 Jiajia Meng1Gan Gu1Pengqin Dang1Xuping Zhang1
Jiajia Meng1Gan Gu1Pengqin Dang1Xuping Zhang1 Weixuan Wang1Jungui Dai2Yang Liu3
Weixuan Wang1Jungui Dai2Yang Liu3 Daowan Lai1*
Daowan Lai1* Ligang Zhou1*
Ligang Zhou1*- 1Department of Plant Pathology, College of Plant Protection, China Agricultural University, Beijing, China
- 2State Key Laboratory of Bioactive Substance and Function of Natural Medicines, Institute of Materia Medica, Peking Union Medical College, Chinese Academy of Medical Science, Beijing, China
- 3Institute of Food Science and Technology, Chinese Academy of Agricultural Sciences, Beijing, China
Ustilaginoidea virens, the causal fungus of rice false smut, was found in previous studies to produce two types of metabolites, ustiloxins and ustilaginoidins. However, genome sequencing of U. virens revealed a plethora of secondary-metabolites-biosynthetic core genes that were capable to biosynthesize unreported metabolites. A large-scale fermentation of U. virens was thus performed, and the fungal extract was chemically re-investigated. After removing the known metabolites, we found a fraction containing unknown phytotoxic substances. Fractionation of this part has led to the isolation of six new sorbicillinoids, namely ustisorbicillinols A~F (1~6), and two new sorbicillinoid-related pyrones, named ustilopyrones A (7) and B (8), together with nine known cogeners (9~17). The structures of the new compounds were elucidated by analysis of their NMR, HRMS, and CD spectra, while ECD, 13C NMR and optical rotation calculations were additionally used for configurational assignments. Plausible biosynthetic pathways for the new compounds were proposed. Phytotoxicity assays revealed that the major sorbicillinoids (12~14, and 16) showed strong inhibition against the radicle and germ elongation of rice and lettuce seeds, with compound 12 displaying the strongest inhibition. The isolated compounds were also evaluated for their cytotoxic, antibacterial, and antifungal activities. Compounds 10, and 12~14 showed moderate cytotoxicities against the tested cell lines with IC50s of 8.83~74.7 μM. Compounds 2, and 10~13 were active against the tested bacteria (MICs 4~128 μg/mL), while compounds 11~13 displayed moderate antifungal activities.
Introduction
Plant pathogenic fungi can produce toxic substances, known as phytotoxins, that adversely affect the host plants, and might play a role in the development of plant disease (Strange, 2007; Varejao et al., 2013). Understanding their roles in pathogenesis or virulence will help to find strategies to control diseases, such as breeding disease resistant crops (Strange, 2007). Meanwhile, the great structural diversity and potency of phytotoxins (Cimmino et al., 2015) offered remarkable potential for developing natural herbicides with new modes of action (Duke and Dayan, 2011; Dayan and Duke, 2014).
Ustilaginoidea virens (teleomorph: Villosiclava virens), the causal fungus of rice false smut, which is a destructive disease in the rice cultivation area worldwide (Zhang et al., 2014; Fan et al., 2016), uniquely infects the spikelets of rice panicles, and results in the formation of smut balls around the panicles. So far, two known types of phytotoxins, including the ustiloxins (13-membered-ring cyclopeptides), and ustilaginoidins (9,9′-linked bis-naphtho-γ-pyrones), have been found in the smut balls as well as in the cultures of the pathogen (Lu et al., 2015; Sun et al., 2017; Wang et al., 2017). However, genome sequencing of the pathogen U. virens revealed a plethora of secondary-metabolites-biosynthetic core genes (Zhang et al., 2014), including those encoding NRPS, PKS, and terpene synthases, indicating the great potential of this fungus to produce unknown secondary metabolites.
As part of our continuing interest in the secondary metabolites, especially the phytotoxins, of U. virens (Lu et al., 2015; Sun et al., 2017; Lai et al., 2019), we fermented this fungus on rice medium in a large scale (22.0 kg of rice). After removing the two known types of compounds mentioned above by size-exclusion chromatography over Sephadex LH-20, a fraction containing the third type of compounds that showed their maximal UV absorptions at about 370 nm was found, which indicated the presence of sorbicillinoids (Andrade et al., 1992; Lai et al., 2019). This fraction also exhibited interesting phytotoxic activities against the rice seeds (data not shown). A detailed chemical investigation of this part was performed, which led to the isolation of 17 sorbicillinoids, including eight new compounds. Herein, we report the isolation, structural elucidation of the new sorbicillinoids, as well as the biological activities of the isolated compounds. Their biosynthetic pathway was also discussed.
Materials and Methods
General Experimental Procedures
Optical rotations were recorded on a Rudolph Autopol III automatic polarimeter (Rudolph Research Analytical, Hackettstown, New Jersey). Ultraviolet (UV) spectra were recorded on a TU-1810 UV/vis spectrophotometer (Beijing Persee General Instrument Co., Ltd., Beijing, China). Circular dichroism (CD) spectra were recorded on a JASCO J-815 CD spectrometer (JASCO Corp., Tokyo, Japan). Infrared (IR) spectra were measured on a Thermo Nicolet Nexus 470 FT-IR spectrometer (Thermo Electron Scientific Instrument Crop., Madison, Wisconsin). High-resolution electrospray ionization mass spectrometry (HRESIMS) spectra were recorded on a LC 1260/Q-TOF-MS 6520 instrument (Agilent Technologies, Santa Clara, CA). 1H, 13C, and 2D NMR (1H-1H COSY, HSQC/HMQC, HMBC, and NOESY) spectra were measured on Bruker Avance 600 or 400 NMR spectrometers (Bruker BioSpin, Zürich, Switzerland). 1H and 13C NMR chemical shifts were expressed in δ (ppm) referring to the inner standard tetramethylsilane (TMS), and coupling constants in Hertz. HPLC-DAD analysis of the EtOAc extracts was performed on a Shimadzu LC-20A instrument equipping with a SPD-M20A photodiode array detector (Shimadzu Corp., Tokyo, Japan) using an analytical C18 column (250 × 4.6 mm i.d., 5 μm; Phenomenex Inc., Torrance, California). Semipreparative HPLC separation was carried out on a Lumtech instrument (Lumiere Tech. Ltd., Beijing, China) equipped with a K-501 pump (flow rate: 3 mL/min) and a K-2501 UV detector (detection was set at 370 nm) using a Luna-C18 column (250 × 10 mm i.d., 5 μm, Phenomenex Inc., Torrance, California) eluting with a mixture of MeOH and water (containing 0.02% TFA).
Fungal Material
The fungal strain U. virens UV8b (Zhang et al., 2014) was kindly provided by Prof. Wenxian Sun (College of Plant Protection, China Agricultural University, China).
Fermentation, Extraction, and Isolation
The strain U. virens UV8b was grown on potato dextrose agar (PDA, potato 200 g/L, dextrose 20 g/L, and agar 20 g/L) at 25°C for 10 days. Then, several agar plugs (0.5 × 0.5 cm) containing mycelia were added into a 250 mL Erlenmeyer flask in which 100 mL of potato dextrose broth (PDB, potato 200 g/L, and dextrose 20 g/L) was filled. The liquid culture was incubated in a rotatory shaker for 10 days at 150 rpm and 28°C to produce the seed culture, which was used to inoculate the rice medium (100 g of rice, 110 mL of water, in each 1,000 mL flask). The fermentation was carried out using a total of 22.0 kg of rice at 28°C under static condition in the dark for 60 days. After harvest, the culture was extracted with EtOAc for four times. The EtOAc extract was combined and condensed under vacuum using a rotatory evaporator to yield a brownish residue (220 g). The extract was divided into 40 portions, and each portion was subjected to gel permeation chromatography over Sephadex LH-20 (i.d. 4.0 × 80 cm) eluting with CH2Cl2/MeOH (1:1) to obtain three fractions, the second of which was the sorbicillinoid-containing fraction (SF).
The SF (60 g) was subjected to vacuum liquid chromatograph over silica gel (200~300 mesh, i.d. 8 × 16 cm), eluting with a gradient of CH2Cl2/MeOH (100:0~0:100), to yield six fractions (Frs. A~F).
Fr. B was further fractionated by medium pressure liquid chromatography over silica gel (i.d. 4.0 × 50 cm) to obtain ten subfractions (Frs. B1~B10). Compound 17 (6.3 mg) was purified from Fr. B9 by semi-preparative HPLC eluting with 85% MeOH/H2O.
Fr. C was subjected to column chromatography (i.d. 3.0 × 60 cm) over Sephadex LH-20 (CH2Cl2/MeOH, 1:1) to yield seven subfractions (Frs. C1~C7), among which Fr. C5 was purified by semi-preparative HPLC (85% MeOH/H2O) to give oxosorbicillinol (11, 6.0 mg). Fr. C6 was further chromatographed (i.d. 4.0 × 40 cm) over RP-18 eluting with a mixture of MeOH/H2O (from 60:40 to 100:0, increased stepwisely) to afford six subfractions (Frs. C6-1~C6-6), among which Fr. C6-1 was purified by semi-preparative HPLC (70% MeOH/H2O) to yield 5 (2.6 mg) and 6 (1.0 mg).
Fr. D was fractionated by column chromatography (i.d. 4.0 × 40 cm) on RP-18 eluting with MeOH/H2O (gradually increased from 55:45 to 100:0) to obtain 12 subfractions (Frs. D1~D12). Fr. D7 was further chromatographed (i.d. 3.0 × 60 cm) over Sephadex LH-20 (CH2Cl2/MeOH, 1:1) to afford six fractions (Frs. D7.1~D7.6). Compounds 3 (2.2 mg), and 4 (2.7 mg) were obtained from Fr. D7.3, while compounds 10 (5.1 mg) and 15 (2.0 mg) were isolated from Fr. D7.5, by semi-preparative HPLC using 75% MeOH/H2O as the mobile phase. Likewise, compound 16 (20.1 mg) was purified from Fr. D7.6 (eluting with 80% MeOH/H2O). Fr. D8 was subjected to chromatography (i.d. 3.0 × 60 cm) over Sephadex LH-20 (CH2Cl2/MeOH, 1:1), followed by semi-preparative HPLC (80% MeOH/H2O) to give 1 (4.4 mg) and 2 (8.0 mg). Similarly, compounds 12 (22.3 mg), 13 (13.3 mg), and 14 (40.2 mg) were obtained by semi-preparative HPLC purification (85% MeOH/H2O) from Fr. D9 (for 13 and 14), and Fr. D10 (for 12), respectively.
Fr. E was chromatographed (i.d. 4.0 × 40 cm) over ODS eluting with a gradient increase of MeOH in water (from 30:70 to 100:0) to obtain six subfractions (Frs. E1~E6). The 2-pyrones, 7 (8.2 mg), 8 (1.7 mg) and 9 (3.9 mg) were isolated from Fr. E2 by semi-preparative HPLC using 45% MeOH/H2O as the mobile phase.
Ustisorbicillinol A (1). Yellow amorphous powder; −356 (c 0.1, MeOH); UV(MeOH) λmax (log ε) 220 (3.90), 271 (4.05), 379 (3.88) nm; ECD (c = 4.01 × 10−4 M, MeOH) λ (Δε) 372 (−7.92), 298 (+5.82), 290 (+5.65), 272 (+8.16), 218 (−6.93) nm; IR νmax 3420, 2926, 2852, 1714, 1681, 1646, 1623, 1538, 1517, 1453, 1383, 1343, 1228, 1137, 1019, 903, 845, 675, 578 cm−1; 1H NMR (CD3OD, 400 MHz), 13C NMR (CD3OD, 100 MHz) see Table 1; HRESIMS m/z 497.2193 [M-H]− (calcd for C28H33O8, 497.2181).
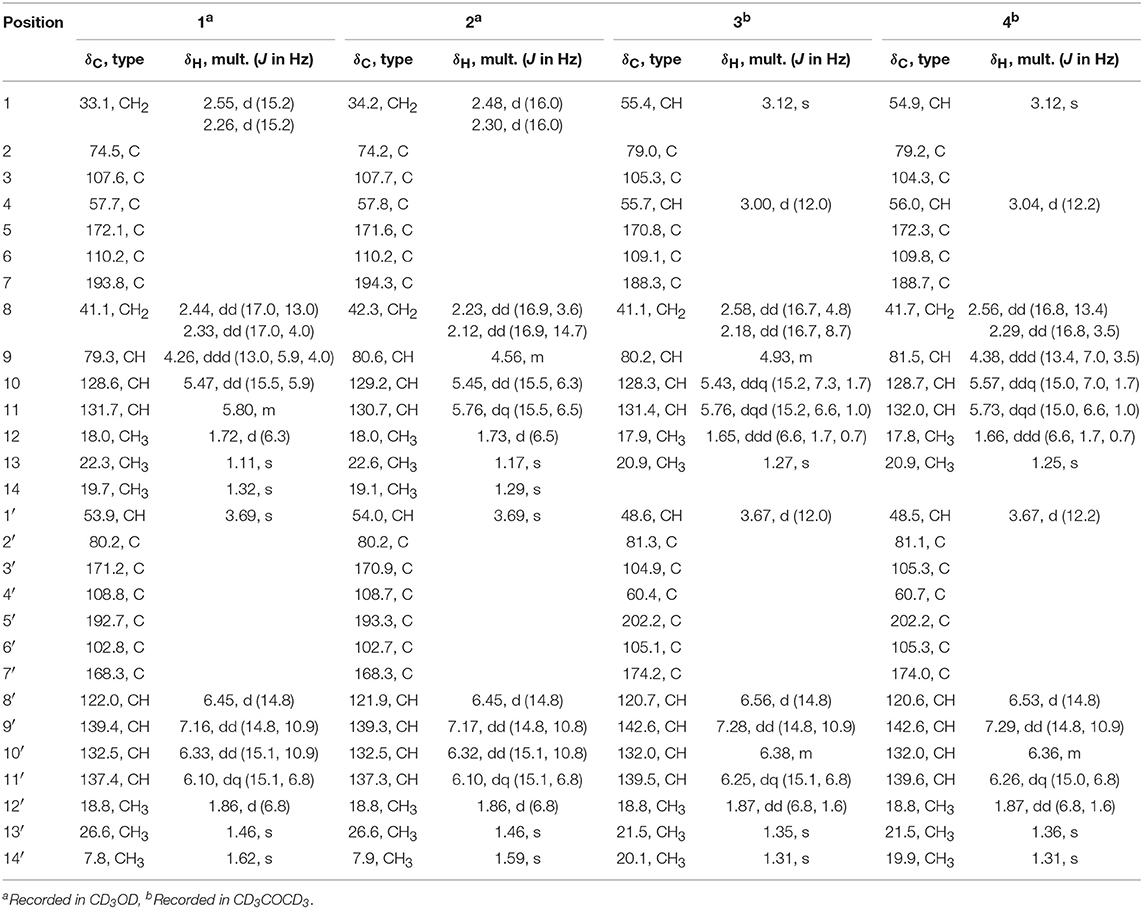
Table 1. 1H and 13C NMR data of 1~4.
Ustisorbicillinol B (2). Yellow amorphous powder; −188 (c 0.1, MeOH); UV(MeOH) λmax (log ε) 219 (3.94), 270 (4.05), 381 (3.93) nm; ECD (c = 4.01 × 10−4 M, MeOH) λ (Δε) 369 (−8.08), 317 (+2.91), 270 (+10.86), 219 (−7.69) nm; IR νmax 3390, 2932, 1713, 1681, 1646, 1623, 1566, 1517, 1472, 1384, 1229, 1209, 1137, 1019, 903, 847, 759, 678, 578 cm−1; 1H NMR (CD3OD, 400 MHz), 13C NMR (CD3OD, 100 MHz) see Table 1; HRESIMS m/z 497.2195 [M-H]− (calcd for C28H33O8, 497.2181).
Ustisorbicillinol C (3). Yellow amorphous powder; +28 (c 0.1, MeOH); UV(MeOH) λmax (log ε) 214 (3.69), 278 (3.89), 360 (3.72) nm; ECD (c = 4.15 × 10−4 M, MeOH) λ (Δε) 368 (−0.26), 310 (+5.40), 272 (−4.78), 227 (+0.06) nm; IR νmax 3726, 3421, 2922, 2852, 1734, 1717, 1695, 1653, 1617, 1576, 1455, 1442, 1384, 1306, 1199, 1131, 1019, 913, 840, 667, 579 cm−1; 1H NMR (CD3COCD3, 600 MHz), 13C NMR (CD3COCD3, 150 MHz) see Table 1; HRESIMS m/z 483.2006 [M+H]+ (calcd for C27H31O8, 483.2013).
Ustisorbicillinol D (4). Yellow amorphous powder; −32 (c 0.1, MeOH); UV(MeOH) λmax (log ε) 214 (3.63), 279 (3.88), 345 (3.79) nm; ECD (c = 4.15 × 10−4 M, MeOH) λ (Δε) 310 (+4.31), 270 (−9.06), 240 (−0.52), 228 (−0.85), 218 (−0.30) nm; IR νmax 3727, 3385, 2936, 1654, 1616, 1439, 1383, 1305, 1256, 1192, 1128, 1050, 1006, 913, 862, 843, 580 cm−1; 1H NMR (CD3COCD3, 600 MHz), 13C NMR (CD3COCD3, 150 MHz) see Table 1; HRESIMS m/z 483.2006 [M+H]+ (calcd for C27H31O8, 483.2013).
Ustisorbicillinol E (5). Yellow amorphous powder; +116 (c 0.1, MeOH); UV(MeOH) λmax (log ε) 214 (3.46), 330 (3.66) nm; ECD (c = 6.02 × 10−4 M, MeOH) λ (Δε) 347 (+10.62), 308 (−12.16), 243 (−0.55), 214 (−3.01) nm; IR νmax 3726, 3625, 3599, 3421, 2921, 2851, 1774, 1735, 1652, 1616, 1576, 1454, 1384, 1201, 1025, 912, 876, 580 cm−1; 1H NMR (CDCl3, 600 MHz), 13C NMR (CDCl3, 150 MHz) see Table 2; HRESIMS m/z 331.1201 [M-H]− (calcd for C18H19O6, 331.1187).
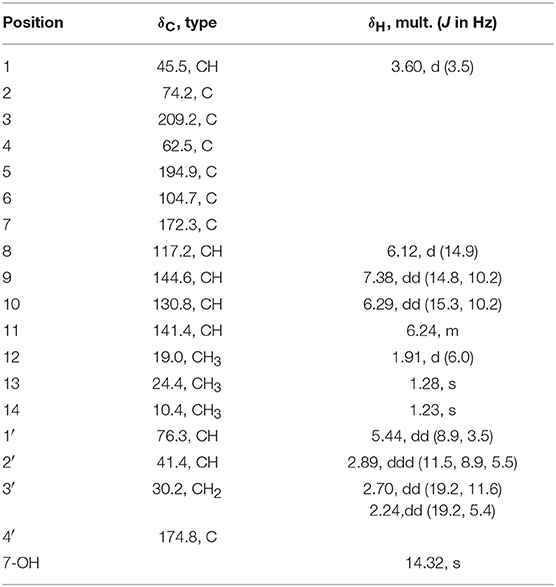
Table 2. 1H (600MHz) and 13C (150MHz) NMR data of 5 (CDCl3).
Ustisorbicillinol F (6). Yellow amorphous powder; −24 (c 0.05, MeOH); UV(MeOH) λmax (log ε) 223 (3.53), 293 (3.40) nm; IR νmax 3720, 3648, 3614, 3445, 2918, 2850, 1750, 1717, 1682, 1649, 1566, 1541, 1517, 1488, 1454, 1432, 1384, 1159, 1018, 900, 842, 677, 578 cm−1; 1H NMR (CDCl3, 600 MHz), 13C NMR (CDCl3, 150 MHz) see Table 3; HRESIMS m/z 311.0892 [M+Na]+ (calcd for C16H16NaO5, 311.0890).
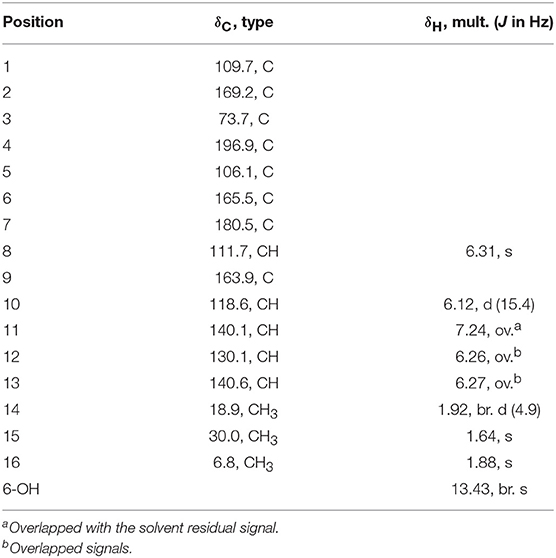
Table 3. 1H (600MHz) and 13C (150MHz) NMR data of 6 (CDCl3).
Ustilopyrone A (7). Yellow amorphous powder; UV(MeOH) λmax (log ε) 227 (3.97), 296 (3.74) nm; IR νmax 3647, 3614, 3363, 2933, 1712, 1646, 1623, 1472, 1363, 1239, 1136, 1017, 909, 848, 756, 678, 578, 530 cm−1; 1H NMR (CD3OD, 400 MHz), 13C NMR (CD3OD, 100 MHz) see Table 4; HRESIMS m/z 221.0823 [M-H]− (calcd for C12H13O4, 221.0819).
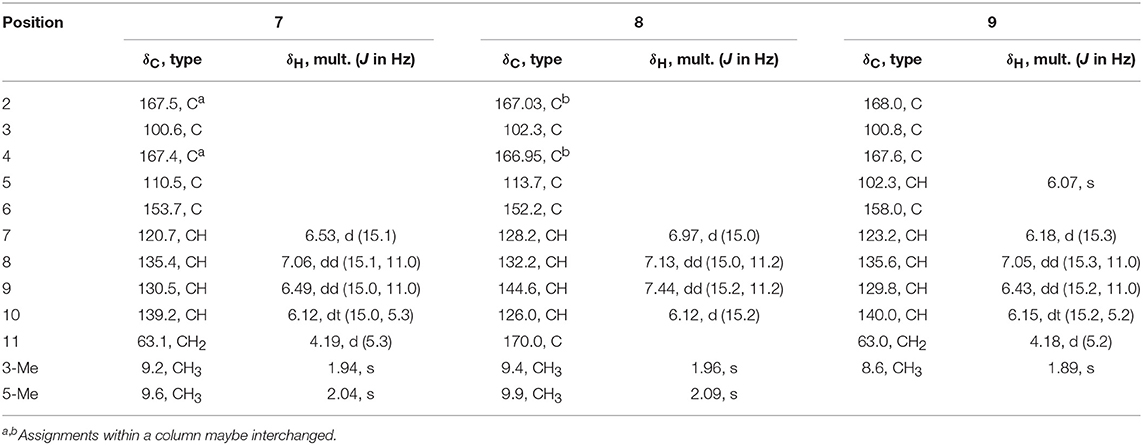
Table 4. 1H (400 MHz) and 13C (100 MHz) NMR data of 7~9 (CD3OD).
Ustilopyrone B (8). Yellow amorphous powder; UV(MeOH) λmax (log ε) 226 (3.71), 261 (3.91), 342 (3.64) nm; IR νmax 3732, 3421, 2919, 2850, 1719, 1652, 1617, 1576, 1542, 1455, 1435, 1384, 1270, 1154, 1020, 835, 732, 579 cm−1; 1H NMR (CD3OD, 400 MHz), 13C NMR (CD3OD, 100 MHz) see Table 4; HRESIMS m/z 235.0617 [M-H]− (calcd for C12H11O5, 235.0612).
5-Demethylustilopyrone A (9). Yellow amorphous powder; UV(MeOH) λmax (log ε) 249 (4.02), 335 (3.76) nm; IR νmax 3252, 2935, 1712, 1646, 1623, 1472, 1384, 1363, 1234, 1135, 1017, 756, 580 cm−1; 1H NMR (CD3OD, 400 MHz), 13C NMR (CD3OD, 100 MHz) see Table 4; HRESIMS m/z 207.0665 [M-H]− (calcd for C11H11O4, 207.0663).
ECD Calculation
The Molecular Merck force field (MMFF) conformational search, geometry optimization and frequency calculations at the B3LYP 6-31G(d) level in vacuo, and TDDFT ECD calculations of the dominant conformers (>1%) at the PBE0/TZVP level with the polarizable continuum model (PCM) for MeOH, were performed as described previously (Lai et al., 2016). ECD spectrum of each conformer was plotted by the program SpecDis (Bruhn et al., 2013) using the dipole-length computed rotational strengths with Gauss curves and exponential half-width (σ) of 0.3 and 0.2 eV, for 1 and 5, respectively. The equilibrium population of each conformer at 298.15 K was calculated from its relative Gibbs free energies using Boltzmann statistics. The Boltzmann-averaged ECD spectra for (2S, 3R, 4R, 9R, 1′R, 2′S)-1, and (1R, 2S, 4R, 1′R, 2′R)-5 were generated according to the Boltzmann distributions of the lowest energy conformers for each structure. The calculated ECD spectra were then compared with the experimental one to determine the absolute configuration. The calculated ECD spectra for 1 and 5 were scaled by 0.25 (y-axes) for a better comparison with the experimental data.
13C NMR Calculation
For the calculations of 13C NMR chemical shifts, B3LYP/6-31G(d,p) method was used to optimize the selected conformers. For all optimized structures, vibrational spectra were calculated to ensure that no imaginary frequencies for energy minimum were obtained. NMR calculations were performed at the level of mPW1PW91/6-31G(d,p) with the gauge-independent atomic orbital (GIAO) method (Forsyth and Sebag, 1997). The solvent effect was considered by using the PCM model (CHCl3 for 6). The calculated 13C NMR chemical shifts for (S)-6a and (S)-6b were analyzed by subtracting the isotopic shifts for TMS calculated with the same method. The 13C NMR chemical shifts in each compound were considered as the average value of the same atom in different conformers according to their Boltzmann distributions, using the relative Gibbs free energies as weighting factors. The differences Δδ values were determined by subtracting the experimental chemical shifts (δexp) from the calculated chemical shifts (δcal). The results were evaluated in terms of the average absolute deviation, and maximum absolute deviation.
Optical Rotation Calculation
The b3lyp/6-31g(d,p)-optimized conformers of (S)-6a were used to calculate optical rotations (OR). The OR calculations were carried out by means of time-dependent DFT methods at the B3LYP/6-31+G (d,p) level (Mazzeo et al., 2010), with the PCM model for methanol, by Gaussian 09. Boltzmann statistics analysis was employed to calculate the overall OR.
Phytotoxicity Assay
The major compounds (12~14, and 16) were tested for their phytotoxic activities against lettuce (Lactuca sativa L. var. ramose Hort.) and rice (Oryza sativa L. variety Zhonghua11) as described previously (Lu et al., 2015). Glyphosate was used as the positive control. After treatment, the length of the radicle or plumule of each seed was measured. The inhibition ratio (%) was calculated by comparison with the blank control.
Cytotoxicity Assay
Cytotoxicities of the isolated compounds were tested against human carcinoma cells using the microculture tetrazolium (MTT) assay as described previously (Sun et al., 2017). The tested cell lines included NCI-H460 and A549 (lung carcinoma), BGC-823 (gastric cancer), Daoy (desmoplastic cerebellar medulloblastoma), HepG2 (liver carcinoma), HCT116 (colon cancer), A375 (skin malignant melanoma), MCF-7 (breast cancer), and Capan2 (pancreatic carcinoma). Taxol was used as the positive control. The major compounds (11~14, 16) were screened against A375, A549, MCF-7, Capan2, and HCT116 cells, while the minor compounds (i.e., 1~10, 15, 17) were tested against NCI-H460, BGC823, Daoy, HepG2, and HCT116 cell lines.
Antibacterial Assay
The isolated compounds were evaluated for antibacterial activities against six human/plant pathogenic bacteria, including Bacillus subtilis ATCC 11562, Staphylococcus haemolyticus ATCC 29970, Ralstonia solanacearum ATCC11696, Xanthomonas vesicatoria ATCC 11633, Agrobacterium tumefaciens ATCC 11158, and Pseudomonas lachrymans ATCC 11921, by the modified broth dilution colorimetric assay (Shan et al., 2014). The minimum inhibitory concentration (MIC) and half maximum inhibitory concentration (IC50) were determined. Streptomycin sulfate was used as the positive control.
Antifungal Assay
The antifungal activities of compounds 2, 7, 10~14, 16, and 17 were evaluated against the spore germination of the rice blast pathogen Magnaporthe oryzae as described previously (Shan et al., 2014). Carbendazim was used as the positive control.
Results
Identification of New Compounds
The crude EtOAC extract was repeatedly fractionated over sephadex LH-20, silica gel, and RP-18, and purified by semi-preparative HPLC to give 12 bisorbicillinoids, and five sorbicillinoid-like metabolites (Figure 1).
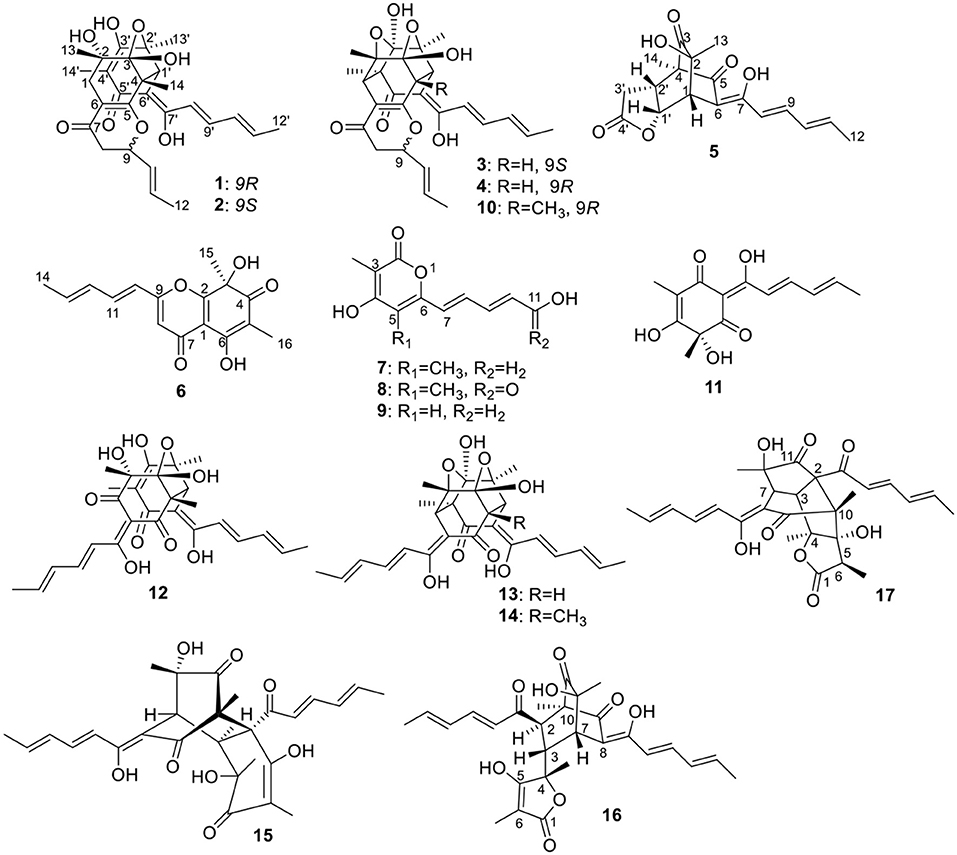
Figure 1. Structures of the isolated sorbicillinoids.
Compound 1 was isolated as a yellow amorphous powder. It showed a prominent deprotonated peak at m/z 497.2193 [M-H]− in the negative HRESIMS spectrum, indicating a molecular formula of C28H33O8. Its UV spectrum exhibited maximum absorptions at 220, 271, and 379 nm, which were similar to those of bisvertinols (Trifonov et al., 1986). The IR spectrum indicated the presence of hydroxy (3,420 cm−1), ketone (1,714, 1,681 cm−1), and double bond (1,646, 1,623 cm−1) groups. The 13C NMR spectrum displayed a total of 28 carbon resonances, which were assignable to two carbonyls (δC 193.8, 192.7), 12 sp2 hybridized carbons that were incorpated into six double bonds, four quarternary carbons (δC 107.6, 80.2, 74.5, 57.7), two methines (δC 79.3, 53.9), two methylenes (δC 41.1, 33.1), as well as six methyl (δC 26.6, 22.3, 19.7, 18.8, 18.0, 7.8) groups, by the aid of HMQC spectrum. Accordingly, the 1H NMR spectrum showed resonances for six olefinic protons (δH 5.47~7.16), two methines (δH 4.26, 3.69), two methylenes (δH 2.55, 2.44, 2.33, 2.26), and six methyls (δH 1.86, 1.72, 1.62, 1.46, 1.32, 1.11). These data revealed the bisorbicillinoid nature of 1. The planar structure of 1 was established by a combined analysis of the 1D, and 2D NMR data.
A long spin system was easily recognized by anlysis of the coupling constants and 1H-1H COSY spectrum, in which cross-peaks were observed between CH3-12′ (δH 1.86, d)/H-11′ (δH 6.10, dq), H-11′/H-10′ (δH 6.33, dd), H-10′/H-9′ (δH 7.16, dd), and H-9′/H-8′ (δH 6.45, d). The HMBC correlations from H-9′ to C-7′ (δC 168.3), as well as from H-8′ to C-6′ (δC 102.8) and C-7′ established an enol-sorbyl chain (box A, Figure 2). Meanwhile, the HMBC correlations from CH3-14′ (δH 1.62, s) to C-3′ (δC 171.2), C-4′ (δC 108.8), and C-5′ (δC 192.7), CH3-13′ (δH 1.46, s) to C-1′ (δC 53.9), C-2′ (δC 80.2), and C-3′, and from H-1′ (δH 3.69, s) to C-5′, C-6′, and C-7′, indicated the presence of a cyclohexenone ring, in which the enol-sorbyl chain was attached to C-6′. Hence, the first monomeric unit (A) was constructed as shown in Figure 2.
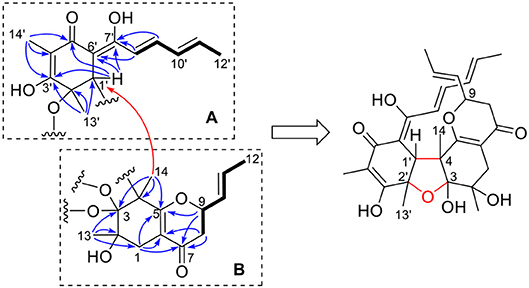
Figure 2. Selected 1H-1H COSY (bold) and HMBC (H→ C, arrow) correlations of two monomeric units (A,B) of 1.
The second monomeric unit (box B, Figure 2) was likewise deduced by analysis of the 1H-1H COSY and HMBC spectra. A spin system starting from H2-8 (δH 2.44, 2.33, each dd), terminating at CH3-12 (δH 1.72, d) was established by inspecting the 1H-1H COSY spectrum. The HMBC correlations from H-9 (δH 4.26, ddd), and H2-8 to the carbonyl (δC 193.8, C-7) established a 3-hydroxyhex-4-enoyl side chain. The correlations from CH3-13 (δH 1.11, s) to C-1 (δC 33.1), C-2 (δC 74.5), and C-3 (δC 107.6), from CH3-14 (δH 1.32, s) to C-3, C-4 (δC 57.7), and C-5 (δC 172.1), from H2-1 (δH 2.55, 2.26, each d) to C-5, and C-6 (δC 110.2), allowed the establishment of a cyclohexene ring. Further correlations from H2-8 to C-6, H2-1 to C-7, indicated that the side chain was attached to C-6 of the cyclohexene ring, while correlation from H-9 to C-5 suggested C-5 and C-9 was connected via an ether bond.
The linkage of the two units was deduced by analysis of the HMBC spectrum. A key HMBC correlation from CH3-14 to C-1′ indicated a direct sigma bond between C-1′ and C-4. Considering the molecular formula and the required degrees of unsaturation, one additional ether ring had to be formed. Then, a C-2′-O-C-3 ether was deduced by analysis of the chemical shifts of C-2′ (δC 80.2) and C-3 (δC 107.6), which was consistent with those of the reported bisvertinols (Trifonov et al., 1986). Thus, the planar structure of 1 was established as shown in Figure 2.
The stereochemistry of 1 was determined by analysis of the 1H-1H coupling constants and the NOESY correlations. The large coupling constants between H-10/H-11 (15.5 Hz), H-8′/H-9′ (14.8 Hz), H-10′/H-11′ (15.1 Hz), defined the E-geometry for all these double bonds. The NOESY correlations seen between CH3-13/CH3-14, CH3-14/H-1′, and H-1′/CH3-13′ suggested these protons were close in space (Figure 3), while the cross-space correlation between H-9 and CH3-14′ was crucial to assign a 9R* configuration.
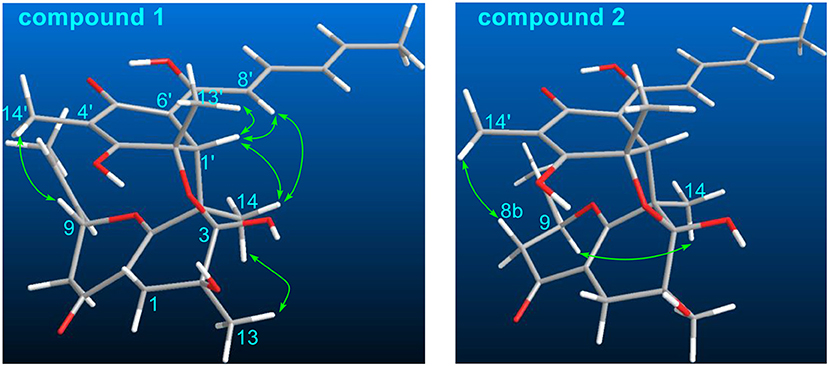
Figure 3. Selected NOESY correlations of 1 and 2.
Structurally, 1 was related to bisvertinol (Andrade et al., 1992), and they only differed at the side chain, in which a 2-propenyl-dihydropyran-4-one in 1 replaced a sorbyl in bisvertinol. The CD spectrum of 1 was similar to that of bisvertinol (Andrade et al., 1992), indicating the absolute configuration should be retained for all the chiral centers except for C-9, though 1 was of smaller magnitudes and hypsochromically shifted. In order to confirm this deduction, the ECD spectrum of 1 was calculated using TDDFT ECD computations, which has been a powerful tool to establish the absolute configuration of natural products (Bringmann et al., 2009). A randomly selected configuration of 1 was used for calculation, which included molecular Merck force field (MMFF) conformational search, geometry optimization using the DFT method at the B3LYP/6-31G(d) level, and ECD calculation of the low-energized conformers (see Table S1 and Figure S1 in the Supplementary Material) at the pbe0/TZVP level with the polarizable continuum model (PCM) in MeOH, successively. The calculated ECD spectrum of (2S, 3R, 4R, 9R, 1′R, 2′S)-1 matched well with the experimental one (Figure 4), supporting the 2S, 3R, 4R, 9R, 1′R, 2′S configuration of 1. Compound 1 was designated as ustisorbicillinol A (1).
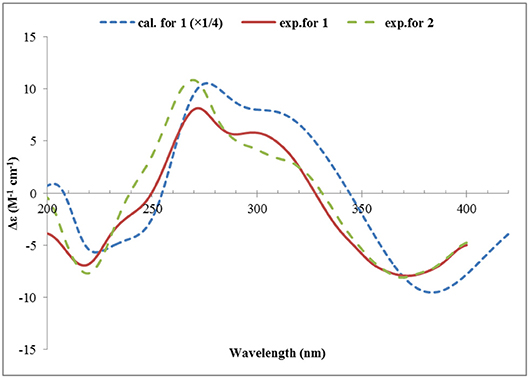
Figure 4. Experimental ECD spectra of 1 and 2, and the calculated ECD spectrum of (2S, 3R, 4R, 9R, 1′R, 2′S)-1.
Ustisorbicillinol B (2) was isolated as a yellow amorphous powder, and its molecular formula was determined as C28H34O8 by analysis of the HRESIMS spectrum, which was the same as that of 1. A comparison of the NMR data (Table 1) revealed their great similarities. The notable differences were found at C-8 and C-9, in which C-9 and C-8 of 2 were shifted to downfield (+1.3 and +1.2 ppm, respectively), and H-9 was downfield shifted (+0.30 ppm), while both H2-8 were upfield shifted (−0.21, −0.21 ppm), comparing to those of 1. This implied that 2 differed from 1 only in having a different configuration at C-9. This was confirmed by analysis of the NOESY spectrum, in which cross-peaks were seen between H-9 (δH 4.56, m) and CH3-14 (δH 1.29, s), while CH3-14′ (δH 1.59, s) showed correlation to H-8b (δH 2.12, dd), but not to H-9 (Figure 3). Moreover, compound 2 showed similar CD spectrum as that of 1 (Figure 4), indicating their absolute configurations were the same, expect that C-9 had an opposite configuration. Thus, compound 2 was determined as the 9-epimer of 1.
Ustisorbicillinol C (3) had a molecular formula of C27H30O8, as determined by HRESIMS. Its NMR data (Table 1) were similar to those reported for dihydrotrichodimer ether B (Zhai et al., 2016), except that one methyl singlet was missing, while one additional methine replaced one quaternary carbon of the later compound, suggesting that compound 3 was a demethyl derivative of the later when taking into consideration their molecular formulae. This new methine (δH 3.00, d; δC 55.7) was positioned at C-4, as it coupled to H-1′ (δH 3.67, d) (3J = 12.0 Hz) and showed COSY correlation to H-1′. The HMBC correlation from this methine to C-5 (δC 170.8), and C-6 (δC 109.1), as well as the NOESY correlation between this methine and H-1′, unequivocally supported this assgiment. Similar NOE correlations between H-1/CH3-14′, H-1/CH3-13, H-1′/H-4, H-1′/CH3-13′, and H-1′/H-8′, and similar 3JH values of the vinicial protons of the side chains, indicated a similar relative stereochemistry was retained in 3. In addition, the CD spectrum of 3 displayed cotton effects at 310 (+5.40), 272 (−4.78) nm (Figure 5), which resembled that of dihydrotrichodimer ether B (Zhai et al., 2016), implying a same absolute configuration for the cage structure. In literature, the absolute configuration of dihydrotrichodimer ether B was determined by comparing its CD spectrum with that of tetrahydrotrichodimer ether, whose configuration had been determined by X-ray diffraction analysis (Zhai et al., 2016). Thus, the structure of ustisorbicillinol C (3) was determined as shown in Figure 1.
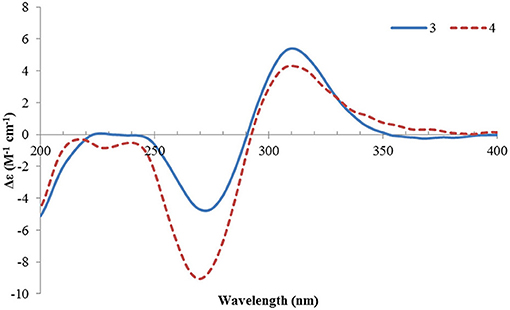
Figure 5. ECD spectra of 3 and 4.
Ustisorbicillinol D (4) was isolated as an isomer of ustisorbicillinol C (3), as they shared the same molecular formula. The NMR data of 4 closely resembled those of 3 (Table 1), and the notable differences were found for the hydroxyl-bearing methine (CH-9: δH 4.38/δC 81.5 in 4 vs. δH 4.93/δC 80.2 in 3), suggesting they contained the same skeleton but with different configuration for C-9. This was also reflected by the coupling constants found between H-9 and H2-8 (3JH−9/H−8a and 3JH−9/H−8b: 13.4, 3.5 in 4, vs. 4.8, 8.7 in 3). Taken together with the CD spectra as shown in Figure 5, ustisorbicillinol D (4) was thus determined to be the 9-epimer of ustisorbicillinol C (3).
Ustisorbicillinol E (5), a minor sorbicillinoid, displayed a deprotonated peak at m/z 331.1201 [M-H]− in the HRESIMS spectrum, suggesting a molecular formula of C18H20O6. Inspection of the 13C NMR spectrum disclosed 18 carbon signals (Table 2), which were attributable to two keto groups (δC 209.2, 194.9), one ester carbonyl (δC 174.8), six sp2-hybridized carbons (δC 172.3, 144.6, 141.4, 130.8, 117.2, 104.7), two quaternary sp3 carbons (δC 74.2, 62.5), three methines (δC 76.3, 45.5, 41.4), one methylene (δC 30.2), and three methyl groups (δC 19.0, 24.4, 10.4), as aided by an HSQC experiment. Apparently, 14 of those carbons comprised a sorbicillinol-like unit, which included one sorbyl chain (C7~12), two keto (C3 and C5), three non-protonated carbons (C2, C4, and C6), one methine (C1), and two quaternary carbon-attaching methyls (CH3-13, CH3-14), as revealed by analysis of the 3JH values, 1H-1H COSY and HMBC spectra (Figure 6A). Apart from these, four additional carbons remained to be assigned. A spin system involving C-1′~C-3′ was disclosed by inspecting the COSY correlations, while the HMBC correlations from H-1′ (δH 5.44, dd), H-2′ (δH 2.89, ddd), and H2-3′ (δH 2.70, 2.24, each dd) to the carbonyl (δC 174.8, C-4′) indicated the presence of a γ-lactone unit (Figure 6A). The connection between both units was established by the key HMBC correlations from CH3-14 (δH 1.23, s) to C-2′ (δC 41.4), and from H-1′ to C-6 (δC 104.7), and the 1H-1H COSY correlation between H-1′ and H-1 (δH 3.60, d). Thus, these two units have to be linked via C-2′ /C-4, and C-1′ /C-1 to form a bridge structure (Figure 1).
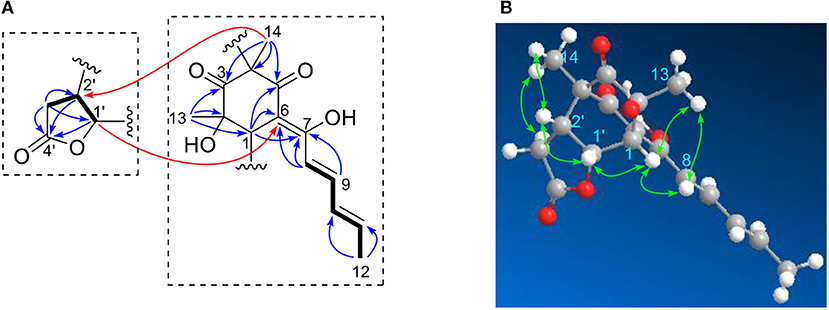
Figure 6. Selected 1H-1H COSY, and HMBC (A), and NOESY (B) correlations of 5.
The relative configuration of 5 was established by analysis of the 1H-1H coupling constants and NOESY correlations. The large 3JH values between H-8/H-9 (14.9 Hz), and H-10/H-11 (15.3 Hz) revealed the E-configuration of these double bonds. The NOESY correlations between H-1 and H-8 (δH 6.12, d) indicated the Z-configuration of the C6/C7 double bond. The NOESY correlations of H-1 (δH 3.60, d)/CH3-13 (δH 1.28, s), H-1/H-1′ (δH 5.44, dd), and H-1′/H-2′ (δH 2.89, ddd) suggested these protons were close in space, while the observed correlations of H-3′b (δH 2.24, dd)/CH3-14 (δH 1.23, s), and H-2′/CH3-14 were consistent with the equtorial orientation of CH3-14, as shown in Figure 6B. The absolute configuration of 5 was determined by TDDFT ECD calculation. MMFF conformational search of randomly selected (1R, 2S, 4R, 1′R, 2′R)-5 resulted in 9 conformers within a 21 kJ/mol energy window, which were optimized at the B3LYP/6-31G(d) level in vacuo to yield one prodominant conformer (99.1%) (Table S2 and Figure S2). Subsequent ECD calculation with this conformer was performed at the Pbe0/TZVP level with the PCM solvent model for MeOH. The calculated ECD spectrum was in good agreement with the measured spectrum (Figure 7), thus the absolute configuration of 5 was determined to be 1R, 2S, 4R, 1′R, 2′R. Interestingly, compound 5 was similar to rezishanone A that was isolated from Penicillium notatum, and they only differed at C-2′, where a hydroxymethyl group, instead of a proton, was present in rezishanone A (Maskey et al., 2005).
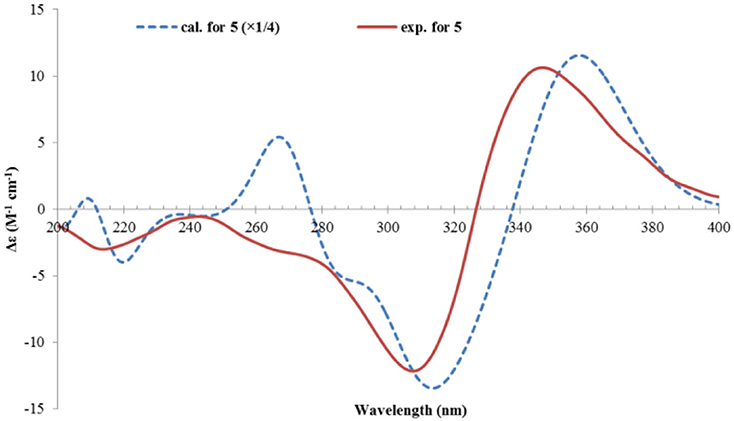
Figure 7. Experimental ECD spectrum of 5, and the calculated ECD spectrum of (1R, 2S, 4R, 1′R, 2′R)-5.
Ustisorbicillinol F (6) was isolated as a homolog of oxosorbicillinol (Abe et al., 2000), and had a molecular formula of C16H16O5, with nine degrees of unsaturation. The NMR data of 6 (Table 3) were similar to those of oxosorbicillinol (Abe et al., 2000), however, two more sp2-hybridized carbons (δC 111.7, CH/δH 6.31, s; 163.9, C) that comprised a double bond appeared. A detailed interpretation of the 2D NMR data, allowed the placement of this double bond in the sorbyl chain, as key HMBC correlations were observed from H-10 (δH 6.12, d) to C-9 (δC 163.9), and C-8 (δC 111.7), and from H-11 (δH 7.24) to C-9, as well as from H-8 (δH 6.31, s) to C-7 (δC 180.5), and C-1 (δC 109.7) (Figure 8). To fulfill the required degrees of unsaturation, one additional ring had to be formed via either C9-O-C2 (6a) or C9-O-C6 (6b) (Figure 8). However, these two possible structures could not be distinguished by NOESY experiments, as no correlations were observed between H-10, CH3-15, or CH3-16. So 13C NMR calculations were performed to determine the structure of 6 (Table S3 and Figure S3). The calculated data for (S)-6a were in good agreement with the measured values, with average absolute deviation of 2.60 ppm, and maximum absolute deviation of 5.9 ppm, while those of 4.61, 16.9 ppm, respectively, were found for (S)-6b (Table S4). Therefore, ustisorbicillinol F (6) had a planar structure as shown in 6a. The calculated optical rotation value for (S)-6a was +60.3 at the b3lyp/6-31+g(d, p) level with PCM model for methanol, while the measured value for 6 was−24 (c 0.05, MeOH). Therefore, the absolute configuration for 6 was determined to be R.
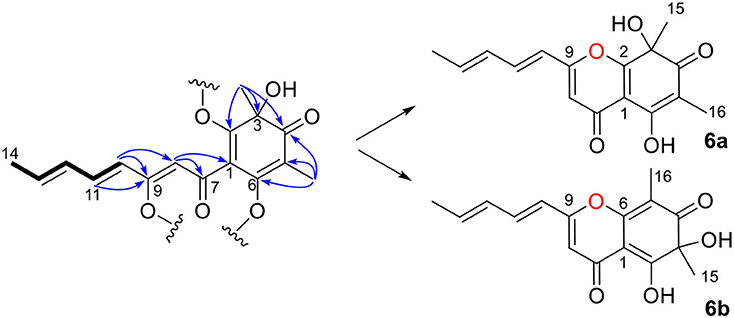
Figure 8. Selected HMBC correlations of 6, and its two possible structures (6a and 6b).
Ustilopyrone A (7), was identified as a 2-pyrone derivative, as inferred from its UV spectrum. The NMR data (Table 4) were similar to those of saturnispol H (Meng et al., 2018), except that an olefinic methyl (δC 9.6; δH 2.04, s) of 7 replaced the olefinic proton (H-5) of the latter. This was supported by the HMBC experiment, as correlations were found from 5-CH3 (δH 2.04, s) to C-4 (δC 167.4), C-5 (δC 110.5), and C-6 (δC 153.7).
Additionally, a new analog of ustilopyrone A (7), named ustilopyrone B (8) was isolated. Its molecular formula was determined as C12H12O5, as revealed by HRESIMS, bearing one more oxygen atom but two less protons than that of 7. Detailed comparison of the NMR data (Table 4) indicated that they were quite similar, and the differences were found in the side chain, especially at C-11, where a carboxylate group (δC 170.0) replaced the hydroxymethyl group of 7. This could explain the differences in molecular formula, as well as the obvious downfield shifts for H-9 (Δδ +0.95 ppm), H-7 (Δδ +0.44 ppm), C-9 (+14.1 ppm), and C-7 (+7.5 ppm), while upfield shift for C-10 (-13.2 ppm). Further evidences were obtained from the HMBC spectrum, as cross-peaks were found from H-9 (δH 7.44, dd), and H-10 (δH 6.12, d) to this carboxylate group. Thus, ustilopyrone B (8) was determined as the 11-oxidized derivative of 7.
The other isolated compounds were identified by comparing the spectroscopic data with those published in the literature, and included 5-demethylustilopyrone A (also named saturnispol H, 9) (Meng et al., 2018) and dihydrotrichodimer ether A (10) (Zhai et al., 2016), oxosorbicillinol (11) (Abe et al., 2000), bisvertinolone (12) (Trifonov et al., 1986), demethyltrichodimerol (13) (Abe et al., 1998a), trichodimerol (14) (Andrade et al., 1992), bisorbicillinol (15) (Abe et al., 1998a), bislongiquinolide (also named trichotetronine) (16) (Andrade et al., 1997; Shirota et al., 1997), and bisorbicillinolide (17) (Abe et al., 1998b).
Biological Activity
The major bisorbicillinoids 12~14, and 16 were investigated for their phytotoxic effect against the radicle and germ elongation of rice seeds and lettuce seeds (Table 5). Among them, compound 12 showed the strongest and dose-dependent inhibition in the case of rice seeds (Figure 9A), with 50% inhibitory concentration <50 μg/mL, which was comparable to the positive control glyphosate. At the tested concentration of 200 μg/mL, it showed 98.33% inhibitory rate, which was even better than that of glyphosate (95.80%). The other compounds exhibited much weaker activity, and when tested at 400 μg/mL, only compounds 13 and 16 displayed more than 50% inhibition. With regard to the inhibition of the germ elongation of rice seeds, compound 12 was still the most active one, and it displayed 51.92% inhibitory rate at 200 μg/mL, though weaker than the positive control, whereas the other compounds failed to reach 40% inhibition even at 400 μg/mL. Generally, the germ of rice seeds was less susceptible to the tested substances (including the positive control), comparing to the radicle. However, when tested against the lettuce seeds, different inhibition profiles were observed. When tested at the concentration of 400 μg/mL, the growth of the radicle and germ was completed inhibited by compounds 12, 13, and 16, which was much better than the effect of glyphosate (Table 5). At this concentration, compound 14 displayed >50% inhibition against the elongation of radicle and germ, though it was the least active one. Compound 12 also exhibited the strongest inhibition amongst the tested compounds (Figure 9B). It was worth noting that the germ was equal or slightly more sensitive to the tested substances at lower concentration (<200 μg/mL) than the radicle, in general, while at higher concentrations this trend was reversed.
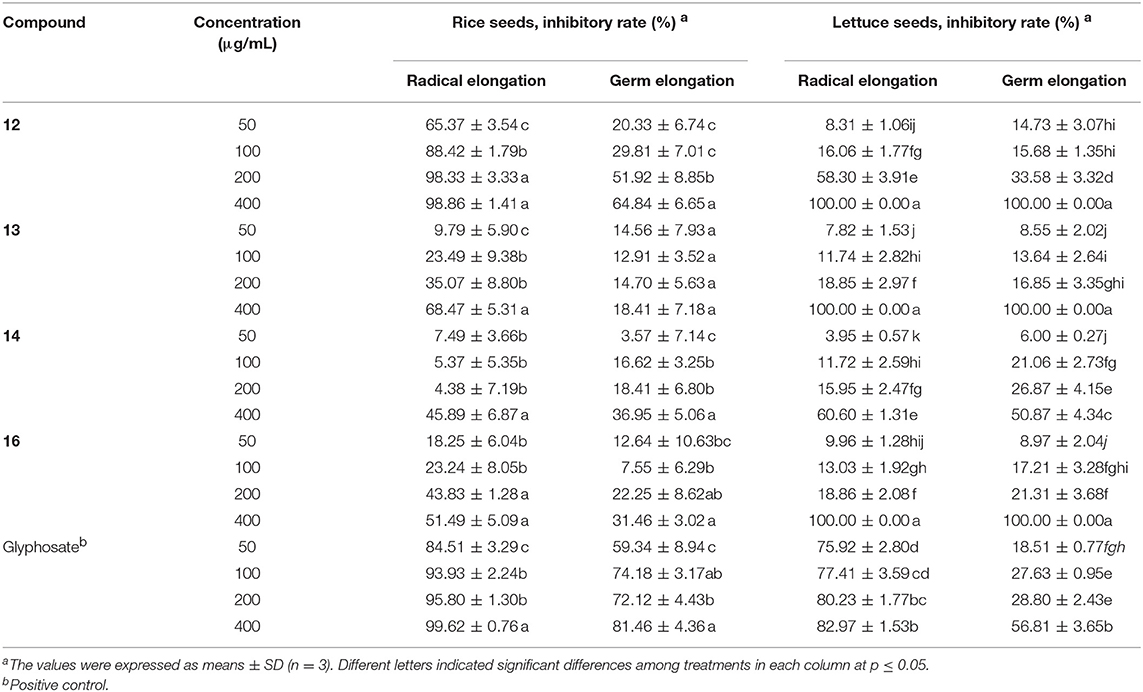
Table 5. Phytotoxicity of the isolated compounds against the radicle and germ elongation of rice and lettuce seeds.

Figure 9. Inhibitory activity of 12 on the radicle and germ elongation of rice seeds (A) and lettuce seeds (B).
When compared the inhibitory activity of each tested compound at each tested concentration against both type of seeds, a general potency of 12>16≥13>14 was found. More structures should be evaluated in order to gain a better understanding of the structure-activity relationship.
The isolated compounds were also evaluated for their cytotoxicities against nine human carcinoma cell lines including NCI-H460, BGC823, Daoy, HepG2, HCT116, A375, A549, MCF-7, and Capan2 (Table 6). Compounds 10, 12, 13, and 14 showed moderate cytotoxicities against the tested cell lines, whereas the other isolated compounds were inactive (IC50 > 50 μM). Among the active compounds, compound 10 exhibited the strongest inhibition with IC50 in the range of 8.83~38.2 μM, though less potent than the positive control. It was interesting that all the active compounds were bisorbicillinols with at least one ether bridge. The methyl group at C-14 might positively correlate to the cytotoxicity, as in the case of 10 vs. 4. However, the structure-activity relationship remained unclear.
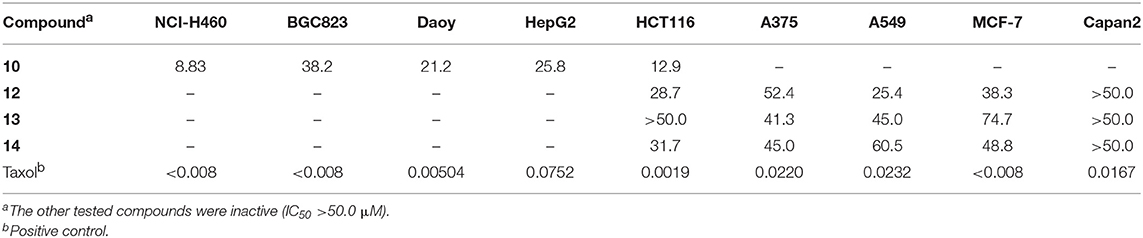
Table 6. Cytotoxicity of the isolated compounds (IC50, μM).
The antibacterial activities of the isolated compounds were also tested (Table 7). Compounds 2, and 10~13 were active against the tested pathogenic bacteria including A. tumefaciens, B. subtilis, P. lachrymans, R. solanacearum, S. haemolyticus, and X. vesicatoria, with MIC values in the range of 4~128 μg/mL, whereas the other compounds were inactive (MIC > 128 μg/mL). Compound 12 was the most potent one, with MIC values ≤8 μg/mL, which were comparable to those of the positive control (streptomycin sulfate), and this was followed by compound 13 (MICs ≤ 32 μg/mL). It is interesting that except the monomeric compound, oxosorbicillinol (11), the other active compounds were bisorbicillinols (2, 10, 12, 13) with one or two ether bridges.
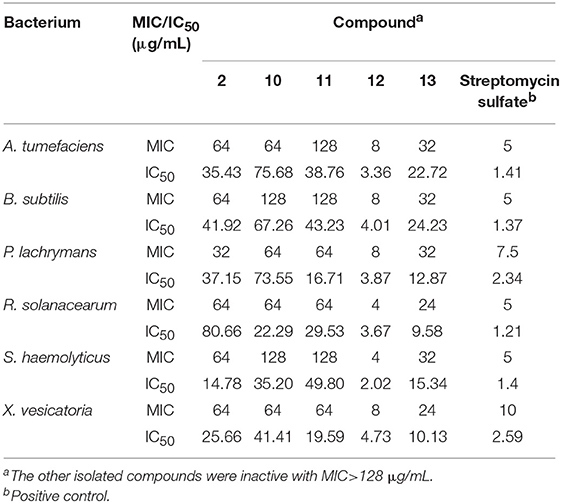
Table 7. Antibacterial activity of the isolated compounds.
Additionally, compounds 2, 7, 10~14, 16, and 17 were tested for inhibition against the germination of the spores of rice blast fungus M. oryzae. Compounds 11~13 displayed moderate effect with IC50 values of 40.88, 36.70, and 55.99 μg/mL, respectively, compared to the positive control carbendazim (IC50 6.86 μg/mL). This implied that the bisorbicillinoids 12 and 13 were more potent than the monomeric compound 11, when taking into consideration of their molar units. The other tested compounds did not exhibited strong inhibition (IC50 > 200 μg/mL). No clear structure-activity relationship could be drawn.
Discussion
In the present study, we isolated 17 sorbicillinoids including eight new compounds from the pathogenic fungus U. virens. These isolated compounds could be classified as monomeric sorbicillinoid (11), dimeric sorbicillinoids (1~5, 10, 12~17), and related compounds (6~9).
The biosynthesis of sorbicillinol has been reported in Penicillium chrysogenum (Fahad et al., 2014) that involved SorA, a highly-reducing iterative PKS (HRiPKS), and SorB, a non-reducing iterative PKS (NRiPKS). SorA combined three acetate units, and modified the forming triketide with the ketoacyl reductase and dehydratase domains to produce the sorbyl-containing thioester (S2a), which was passed to SorB to appendix three further acetate units and two methyl groups, resulting in the formation of sorbicillin. Further oxidation of sorbicillin generated sorbicillinol, and oxosorbicillinol (11), by the FAD-dependent monooxygenase SorC, and oxidoreductase SorD, respectively (Figure 10A) (Guzmán-Chávez et al., 2017). Similarly, the biosynthesis of the analogs of sorbicillinol, S1b, and S1c, with a 3-hydroxy-4-hexenoyl side chain, were proposed. It was speculated that there was a 3-hydroxy-4-hexenoyl thioester intermediate (S1a) before the formation of the sorbyl thioester (S2a), though this has not been reported yet. The addition of three malonyl CoA, and one or two methyl group from S-adenosylmethionine (SAM) could produce the sorbicillin analogs with S1-type of side chain, which were then oxidized to give the sorbicillinol analogs, 9-hydroxydihydrosorbicillinol (S1c), and 3-demethyl-9-hydroxydihydrosorbicillinol (S1b). Likewise, the biosynthesis of sorbicillinol related compound, 6, was envisaged to involve one more acetate unit when appended to S2a, resulting in the formation of a sorbicillin analog with a 8C side chain. Subsequent oxidation, and keto-tautomerism of this analog would give the precursor 6p, which could lose one molecule of water to yield the C2-O-C9 cyclic ether (i.e., 6) (Figure 10A). With regard to the biosynthesis of 2-pyrones, it was hypothesized that two acetate units, and one or two methyl groups should add to the thioester S2a, following by keto-enol tautomerism and lactonization to give the 2-pyrone products with the S2-type side chain. The oxidation at the end of this chain (ω-oxidation) would give rise to 7~9 as depicted in Figure 10A. Taken together the above mentioned pathways, the PKS, especially the SorB-like NRiPKS, of this fungus showed a remarkable plasticity, as it added 2~4 units of malonyl CoA to the triketide produced by the HRiPKS, resulting in a different type of products.
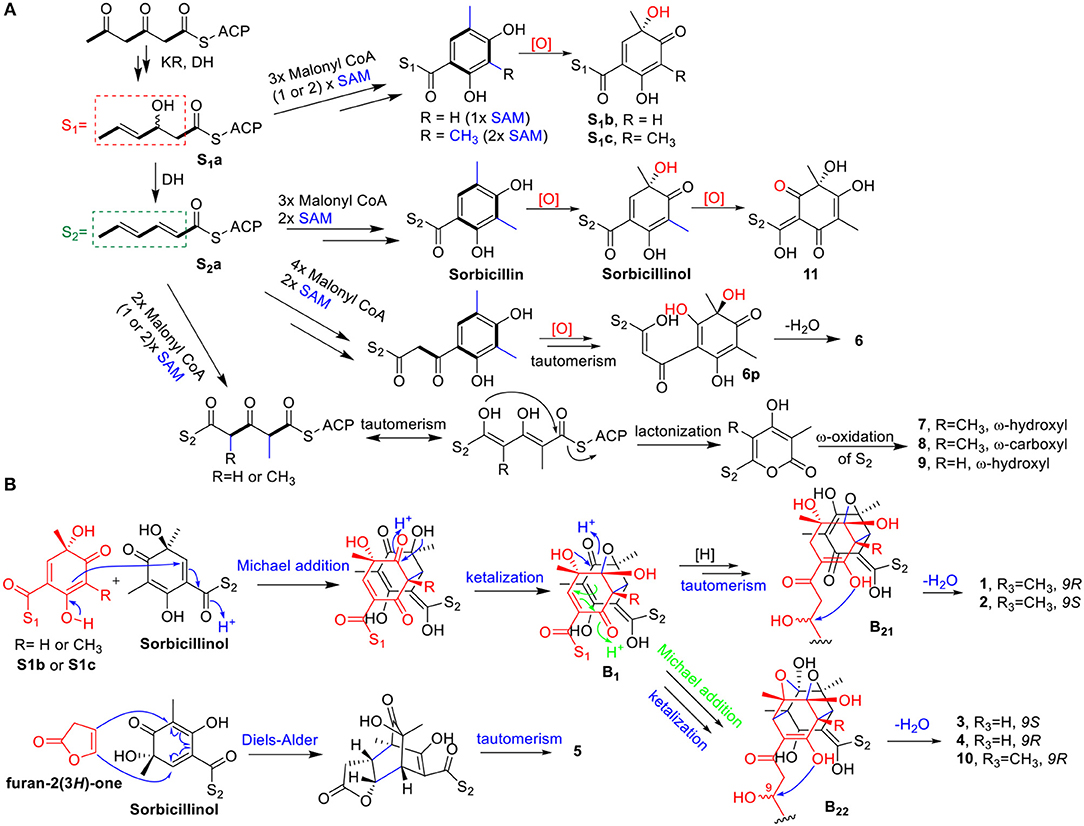
Figure 10. Proposed biosynthetic route for the monomeric sorbicillinoid and related metabolites (6~9, 11) (A), and the dimeric sorbicillinoids (1~5, 10) (B).
The biosynthetic pathway for bisorbicillinols 1~5, and 10 was proposed in Figure 10B. Sorbicillinol was reported to be a key intermediate for the biosynthesis of bisorbicillinols (Harned and Volp, 2011; Meng et al., 2016). The Michael addition of sorbicillinol and its analog S1b or S1c, followed by ketalization, could produce the intermediates as shown in B1. These intermediates could be hydrogenated and keto-enol tautomerized to form another intermediates (B21) that underwent dehydration in the side chain of S1 to yield 1 and 2. On the other hand, intramolecular Michael addition and ketalization of B1 could generate the intermediates (B22), which were dehydrated to form the bisorbicillinols 3, 4, and 10. Whereas, the formation of 5, a heterodimer, should involve the Diels-Alder reaction of sorbicillinol and furan-2(3H)-one, followed by a keto-enol tautomerism step.
Compounds 1-4 and 10, featuring a dihydro-γ-pyrone fused cyclohexene ring, were interesting members of the bisorbicillinoids. Previously, this type of structures has only been reported once, and included tetrahydrotrichodimer ether, and dihydrotrichodimer ethers A and B (Zhai et al., 2016). It is of interest that both epimers at C-9 were co-isolated in this study, as well as in the literature (Zhai et al., 2016). The possibility that an acid-mediated epimerism could occur can be ruled out, as these compounds were detectable in the crude extract. In addition, incubation of 2 in 80%MeOH/H2O (containing 0.02% TFA) at RT for 1 week, the same condition employed for separation, did not lead to the production of any decomposed product.
Sorbicillinoids were reported to have diverse biological activities, such as cytotoxic, antimicrobial, and antioxidant activity (Harned and Volp, 2011; Meng et al., 2016). In this study, the isolated compounds were tested for their cytotoxic, antibacterial, and antifungal activities. The bisorbicillinoids 10, and 12~14 showed moderate cytotoxicities against the tested cell lines. These cytotoxic compounds (except 14), along with compounds 2 and 11, displayed antibacterial activity. In addition, the bisorbicillinoids 12 and 13, as well as the monomer 11 displayed moderate antifungal activities. Among the antimicrobial compounds, 12 was the most effective. Meanwhile, the isolated compounds were screened for their phytotoxicities, and the results revealed that bisorbicillinoids were potent phytotoxins. Such activity has not been reported for sorbicillinoids so far. Whether these phytotoxins play a role in the pathogenesis of U. virens are yet to be discovered.
Conclusion
In conclusion, 17 sorbicillinoids were isolated from the fermentation of U. virens, which included five new bisorbicillinols, ustisorbicillinols A~E (1~5), and one new oxosorbicillinol-related compound, ustisorbicillinol F (6), together with two new 2-pyrones (7 and 8). The structures of the new compounds were elucidated by a combined analysis of the HRESIMS, NMR, and CD spectra, and by quantum chemical calculations of ECD, 13C NMR, and OR. Their biosynthetic pathways were proposed. The isolated compounds were evaluated for their phytotoxic, cytotoxic, antibacterial, and antifungal activities. The major bisorbicillinoids 12~14, and 16 were phytotoxic against radicle or germ growth of rice and lettuce seeds, with 12 displaying the strongest inhibition. Compound 12 could be a promising lead for the development of natural herbicides. The phytotoxicity of sorbicillinoids has not been reported previously. Compounds 10, and 12~14 showed moderate cytotoxicities against the tested cell lines with IC50s of 8.83~74.7 μM. Compounds 2, and 10~13 were active against the tested bacteria (MICs 4~128 μg/mL). Meanwhile, compounds 11~13 displayed moderate antifungal activities.
Data Availability
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
LZ, DL, and JM conceived and designed the experiments. JM was responsible for the isolation of compounds. DL and JM elucidated the structures. JD tested cytotoxicity of the compounds. JM, GG, PD, XZ, and WW performed the experiments of phytotoxic and antimicrobial activities. DL, LZ, and JM interpreted the data and wrote the paper. YL and JD revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Key R&D Program of China (2017YFC1600905 and 2017YFD0201105) and the National Natural Science Foundation of China (31471729 and 31271996).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00435/full#supplementary-material
References
Abe, N., Murata, T., and Hirota, A. (1998a). Novel DPPH radical scavengers, bisorbicillinol and demethyltrichodimerol, from a fungus. Biosci. Biotechnol. Biochem. 62, 661–666. doi: 10.1271/bbb.62.661
Abe, N., Murata, T., and Hirota, A. (1998b). Novel oxidized sorbicillin dimers with 1,1-diphenyl-2-picrylhydrazyl radical scavenging activity from a fungus. Biosci. Biotechnol. Biochem. 62, 2120–2126. doi: 10.1271/bbb.62.2120
Abe, N., Yamamoto, K., and Hirota, A. (2000). Novel fungal metabolites, demethylsorbicillin and oxosorbicillinol, isolated from Trichoderma sp. USF-2690. Biosci. Biotechnol. Biochem. 64, 620–622. doi: 10.1271/bbb.64.620
Andrade, R., Ayer, W. A., and Mebe, P. P. (1992). The metabolites of Trichoderma longibrachiatum. Part 1. Isolation of the metabolites and the structure of trichodimerol. Can. J. Chem. 70, 2526–2535. doi: 10.1139/v92-320
Andrade, R., Ayer, W. A., and Trifonov, L. S. (1997). The metabolites of Trichoderma longibrachiatum. III. Two new tetronic acids: 5-hydroxyvertinolide and bislongiquinolide. Aust. J. Chem. 50, 255–257. doi: 10.1071/C96103
Bringmann, G., Bruhn, T., Maksimenka, K., and Hemberger, Y. (2009). The assignment of absolute stereostructures through quantum chemical circular dichroism calculations. Eur. J. Org. Chem. 2009, 2717–2727. doi: 10.1002/ejoc.200801121
Bruhn, T., Schaumlöeffel, A., Hemberger, Y., and Bringmann, G. (2013). SpecDis: quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality 25, 243–249. doi: 10.1002/chir.22138
Cimmino, A., Masi, M., Evidente, M., Superchi, S., and Evidente, A. (2015). Fungal phytotoxins with potential herbicidal activity: chemical and biological characterization. Nat. Prod. Rep. 32, 1629–1653. doi: 10.1039/C5NP00081E
Dayan, F. E., and Duke, S. O. (2014). Natural compounds as next-generation herbicides. Plant Physiol. 166, 1090–1105. doi: 10.1104/pp.114.239061
Duke, S. O., and Dayan, F. E. (2011). Modes of action of microbially-produced phytotoxins. Toxins 3, 1038–1064. doi: 10.3390/toxins3081038
Fahad, A.a., Abood, A., Fisch, K. M., Osipow, A., Davison, J., Avramovic, M., et al. (2014). Oxidative dearomatisation: the key step of sorbicillinoid biosynthesis. Chem. Sci. 5, 523–527. doi: 10.1039/C3SC52911H
Fan, J., Yang, J., Wang, Y.-Q., Li, G.-B., Li, Y., Huang, F., et al. (2016). Current understanding on Villosiclava virens, a unique flower-infecting fungus causing rice false smut disease. Mol. Plant Pathol. 17, 1321–1330. doi: 10.1111/mpp.12362
Forsyth, D. A., and Sebag, A. B. (1997). Computed 13C NMR chemical shifts via empirically scaled GIAO shieldings and molecular mechanics geometries. conformation and configuration from 13C Shifts. J. Am. Chem. Soc. 119, 9483–9494. doi: 10.1021/ja970112z
Guzmán-Chávez, F., Salo, O., Nygård, Y., Lankhorst, P. P., Bovenberg, R. A. L., and Driessen, A. J. M. (2017). Mechanism and regulation of sorbicillin biosynthesis by Penicillium chrysogenum. Microb. Biotechnol. 10, 958–968. doi: 10.1111/1751-7915.12736
Harned, A. M., and Volp, K. A. (2011). The sorbicillinoid family of natural products: Isolation, biosynthesis, and synthetic studies. Nat. Prod. Rep. 28, 1790–1810. doi: 10.1039/c1np00039j
Lai, D., Mao, Z., Xu, D., Zhang, X., Wang, A., Xie, R., et al. (2016). Hyalodendriellins A-F, new 14-membered resorcylic acid lactones from the endophytic fungus Hyalodendriella sp. Ponipodef12. RSC Adv. 6, 108–109. doi: 10.1039/C6RA24009G
Lai, D., Meng, J., Zhang, X., Xu, D., Dai, J., and Zhou, L. (2019). Ustilobisorbicillinol A, a cytotoxic sorbyl-containing aromatic polyketide from Ustilaginoidea virens. Org. Lett. 21, 1311–1314. doi: 10.1021/acs.orglett.8b04101
Lu, S., Sun, W., Meng, J., Wang, A., Wang, X., Tian, J., et al. (2015). Bioactive bis-naphtho-γ-pyrones from rice false smut pathogen Ustilaginoidea virens. J. Agric. Food Chem. 63, 3501–3508. doi: 10.1021/acs.jafc.5b00694
Maskey, R. P., Grüen-Wollny, I., and Laatsch, H. (2005). Sorbicillin analogues and related dimeric compounds from Penicillium notatum. J. Nat. Prod. 68, 865–870. doi: 10.1021/np040137t
Mazzeo, G., Giorgio, E., Zanasi, R., Berova, N., and Rosini, C. (2010). Absolute configuration through the DFT simulation of the optical rotation. Importance of the correct selection of the input geometry: a caveat. J. Org. Chem. 75, 4600–4603. doi: 10.1021/jo100401w
Meng, J., Cheng, W., Heydari, H., Wang, B., Zhu, K., Konuklugil, B., et al. (2018). Sorbicillinoid-based metabolites from a sponge-derived fungus Trichoderma saturnisporum. Marine Drugs 16:226. doi: 10.3390/md16070226
Meng, J., Wang, X., Xu, D., Fu, X., Zhang, X., Lai, D., et al. (2016). Sorbicillinoids from fungi and their bioactivities. Molecules 21:715. doi: 10.3390/molecules21060715
Shan, T., Tian, J., Wang, X., Mou, Y., Mao, Z., Lai, D., et al. (2014). Bioactive spirobisnaphthalenes from the endophytic fungus Berkleasmium sp. J. Nat. Prod. 77, 2151–2160. doi: 10.1021/np400988a
Shirota, O., Pathak, V., Faiz Hossain, C., Sekita, S., Takatori, K., and Satake, M. (1997). Structural elucidation of trichotetronines: polyketides possessing a bicyclo[2.2.2]octane skeleton with a tetronic acid moiety isolated from Trichoderma sp. J. Chem. Soc. Perkin Trans. 1, 2961–2964. doi: 10.1039/a704963c
Strange, R. N. (2007). Phytotoxins produced by microbial plant pathogens. Nat. Prod. Rep. 24, 127–144. doi: 10.1039/B513232K
Sun, W., Wang, A., Xu, D., Wang, W., Meng, J., Dai, J., et al. (2017). New ustilaginoidins from rice false smut balls caused by Villosiclava virens and their phytotoxic and cytotoxic activities. J. Agric. Food Chem. 65, 5151–5160. doi: 10.1021/acs.jafc.7b01791
Trifonov, L. S., Hilpert, H., Floersheim, P., Dreiding, A. S., Rast, D. M., Skrivanova, R., et al. (1986). Bisvertinols: a new group of dimeric vertinoids from Verticillium intertextum. Tetrahedron 42, 3157–3179. doi: 10.1016/S0040-4020(01)87382-8
Varejao, E. V. V., Demuner, A. J., Barbosa, L. C. A., and Barreto, R. W. (2013). The search for new natural herbicides - Strategic approaches for discovering fungal phytotoxins. Crop Prot. 48, 41–50. doi: 10.1016/j.cropro.2013.02.008
Wang, X., Wang, J., Lai, D., Wang, W., Dai, J., Zhou, L., et al. (2017). Ustiloxin G, a new cyclopeptide mycotoxin from rice false smut balls. Toxins 9:54. doi: 10.3390/toxins9020054
Zhai, M.-M., Qi, F.-M., Li, J., Jiang, C.-X., Hou, Y., Shi, Y.-P., et al. (2016). Isolation of secondary metabolites from the soil-derived fungus Clonostachys rosea YRS-06, a biological control agent, and evaluation of antibacterial activity. J. Agric. Food Chem. 64, 2298–2306. doi: 10.1021/acs.jafc.6b00556
Keywords: rice false smut disease, Ustilaginoidea virens, sorbicillinoids, phytotoxic activity, cytotoxic activity
Citation: Meng J, Gu G, Dang P, Zhang X, Wang W, Dai J, Liu Y, Lai D and Zhou L (2019) Sorbicillinoids From the Fungus Ustilaginoidea virens and Their Phytotoxic, Cytotoxic, and Antimicrobial Activities. Front. Chem. 7:435. doi: 10.3389/fchem.2019.00435
Received: 10 March 2019; Accepted: 28 May 2019;
Published: 12 June 2019.
Edited by:
Zhendong Jin, The University of Iowa, United StatesReviewed by:
Zhen Liu, Heinrich Heine Universität Düsseldorf, GermanyXiuping Lin, Key Laboratory of Tropical Marine Bio-resources and Ecology, South China Sea Institute of Oceanology (CAS), China
Fernando Reyes, Fundación MEDINA, Spain
Copyright © 2019 Meng, Gu, Dang, Zhang, Wang, Dai, Liu, Lai and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daowan Lai, ZHdsYWlAY2F1LmVkdS5jbg==; Ligang Zhou, bGd6aG91QGNhdS5lZHUuY24=