Abstract
Alkylative ring-opening of bicyclic aziridinium ion generated from 4-hydroxybutylaziridine with organocopper reagent was achieved successfully to afford 2-alkylsubstituted piperidine in high or moderate yield. This method allowed carbon-carbon bond formation of “non-activated” aziridine via aziridinium ion ring-opening in regio- and stereo-selective manner for the first time. This newly developed reaction was applied for an efficient synthesis of alkaloid with the representative example of conine and epiquinamide.
Graphical Abstract
Introduction
Aziridine as a nitrogen-containing three membered ring has high ring-strain described as a “spring-loaded” compound by Yudin in his book (Yudin, 2006). This ring strain has two different aspects as unstability and high reactivity. Comparing to other popular three membered rings such as cyclopropane and oxirane (epoxide), aziridine has an extra-commander to control of two different aspects by the change of the characteristics of aziridine ring-nitrogen (Yudin, 2006). When the aziridine ring-nitrogen has electron-attracting substituent we call it “activated aziridine” with instability and high activity. Most of these aziridines are quite reactive toward the aziridine-ring opening with the most nucleophiles including carbon nucleophiles (Ghorai et al., 2011). However, so called “non-activated aziridine” (D'hooghe and Ha, 2016) with strong basic ring nitrogen having electron-donating substituents such as alkyl groups are not reactive toward either common alkyl nucleophiles or quite reactive heteroatom nucleophiles such as azide, acetate, alkoxide. For the successful reactions with these heteroatom nucleophiles, the aziridine ring should be activated as aziridinium ion in the presence of acids (Kim et al., 2006; Stanković et al., 2012). In past few years we succeeded in generation of alkyl aziridinium ions as their stable forms in inter- and intramolecular manner and their successful ring opening with various heteroatom nucleophiles to give multi-substituted nitrogen-containing valuables in high regio- and stereo-specific manner (Kim et al., 2008; Dolfen et al., 2016; Boydas et al., 2018). However, limited number of alkylative aziridine-ring opening was succeed only after N-methylation by highly reactive methyl triflate (Yoon et al., 2012).
In this communication we would like to report generation of 1-azoniabicyclo[4.1.0]heptanes tosylate as one of aziridinium ions and its regio- and stereo-specific ring opening for the synthesis of chain extended 2-alkylpiperidine. This chemistry has been used for efficient synthesis of alkaloids such as conine and epiquinamide as representative examples (Graphical Abstract). Efficient and stereoselective construction of aza-heterocycles have always been a challenge for synthetic organic chemists because they played a very important role in the field of pharmaceutical industry (Bailey et al., 1998; Husson and Royer, 1999; Passarella et al., 2002; Carry et al., 2013; Yadav et al., 2016). Among them piperidines as a six-membered aza-heterocycles are present in large number of biologically active natural products especially in the class of alkaloids. Many novel synthetic approaches have been developed for aza-heterocycles in an efficient manner to encounter regio-and stereoselective demand for the drug specification (Nicolaou et al., 1995; Masse et al., 2000; Kumar and Bodas, 2005; Trost et al., 2007; Srivastava and Panda, 2008; Chavan et al., 2015). However, no synthetic method is available to construct piperidine ring with concomitant introduction of the proper alkyl group at the alpha-position of the piperidine ring. Recently, we succeeded in regio- and stereoselective ring opening reaction of stable bicyclic aziridinium ion by diverse heteroatom nucleophiles to get various pipiridines and azepane in good to excellent yields (Ji et al., 2014; Eum et al., 2015; Dolfen et al., 2016; Choi et al., 2017; Yadav and Ha, 2018; Macha et al., 2019). In continuation of this chemistry, we herein report the alkylative, regio-, and stereo-selective ring-opening of 1-azoniabicyclo[4.1.0]heptanes tosylate by various organocopper reagents to afford piperidines with carbon-chain extension at C2 position. This selective transformation allow us to access various biologically important natural products with representative examples including (+)-conine (Hattori and Yamamoto, 1993; Hirai and Nagatsu, 1994; Munchhof and Meyers, 1995; Reding and Buchwald, 1998), and (+)-epiquinamide (Suyama and Gerwick, 2006; Tong and Barker, 2006; Srivastava et al., 2009; Airiau et al., 2010) in highly efficient manner.
Results and Discussion
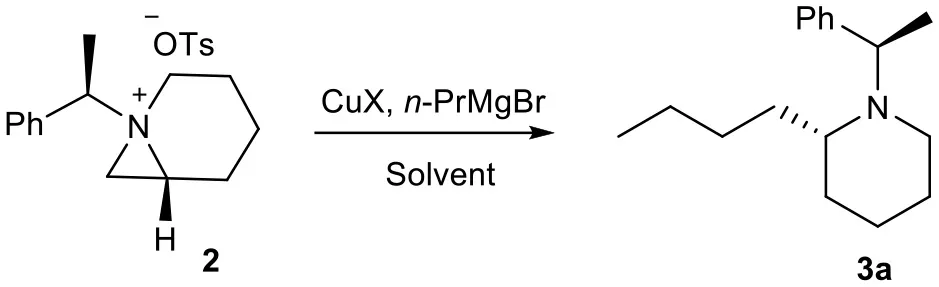
The bioavailability and importance of 2-alkyl piperidines as key intermediate for synthesis of various bioactive compounds encourage us to develop an efficient method for these systems from chiral aziridines. Our recent report on formation of stable 1-azoniabicyclo[4.1.0]heptanes tosylate 2 and its subsequent ring opening by the heteroatom nucleophile to give heteroatom-substituted piperidine and azepanes, promoted us to explore carbon-chain extension with concomitant highly strained aziridine-ring expansion with carbon nucleophile. Initially, we have treated freshly prepared bicyclic aziridinium ion 2 with n-PrMgBr in dry THF but the desired ring expanded product 3 was not formed. This may be due to hardness of the nucleophile generated from Grignard reagent (Kozlowski, 1991). Then we added CuI as reagent for in situ generation of organocopper reagents, which can changed the nature of nucleophile (Kozlowski, 1991) and we obtained the required product in 50% yields (Scheme 1). Furthermore, the reaction is highly regioselective and ring-opening proceeded by nucleophilic attack at C2 of aziridinium ion 2 to give piperidine 3a along with formation of traces of azepane compound 4 by nucleophilic attack at C3.
Scheme 1
Once we isolated and characterized compound 3a, we tried to improve the regioselectivity and yields. We have screened several different Cu(I) salts and solvents to carry the reaction. We found that the reaction of bicyclic aziridinium tosylate 2 with n-PrMgBr in anhydrous 1,4-dioxane in the presence of CuI gave the desired product 3a in 70% yields (Table 1).
Table 1
 | |||||
|---|---|---|---|---|---|
| Entrya | Reagentb | Solvent | Temp. (°C) | Time (h) | Yield (%)c |
| a | CuCN | THF | 0–25 | 1.0 | 15 |
| b | CuCN | 1,4-dioxane | 0–25 | 1.0 | 24 |
| c | CuBr | THF | 0–25 | 1.0 | 20 |
| d | CuI | 1,4-dioxane | 0–25 | 1.0 | 70 |
| e | CuBr | 1,4-dioxane | 0–25 | 1.0 | 32 |
| f | CuI | THF | 0–25 | 1.0 | 50 |
Optimization of reaction conditions for ring expansion of aziridinium ion with in situ generated organocopper reagents.
Reaction was performed at 0.5 mmol.
Copper reagent 1.5 equiv., Grignard 3.0 equiv.
Yield refers to pure products.
Once we have optimized reaction condition for alkylative ring opening of aziridinium ion by organocopper reagents in hand, we used various reagents generated in situ from Grignard reagents and CuI in 1,4-dioxane to get various piperidine 3a–l as shown in Table 2. Reagents having non-hindered alkyl group such as n-propyl 3a, methyl 3f, ethyl 3b, and allyl 3e resulted into the expected product in good yields (>50%), while those having hindered and bulky alkyl groups such as benzyl 3c, n-nonyl 3d, phenyl 3g, iso-propyl 3i, p-chlorophenyl 3k, and cyclopentyl 3l gave poor yields (25–38%) due to possible hindrance for the alkyl nucleophiles to approach the aziridinium ions.
Table 2

Synthesis of 2-alkyl piperidines from aziridine.
After successful development of a new method to prepare 2-alkyl substituted piperidines from chiral aziridines, our attention was to apply this reaction for a short and efficient synthetic route for few biologically active molecules having piperidine skeleton such as (+)-conine 6 and (+)-epiquinamide 7 (Figure 1).
Figure 1
Optically active conine, the poisonous hemlock alkaloid is a very important among piperidines alkaloids (Hattori and Yamamoto, 1993; Hirai and Nagatsu, 1994; Munchhof and Meyers, 1995; Reding and Buchwald, 1998; Jo et al., 1999; Pachamuthu and Vankar, 2001; Chacko and Ramapanicker, 2015). Using our strategy, we were able to synthesized N-Boc-conine 5 in optically pure form in few steps from 1-azoniabicyclo[4.1.0]heptane tosylate 2, which was easily access from commercially available chiral aziridine (Choi et al., 2017) (Scheme 2).
Scheme 2
Synthesis of N-Boc-conine 5 was initiated by the preparation of bicyclic aziridinium ion 2 (Choi et al., 2017) followed by its ring expansion with EtMgBr and CuI in 1,4-dioxane as solvent to yield the compound 3b in 64% yield in two steps. One-pot debenzylation followed by Boc-protection furnished N-Boc-conine 5 in 70% yield whose Boc group would be removed by the known procedure yielding the natural product conine 6. Analytical data for compound 5 were found in good agreement with reported value in literature (Hirai and Nagatsu, 1994; Jo et al., 1999).
The synthesis of (+)-epiquinamide 7 was the next task by use of our newly developed protocol with the chain extension of C2 of piperidine. Epiquinamide 7 is a quinolizidine alkaloid isolated from skin of Epipedobates tricolor, an Ecuadorian poisonous frog. It has been found to be highly selective toward β2 nicotinic acetylcholine receptors (nAChRs) involving in cellular signaling in both the central (CNS) and peripheral nervous systems (PNS). The importance of this receptor encourages us to utilize our strategy for its synthesis. The formal synthesis of (+)-epiquinamide 7 began with our previously prepared hydroxybutyl aziridine 9 from aziridine carboxylate 8 (Scheme 3) (Choi et al., 2017).
Scheme 3
Tosylation of compound 9 with p-toluenesulphonic anhydride and triethylamine in dry CH2Cl2 gives corresponding tosylate 10 in quantitative yields. Compound 10 was kept in dry CH3CN under N2 atmosphere for 24 h to attain complete conversion to bicyclic aziridinium ion 11, which was confirmed by NMR spectrum. Aziridine ring expansion of 11 with Gilman's reagent generated from Grignard reagent 12 and CuI gives piperidine 13 in 57% yields.
Compound 13 was treated under acidic condition to retrieve an aldehyde from its protected acetal functionality which was subjected to one-pot reductive amination by catalytic hydrogenation under atmospheric H2 to get the bicyclic compound 14 in 59% yield which is able to yield epiquinamide (Scheme 4).
Scheme 4
Conclusion
In conclusion, we have developed a new and highly efficient method for the selective formation of 2-alkylpiperidine by the alkylative, regio- and stereospecific ring opening of highly strained bicyclic aziridinium ion with organocopper reagent. This method was applied for the synthesis of important alkaloids including conine and (+)-epiquinamide in highly efficient manner.
Experimental Section
Materials and Methods
Chiral aziridine-2-carboxylates are available as their menthyl ester from Sigma-Aldrich as reagents and from Imagene Co., Ltd. (http://www.imagene.co.kr/) in bulk quantities. Their corresponding ethyl esters were also available either from transesterification of menthyl ester or from Imagene. Reagents are commercially available. All commercially available reagents were used as received unless stated otherwise. Acetonitrile used to make bicyclic aziridinium ion and 1,4-dioxane used in next reactions are purified and used using calcium hydride. All reactions were carried out under an atmosphere of nitrogen in oven-dried glassware with magnetic stirrer. Reactions were monitored by thin layer chromatography (TLC) with 0.25 mm E. Merck pre-coated silica gel plates (60 F254). Visualization was accomplished with either UV light, or by immersion in solutions of ninhydrin, p-anisaldehyde, or phosphomolybdic acid (PMA) followed by heating on a hot plate for about 10 sec. Flash column chromatography was carried out using Intertec silica gel (Particle size: 70–230 mesh). 1H-NMR and 13C-NMR spectra were obtained using Varian unity lNOVA 400WB (400 MHz) or Bruker AVANCE III HD (400 MHz) spectrometer. Chemical shifts are reported relative to chloroform (δ = 7.26) for 1H NMR and chloroform (δ = 77.2) for 13C NMR, acetonitrile (δ = 1.94) for 1H NMR and acetonitrile (δ = 1.32) for 13C NMR, dimethyl sulfoxide (δ = 3.33) for 1H NMR and dimethyl sulfoxide (δ = 39.5) for 13C NMR. Data are reported as (br = broad, s = singlet, d = doublet, t = triplet, q = quartet, p = quintet, m = multiplet). Coupling constants are given in Hz. Ambiguous assignments were resolved on the basis of standard one dimensional proton decoupling experiments. Optical rotations were obtained using Rudolph Autopol III digital polarimeter and JASCO P-2000.Optical rotation data was reported as follows: [α]20 (concentration c = g/100 mL, solvent). High resolution mass spectra were recorded on a 4.7 Tesla IonSpec ESI-TOFMS, JEOL (JMS-700) and AB Sciex 4800 Plus MALDI TOFTM (2,5-dihydroxybenzoic acid (DHB) matrix was used to prepare samples for MS. Data was obtained in the reflector positive mode with internal standards for calibration) (Supplementary Materials).
(6R)-1-[(R)-1-Phenylethyl)-1-Azoniabicyclo[4.1.0]Heptane Tosylate (2)
p-Toluenesulfonic anhydride (489 mg, 1.50 mmol) was added to a stirring solution of alcohol 1 (300 mg, 1.36 mmol) and Et3N (0.42 mL, 3.0 mmol) in CH2Cl2 at 0°C. The resulting mixture was stirred for 45 min, warmed to RT, and stirred it for a further additional 30 min. The reaction mixture was quenched with water (15 mL), extracted with CH2Cl2 (3 × 50 mL), dried over anhydrous Na2SO4, and concentrated under vacuum to afford the crude tosylate, purified by column chromatography to give pure tosylate as a viscous liquid (486 mg, 95% yield).
Freshly prepared tosylate was stored in CD3CN for 24 h to allow complete conversion into azoniabicycle 2, which was characterized by NMR spectroscopy.
1H NMR (400 MHz, CD3CN) δ 7.61 (d, J = 8.1 Hz, 2H), 7.63–7.48 (m, 5H), 7.15 (d, J = 7.8 Hz, 2H), 4.11 (q, J = 6.9 Hz, 1H), 3.81–3.73 (m, 1H), 3.49 (dt, J = 13.3, 5.3 Hz, 1H), 3.23 (dd, J = 9.0, 4.2 Hz, 1H), 3.15 (ddd, J = 13.4, 10.3, 4.9 Hz, 1H), 3.03 (dd, J = 7.9, 4.2 Hz, 1H), 2.33 (s, 3H), 2.12–2.04 (m, 2H), 1.72 (d, J = 7.0 Hz, 3H), 1.53–1.44 (m, 1H), 1.37–1.29 (m, 2H), 0.94–0.82 (m, 1H); 13C NMR (CD3CN, 101 MHz): δ 146.6, 139.6, 134.7, 131.1, 130.2, 129.5, 129.3, 126.6, 71.7, 49.7, 49.4, 43.1, 21.2, 20.8, 20.7, 15.5, 14.1.
Typical Procedure for the Synthesis of Various Piperidine (3)
To a stirred solution of alcohol 1 (200 mg, 0.913 mmol) and Et3N (0.28 mL, 0.570 mmol) in CH2Cl2 (4 mL) at 0°C was added p-toluenesulfonic anhydride (328 mg, 1.00 mmol) and the resulting mixture was stirred for another 45 min and then warmed to rt and allowed to stir for 30 min. Reaction mixture was concentrated under vacuums and crude tosylate was dissolved in MeCN (4.0 mL) and allowed for complete conversion to azoniabicycle 2 for 24 h. Removal of MeCN solvent from tosylated azonia 2, added dry 1,4-dioxane (5 mL) followed by CuI (1.5 eq) and Grignard reagent (3.0 eq) at 0°C. The reaction mixture was allowed to stir for appropriate time. After completion of starting material as confirmed by TLC, the reaction mixture was quenched with water (5 mL). Combined reaction mixture were filtered over Celite, followed by extracted with EtOAc (2 × 15 mL), dried over anhydrous Na2SO4, and concentrated in vacuo to get crude product, which was easily purified by silica gel chromatography to get pure alkylated compound 3.
(S)-2-Butyl-1-((R)-1-Phenylethyl)Piperidine (3a)
[α]20D = +51.2 (c = 0.7, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.33–7.16 (m, 5H), 4.04 (q, J = 6.8 Hz, 1H), 2.74 (ddd, J = 11.5, 5.6, 2.1 Hz, 1H), 2.41–2.32 (m, 1H), 2.25 (ddd, J = 16.7, 10.2, 6.8 Hz, 1H), 1.66–1.51 (m, 5H), 1.41–1.33 (m, 4H), 1.31–1.19 (m, 5H), 1.14–1.01 (m, 1H), 0.92–0.84 (t, J = 7.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 143.57, 128.03, 126.64, 57.51, 56.06, 44.56, 29.41, 28.27, 28.01, 25.78, 23.31, 21.88, 21.04, 14.33; HRMS-MALDI (m/z): calcd. For C17H27N [M+H]+ 246.1426; found:246.1422.
(S)-1-((R)-1-Phenylethyl)-2-Propylpiperidine (3b)
[α]20D = +48.8 (c = 0.45, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.32–7.22 (m, 5H), 4.03 (q, J = 6.8 Hz, 1H), 2.78–2.67 (m, 1H), 2.44–2.35 (m, 1H), 2.32–2.22 (m, 1H), 1.66–1.41 (m, 6H), 1.40–1.33 (m, 3H), 1.31–1.19 (m, 3H), 1.11 (dddd, J = 20.2, 13.1, 8.3, 5.0 Hz, 1H), 0.87 (t, J = 7.3 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 143.71, 128.02, 126.64, 57.54, 55.77, 44.47, 30.57, 29.32, 25.73, 21.80, 21.07, 19.29, 14.68; HRMS-MALDI (m/z): calcd. For C16H25N [M+H]+ 232.1310; found:232.1319.
(R)-2-Phenethyl-1-((R)-1-Phenylethyl)Piperidine (3c)
[α]20D = + 96.3 (c = 0.65, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.36–7.11 (m, 10H), 4.02 (q, J = 6.8 Hz, 1H), 2.71 (m, 1H), 2.61–2.50 (m, 2H), 2.44–2.31 (m, 2H), 2.01–1.80 (m, 2H), 1.61 (m, 3H), 1.51–1.38 (m, 2H), 1.36 (d, J = 6.8 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 143.82, 143.18, 128.45, 128.42, 128.12, 127.94, 126.71, 125.74, 57.91, 55.37, 44.29, 32.54, 30.19, 29.01, 25.46, 21.53, 21.28; HRMS-MALDI (m/z): calcd. For C21H27N [M+H]+ 294.1345; found:294.1346.
(S)-2-Decyl-1-((R)-1-Phenylethyl)Piperidine (3d)
[α]20D = +68.1 (c = 0.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.37–7.26 (m, 5H), 4.06 (q, J = 6.8 Hz, 1H), 2.75 (m, 1H), 2.39 (s, 1H), 2.34–2.23 (m, 1H), 1.63–1.55 (m, 5H), 1.39 (d, J = 6.7 Hz, 3H), 1.31 (m, 18H), 1.13 (m, 1H), 0.94–0.86 (t, J = 6.8 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 143.57, 128.09, 126.67, 56.10, 44.61, 32.08, 30.26, 29.81, 29.52, 28.33, 25.98, 25.81, 22.85, 21.94, 21.00, 14.26; HRMS-MALDI (m/z): calcd. For C23H39N [M+H]+ 330.2336; found:330.2334.
(R)-2-(But-3-En-1-Yl)-1-((R)-1-Phenylethyl)Piperidine (3e)
[α]20D = +58.8 (c = 1.2, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.32–7.19 (m, 5H), 5.87–5.70 (m, 1H), 5.04–4.85 (m, 2H), 4.04–3.95 (m, 1H), 2.77–2.64 (m, 1H), 2.51–2.42 (m, 1H), 2.32 (ddd, J = 15.9, 9.4, 5.9 Hz, 1H), 2.07–1.95 (m, 1H), 1.92–1.81 (m, 1H), 1.80–1.68 (m, 1H), 1.66–1.38 (m, 5H), 1.35 (d, J = 6.8 Hz, 3H), 1.34–1.23 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 144.00, 139.46, 128.20, 128.07, 126.81, 114.30, 57.95, 55.34, 44.42, 30.53, 29.02, 27.55, 25.52, 21.68, 21.31; HRMS-MALDI (m/z): calcd. For C17H25N [M+H]+ 244.1270; found:244.1272.
(S)-2-Ethyl-1-((R)-1-Phenylethyl)Piperidine (3f)
[α]20D = +63.4 (c = 0.62, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.34–7.18 (m, 5H), 4.06 (q, J = 6.8 Hz, 1H), 2.79–2.71 (m, 1H), 2.32–2.15 (m, 2H), 1.72–1.43 (m, 6H), 1.38 (d, J = 6.8 Hz, 3H), 1.27–1.13 (m, 2H), 0.79 (t, J = 7.5 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 143.14, 128.10, 127.99, 126.65, 57.45, 57.27, 44.73, 29.03, 25.88, 22.14, 21.33, 20.74, 10.23; HRMS-MALDI (m/z): calcd. For C15H23N [M+H]+ 218.1153; found:218.1156.
(R)-2-Benzyl-1-((R)-1-Phenylethyl)Piperidine (3g)
[α]20D = −8.8 (c = 1.05, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.36 (dt, J = 14.9, 4.5 Hz, 4H), 7.29–7.22 (m, 1H), 7.19–7.06 (m, 3H), 6.85 (d, J = 7.0 Hz, 2H), 4.02 (q, J = 6.6 Hz, 1H), 3.04 (dd, J = 12.7, 3.3 Hz, 1H), 2.79–2.58 (m, 4H), 1.65–1.60 (m, 2H), 1.39 (d, J = 6.6 Hz, 3H), 1.36–1.29 (m, 2H), 1.28–1.22 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 144.79, 141.17, 129.30, 128.44, 128.31, 127.93, 126.98, 125.70, 59.67, 57.57, 44.25, 31.71, 27.91, 26.29, 22.14, 20.51; HRMS-MALDI (m/z): calcd. For C20H25N [M+H]+ 280.1245; found:280.1240.
(S)-2-Pentyl-1-((R)-1-Phenylethyl)Piperidine (3h)
[α]20D = +53.2 (c = 0.65, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.33–7.19 (m, 5H), 4.04 (q, J = 6.8 Hz, 1H), 2.72 (m, 1H), 2.39–2.34 (m, 1H), 2.32–2.23 (m, 1H), 1.62–1.50 (m, 4H), 1.36 (d, J = 6.8 Hz, 3H), 1.33–1.06 (m, 10H), 0.87 (t, J = 7.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 143.62, 128.06, 128.03, 126.66, 57.62, 56.09, 44.58, 32.49, 29.42, 28.25, 25.80, 25.69, 22.82, 21.89, 21.03, 14.22; HRMS-MALDI (m/z): calcd. For C18H29N [M+H]+ 260.1593; found:260.1594.
(R)-2-Isobutyl-1-((R)-1-Phenylethyl)Piperidine (3i)
[α]20D = +41.2 (c = 1.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.36–7.17 (m, 5H), 3.97 (q, J = 6.7 Hz, 1H), 2.69–2.60 (m, 2H), 2.46 (m, 1H), 1.70–1.40 (m, 6H), 1.39–1.23 (m, 6H), 0.87 (d, J = 6.2 Hz, 3H), 0.69 (d, J = 6.1 Hz); 13C NMR (101 MHz, CDCl3) δ 145.15, 128.11, 127.82, 126.61, 58.30, 52.98, 43.73, 35.98, 28.45, 25.28, 25.24, 24.19, 21.99, 21.93, 20.81; HRMS-MALDI (m/z): calcd. For C17H27N [M+H]+ 246.1476; found:246.1479.
(R)-2-Isopentyl-1-((R)-1-Phenylethyl)Piperidine (3j)
[α]20D = +52.3 (c = 2.35, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.31–7.19 (m, 5H), 4.03 (q, J = 6.8 Hz, 1H), 2.77–2.69 (m, 1H), 2.38–2.24 (m, 2H), 1.65–1.37 (m, 7H), 1.36 (d, J = 6.8 Hz, 3H), 1.30–1.19 (m, 2H), 1.12 (dddd, J = 16.7, 11.9, 6.8, 5.2 Hz, 1H), 0.96 (tdd, J = 9.1, 5.0, 4.2 Hz, 1H), 0.88 (dd, J = 6.7, 2.7 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ 143.60, 128.02, 126.64, 57.68, 56.28, 44.60, 35.21, 29.40, 28.61, 25.87, 25.81, 22.94, 22.74, 21.86, 21.01; HRMS-MALDI (m/z): calcd. For C18H29N [M+H]+ 260.1523; found:260.1526.
(R)-2-(4-Chlorobenzyl)-1-((R)-1-Phenylethyl)Piperidine (3k)
[α]20D = +3.6 (c = 1.28, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.40–7.23 (m, 5H), 7.16–7.08 (m, 2H), 6.75 (d, J = 8.3 Hz, 2H), 3.97 (q, J = 6.6 Hz, 1H), 2.95 (m, 1H), 2.77–2.56 (m, 4H), 1.68–1.53 (m, 3H), 1.38 (d, J = 6.6 Hz, 3H), 1.28–1.13 (m, 3H); 13C NMR (101 MHz, CDCl3) δ 144.66, 139.56, 131.36, 130.49, 128.43, 128.33, 127.80, 127.00, 59.77, 57.34, 44.13, 30.85, 27.63, 26.06, 22.13, 20.28; HRMS-MALDI (m/z): calcd. For C20H24ClN [M+H]+ 313.9880; found:313.9882.
(R)-2-(Cyclopentylmethyl)-1-((R)-1-Phenylethyl)Piperidine (3l)
[α]20D = +96.7 (c = 0.395, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.33–7.19 (m, 5H), 3.98 (q, J = 6.7 Hz, 1H), 2.73–2.55 (m, 2H), 2.51–2.41 (m, 1H), 1.82–1.38 (m, 16H), 1.33 (d, J = 6.7 Hz, 3H) 1.15–1.02 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 144.98, 128.10, 127.83, 126.63, 58.37, 54.33, 43.90, 37.37, 33.99, 33.09, 32.05, 28.57, 25.39, 25.20, 25.00, 21.85, 20.96; HRMS-MALDI (m/z): calcd. For C19H29N [M+H]+ 272.0563; found:272.0562.
(S)-Tert-Butyl 2-Propylpiperidine-1-Carboxylate: (+)-Boc-Coniine (5)
To a solution of compound 3b (0.3g, 1.296 mmol) in MeOH. Pd(OH)2 (0.455 g, 2.24 mmol) and Boc2O (0.566 g, 2.59 mmol) were added. The reaction mixture was allowed to stir under hydrogenation atmospheric pressure of hydrogen for 12 h. The reaction mixture was filtered over Celite pad by using MeOH as solvent, concentrated in vacuo to get crude product which was purified by column chromatography to give pure product 5 (206 mg, 70 %).
[α]20D = +16.072 (c = 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 4.21 (s, 1H), 3.96 (d, J = 11.9 Hz, 1H), 2.74 (m, 1H), 1.65–1.54 (m, 5H), 1.45 (s, 9H), 1.43–1.20 (m, 5H), 0.92 (t, J = 7.3 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 155.36, 79.09, 50.32, 38.83, 32.10, 28.67, 27.59, 25.88, 19.66, 19.23, 14.24; HRMS-MALDI (m/z): calcd. For C13H25NO2 [M+Na]+ 250.0227; found:250.0229.
(R)-4-(Benzyloxy)-4-((S)-1-((R)-1-Phenylethyl)Aziridin-2-yl)Butyl-4-Methylbenzenesulfonate (10)
p-Toluenesulfonic anhydride (300 mg, 0.92 mmol) was added to a stirring solution of 9 (330 mg, 1.01 mmol) and triethylamine (0.28 mL, 2.02 mmol) in CH2Cl2 at 0°C. The resulting mixture was stirred for 45 min, warmed to rt, and stirred for a further 30 min. The reaction mixture was quenched with water, extracted with CH2Cl2, dried over sodium sulfate, and concentrated under vacuum to afford the crude tosylate, which was purified by chromatography to give pure tosylate as a viscous liquid 10 (490 mg, 95% yield).
1H NMR (400 MHz, CD3CN) δ 7.79–7.64 (m, 2H), 7.42–7.39 (m, 2H), 7.36–7.17 (m, 10H), 4.53 (m, 1H), 4.31 (d, J = 11.7 Hz, 1H), 3.71–3.61 (m, 2H), 2.75 (td, J = 7.2, 3.5 Hz, 1H), 2.43 (s, 3H), 2.42–2.37 (m, 1H), 1.77 (d, J = 3.3 Hz, 1H), 1.51 (d, J = 6.2 Hz, 1H), 1.45–1.36 (m, 2H), 1.34 (d, J = 6.5 Hz, 3H), 1.31–1.16 (m, 2H), 1.03–0.87 (m, 1H); 13C NMR (101 MHz, CD3CN) δ 145.86, 145.52, 139.90, 133.73, 130.72, 129.79, 129.43, 128.92, 128.41, 128.30, 128.04, 127.88, 80.00, 71.72, 71.37, 70.39, 40.84, 33.76, 29.32, 24.48, 22.66, 21.42.
(5R, 6S)-5-(Benzyloxy)-1-((R)-1-Phenylethyl)-1-Azoniabicyclo[4.1.0] Heptane Tosylate (11)
Freshly prepared tosylate 10 was stored in CD3CN for 24 h to allow complete conversion into azoniabicycle 11, which was characterized by NMR spectroscopy.
1H NMR (400 MHz, CD3CN) δ 7.68–7.63 (m, 2H), 7.61–7.57 (m, 2H), 7.47–7.27 (m, 9H), 7.15 (dd, J = 8.4, 0.6 Hz, 2H), 4.61 (d, J = 11.6 Hz, 1H), 4.53 (d, J = 11.6 Hz, 1H), 4.31 (q, J = 7.0 Hz, 1H), 4.15–4.04 (m, 2H), 3.50–3.39 (m, 2H), 3.28–3.14 (m, 2H), 2.30 (s, 3H), 1.69 (d, J = 7.0 Hz, 3H), 1.65–1.47 (m, 2H), 1.34–1.13 (m, 2H); 13C NMR (101 MHz, CD3CN) δ 146.34, 139.76, 138.97, 134.48, 130.93, 129.90, 129.68, 129.37, 129.27, 128.58, 128.50, 126.61, 71.51, 69.58, 53.04, 48.70, 42.50, 21.85, 21.25, 15.72, 14.18.
(2S,3R)-2-(3-(1,3-Dioxan-2-Yl)Propyl)-3-(Benzyloxy)-1-((R)-1-Phenylethyl)Piperidine (13)
After removal of MeCN solvent from tosylated azonia 11, dry 1,4-dioxane (10 mL) followed by CuI (1.5 eq) and Grignard reagent 12 (3.0 eq) were added and allowed to stir at 0°C for 15 min. followed by additional 40 min. at room temperature. After completion of starting material as confirmed by TLC, the reaction mixture was quenched with water (5 mL). The Combined reaction mixture were filtered over Celite, followed by extracted with EtOAc (2 × 10 mL), dried over anhydrous Na2SO4, and concentrated in vacuo to get crude product, which was easily purified by silica gel chromatography to get pure alkylated compound 13 (104 mg, 57 %).
[α]20D = +24.012 (c = 0.20, CHCl3);1H NMR (400 MHz, CDCl3) δ 7.38–7.26 (m, 10H), 4.66 (d, J = 10.6 Hz, 1H), 4.60–4.45 (m, 2H), 4.46–4.35 (m, 1H), 4.17–4.07 (m, 1H), 3.97–3.73 (m, 3H), 3.54–3.40 (m, 1H), 2.97–2.83 (m, 2H), 2.81–2.63 (m, 1H), 2.29–2.17 (m, 1H), 2.16–1.77 (m, 4H), 1.66–1.51 (m, 2H), 1.49 (d, J = 6.8 Hz, 3H), 1.44–1.22 (m, 4H); 13C NMR (101 MHz, CDCl3) δ 142.08, 138.70, 128.42, 128.26, 128.21, 127.93, 127.56, 127.06, 127.51, 127.48, 127.10, 102.37, 74.93, 71.02, 66.90, 60.12, 58.02, 57.13, 43.56, 27.52, 25.88, 20.49, 19.39.
(1R, 9aS)-Octahydro-1H-Quinolizin-1-Ol (14)
To a stirred solution of 13 (100 mg, 0.236 mmol)in water (2 ml) treated with catalytic amount of sulfuric acid and stir it for 2 h, followed by quenched with NaHCO3. The aqueous phase was extracted with CH2Cl2. The combined organic layers were dried over anhydrous Na2SO4, concentrated in vacuo to get crude product, which was dissolved in MeOH (2 ml) followed by Pd(OH)2 (98 mg, 0.709 mmol) was added and the mixture was hydrogenated under an atmospheric pressure of hydrogen for 12 h. The reaction mixture was diluted with MeOH and filtered on a pad of Celite using MeOH as solvent. The pure product was obtained by column chromatography as viscous liquid 14 (57 mg, 59%).
[α]20D = −20.307 (c = 0.35, CHCl3);1H NMR (400 MHz, CDCl3) δ 4.53 (s, 1H), 3.39 (ddd, J = 11.1, 9.0, 4.5 Hz, 1H), 2.96 (d, J = 11.2 Hz, 1H), 2.84 (d, J = 11.4 Hz, 1H), 2.25–1.98 (m, 4H), 1.88–1.59 (m, 6H), 1.37–1.22 (m, 3H); 13C NMR (101 MHz, CDCl3) δ 72.76, 68.92, 56.25, 56.01, 34.12, 28.82, 25.79, 24.28, 23.45; HRMS-MALDI (m/z): calcd. For C9H17NO [M+H]+ 156.0543; found:156.0546.
Statements
Data availability statement
All datasets generated for this study are included in the manuscript and/or the Supplementary Files.
Author contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by the National Research Foundation of Korea (NRF-2012M3A7B4049645 and 2014R1A5A1011165 with Centre for New Directions in Organic Synthesis).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00460/full#supplementary-material
References
1
AiriauE.SpangenbergT.GirardN.BreitB.MannA. (2010). Short access to (+)-lupinine and (+)-epiquinamide via double hydroformylation. Org. Lett.12, 528–531. 10.1021/ol902718q
2
BaileyP. D.MillwoodP. A.SmithP. D. (1998). Asymmetric routes to substituted piperidines. Chem. Commun.633–640. 10.1039/a709071d
3
BoydasE. B.TanriverG.D'hoogheM.HaH.-J.Van SpeybroeckV.CatakS. (2018). Theoretical insight into the regioselective ring-expansions of bicyclic aziridinium ions. Org. Biomol. Chem. 16, 796–806. 10.1039/C7OB02253K
4
CarryJ. C.BrohanE.PerronS.BardouilletP. E. (2013). Chiral supercritical fluid chromatography in the preparation of enantiomerically pure (S)-(+)-tert-Butyl-3-hydroxyazepane-1-carboxylate. Org. Process Res.Dev.17, 1568–1571. 10.1021/op400274b
5
ChackoS.RamapanickerR. (2015). Proline catalyzed, one-pot three component Mannich reaction and sequential cyclization toward the synthesis of 2-substituted piperidine and pyrrolidine alkaloids. Tetrahedron Lett. 56, 2023–2026. 10.1016/j.tetlet.2015.03.001
6
ChavanS. P.KhairnarL. B.PawarK. P.ChavanP. N.KawaleS. A. (2015). Enantioselective syntheses of (R)-pipecolic acid, (2R, 3R)-3-hydroxypipecolic acid, β-(+)-conhydrine and (–)-swainsonine using an aziridine derived common chiral synthon. RSC Adv.5, 50580–50590. 10.1039/C5RA06429E
7
ChoiJ.YadavN. N.HaH. J. (2017). Preparation of a stable bicyclic aziridinium ion and its ring expansion toward piperidines and azepanes. Asian J. Org. Chem.6, 1292–1307. 10.1002/ajoc.201700080
8
D'hoogheM.HaH.-J. (eds). (2016). Synthesis of 4- to 7-Membered Heterocycles by Ring Expansion: Aza-, Oxa-and Thiaheterocyclic Small-Ring Systems. Heidelberg: Springer.
9
DolfenJ.YadavN. N.De KimpeN.D'hoogheM.HaH. J. (2016). Bicyclic aziridinium ions in azaheterocyclic chemistry – preparation and synthetic application of 1-Azoniabicyclo [n.1.0]alkanes. Adv. Synth. Catal.358, 3483–3511. 10.1002/adsc.201601081
10
EumH.ChoiJ.ChoC.-G.HaH.-J. (2015). Regiochemistry-Directed syntheses of polyhydroxylated alkaloids from chiral aziridines. Asian J. Org. Chem. 4, 1325–1340. 10.1002/ajoc.201500380
11
GhoraiM. K.NanajiY.YadavA. K. (2011). Ring opening/C–N cyclization of activated aziridines with carbon nucleophiles: highly diastereo- and enantioselective synthesis of tetrahydroquinolines. Org. Lett.13, 4256–4259. 10.1021/ol2016077
12
HattoriK.YamamotoH. (1993). Asymmetric aza-Diels-Alder reaction: enantio- and diastereoselective reaction of imine mediated by Chiral Lewis acid. Tetrahedron49, 1749–1760. 10.1016/S0040-4020(01)80532-9
13
HiraiY.NagatsuM. (1994). Construction of chiral 2-functionalized piperidine via enzymatic resolution and palladium-catalyzed N-alkylation. Chem. Lett. 23, 21–22. 10.1246/cl.1994.21
14
HussonH.-P.RoyerJ. (1999). Chiral non-racemic N-cyanomethyloxazolidines: the pivotal system of the CN (R,S) method. Chem. Soc. Rev.28, 383–394. 10.1039/a900153k
15
JiM.-K.HertsenD.YoonD.-H.EumH.GoossensH.WaroquierM.et al. (2014). Nucleophile-dependent regio- and stereoselective ring opening of 1-Azoniabicyclo-[3.1.0]hexane tosylate. Chem. Asian. J. 9, 1060–1067. 10.1002/asia.201301551
16
JoE.NaY.ChangS. (1999). A highly efficient synthesis of (S)-(+)-N-Boc-coniine using ring-closing olefin metathesis (RCM). Tetrahedron Lett. 40, 5581–5582. 10.1016/S0040-4039(99)01081-3
17
KimY.HaH.-J.YunS. Y.LeeW. K. (2008). The preparation of stable aziridinium ions and their ring-openings. Chem. Commun. 4363–4365. 10.1039/b809124b
18
KimY.HaH. J.YunH.LeeB. K.LeeW. K. (2006). Ring opening of 2-acylaziridines by acid chlorides. Tetrahedron62, 8844–8849. 10.1016/j.tet.2006.06.025
19
KozlowskiJ. A. (1991). Organocuprates in the conjugate addition reaction. Compr. Org. Synth. 4, 169–198. 10.1016/B978-0-08-052349-1.00093-7
20
KumarP.BodasM. S. (2005). Asymmetric synthesis of both the enantiomers of trans-3-Hydroxypipecolic acid. J. Org. Chem. 70, 360–363. 10.1021/jo0485381
21
MachaL.D'hoogheM.HaH.-J. (2019). Deployment of aziridines for the synthesis of alkaloids and their derivatives. Synthesis51, 1491–1515. 10.1055/s-0037-1611715
22
MasseC. E.MorganA. J.PanekJ. S. (2000). An asymmetric aminohydroxylation approach to the azepine core of (–)-balanol. Org. Lett. 2, 2571–2573. 10.1021/ol0061034
23
MunchhofM. J.MeyersA. I. (1995). An asymmetric route to chiral, nonracemic 2-substituted piperidines. Synthesis of (-)-Pipecoline, (+)-Coniine, and (-)-Coniceine. J. Org. Chem. 60, 7084–7085. 10.1021/jo00127a005
24
NicolaouK. CKoideK.BunnageM. E. (1995). Total synthesis of balanol and designed analogues. Chem. Eur. J.1, 454–466. 10.1002/chem.19950010711
25
PachamuthuK.VankarY. D. (2001). Synthesis of (–)-coniine and (–)-pipecoline using ruthenium catalyzed ring closing metathesis. J. Organomet. Chem.624, 359–363. 10.1016/S0022-328X(00)00930-X
26
PassarellaD.AngoliM.GiardiniA.LesmaG.SilvaniA.DanieliB. (2002). Concise total synthesis of (±)-Aloperine and 6-epi-Aloperine. Org. Lett.4, 2925–2928. 10.1021/ol0263144
27
RedingM. T.BuchwaldS. L. (1998). Short enantioselective total syntheses of the piperidine Alkaloids (S)- Coniine and (2R,6R)-trans-solenopsin A via catalytic asymmetric imine hydrosilylation. J. Org. Chem.63, 6344–6347. 10.1021/jo980808q
28
SrivastavaA. K.DasS. K.PandaG. (2009). An approach towards the total synthesis of (+)-epiquinamide and (+)-α-conhydrine from Garner aldehyde. Tetrahedron65, 5322–5327. 10.1016/j.tet.2009.04.074
29
SrivastavaA. K.PandaG. (2008). Total synthesis of (–)-balanol, all stereoisomers, their N- tosyl analogues, and fully protected ophiocordin: an easy route to hexahydroazepine cores from garner aldehydes. Chem. Eur. J.14, 4675–4688. 10.1002/chem.200701991
30
StankovićS.D'hoogheM.CatakS.EumH.WaroquierM.Van SpeybroeckV.et al. (2012). Regioselectivity in the ring opening of non-activated aziridine. Chem. Soc. Rev. 41, 643–665. 10.1039/C1CS15140A
31
SuyamaT. L.GerwickW. H. (2006). Practical total syntheses of epiquinamide enantiomers. Org. Lett.08, 4541–4543. 10.1021/ol061736p
32
TongS. T.BarkerD. (2006). A concise synthesis of (±) and a total synthesis of (+)-epiquinamide. Tetrahedron Lett. 47, 5017–5020. 10.1016/j.tetlet.2006.05.092
33
TrostB. M.FandrickD. R.BrodmannT.StilesD. T. (2007). Dynamic kinetic asymmetric allylic amination and acyl migration of vinyl aziridines with imido carboxylates. Angew. Chem. Int. Ed.46, 6123–6125. 10.1002/anie.200700835
34
YadavN. N.ChoiJ.HaH. J. (2016). Asymmetric synthesis of 2, 6-cis-disubstituted piperidine alkaloids from chiral aziridine. Org. Biomol. Chem. 14, 6426–6434. 10.1039/C6OB00806B
35
YadavN. N.HaH.-J. (2018). Preparation of stable bicyclic aziridinium ions and their ring-opening for the synthesis of azaheterocycles. J. Vis. Exp.e57572. 10.3791/57572
36
YoonD.-H.KangP.LeeW. K.KimY.HaH.-J. (2012). Alkylative ring opening of N-methyl aziridinium ions and a formal synthesis of tyroscherin. Org. Lett. 14, 429–431. 10.1021/ol202683k
37
YudinA. K. (2006). Aziridines and Epoxides in Organic Synthesis. Weinheim: Wiley-VCH.
Summary
Keywords
aziridine, bicyclic aziridinium ions, alkylation, ring-expansion, alkaloid synthesis
Citation
Yadav NN, Lee Y-G, Srivastava N and Ha H-J (2019) Alkylative Ring-Opening of Bicyclic Aziridinium Ion and Its Application for Alkaloid Synthesis. Front. Chem. 7:460. doi: 10.3389/fchem.2019.00460
Received
03 May 2019
Accepted
11 June 2019
Published
27 June 2019
Volume
7 - 2019
Edited by
Andrea Basso, University of Genoa, Italy
Reviewed by
Won Koo Lee, Sogang University, South Korea; Andrea Trabocchi, University of Florence, Italy
Updates
Copyright
© 2019 Yadav, Lee, Srivastava and Ha.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hyun-Joon Ha hjha@hufs.ac.kr
This article was submitted to Organic Chemistry, a section of the journal Frontiers in Chemistry
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.