 500 nm), using Ru(pic) demonstrated faster CO formation (TOFCO = 6.7 min−1) and larger TONCO (2347) than Ru(dmb) (TOFCO = 3.6 min−1; TONCO = 2100). These results indicate that Ru(pic) is a superior redox photosensitizer over a wider wavelength range of visible-light absorption.
Introduction
Redox photosensitizers, which absorb visible light and facilitate the electron transfer process, play a key role in various photochemical reactions, such as CO2 reduction (Takeda et al., 2017; Tamaki and Ishitani, 2017), water oxidation (Fukuzumi et al., 2016), hydrogen evolution (Schulz et al., 2012), and organic synthesis (Prier et al., 2013). Effective photosensitizers should be endowed with three important properties, including (1) visible-light absorption, (2) a long lifetime in the excited state to initiate the electron transfer process, and (3) reducing and/or oxidizing power that is strong enough to donate electrons or holes to the catalyst. In particular, the utilization of visible-light over a wider range of wavelengths is important both to utilize sunlight efficiently and avoid the internal filter effect and side reactions that are commonly caused by the light-absorption of catalysts and/or electron donor/acceptor. Ru(II) complexes coordinated with three diimine ligands, [Ru(N∧N)3]2+ (N∧N = diimine ligand) are the most widely used redox photosensitizers in various photochemical redox reactions because these types of complexes exhibit strong absorption in the visible-light region and have a long lifetime in their triplet metal-to-ligand charge-transfer (3MLCT) excited states (Juris et al., 1988; Thompson et al., 2013).
However, one of the disadvantages of [Ru(N∧N)3]2+-type photosensitizers is the limited access to the wavelength region of visible light, e.g., λabs < 560 nm in the cases of N∧N = 2,2′-bipyridine (bpy) and 4,4′-dimethyl-2,2′-bipyridine (dmb), and these complexes cannot utilize visible light having lower energy (λ > 560 nm). To overcome this, ligand-modified Ru(II) photosensitizers have been reported. For example, Ru(II) complexes have an extended π-system for photodynamic therapy (Zhang et al., 2017) and multinuclear Ru(II) complexes by conjugated bridging ligand are used for hydrogen evolution (Tsuji et al., 2018). However, these modifications lower the reducing power of photosensitizers and limit the choice of catalyst especially for the reduction of CO2. On the other hand, we have reported an osmium(II) analog, i.e., [Os(N∧N)3]2+, which could function as a redox photosensitizer utilizing a much wider wavelength range of visible light (λabs < 700 nm) due to its singlet-to-triplet direct excitation (S-T absorption) and drive photocatalytic CO2 reduction by red-light irradiation (λex > 620 nm) in the combination with rhenium(I) catalyst unit (Tamaki et al., 2013b), whereas the high toxicity of OsVIIIO4 inhibits the wider application of osmium complexes.
Therefore, we developed a novel ruthenium(II) redox photosensitizer that can utilize a wider wavelength range of visible light than [Ru(N∧N)3]2+. In the photocatalytic system for CO2 reduction, a photosensitizer mediates an electron from a sacrificial electron donor to a catalyst. Since the positive shift of the LUMO level of redox photosensitizer should limit the choice of a catalyst for reducing CO2, for the expansion of the useable wavelength range, we try to decrease the energy-gap between HOMO and LUMO by the negative shift of the HOMO level, while maintaining the LUMO level. We introduced anionic electron-donating picolinate instead of a diimine ligand into a ruthenium complex (Norrby et al., 1997; Couchman et al., 1998). [Ru(dmb)2(pic)]+ (Ru(pic); Hpic = picolinic acid) was synthesized, and we investigated its photophysical properties and functions as a redox photosensitizer using [Ru(dmb)3]2+ (Ru(dmb)) as a reference redox photosensitizer and Re(dmb)(CO)3Br (Re) as a catalyst for the reduction of CO2 (Hawecker et al., 1983; Gholamkhass et al., 2005; Tamaki et al., 2016). Chart 1 shows structures and abbreviations of the metal complexes used.
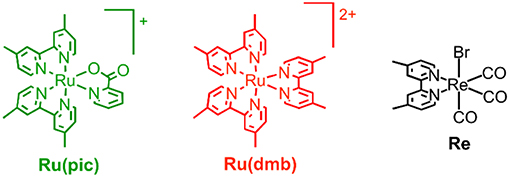
Chart 1. Structures and abbreviations of complexes used.
Results and Discussion
Figure 1 displays UV-vis absorption spectra of Ru(pic), Ru(dmb), and Re measured in N,N-dimethylacetamide (DMA). Ru(pic) exhibited a broad singlet MLCT absorption band at λabs = 450–640 nm, with molar absorptivity at an absorption maximum (λmax = 498 nm) of 1.04 × 104 M−1cm−1, which was red-shifted in wavelength compared to that of Ru(dmb) (λabs = 420–550 nm). The absorption band attributed to the π-π* transition of dmb ligands was observed at 294 nm. According to this result, Ru(pic) have the potential to utilize visible light over a wider range of wavelengths (λabs < 670 nm) than Ru(dmb) (λabs < 560 nm). This expected red-shift of the MLCT band should be induced by the stronger electron-donating ability of the picolinate ligand to negatively shift the energy level of HOMO.
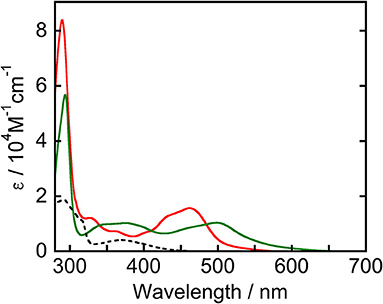
Figure 1. UV-vis absorption spectra of Ru(pic) (green line), Ru(dmb) (red line), and Re (broken line) measured in a DMA solution.
Ru(pic) exhibited phosphorescence from its 3MLCT excited state (Figure 2) with a quantum yield of Φem = 0.8% and a lifetime of τem = 66 ns. Emission spectrum of Ru(pic) (λem = 734 nm) was also red-shifted compared to that of Ru(dmb) (λem = 638 nm). The quantum yield and lifetime of Ru(pic) were smaller and shorter than those of Ru(dmb) (Φem = 9.1%, τem = 741 ns) due to the 12-times faster non-radiative deactivation process (Ru(pic): knr = 1.5 × 107 s−1; Ru(dmb): knr = 1.2 × 106 s−1), which is a reasonable behavior from energy-gap law. Table 1 summarizes photophysical properties of Ru(pic) along with those of Ru(dmb) and Re.
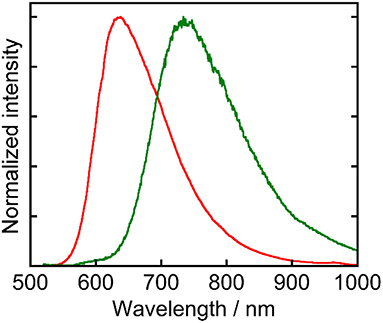
Figure 2. Normalized emission spectra of Ru(pic) (green line) and Ru(dmb) (red line) measured in a DMA solution. The excitation wavelength was 480 nm.
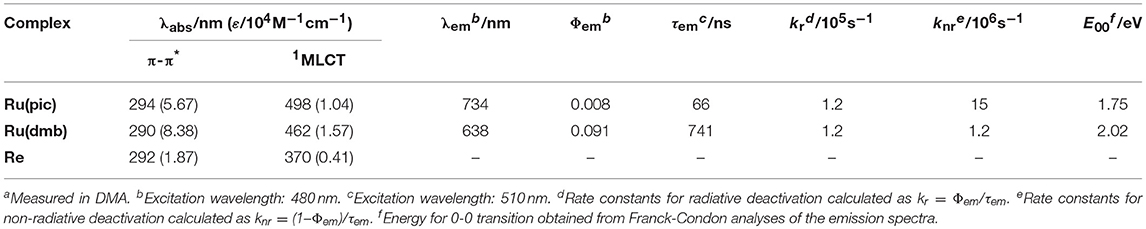
Table 1. Photophysical properties of Ru(pic), Ru(dmb), and Re.a.
Figure 3 shows the cyclic voltammograms of Ru(pic) and Ru(dmb) and their redox potentials are summarized in Table 2 along with that of Re. Ru(pic) displayed two reversible reduction waves and a reversible oxidation wave, which are attributable to the subsequent reduction of two dmb ligands and the oxidation couple of RuIII/II, respectively. Both the first reduction ( = −1.86 V vs. Ag/AgNO3) and oxidation ( = 0.41 V) waves were observed at more negative potentials than those of Ru(dmb) ( = −1.74 V and = 0.77 V), which should be induced by the stronger electron-donating ability of the picolinate ligand. The stronger reducing power of one-electron reduced species (OERS) of Ru(pic) ( = −1.86 V) facilitates an increase in the number of choices of applicable catalyst because the electron transfer from OERS of Ru(pic) to a catalyst must occur during photocatalysis in the case of reductive quenching mechanisms. When using Ru(pic) as a photosensitizer and Re as a catalyst, the electron transfer process from OERS of Ru(pic) to Re ( = −1.76 V) occurs exothermically.
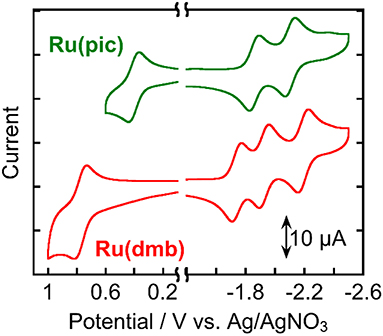
Figure 3. Cyclic voltammograms of Ru(pic) and Ru(dmb) measured in a DMA solution containing Et4NBF4 (0.1 M) as a supporting electrolyte with a Ag/AgNO3 (10 mM) reference electrode.
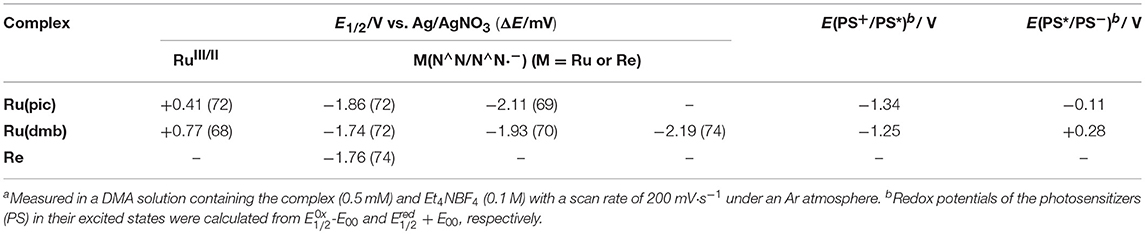
Table 2. Electrochemical properties of the metal complexes in DMAa.
These results indicated that Ru(pic) had some advantages with respect to its function as a redox photosensitizer compared with Ru(dmb), including its wider wavelength range of visible-light absorption and stronger reducing power of OERS, which is effective in the electron transfer to the catalyst. However, certain unfavorable properties were also observed, i.e., a shorter lifetime (τem = 66 ns) and weaker oxidizing power in its excited state (ΔE = E(Ru(dmb)*/Ru(dmb)−)–E(Ru(pic)*/Ru(pic)−) = 0.28–(−0.11) = 0.39 V). In the reductive quenching process, an excited photosensitizer accepts an electron from a sacrificial electron donor. Weaker oxidation power in the excited state of a photosensitizer should decrease the driving force of this electron transfer process. In addition, since this process competes with the radiative and non-radiative deactivation processes from the excited state of a photosensitizer by itself, the shorter lifetime results in less opportunity of the reductive quenching process to occur. To evaluate whether reductive quenching occurs, the emission intensity from Ru(pic) was compared in the presence of five different concentrations of a sacrificial electron donor, 1,3-dimethyl-2-phenyl-2,3-dihydro-1H-benzo[d]imidazole (BIH) (Tamaki et al., 2013a; Hasegawa et al., 2015) in DMA-triethanoamine (TEOA; 5:1 v/v). As shown in Figure 4, the emission intensities from the 3MLCT excited state of Ru(pic) decreased at higher concentrations of BIH, which indicated that the excited Ru(pic) was quenched by BIH. The quenching rate constant was determined to be kq = 1.7 × 108 M−1s−1 from the Stern-Volmer plot (Figure S1) and the lifetime of the emission (τem = 66 ns), which was 8-times slower than that of Ru(dmb) (kq = 1.4 × 109 M−1s−1) as expected from the weaker oxidizing power in the 3MLCT excited state of Ru(pic). In the photocatalytic reaction condition, i.e., [BIH] = 0.2 M, 69% of the excited Ru(pic) was estimated to be quenched by BIH, which should be enough to initiate a photocatalytic reaction.
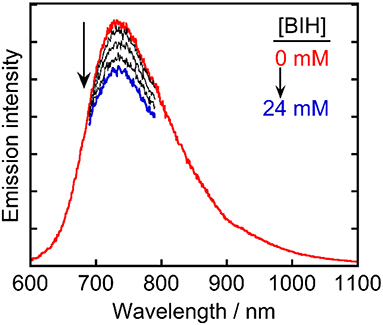
Figure 4. Emission spectra of Ru(pic) in Ar-saturated DMA-TEOA (5:1 v/v) containing five different concentrations of BIH (0–24 mm).
To clarify the produced species as a result of the quenching of excited Ru(pic) by BIH, UV-vis absorption spectral change was observed during photo-irradiation of Ru(pic) in the presence of BIH (Figure 5). Irradiation by light at λex = 480 nm caused spectral changes and new absorption bands appeared at λabs = 420 and 547 nm. The shape of differential absorption spectra before and after irradiation (Figure 5B) were quite similar to that of OERS of Ru(pic) obtained by electrochemical spectroscopy (Figure S2). These results indicate that the reductive quenching of the 3MLCT excited state of Ru(pic) by BIH proceeded successfully to give OERS of Ru(pic) (Equation 1) and Ru(pic) can be expected to function as a redox photosensitizer over the wide-range absorption of visible light.
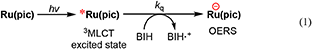
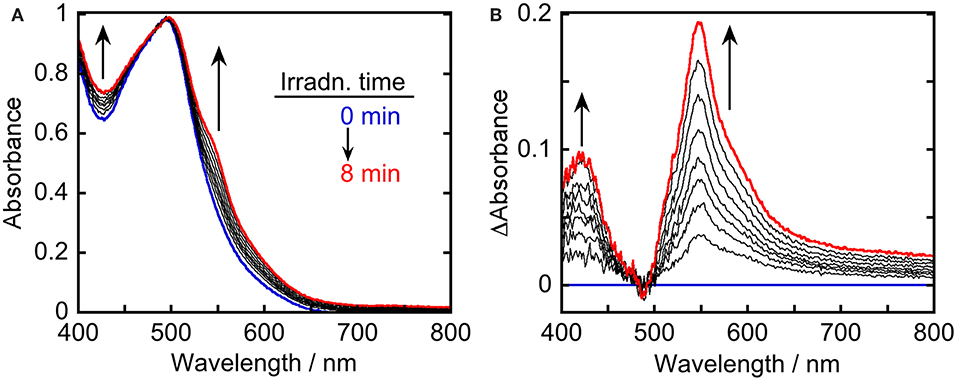
Figure 5. UV-vis (A) absorption and (B) differential absorption spectral change of a DMA-TEOA (5:1 v/v, 4 mL) solution containing Ru(pic) (0.1 mM) and BIH (0.2 M) during irradiation using light at λex = 480 nm (0–8 min at 1-min intervals). The incident light intensity was 5.0 × 10−9 einstein·s−1. Blue and red lines represent spectra at 0 and 8-min irradiation, respectively.
The results of photocatalytic reactions for the reduction of CO2 are summarized in Table 3. In a typical run of photocatalytic reactions, a mixed solution of DMA-TEOA (5:1 v/v) containing Ru(pic) (50 μM), Re (50 μM), and BIH (0.2 M) as a sacrificial electron donor was irradiated under a CO2 atmosphere using light at λex > 620 nm. CO production proceeded linearly and selectively and the turnover number for CO production (TONCO) was 235 after 36 h of irradiation (Figure 6A). The quantum yield for CO formation (ΦCO) was determined to be ΦCO = 8% using λex = 600-nm light (light intensity: 6.0 × 10−9 einstein·s−1). By contrast, when using Ru(dmb) as a redox photosensitizer instead of Ru(pic), no photocatalysis proceeded (Figure 6A) because Ru(dmb) does not absorb lower-energy light at λex > 620 nm (Figure 1). To compare the function as a redox photosensitizer, the photocatalytic reactions were also conducted under photo-irradiation condition, where both Ru(pic) and Ru(dmb) absorb incident light (λex > 480 nm). In this condition, both systems photocatalytically produced CO with high selectivity. Figure 6B shows the time course of photocatalytic CO production using light at λex > 500 nm, and the system using Ru(pic) formed CO faster (TOFCO = 6.7 min−1) than Ru(dmb) (TOFCO = 3.6 min−1) in the initial stage of photocatalysis. TONCO reached 2347 and 2100 after 36 h of irradiation using Ru(pic) and Ru(dmb), respectively. The values of ΦCO using light at λex = 480 nm (light intensity: 6.0 × 10−9 einstein·s−1) were 10% and 44% in the cases using Ru(pic) and Ru(dmb), respectively. The Ru(pic) system demonstrated similar ΦCO values in both irradiation conditions (λex = 600 and 480 nm). These results indicated that Ru(pic) has a clear advantage of a wider wavelength range of utilizable visible light compared to Ru(dmb), even for the photocatalytic condition of λex > 480 nm. Since Ru(pic) displays larger molar absorptivity in the λabs > 480-nm region and a wider wavelength range than Ru(dmb) (Figure 1), Ru(pic) absorbs a much larger number of photons at λex > 480-nm, which leads to a faster TOFCO and larger TONCO, even though the quantum yields for CO production were lower.
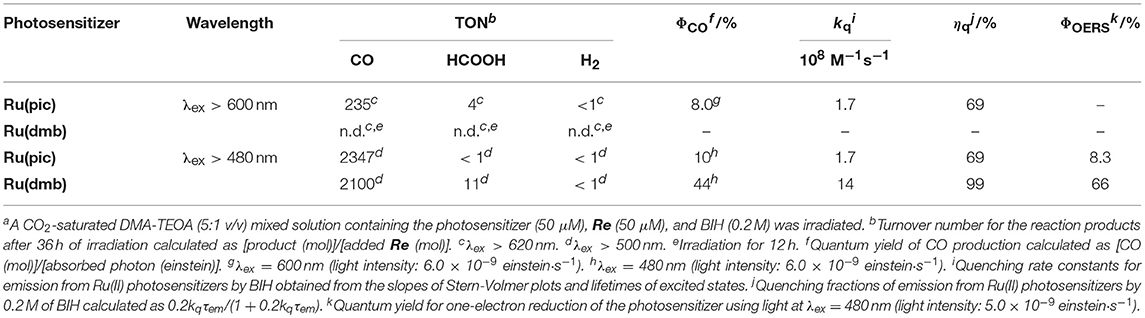
Table 3. Photocatalytic properties using the mixed system of the Ru(II) photosensitizer and Rea.
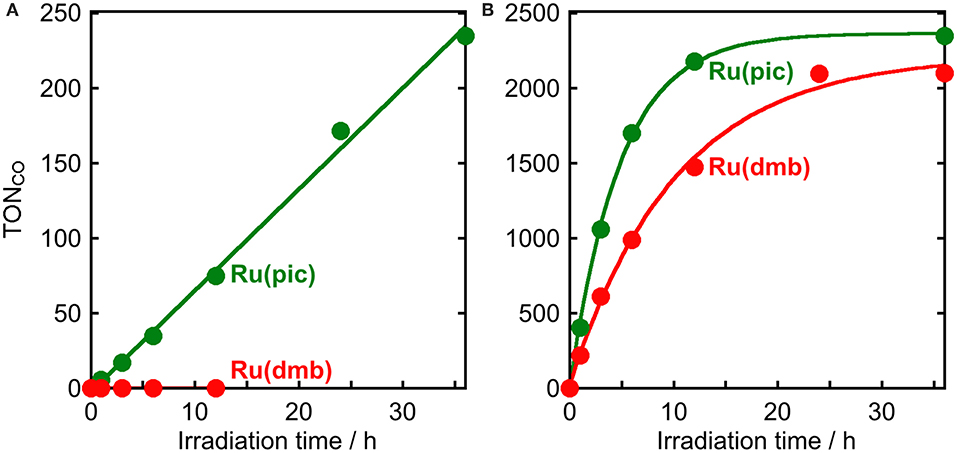
Figure 6. Photocatalytic production of CO as a function of irradiation time using Ru(pic) ( ) or Ru(dmb) (
) or Ru(dmb) ( ) as a photosensitizer: CO2-saturated DMA-TEOA (5:1 v/v, 2 mL) solutions containing Ru(II) photosensitizer (50 μM), Re (50 μM), and BIH (0.2 M) were irradiated at (A) λex > 620 nm or (B) λex > 500 nm.
) as a photosensitizer: CO2-saturated DMA-TEOA (5:1 v/v, 2 mL) solutions containing Ru(II) photosensitizer (50 μM), Re (50 μM), and BIH (0.2 M) were irradiated at (A) λex > 620 nm or (B) λex > 500 nm.
The quantitative analyses of BIH and its oxidized compound during photocatalysis were conducted in the system using 0.1 M of BIH to simplify the HPLC analyses. As the only oxidized compound of BIH, two-electron oxidized and deprotonated BIH (BI+) was observed (Equation 2).
Figure 7 shows the change in the amounts of both BIH and BI+ during photocatalytic reaction along with the amount of CO produced. The amount of produced BI+ was fairly similar to that of CO. For example, after 20 h of irradiation, 205 μmol of BI+ and 203 μmol of CO formed. CO is the two-electron reduced compound of CO2, and BIH supplies two electrons per molecule to give BI+ as a oxidized form. These results clearly indicate that BIH acted as a two-electron donor in the photocatalytic reactions using Ru(pic) as a redox photosensitizer (Equation 3).
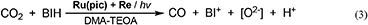
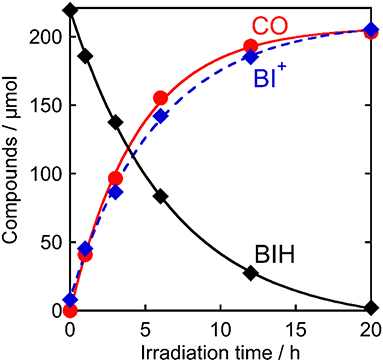
Figure 7. Photocatalytic production of CO () and BI+ ( ) and consumption of BIH (♦): CO2-saturated DMA-TEOA (5:1 v/v) solutions containing Ru(pic) (50 μM), Re (50 μM), and BIH (0.1 M) were irradiated at λex > 500 nm.
) and consumption of BIH (♦): CO2-saturated DMA-TEOA (5:1 v/v) solutions containing Ru(pic) (50 μM), Re (50 μM), and BIH (0.1 M) were irradiated at λex > 500 nm.
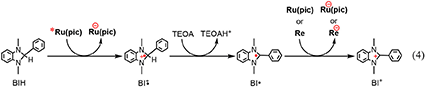
The reaction mechanisms of the photocatalytic reactions using Ru(pic) and Re were investigated. Since Re does not absorb light at λex > 460 nm, as shown in Figure 1, Ru(pic) should absorb the irradiated photon selectively under photocatalytic reaction conditions, i.e., λex > 600 nm or > 480 nm. The photon absorption by Ru(pic) gives its OERS via the reductive quenching process of its 3MLCT excited state by BIH, as described above (Equation 1). The reducing power of OERS of Ru(pic) ( = −1.86 V) is strong enough to trigger electron transfer to Re ( = −1.76 V), which functions as a catalyst for the reduction of CO2. The process of two-electron supply using BIH has already been reported in the photocatalytic reaction system using a Ru(II)-Re(I) supramolecular photocatalyst (Tamaki et al., 2013a). The initial process of the photocatalysis is also a photoinduced electron transfer from BIH to the Ru(II) tris-diimine type photosensitizer unit, forming OERS of the photosensitizer unit and one-electron oxidized BIH (BIH·+). BIH·+ is rapidly deprotonated by TEOA to give BI·. TEOA functioned only as a base, but not as a sacrificial electron donor to quench the excited photosensitizer unit. BI· has a strong reducing power ( = −1.95 V) (Zhu et al., 2008) enough to provide one more electron to the supramolecular photocatalyst to be converted to BI+. In other words, BIH works as a two-electron donor by one-photon excitation of the photocatalyst via the ECE mechanism. Similar processes should also proceed in the photocatalytic system using Ru(pic) and Re because both Ru(pic) ( = −1.86 V) and Re ( = −1.76 V) have a lower reduction potential than BI· ( = −1.95 V). Based on this investigation, the electron-supply processes of BIH are presumed, as depicted in Equation 4.
Photocatalysis using Ru(pic) displayed an advantages of a wider wavelength region of visible-light absorption, which achieved both red-light driven CO2 reduction (λex > 620 nm) and faster CO production than the system using Ru(dmb) (λex > 500 nm), whereas the quantum yield for CO formation using Ru(pic) (ΦCO = 10%) was 1/4 the value when Ru(dmb) (ΦCO = 44%) was used. The main reason for smaller ΦCO should be the smaller quantum yield of one-electron reduction (ΦOERS) of Ru(pic). ΦOERS of Ru(pic) using light at λex = 480 nm (light intensity: 5.0 × 10−9 einstein·s−1) was determined to be 8.3%, which was 1/8 that of Ru(dmb) (ΦOERS = 66%). The elementary processes of one-electron reduction of Ru(pic) is displayed in Scheme 1. The reductive quenching of the 3MLCT excited state of Ru(pic) by BIH gives an ion pair, [Ru(pic)−···BIH·+]. If the ion pair dissociate, free OERS and BIH·+ are obtained. The charge-recombination processes from the ion pair or by the re-collision of OERS of Ru(pic) and BIH·+ should form Ru(pic) and BIH. The differences in properties between Ru(pic) and Ru(dmb), i.e., the cationic valence and the reducing power of OERS, should affect each elementary process and consequently the quantum yield for one-electron reduction. Since OERS of Ru(dmb) is a monovalent cation, the ion pair with BIH·+ involves cationic repulsion, which should accelerate the dissociation process. On the other hand, OERS of Ru(pic) is zero-valent, which provides no repulsion between BIH·+, and therefore, the dissociation process should become slower when using Ru(pic) (smaller kesc). In addition, since the reducing power of OERS of Ru(pic) ( = −1.86 V) is stronger than that of Ru(dmb) ( = −1.74 V), the driving forces for the charge-recombination processes become larger when Ru(pic) is used (larger krec1, krec2). Consequently, the smaller ΦOERS using Ru(pic) should be induced by the slower dissociation process of the ion pair and the faster charge-recombination processes. The quantitative analyses of the factors controlling ΦOERS of photosensitizing complexes are in progress and will be reported elsewhere.
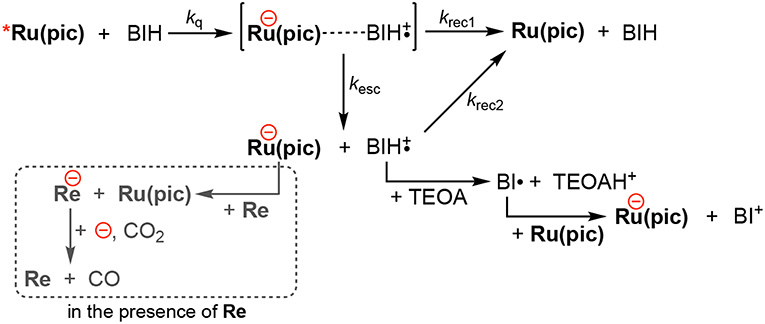
Scheme 1. The one-electron reduction processes of Ru(pic).
In the photocatalytic reaction conditions, the electron-consuming process for CO2 reduction via the electron transfer to Re (the broken box in Scheme 1) will compete against the charge-recombination by the re-collision of OERS and BIH·+. Therefore, since ΦOERSs were determined in the absence of Re, ΦCO (10%) was larger than the expected value from half of ΦOERS (8.3/2 = 4.2%), which was derived from the fact that the reduction of CO2 to CO is a two-electron reduction process. Higher reduction potential of Ru(pic) should operate in favor of the electron transfer to Re. Therefore, the ratio of quantum yields for CO2 reduction between using Ru(pic) and Ru(dmb), i.e., ΦCO(Ru(pic))/ΦCO(Ru(dmb)) = 10/44 = 0.23, became larger than that for one-electron reduction (ΦOERS(Ru(pic))/ΦOERS(Ru(dmb)) = 8.3/66 = 0.13). In other words, Ru(pic) has another advantage of faster electron transfer to Re in the photocatalysis.
Experiments
General Procedures
1H NMR spectra were measured using a JEOL ECA400II (400 MHz) system in solutions of acetone-d6. The residual protons of acetone-d6 were used as an internal standard for measurements. Electrospray ionization-mass spectroscopy (ESI-MS) was performed using a Shimadzu LCMS-2010A system with acetonitrile as the mobile phase. UV-vis absorption spectra were measured with a JASCO V-565 spectrophotometer. Emission spectra were measured using a Horiba Fluorolog-3-21 spectrofluorometer equipped with a NIR-PMT R5509-43 near infrared detector. A Horiba FluoroCube time-correlated single-photon counting system was used to obtain emission lifetimes. The excitation light source was a NanoLED-515L pulse lamp (510 nm). A HAMAMATSU absolute PL quantum yield spectrometer C9920-02 was used to determine emission quantum yields. The samples were degassed by Ar-bubbling of solutions for 30 min prior to measuring emissions. Emission quenching experiments were performed on solutions containing the complexes and five different concentrations of BIH. The quenching rate constants kq were calculated from linear Stern-Volmer plots for the emission from the 3MLCT excited state of the photosensitizing complexes and their lifetimes. The redox potentials of the complexes were measured in an Ar-saturated DMA solution containing Et4NBF4 (0.1 M) as a supporting electrolyte using cyclic voltammetric techniques performed with an ALS CHI-720Dx electrochemical analyzer with a glassy carbon disk working electrode (3 mm diameter), a Ag/AgNO3 (10 mM) reference electrode, and a Pt counter electrode. The supporting electrolyte was dried under vacuum at 100°C for 1 day prior to use. The scan rate was 200 mV·s−1.
Photocatalytic Reactions
Photocatalytic reactions were performed in DMA–TEOA (5:1 v/v) solutions containing the photosensitizer (50 μM), Re (50 μM), and BIH (0.2 M). After the solution was purged with CO2 for 20 min, the solution was irradiated. For TON measurements, the mixed solution (2 mL) in an 11 mL test tube (i.d. 8 mm) was irradiated in a merry-go-round apparatus using λex > 620 nm light from a halogen lamp equipped with a Rhodamin B (0.2% w/v, d = 1 cm) solution filter or λex > 500 nm light from a high-pressure Hg lamp equipped with a uranyl glass and a K2CrO4 (30% w/w, d = 1 cm) solution filter. During irradiation, the temperature of the solution was maintained at 25°C using an EYELA CTP-1000 constant-temperature system. For quantum yield measurements, the mixed solution in a quartz cubic cell (11 mL, light pass length: 1 cm) was irradiated in a Shimadzu photoreaction quantum yield evaluation system QYM-01 using 600 nm or 480 nm light from a 300 W Xe lamp equipped with a 600 nm or 480 nm (FWHM: 10 nm) bandpass filters. The temperature of the solution was controlled during irradiation at 25 ± 0.1°C using an IWAKI CTS-134A constant-temperature system. The gaseous products of photocatalysis, i.e., CO and H2, were analyzed by GC-TCD (GL science GC323). A capillary electrophoresis system (Agilent 7100) was used to analyze HCOOH. HPLC analyses for BIH and BI+ were conducted using a JASCO 880-PU pump, a Develosil ODS-UG-5 column (250 × 4.6 mm), a JASCO 880–51 degasser, and a JASCO UV-2070 detector. The column temperature was maintained at 30°C using a JASCO 860-CO oven. The mobile phase was a 6:4 (v/v) mixture of acetonitrile and a NaOH–KH2PO4 buffer solution (50 mM, pH 7) with a flow rate of 0.5 mL·min−1.
Electrochemical Spectroscopy
Electrochemical spectroscopy to determine the molar absorptivity of OERS was performed using a JASCO PU-980 pump and an EC Frontier flow-type electrolysis cell VF-2 equipped with a carbon felt working electrode (18 mm diameter), a Ag/AgNO3 (10 mM) reference electrode, and a Pt wire counter electrode in an Ar-saturated acetonitrile solution of Ru(pic) (0.5 mM) and Et4NBF4 (0.1 M) as a supporting electrolyte. Applied potential was controlled using an ALS CHI-720Dx electrochemical analyzer and UV-vis absorption spectra were measured using a Photal MCPD-9800 spectrometer (Otsuka Electronics) and a flow-type transmission cell (light pass length: 1.5 mm) (Ishitani et al., 1994).
Quantum Yields for One-Electron Reduction of Photosensitizers
A 4-mL DMA–TEOA (5:1 v/v) solution of the photosesnsitizer (0.1 mM) and BIH (0.2 M) in a quartz cubic cell (light pass length: 1 cm) was purged with Ar for 20 min, and then irradiated with the 500-W Xe lamp combined with a 480-nm (FWHM = 10 nm) bandpass filter (Asahi Spectra Co.), ND filter, and a 5-cm-long H2O solution filter. UV–vis absorption spectral changes during irradiation were measured using a Photal MCPD-9800 spectrometer (Otsuka Electronics). The light intensity was determined as 5.0 × 10−9 einstein·s−1 using a K3Fe(C2O4)3 actinometer.(Hatchard and Parker, 1956) The amount of OERS of Ru(pic) was calculated using the molar absorption coefficient of OERS (500–700 nm) obtained by electrochemical spectroscopy.
Materials
DMA was dried over molecular sieves 4A, distilled under reduced pressure (~10 mmHg) and used in a week. TEOA was distilled under reduced pressure (<1 mmHg) and used in a month. Both solvents were kept under Ar in the dark. All other reagents were of reagent-grade quality and used without further purification.
Synthesis
Ru(dmb) (Sullivan et al., 1978), Re (Morimoto et al., 2013), and BIH (Hasegawa et al., 2005; Zhu et al., 2008) were prepared according to the methods reported in the literatures. Ru(pic) was synthesized using a method similar to the synthesis of [Ru(bpy)2(pic)](PF6) (bpy = 2,2′-bipyridine) (Norrby et al., 1997; Couchman et al., 1998), except for using dmb instead of bpy. [Ru(dmb)2(pic)](PF6) (Ru(pic)): 1H NMR (acetone-d6) δ/ppm: 8.81 (d, J = 5.6 Hz, 1H), 8.65 (s, 1H), 8.63 (s, 1H) 8.60 (s, 1H), 8.55 (s, 1H), 8.14 (dd, J = 5.6, 0.8 Hz, 1H), 8.03 (dd, J = 6.4, 2.4 Hz, 1H), 7.94 (d, J = 5.6 Hz, 1H), 7.91 (d, J = 5.6 Hz, 1H), 7.77 (d, J = 5.6 Hz, 1H), 7.70 (dd, J = 5.6, 0.8 Hz, 1H), 7.64 (d, J = 5.6 Hz, 1H), 7.50 (dd, J = 6.4, 2.4 Hz, 1H), 7.43 (dd, J = 5.6, 1.2 Hz, 1H), 7.26 (dd, J = 5.6, 1.2 Hz, 1H), 7.21 (dd, J = 5.6, 1.2 Hz, 1H), 2.67 (s, 3H), 2.58 (s, 3H), 2.55 (s, 3H), 2.49 (s, 3H). ESI-MS (in acetonitrile) m/z: 592 ([M–PF]+). Anal. calcd for C30H28F6N5O2PRu·H2O: C, 47.75; H, 4.01; N, 9.28. Found: C, 47.72; H, 3.75; N, 9.40.
Conclusion
Ruthenium(II) picolinate complex, Ru(pic), successfully functioned as a redox photosensitizer with a much wider wavelength range of visible-light absorption (λabs < 670 nm) compared with a fairly typical Ru(dmb) (λabs < 560 nm). The system using Ru(pic) as a photosensitizer and Re as a catalyst photocatalyzed the reduction of CO2 to CO by red-light irradiation (λex > 620 nm). TONCO reached 235 and ΦCO was 8.0%. Even in the irradiation conditions where Ru(dmb) also absorbed light, i.e., λex > 500 nm, the system using Ru(pic) demonstrated faster CO formation (TOFCO = 6.7 min−1) and larger TONCO (2347) than that using Ru(dmb) (TOFCO = 3.6 min−1, TONCO = 2100).
Author Contributions
KT, DS, YY, and YT performed all experiments. YU and OI designed this project. YT wrote the manuscript.
Funding
This work was supported by JSPS KAKENHI Grant Numbers JP17H06440 in Scientific Research on Innovative Areas Innovations for Light-Energy Conversion (I4LEC), JP18K14238, JP17K14526.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00327/full#supplementary-material
References
Couchman, S. M., Dominguez-Vera, J. M., Jeffery, J. C., McKee, C. A., Nevitt, S., Pohlman, M., et al. (1998). Structures, electrochemical and spectroscopic properties of ternary ruthenium(II)-polypyridyl complexes with additional carboxylate, biguanide or sulfonamide donors. Polyhedron 17, 3541–3550. doi: 10.1016/S0277-5387(98)00145-4
Fukuzumi, S., Jung, J., Yamada, Y., Kojima, T., and Nam, W. (2016). Homogeneous and heterogeneous photocatalytic water oxidation by persulfate. Chemist. Asian J. 11, 1138–1150. doi: 10.1002/asia.201501329
Gholamkhass, B., Mametsuka, H., Koike, K., Tanabe, T., Furue, M., and Ishitani, O. (2005). Architecture of supramolecular metal complexes for photocatalytic co2 reduction: ruthenium-rhenium bi- and tetranuclear complexes. Inorg. Chem. 44, 2326–2336. doi: 10.1021/ic048779r
Hasegawa, E., Ohta, T., Tsuji, S., Mori, K., Uchida, K., Miura, T., et al. (2015). Aryl-substituted dimethylbenzimidazolines as effective reductants of photoinduced electron transfer reactions. Tetrahedron 71, 5494–5505. doi: 10.1016/j.tet.2015.06.071
Hasegawa, E., Seida, T., Chiba, N., Takahashi, T., and Ikeda, H. (2005). Contrastive photoreduction pathways of benzophenones governed by regiospecific deprotonation of imidazoline radical cations and additive effects. J. Org. Chem. 70, 9632–9635. doi: 10.1021/jo0514220
Hatchard, C. G., and Parker, C. A. (1956). A new sensitive chemical actinometer. II. Potassium ferrioxalate as a standard chemical actinometer. Proc. R. Soc. London Ser. A. Mathemat. Phys. Sci. 235, 518–536. doi: 10.1098/rspa.1956.0102
Hawecker, J., Lehn, J.-M., and Ziessel, R. (1983). Efficient photochemical reduction of CO2 to CO by visible light irradiation of systems containing Re(bipy)(CO)3X or Ru(bipy)32+ -Co2+ combinations as homogeneous catalysts. J.Chem. Soc. Chem. Commun. 9, 536–538. doi: 10.1039/c39830000536
Ishitani, O., George, M. W., Ibusuki, T., Johnson, F. P. A., Koike, K., Nozaki, K., et al. (1994). Photophysical behavior of a new co2 reduction catalyst, Re(CO)2(bpy){P(OEt)3}2+. Inorg. Chem. 33, 4712–4717.
Juris, A., Balzani, V., Barigelletti, F., Campagna, S., Belser, P., and von Zelewsky, A. (1988). Ru(II) polypyridine complexes: photophysics, photochemistry, eletrochemistry, and chemiluminescence. Coord. Chem. Rev. 84, 85–277. doi: 10.1016/0010-8545(88)80032-8
Morimoto, T., Nakajima, T., Sawa, S., Nakanishi, R., Imori, D., and Ishitani, O. (2013). CO2 Capture by a rhenium(I) complex with the aid of triethanolamine. J. Am. Chem. Soc. 135, 16825–16828. doi: 10.1021/ja409271s
Norrby, T., Börje, A., Åkermark, B., Hammarström, L., Alsins, J., Lashgari, K., et al. (1997). Synthesis, structure, and photophysical properties of novel ruthenium(II) carboxypyridine type complexes. Inorg. Chem. 36, 5850–5858. doi: 10.1021/ic9705812
Prier, C. K., Rankic, D. A., and MacMillan, D. W. (2013). Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev. 113, 5322–5363. doi: 10.1021/cr300503r
Schulz, M., Karnahl, M., Schwalbe, M., and Vos, J. G. (2012). The role of the bridging ligand in photocatalytic supramolecular assemblies for the reduction of protons and carbon dioxide. Coordination Chemis. Rev. 256, 1682–1705. doi: 10.1016/j.ccr.2012.02.016
Sullivan, B. P., Salmon, D. J., and Meyer, T. J. (1978). Mixed phosphine 2,2'-bipyridine complexes of ruthenium. Inorg. Chem. 17, 3334–3341. doi: 10.1021/ic50190a006
Takeda, H., Cometto, C., Ishitani, O., and Robert, M. (2017). Electrons, photons, protons and earth-abundant metal complexes for molecular catalysis of CO2 reduction. ACS Catal. 7, 70–88. doi: 10.1021/acscatal.6b02181
Tamaki, Y., Imori, D., Morimoto, T., Koike, K., and Ishitani, O. (2016). High catalytic abilities of binuclear rhenium(i) complexes in the photochemical reduction of CO2 with a ruthenium(ii) photosensitiser. Dalton Trans. 45, 14668–14677. doi: 10.1039/C6DT00996D
Tamaki, Y., and Ishitani, O. (2017). Supramolecular photocatalysts for the reduction of CO2. ACS Catal. 7, 3394–3409. doi: 10.1021/acscatal.7b00440
Tamaki, Y., Koike, K., Morimoto, T., and Ishitani, O. (2013a). Substantial improvement in the efficiency and durability of a photocatalyst for carbon dioxide reduction using a benzoimidazole derivative as an electron donor. J. Catal. 304, 22–28. doi: 10.1016/j.jcat.2013.04.002
Tamaki, Y., Koike, K., Morimoto, T., Yamazaki, Y., and Ishitani, O. (2013b). Red-light-driven photocatalytic reduction of CO2 using Os(II)–Re(I) supramolecular complexes. Inorg. Chem. 52, 11902–11909. doi: 10.1021/ic4015543
Thompson, D. W., Ito, A., and Meyer, T. J. (2013). [Ru(bpy)3]2+* and other remarkable metal-to-ligand charge transfer (MLCT) excited states. Pure Applied Chemis. 85, 1257–1305. doi: 10.1351/pac-con-13-03-04
Tsuji, Y., Yamamoto, K., Yamauchi, K., and Sakai, K. (2018). Near-infrared light-driven hydrogen evolution from water using a polypyridyl triruthenium photosensitizer. Angew. Chem. Internat. Ed. 57, 208–212. doi: 10.1002/anie.201708996
Zhang, Y., Zhou, Q., Tian, N., Li, C., and Wang, X. (2017). Ru(II)-Complex-based DNA photocleaver having intense absorption in the phototherapeutic window. Inorg. Chem. 56, 1865–1873. doi: 10.1021/acs.inorgchem.6b02459
Zhu, X. Q., Zhang, M. T., Yu, A., Wang, C. H., and Cheng, J. P. (2008). Hydride, hydrogen atom, proton, and electron transfer driving forces of various five-membered heterocyclic organic hydrides and their reaction intermediates in acetonitrile. J. Am. Chem. Soc. 130, 2501–2516. doi: 10.1021/ja075523m
Keywords: redox photosensitizer, CO2 reduction, photocatalyst, Ruthenium(II) complex, wide-band absorption
Citation: Tamaki Y, Tokuda K, Yamazaki Y, Saito D, Ueda Y and Ishitani O (2019) Ruthenium Picolinate Complex as a Redox Photosensitizer With Wide-Band Absorption. Front. Chem. 7:327. doi: 10.3389/fchem.2019.00327
Received: 30 January 2019; Accepted: 24 April 2019;
Published: 14 May 2019.
Edited by:
Hitoshi Ishida, Kitasato University, JapanReviewed by:
Wee Han Ang, École Polytechnique Fédérale de Lausanne, SwitzerlandMahmut Özacar, Sakarya University, Turkey
Copyright © 2019 Tamaki, Tokuda, Yamazaki, Saito, Ueda and Ishitani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Osamu Ishitani, aXNoaXRhbmlAY2hlbS50aXRlY2guYWMuanA=
†Present Address: Department of Materials and Life Science, Seikei University, Tokyo, Japan