 Stylianos Panagiotakis1Georgios Landrou1
Stylianos Panagiotakis1Georgios Landrou1 Vasilis Nikolaou1Anisa Putri2
Vasilis Nikolaou1Anisa Putri2 Renaud Hardré2Julien Massin2Georgios Charalambidis1*Athanassios G. Coutsolelos1*
Renaud Hardré2Julien Massin2Georgios Charalambidis1*Athanassios G. Coutsolelos1* Maylis Orio2*
Maylis Orio2*- 1Laboratory of Bioinorganic Chemistry, Department of Chemistry, University of Crete, Heraklion, Greece
- 2Aix Marseille Université, CNRS, Centrale Marseille, iSm2, Marseille, France
In the following work, we carried out a systematic study investigating the behavior of a thiosemicarbazone-nickel (II) complex (NiTSC-OMe) as a molecular catalyst for photo-induced hydrogen production. A comprehensive comparison regarding the combination of three different chromophores with this catalyst has been performed, using [Ir(ppy)2(bpy)]PF6, [Ru(bpy)3]Cl2 and [ZnTMePy]PCl4 as photosensitizers. Thorough evaluation of the parameters affecting the hydrogen evolution experiments (i.e., concentration, pH, solvent nature, and ratio), has been performed in order to probe the most efficient photocatalytic system, which was comprised by NiTSC-OMe and [Ir(ppy)2(bpy)]PF6 as catalyst and chromophore, respectively. The electrochemical together with the photophysical investigation clarified the properties of this photocatalytic system and allowed us to propose a possible reaction mechanism for hydrogen production.
Introduction
One of the most important challenges of our society, that still lie ahead, is to discover renewable and abundant energy sources (Hosenuzzaman et al., 2015; Hosseini and Wahid, 2016). Solar energy is indeed an attractive and unlimited energy source which nonetheless requires the development of novel as well as efficient storage technologies (Styring, 2012; Tachibana et al., 2012; Faunce et al., 2013). Interestingly, hydrogen could unquestionably be applied for such a purpose: (i) it is the simplest and the most plentiful element on earth, (ii) the energy of the hydrogen-hydrogen bond is high, and (iii) it is considered as a non-polluting fuel (Peel, 2003). Hence, photocatalytic water splitting leading to hydrogen production is a method that without any doubt could be proved as an auspicious solution (Lewis and Nocera, 2006). Photocatalytic hydrogen production can be accomplished by systems containing a photosensitizer, a sacrificial electron donor and a catalyst (Ladomenou et al., 2015; Yuan et al., 2017). Nevertheless, there are plenty unsolved issues that still rest in the field of photocatalytic hydrogen production. Specifically, the development of systems utilizing earth-abundant materials with enhanced efficiency and durability (Wang and Sun, 2010; Du and Eisenberg, 2012). To that end, numerous hydrogen evolution catalysts along with a great number of different photosensitizers have been extensively examined over the last years (Tran et al., 2010, 2012; Du and Eisenberg, 2012; Wang et al., 2012; Sartorel et al., 2013).
Photocatalytic systems involving low-cost molecular catalysts and compounds prepared through easy synthetic approaches have been widely studied over the past decade (Artero et al., 2011; Eckenhoff et al., 2013; Ladomenou et al., 2015). Specifically, cobaloximes (Fihri et al., 2008; Lazarides et al., 2009, 2014; Du and Eisenberg, 2012; Landrou et al., 2016; Panagiotopoulos et al., 2016), and other polypyridine cobalt complexes have been applied as noble-metal-free catalysts (Eckenhoff et al., 2013; Yin et al., 2015; Zee et al., 2015). Although, several of these catalysts are efficient for photocatalytic hydrogen evolution reaction (HER), their stability was greatly limited upon visible light irradiation. Moreover, many researches draw inspiration from Nature trying to replicate the function of the hydrogenase enzymes (Lubitz et al., 2014; Brazzolotto et al., 2016), leading to the design of nickel complexes that were evaluated as molecular catalysts for HER. As a result, plenty nickel catalysts such as nickel bis(diphosphine) (DuBois and DuBois, 2009a,b; Helm et al., 2011; McLaughlin et al., 2011), and pyridinethiolate (Han et al., 2012, 2013; Rao et al., 2016) have been applied in such schemes, since they reproduce the structure of the active site of hydrogenase. Due to the effect of non-innocent ligands, (Han et al., 2012, 2013; Rao et al., 2015, 2016; Inoue et al., 2017) such nickel complexes have displayed excellent efficiency as catalyst reaching around 7,500 TON (Han et al., 2013; Rao et al., 2016). Thiosemicarbazone metal complexes are an emerging class of new HER electrocatalysts (Haddad et al., 2016, 2017; Straistari et al., 2017, 2018a,b) that have already been proved to be redox active (Blanchard et al., 2005; Haddad et al., 2017; Straistari et al., 2017) The presence of S-donors as well as N-atoms in thiosemicarbazone allows the protonation of the ligand and serve as proton relays (Campbell, 1975; DuBois, 2014; Coutard et al., 2016). One of the most essential aspect of light-driven proton reduction is the appropriate choice of the light-harvesting unit (i.e., photosensitizer, Ps). Despite the fact that [Ru(bpy)3]Cl2 remains the most widely employed chromophore in such systems (Khnayzer et al., 2014; Lo et al., 2016), iridium complexes are still the most efficient entities found in several photocatalytic systems (Goldsmith et al., 2005; Andreiadis et al., 2011). Additionally, porphyrins and other tetrapyrrolic derivatives can be effective candidates for HER due to their unique stability, electrochemical properties, and appropriate energy levels (Ladomenou et al., 2015). For this reason, various metalloporphyrins such as Zn(II) or Sn(IV), have been utilized as photosensitizers for photocatalytic HER over the years (Lazarides et al., 2014; Koposova et al., 2016; Landrou et al., 2016; Queyriaux et al., 2018).
Here we will discuss the implications of our findings regarding a novel photoinduced HER scheme using a noble-metal-free bis-thiosemicarbazone nickel (II) complex (Straistari et al., 2017), namely NiTSC-OMe. In this study, three different light harvesting complexes, [Ir(ppy)2(bpy)]PF6 (Ps1), [Ru(bpy)3]Cl2 (Ps2), and [ZnTMePy]PCl4 (Ps3) (Andreiadis et al., 2011; Lazarides et al., 2014; Natali et al., 2014) were used as photosensitizers and trimethylamine (TEA) as the sacrificial electron donor (Figure 1). The efficiency of the resulting photocatalytic system was optimized by studying different concentrations of the catalyst, the effect of solvent mixture, the solvent ratio, and the influence of pH in the buffer solution. The electron transfer processes that occur were examined through fluorescence spectroscopic techniques. To solidify the photochemical stability of our system, regeneration experiments were conducted and the homogeneous nature of our catalytic system was proved using poisoning experiments. Based on the results gathered from these studies we were finally able to propose a possible reaction mechanism for light-driven hydrogen production with our photocatalytic system.
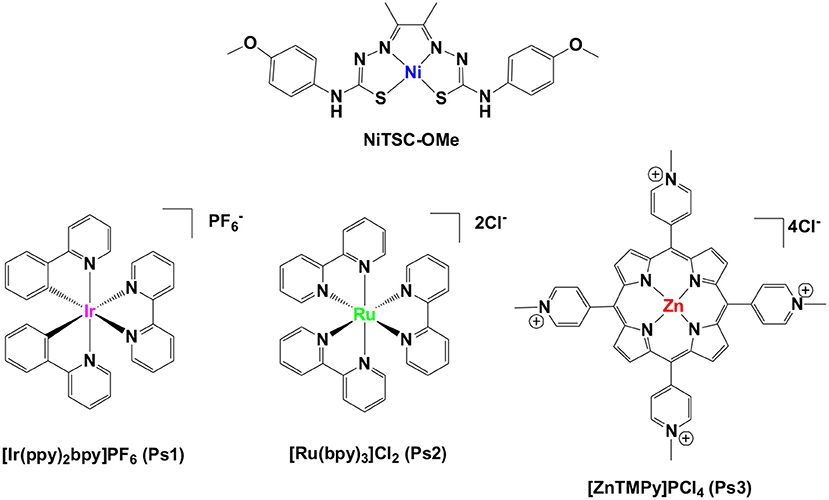
Figure 1. Structures of the molecular catalyst and the photosensitizers used in this work.
Experimental Section
Materials and Methods
Reagents and solvents were purchased as reagent grade from usual commercial sources and were used without further purification, unless otherwise stated. [Ir(ppy)2(bpy)]PF6 and [Ru(bpy)3]Cl2 were purchased from commercial sources and used without further purification. The nickel thiosemicarbazone complex (NiTSC-OMe) (Straistari et al., 2017) and the Zinc (II) meso-tetrakis (1-methylpyridinium-4-yl) porphyrin tetrachloride ([ZnTMePy]PCl4) (Lazarides et al., 2014) were prepared as previously reported.
Photophysical Measurements
UV-Vis absorption spectra were measured on a Shimadzu UV-1700 spectrophotometer using 10 mm path-length cuvettes (Figure S1). The emission spectra were measured by exciting the samples at 337 nm using a JASCO FP-6500 fluorescence spectrophotometer equipped with a red sensitive WRE-343 photomultiplier tube (wavelength range 200–850 nm).
Photocatalytic HER Experiments
For the photoinduced HER studies, each sample was prepared in a 42 mL glass vial with silicone septum. The buffer solutions were prepared by dissolving the sacrificial electron donor [triethylamine (TEA) or ascorbic acid (AA)] in water. It was necessary to add a small amount of acetonitrile in order to obtain a homogeneous solution. The pH was adjusted to the required value using concentrated HCl or NaOH solutions. Then the organic solvent (CH3CN or EtOH) was added in order to obtain the desired ratio. For the sample preparation, the chromophore was dissolved in the buffer solution and consequently a solution of the catalyst in CH3CN or EtOH was added. The final volume of the sample was 5 mL and the mixture was degassed for 10 min using nitrogen. The vials were sealed and the samples irradiated with a white LED lamp (power of 40 W, color temperature of 6,400 K and lumen of 3,800 LM, Figure S2). The amounts of produced hydrogen were determined by gas chromatography (external standard technique) using a Shimadzu GC-2010 plus chromatograph with a TCD detector and a molecular sieve 5 Å column (30–0.53 mm). Gas samples of 100 μL were taken from the headspace and injected immediately into the GC. In all cases, the reported results are the average of three independent experiments. The TONs were calculated using the produced moles of hydrogen vs. the moles of the catalyst. Control experiments were performed under the same conditions after the removal of the catalyst from the hydrogen generating system.
Mercury poisoning experiments were performed, in order to examine the possible formation of metallic nanoparticles or colloids during the hydrogen evolution process. In these studies, an excess of mercury (ca. 40 equiv.) was added to the hydrogen evolution solutions (prepared with the above mentioned procedure).
Results and Discussion
In our recent work, we reported the synthesis of a novel nickel catalyst (NiTSC-OMe, Figure 1) that exhibits high electrocatalytic activity for proton reduction to dihydrogen (Straistari et al., 2017). Based on these encouraging results we wanted to examine the capability of this catalyst toward photochemical hydrogen production. Thus, in the present study three different chromophores (Figure 2) were tested as photosensitizers and combined with the NiTSC-OMe catalyst to determine its ability as an effective photocatalytic system to reduce protons into hydrogen. We have analyzed various parameters, such as the concentration of the catalyst (10−5 – 5 × 10−8 M), the pH of the buffer solution, the effect of the solvent ratio in the photocatalytic mixture and the stability of our system. We carried out several experiments using TEA [5% (v/v)] or AA (0.2 M) as the sacrificial electron donors in various pH buffers (from pH = 2.5 to pH = 10). In addition, different concentrations of the catalyst and the chromophore were tested using varied solvent mixtures. Notably, in all cases no hydrogen production was observed using [Ru(bpy)3]2+ (Ps2) or [ZnTMePy]+ (Ps3) as photosensitizers. However, when [Ir(ppy)2(bpy)]+ (Ps1) was used as photosensitizer and TEA [5% (v/v)] as sacrificial electron donor, a photocatalytic hydrogen evolution of 140 μL from 1 ml of H2O was recorded, highlighting once more that the photosensitizer is an essential component in such photocatalytic HER systems.
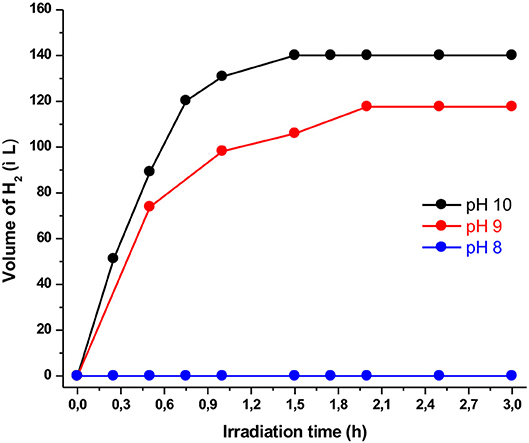
Figure 2. Plot of hydrogen production upon irradiation of solutions (4:1 CH3CN/H2O) containing Ps1 (5 × 10−4 M), NiTSC-OMe (10−5 M), and TEA [5% (v/v)] at different pH values.
In the above mentioned system, the first parameter that we examined was the effect of the protons concentration (pH). In detail, we used three different buffer solutions with pH values of 8, 9, and 10, concluding that the optimum one was at pH = 10. As presented in Figure 2 using the solution with pH = 8, no hydrogen production was observed after 3 h of irradiation. On the contrary, using buffer solutions of pH = 9 and pH = 10 we detected hydrogen production of 105 and 125 TONs, respectively. These findings are consistent with results derived from similar systems in which the optimum pH value is close to the pKa value of the sacrificial electron donor. Namely, in our case since the pKa of TEA is 10.7, the optimum pH of the buffer solution was expected at pH = 10 (Pellegrin and Odobel, 2017). Moreover, at pH values lower than its pKa value TEA is protonated and loses its electron donating ability (Rao et al., 2016).
Furthermore, the performances of the photocatalytic HER systems strongly depend on the catalyst concentration as well as the relative ratio between the photosensitizer and the catalyst. Accordingly, we kept the concentration of the Ps1 photosensitizer constant (5 × 10−4 M), while the concentration of the catalyst varied from 10−5 to 5 × 10−8 M. As illustrated in Figure 3, the volume of the produced H2 was increased when the concentration of the catalyst decreased from 10−5 to 10−6 M, reaching a maximum of 204 μL. In addition, further decrease in the concentration of the catalyst (5 × 10−7–5 × 10−8) resulted in lower catalytic efficiency. On the other hand, the catalytic activity of the system (TON) was increased when the concentration of the catalyst decreased from 10−5 to 5 × 10−8 M (Figure S4). Notably, when the concentration of the NiTSC-OMe was 5 × 10−8 M the system displayed the maximum TON and TOF values, namely 11,333 and 7,971, respectively (see Table S1). These results are in contrast with our previous work (Lazarides et al., 2014; Panagiotopoulos et al., 2016), where the maximum photocatalytic activity was observed when the concentration of the catalyst was in excess compared to that of the photosensitizer. This behavior can be attributed to two possible reasons: (i) the quenching of the excited state of the Ps1 by the NiTSC-OMe complex and (ii) the great difference in the molecular absorptivity (ε) of complexes. Concerning the first hypothesis, since the reductive quenching process of photosensitizer by the catalyst is in competition with the expected reaction with TEA, decreasing the catalyst concentration will possibly favor the suggested reductive pathway. Moreover, regarding the second probable assumption, in our previous work the photosensitizer [ZnTMePy]+ exhibited an absorption coefficient of ε = 1,80,000 M−1.cm−1. In the present study though, the Ps1 exhibits an absorption coefficient of ε = 6000 M−1.cm−1 (Andreiadis et al., 2011), and the catalyst (NiTSC-OMe) displays an absorption band at 470 nm with an epsilon value of ε = 17,000 M−1cm−1 (Straistari et al., 2017). Consequently, the absorption properties of the catalyst can reduce the available photons for the photosensitizer, thus the catalyst concentration should be lower than that of the photosensitizer in order for the system to be more efficient.
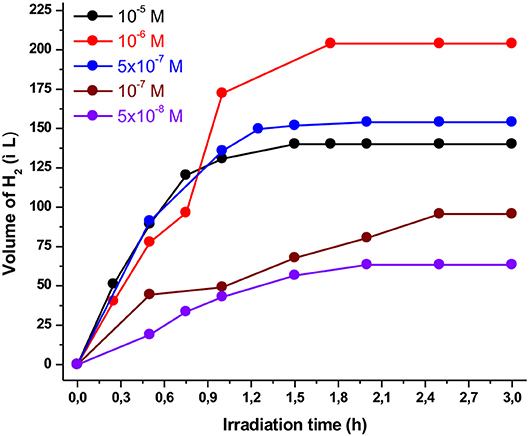
Figure 3. Plot of hydrogen production upon irradiation of different NiTSC-OMe concentrations containing Ps1 (5 × 10−4 M), TEA [5% (v/v)] in a 4:1 CH3CN/H2O solution at pH 10.
The nature as well as the ratio of the solvents definitely plays a significant role in the HER. As a result, we examined two different solvent mixtures, i.e., CH3CN/H2O and Ethanol/H2O, using also different ratio (from 4:1 to 9:1). As shown in Figure 4, the best solvent mixture was found to be CH3CN/H2O in a 4:1 ratio producing a maximum volume of hydrogen of 63 μL after almost 2 h of irradiation. We concluded that the solvent ratio induced critical changes on the hydrogen production, most likely because affecting the solubility properties. These solubility properties can be altered via the dielectric constant and the diffusion coefficient of each solvent (Rao et al., 2015; Pellegrin and Odobel, 2017). When CH3CN/H2O in a 9:1 ratio was utilized as the solvent mixture, the photocatalytic performance dramatically decreased. The smaller water concentration probably leads to lower solubility with direct impact on the photocatalytic activity of the system.
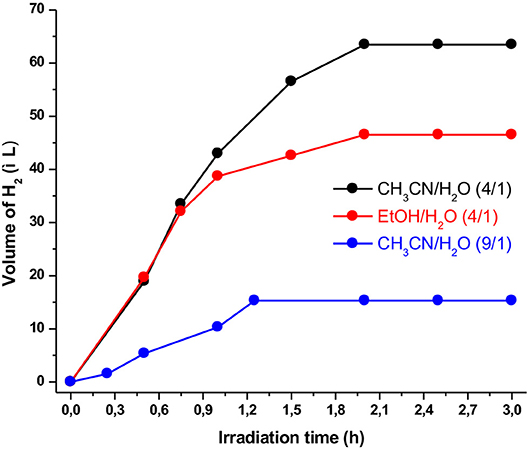
Figure 4. Effect of various solvent mixtures on hydrogen production using the photocatalytic system containing Ps1 (5 × 10−4 M), NiTSC-OMe (5 × 10−8 M), and TEA [5% (v/v)] at pH 10.
In all presented photocatalytic experiments, H2 production stops after almost 2 h of irradiation. Therefore, regeneration and photolysis experiments have been performed in order to examine the stability of our system (Figure 5 and Figure S3). As illustrated in Figure 5 (right part), we performed UV-Vis absorption photolysis experiment in a solution containing Ps1 and NiTSC-OMe. The characteristic absorption bands of our system (416 for Ps1 and 460 nm for NiTSC-OMe) were significantly decreased after 15 min of irradiation, suggesting bleaching of the photolysis solutions. After the addition of either the catalyst or the photosensitizer we didn't observed any hydrogen evolution. However, when both components were added to the reaction mixture, the catalytic system was effectively regenerated, leading to 3,067 TON (Figure 5, left), suggesting that both these components undergo concomitant decomposition after almost 2 h of irradiation.
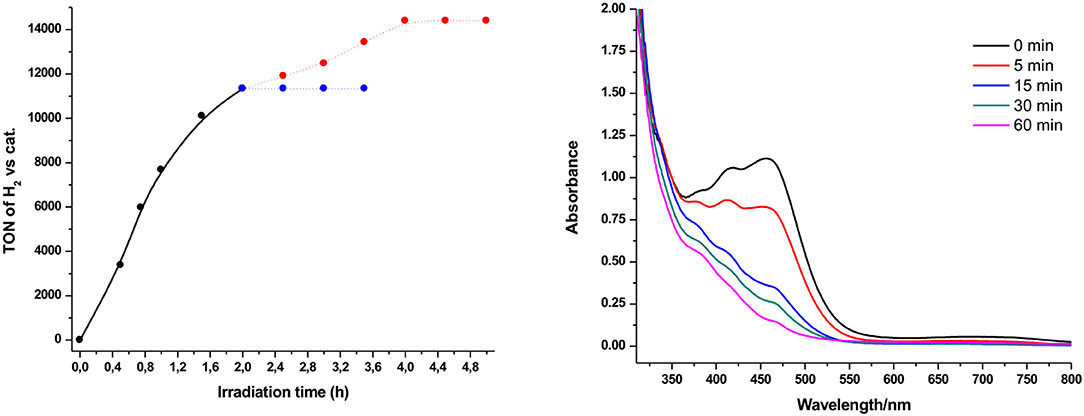
Figure 5. Hydrogen production upon irradiation of solutions (4:1 CH3CN/H2O) containing Ps1 (5 × 10−4 M), NiTSC-OMe (5 × 10−8 M), and TEA [5% (v/v)] at pH = 10. Regeneration of Ps1 and NiTSC-OMe (red dot line). Regeneration of Ps1 or NiTSC-OMe (blue dot line) (left). UV-vis absorption spectra recorded during the course of the photolysis experiment in 10 mm cuvette, containing Ps1 (7 × 10−5 M), NiTSC-OMe (5 × 10−5 M), and TEA [5% (v/v)] at pH = 10 (right).
The degraded compounds can form metallic nanoparticles, which can act as the catalytic species during HER (Lazarides et al., 2014). In order to exclude this possibility we performed mercury poisoning experiments. Photocatalytic experiments in the presence of mercury showed no significant change in the amount of the produced H2 (11,787 TONs), thus confirming the homogeneous nature of our photocatalytic system.
To shed light into the mechanism of photocatalytic system, fluorescence spectroscopy was used. Specifically, emission spectroscopy experiments were carried out using Ps1 (4 × 10−5 M) as photosensitizer upon its excitation at 337 nm in CH3CN solution. While the photocatalytic measurements were performed in a CH3CN/H2O mixture, the photophysical studies were carried out using CH3CN as a solvent. This is due to the fact that TEA was not soluble enough in the high concentrations needed for the Stern Volmer plots in the CH3CN/H2O mixture. Firstly, we examined the quenching process on Ps1 by increasing the concentration of either TEA (0 → 0.36 M)] (Figure S5, left) or NiTSC-OMe (0 → 4.1 × 10−5 M) (Figure S6, left). In addition, we have calculated the Stern-Volmer constant (KSV) based on Stern-Volmer plot (Figures S4, S5, right), using the equation Io/I = 1 + KSV[Q], where Io and I are the fluorescence intensities observed in the absence and in the presence of each quencher, respectively, and [Q] is defined as the quencher concentration (Keizer, 1983). In perfect agreement with previous publications, the KSV constant was higher in the case of the catalyst (KSV = 13,419.5 M−1) compared to the TEA (KSV = 11.7 M−1). Moreover, we calculated the quenching constant for NiTSC-OMe (KQ = 4.99 × 1010 M−1s−1) and for TEA (KQ = 4.35 × 107 M−1s−1) as well, which are derived from the equation: KSV = KQ τ, where τ stands for the excited state lifetime in the absence of the quencher (Han et al., 2013; Yuan et al., 2016). However, under similar conditions to the H2 production experiments in the emission spectra of Ps1, we observe that the characteristic fluorescence peak of the photosensitizer (at 590 nm) decreases only when the sacrificial electron donor (TEA) is added (Figure S7). What is more, there is a great difference in the quenching process, namely 50% in case of TEA and 1% in case of the catalyst. Therefore, even though, the rate constant by the NiTSC-OMe is greater than the rate constant by TEA, the major electron transfer pathway occurs from the excited photosensitizer to TEA, since the concentration of electron donor is bigger than that of the catalyst in the system (Han et al., 2013). In summary, hydrogen production in our system is initiated via reductive quenching of the photosensitizer.
The redox potentials of the reported compounds used in this study are listed in Table 1. Based on these values the thermodynamic driving forces for the different electron transfer processes, ΔGi(PS/Cat), were calculated [(Queyriaux et al., 2017) and references herein, Goldsmith et al., 2005]. The resulting ΔG1(PS/Cat) values turn out to be in agreement with the fluorescence quenching measurements which seems to indicate that the initial step is the reductive quenching of the photosensitizer. Indeed, the calculated ΔG1(PS/Cat) values are positive for a reduction from Ps+/Ps* for Ps1 and Ps2 (+0.44 and +0.18, respectively) whereas they are negative for a reduction from Ps/Ps− (−0.13 and −0.24, respectively). It is also possible to find that whatever the mechanism is, the Ps3 potentials are not negative enough to reduce the nickel catalyst, which would explain the observed lack of photocatalytic activity. The double reduction of the catalyst is also unlikely since the values of ΔG2(PS/Cat) are largely positive (> +0.40 eV) which would not be in favor an EECC mechanism (E corresponds to an electron transfer step and C to a chemical reaction, here protonation). Finally, the difference in activity between Ps1 and Ps2 does not seem to be rationalized by the little difference in driving force of injection (respectively, −0.13 and −0.24 eV), but rather by the difference of driving regeneration force ΔG(SED/PS) which is larger in the case of Ps2 than for Ps1 (respectively, +0.09 and 0 eV).
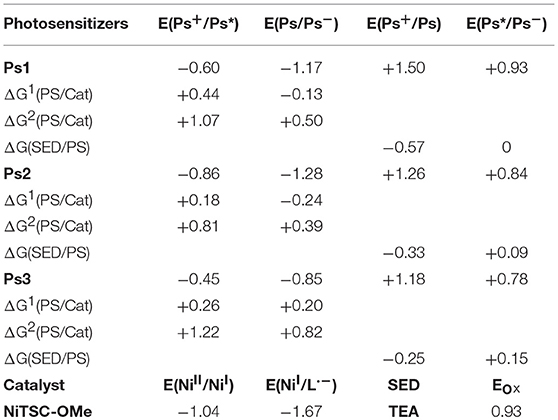
Table 1. Redox potentials (V vs. NHE) of the different compounds employed in this study (Ps* represents the excited state of Ps: Ps+ and Ps−, its oxidized and reduced forms, respectively) together with the thermodynamic driving forces for the different electron transfer processes (ΔG1(PS/Cat), ΔG2(PS/Cat), and ΔG(SED/PS), eV).
Taking into consideration all studies reported in this work, we propose a possible H2 production mechanism as illustrated in Scheme 1. First, [Ir(ppy)2(bpy)]+ (Ps) is excited by visible light irradiation to form the excited state of the photosensitizer (Ps*). Subsequently, the sacrificial electron donor (TEA) transfers an electron to the excited photosensitizer via reductive quenching process forming its oxidized state (TEA+) and the reduced state of the photosensitizer (Ps−) (Scheme 1).
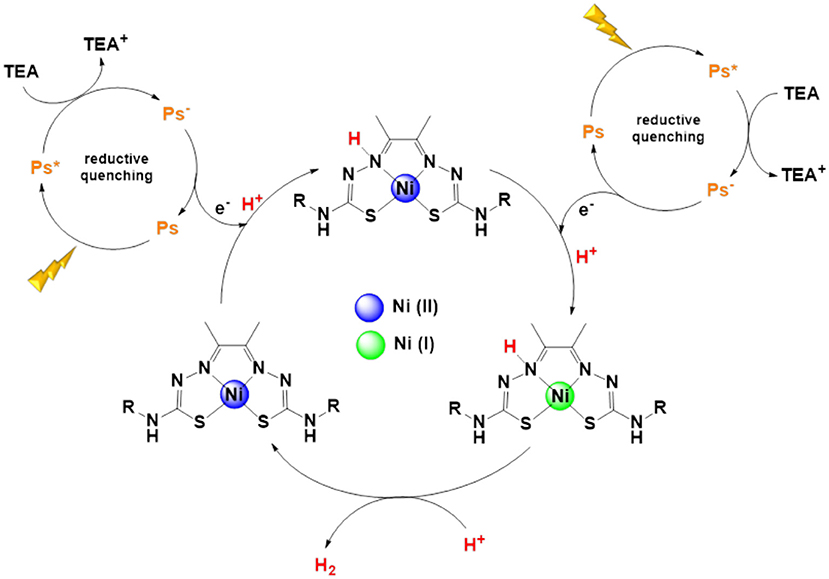
Scheme 1. Proposed mechanism of hydrogen photogeneration.
Then, a protonation of coordinated N-atom of the ligand on nickel complex takes place (Straistari et al., 2017) followed by an electron transfer process from the photosensitizer (Ps−) to the nickel catalyst, creating the nickel (I) complex and forming an hydride intermediate as presented in the scheme bellow. Finally, H2 production from the system through the nickel catalyst occurs together with its regeneration.
Conclusions
In summary, a systematic photocatalytic study toward light-driven hydrogen production is presented herein, using three different chromophores and one bis-thiosemicarbazone nickel complex. When NiTSC-OMe was combined with either Ps2 or Ps3, no hydrogen production was observed under various experimental conditions. However, when Ps1 was utilized as a chromophore, H2 production was detected. In order to estimate the optimal conditions we examined the influence of various parameters: the concentration of the catalyst, the pH value of the buffer solution and the ratio as well as the nature of the solvent mixture. Overall, the highest amount of H2 (204 μL,) was attained using a 4:1 CH3CN/H2O solution containing NiTSC-OMe (10−6 M), Ps1 (5 × 10−4 M), TEA [5% (v/v)] at pH 10. The maximum catalytic activity of the system though, with the highest TON and TOF values (11,333 and 7,971) were observed using NiTSC-OMe (5 × 10−8 M), Ps1 (5 × 10−4 M), TEA [5% (v/v)] at pH 10 in 4:1 CH3CN/H2O solution. All the promising results displayed in this study offer new aspects regarding the combination of chromophores with nickel catalysts for hydrogen production. In addition, such efforts could in general provide new perspectives improving the efficiency and the function of catalytic systems developed for photoinduced hydrogen evolution using water.
Author Contributions
MO and AC designed and directed the study. AP synthesized the catalyst. SP, GL, and VN performed the experiments. RH, JM, GC, AC, and MO analyzed the data. GC, AC, and MO wrote the paper with input from all authors. All authors contributed to the design and implementation of the research, to the analysis of the results and to the writing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
General Secretariat for Research and Technology (GSRT) and Hellenic Foundation for Research and Innovation (HFRI) (project code: 508) are gratefully acknowledged for the financial support of this research. Also, the European Commission's Seventh Framework Program (FP7/2007-2013) under grant agreement no. 229927 (FP7-REGPOT-2008-1, Project BIO-SOLENUTI), and the Special Research Account of the University of Crete.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00405/full#supplementary-material
References
Andreiadis, E. S., Chavarot-Kerlidou, M., Fontecave, M., and Artero, V. (2011). Artificial photosynthesis: from molecular catalysts for light-driven water splitting to photoelectrochemical cells. Photochem. Photobiol. 87, 946–964. doi: 10.1111/j.1751-1097.2011.00966.x
Artero, V., Chavarot-Kerlidou, M., and Fontecave, M. (2011). Splitting water with cobalt. Angewandte Chemie 50, 7238–7266. doi: 10.1002/anie.201007987
Blanchard, S., Neese, F., Bothe, E., Bill, E., Weyhermüller, T., and Wieghardt, K. (2005). Square planar vs tetrahedral coordination in diamagnetic complexes of nickel(II) containing two bidentate pi-radical monoanions. Inorg. Chem. 44, 3636–3656. doi: 10.1021/ic040117e
Brazzolotto, D., Gennari, M., Queyriaux, N., Simmons, T. R., Pecaut, J., Demeshko, S., et al. (2016). Nickel-centred proton reduction catalysis in a model of [NiFe] hydrogenase. Nat. Chem. 8, 1054–1060. doi: 10.1038/nchem.2575
Campbell, M. J. M. (1975). Transition-metal complexes of thiosemicarbazide and thiosemicarbazones. Coord. Chem. Rev. 15, 279–319. doi: 10.1016/S0010-8545(00)80276-3
Coutard, N., Kaeffer, N., and Artero, V. (2016). Molecular engineered nanomaterials for catalytic hydrogen evolution and oxidation. Chem. Commun. 52, 13728–13748. doi: 10.1039/C6CC06311J
Du, P. W., and Eisenberg, R. (2012). Catalysts made of earth-abundant elements (Co, Ni, Fe) for water splitting: Recent progress and future challenges. Energy Environ. Sci. 5, 6012–6021. doi: 10.1039/c2ee03250c
DuBois, D. L. (2014). Development of molecular electrocatalysts for energy storage. Inorg. Chem. 53, 3935–3960. doi: 10.1021/ic4026969
DuBois, M. R., and DuBois, D. L. (2009a). Development of molecular electrocatalysts for CO2 reduction and H-2 production/oxidation. Acc. Chem. Res. 42, 1974–1982. doi: 10.1021/ar900110c
DuBois, M. R., and DuBois, D. L. (2009b). The roles of the first and second coordination spheres in the design of molecular catalysts for H-2 production and oxidation. Chem. Soc. Rev. 38, 62–72. doi: 10.1039/B801197B
Eckenhoff, W. T., Mcnamara, W. R., Du, P. W., and Eisenberg, R. (2013). Cobalt complexes as artificial hydrogenases for the reductive side of water splitting. Biochim. Biophys. Acta 1827, 958–973. doi: 10.1016/j.bbabio.2013.05.003
Faunce, T. A., Lubitz, W., Rutherford, A. W., Macfarlane, D. R., Moore, G. F., Yang, P. D., et al. (2013). Energy and environment policy case for a global project on artificial photosynthesis. Energy Environ. Sci. 6, 695–698. doi: 10.1039/c3ee00063j
Fihri, A., Artero, V., Razavet, M., Baffert, C., Leibl, W., and Fontecave, M. (2008). Cobaloxime-based photocatalytic devices for hydrogen production. Angewandte Chemie 47, 564–567. doi: 10.1002/anie.200702953
Goldsmith, J. I., Hudson, W. R., Lowry, M. S., Anderson, T. H., and Bernhard, S. (2005). Discovery and high-throughput screening of heteroleptic iridium complexes for photoinduced hydrogen production. J. Am. Chem. Soc. 127, 7502–7510. doi: 10.1021/ja0427101
Haddad, A. Z., Cronin, S. P., Mashuta, M. S., Buchanan, R. M., and Grapperhaus, C. A. (2017). Metal-assisted ligand-centered electrocatalytic hydrogen evolution upon reduction of a Bis(thiosemicarbazonato)Cu(II) complex. Inorg. Chem. 56, 11254–11265. doi: 10.1021/acs.inorgchem.7b01608
Haddad, A. Z., Garabato, B. D., Kozlowski, P. M., Buchanan, R. M., and Grapperhaus, C. A. (2016). Beyond metal-hydrides: non-transition-metal and metal-free ligand-centered electrocatalytic hydrogen evolution and hydrogen oxidation. J. Am. Chem. Soc. 138, 7844–7847. doi: 10.1021/jacs.6b04441
Han, Z., Mcnamara, W. R., Eum, M. S., Holland, P. L., and Eisenberg, R. (2012). A nickel thiolate catalyst for the long-lived photocatalytic production of hydrogen in a noble-metal-free system. Angewandte Chemie 51, 1667–1670. doi: 10.1002/anie.201107329
Han, Z., Shen, L., Brennessel, W. W., Holland, P. L., and Eisenberg, R. (2013). Nickel pyridinethiolate complexes as catalysts for the light-driven production of hydrogen from aqueous solutions in noble-metal-free systems. J. Am. Chem. Soc. 135, 14659–14669. doi: 10.1021/ja405257s
Helm, M. L., Stewart, M. P., Bullock, R. M., DuBois, M. R., and DuBois, D. L. (2011). A synthetic nickel electrocatalyst with a turnover frequency above 100,000 s(-1) for H-2 production. Science 333, 863–866. doi: 10.1126/science.1205864
Hosenuzzaman, M., Rahim, N. A., Selvaraj, J., Hasanuzzaman, M., Malek, A. B. M. A., and Nahar, A. (2015). Global prospects, progress, policies, and environmental impact of solar photovoltaic power generation. Renew. Sust. Energy Rev. 41, 284–297. doi: 10.1016/j.rser.2014.08.046
Hosseini, S. E., and Wahid, M. A. (2016). Hydrogen production from renewable and sustainable energy resources: Promising green energy carrier for clean development. Renew. Sust. Energy Rev. 57, 850–866. doi: 10.1016/j.rser.2015.12.112
Inoue, S., Mitsuhashi, M., Ono, T., Yan, Y. N., Kataoka, Y., Handa, M., et al. (2017). Photo- and electrocatalytic hydrogen production using valence isomers of N2S2-type nickel complexes. Inorg. Chem. 56, 12129–12138. doi: 10.1021/acs.inorgchem.7b01244
Keizer, J. (1983). Non-linear fluorescence quenching and the origin of positive curvature in Stern-Volmer plots. J. Am. Chem. Soc. 105, 1494–1498. doi: 10.1021/ja00344a013
Khnayzer, R. S., Thoi, V. S., Nippe, M., King, A. E., Jurss, J. W., El Roz, K. A., et al. (2014). Towards a comprehensive understanding of visible-light photogeneration of hydrogen from water using cobalt(II) polypyridyl catalysts. Energy Environ. Sci. 7, 1477–1488. doi: 10.1039/C3EE43982H
Koposova, E., Liu, X., Pendin, A., Thiele, B., Shumilova, G., Ermolenko, Y., et al. (2016). Influence of meso-substitution of the porphyrin ring on enhanced hydrogen evolution in a photochemical system. J. Phys. Chem. C 120, 13873–13890. doi: 10.1021/acs.jpcc.6b01467
Ladomenou, K., Natali, M., Iengo, E., Charalampidis, G., Scandola, F., and Coutsolelos, A. G. (2015). Photochemical hydrogen generation with porphyrin-based systems. Coord. Chem. Rev. 304, 38–54. doi: 10.1016/j.ccr.2014.10.001
Landrou, G., Panagiotopoulos, A. A., Ladomenou, K., and Coutsolelos, A. G. (2016). Photochemical hydrogen evolution using Sn-porphyrin as photosensitizer and a series of Cobaloximes as catalysts. J. Porphyr. Phthalocyanines 20, 534–541. doi: 10.1142/S1088424616500243
Lazarides, T., Delor, M., Sazanovich, I. V., Mccormick, T. M., Georgakaki, I., Charalambidis, G., et al. (2014). Photocatalytic hydrogen production from a noble metal free system based on a water soluble porphyrin derivative and a cobaloxime catalyst. Chem. Commun. 50, 521–523. doi: 10.1039/C3CC45025B
Lazarides, T., Mccormick, T., Du, P., Luo, G., Lindley, B., and Eisenberg, R. (2009). Making hydrogen from water using a homogeneous system without noble metals. J. Am. Chem. Soc. 131, 9192–9194. doi: 10.1021/ja903044n
Lewis, N. S., and Nocera, D. G. (2006). Powering the planet: Chemical challenges in solar energy utilization. Proc. Natl. Acad. Sci. U. S. A, 103, 15729–15735. doi: 10.1073/pnas.0603395103
Lo, W. K. C., Castillo, C. E., Gueret, R., Fortage, J., Rebarz, M., Sliwa, M., et al. (2016). Synthesis, characterization, and photocatalytic H-2-evolving activity of a family of [Co(N4Py)(X)](n+) complexes in aqueous solution. Inorg. Chem. 55, 4564–4581. doi: 10.1021/acs.inorgchem.6b00391
Lubitz, W., Ogata, H., Rudiger, O., and Reijerse, E. (2014). Hydrogenases. Chem. Rev. 114, 4081–4148. doi: 10.1021/cr4005814
McLaughlin, M. P., Mccormick, T. M., Eisenberg, R., and Holland, P. L. (2011). A stable molecular nickel catalyst for the homogeneous photogeneration of hydrogen in aqueous solution. Chem. Commun. 47, 7989–7991. doi: 10.1039/c1cc12347e
Natali, M., Luisa, A., Lengo, E., and Scandola, F. (2014). Efficient photocatalytic hydrogen generation from water by a cationic cobalt(II) porphyrin. Chem. Commun. 50, 1842–1844. doi: 10.1039/c3cc48882a
Panagiotopoulos, A., Ladomenou, K., Sun, D., Artero, V., and Coutsolelos, A. G. (2016). Photochemical hydrogen production and cobaloximes: the influence of the cobalt axial N-ligand on the system stability. Dalton Transact. 45, 6732–6738. doi: 10.1039/C5DT04502A
Peel, M. (2003). The Hydrogen Economy: The Creation of the Worldwide Energy Web and the Redistribution of Power on Earth. Tls-the Times Literary Supplement, 5227 29–29.
Pellegrin, Y., and Odobel, F. (2017). Sacrificial electron donor reagents for solar fuel production. Comptes Rendus Chimie 20, 283–295. doi: 10.1016/j.crci.2015.11.026
Queyriaux, N., Giannoudis, E., Windle, C. D., Roy, S., Pecaut, J., Coutsolelos, A. G., et al. (2018). A noble metal-free photocatalytic system based on a novel cobalt tetrapyridyl catalyst for hydrogen production in fully aqueous medium. Sustain. Energy Fuels 2, 553–557. doi: 10.1039/C7SE00428A
Queyriaux, N., Wahyuono, R. A., Fize, J., Gablin, C., Wächtler, M., Martinez, E., et al. (2017). Aqueous photocurrent measurements correlated to ultrafast electron transfer dynamics at ruthenium tris diimine sensitized NiO photocathodes. J. Phys. Chem. C 121, 5891–5904. doi: 10.1021/acs.jpcc.6b12536
Rao, H., Wang, Z. Y., Zheng, H. Q., Wang, X. B., Pan, C. M., Fan, Y. T., et al. (2015). Photocatalytic hydrogen evolution from a cobalt/nickel complex with dithiolene ligands under irradiation with visible light. Catal. Sci. Technol. 5, 2332–2339. doi: 10.1039/C4CY01574F
Rao, H., Yu, W. Q., Zheng, H. Q., Bonin, J., Fan, Y. T., and Hou, H. W. (2016). Highly efficient photocatalytic hydrogen evolution from nickel quinolinethiolate complexes under visible light irradiation. J. Power Sourc. 324, 253–260. doi: 10.1016/j.jpowsour.2016.05.095
Sartorel, A., Bonchio, M., Campagna, S., and Scandola, F. (2013). Tetrametallic molecular catalysts for photochemical water oxidation. Chem. Soc. Rev. 42, 2262–2280. doi: 10.1039/C2CS35287G
Straistari, T., Fize, J., Shova, S., Reglier, M., Artero, V., and Orio, M. (2017). A Thiosemicarbazone-nickel(II) complex as efficient electrocatalyst for hydrogen evolution. Chemcatchem 9, 2262–2268. doi: 10.1002/cctc.201600967
Straistari, T., Hardré, R., Fize, J., Shova, S., Giorgi, M., Réglier, M., et al. (2018a). Hydrogen evolution reactions catalyzed by a bis(thiosemicarbazone) cobalt complex: an experimental and theoretical study. Chem. Euro. J. 24, 8779–8786. doi: 10.1002/chem.201801155
Straistari, T., Hardre, R., Massin, J., Attolini, M., Faure, B., Giorgi, M., et al. (2018b). Influence of the metal ion on the electrocatalytic hydrogen production by a thiosemicarbazone palladium complex. Euro. J. Inorg. Chem. 2259–2266. doi: 10.1002/ejic.201800120
Styring, S. (2012). Artificial photosynthesis for solar fuels. Faraday Discus. 155, 357–376. doi: 10.1039/C1FD00113B
Tachibana, Y., Vayssieres, L., and Durrant, J. R. (2012). Artificial photosynthesis for solar water-splitting. Nat. Photonics 6, 511–518. doi: 10.1038/nphoton.2012.175
Tran, P. D., Artero, V., and Fontecave, M. (2010). Water electrolysis and photoelectrolysis on electrodes engineered using biological and bio-inspired molecular systems. Energy Environ. Sci. 3, 727–747. doi: 10.1039/b926749b
Tran, P. D., Wong, L. H., Barber, J., and Loo, J. S. C. (2012). Recent advances in hybrid photocatalysts for solar fuel production. Energy Environ. Sci. 5, 5902–5918. doi: 10.1039/c2ee02849b
Wang, M., Chen, L., and Sun, L. C. (2012). Recent progress in electrochemical hydrogen production with earth-abundant metal complexes as catalysts. Energy Environ. Sci. 5, 6763–6778. doi: 10.1039/c2ee03309g
Wang, M., and Sun, L. C. (2010). Hydrogen production by noble-metal-free molecular catalysts and related nanomaterials. Chemsuschem 3, 551–554. doi: 10.1002/cssc.201000062
Yin, M. C., Ma, S., Wu, C. J., and Fan, Y. T. (2015). A noble-metal-free photocatalytic hydrogen production system based on cobalt(III) complex and eosin Y-sensitized TiO2. Rsc Adv. 5, 1852–1858. doi: 10.1039/C4RA10767E
Yuan, Y. J., Tu, J. R., Lu, H. W., Yu, Z. T., Fan, X. X., and Zou, Z. G. (2016). Neutral nickel(II) phthalocyanine as a stable catalyst for visible-light-driven hydrogen evolution from water. Dalton Transact. 45, 1359–1363. doi: 10.1039/C5DT04311E
Yuan, Y. J., Yu, Z. T., Chen, D. Q., and Zou, Z. G. (2017). Metal-complex chromophores for solar hydrogen generation. Chem. Soc. Rev. 46, 603–631. doi: 10.1039/C6CS00436A
Keywords: light-driven hydrogen production, catalyst, nickel, molecular photosensitizer, photophysics
Citation: Panagiotakis S, Landrou G, Nikolaou V, Putri A, Hardré R, Massin J, Charalambidis G, Coutsolelos AG and Orio M (2019) Efficient Light-Driven Hydrogen Evolution Using a Thiosemicarbazone-Nickel (II) Complex. Front. Chem. 7:405. doi: 10.3389/fchem.2019.00405
Received: 17 December 2018; Accepted: 20 May 2019;
Published: 27 June 2019.
Edited by:
Bunsho Ohtani, Hokkaido University, JapanReviewed by:
Marco Armandi, Polytechnic University of Turin, ItalyMirco Natali, University of Ferrara, Italy
Copyright © 2019 Panagiotakis, Landrou, Nikolaou, Putri, Hardré, Massin, Charalambidis, Coutsolelos and Orio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Georgios Charalambidis, Z2NoYXJhbEB1b2MuZ3I=; Athanassios G. Coutsolelos, YWNvdXRzb2xAdW9jLmdy; Maylis Orio, bWF5bGlzLm9yaW9AdW5pdi1hbXUuZnI=