 Marta Diez-Castellnou
Marta Diez-Castellnou Giovanni Salassa
Giovanni Salassa Fabrizio Mancin3
Fabrizio Mancin3- 1EaStCHEM School of Chemistry, University of St Andrews, St. Andrews, United Kingdom
- 2Département de Chimie Physique, Université de Genève, Genève, Switzerland
- 3Dipartimento di Scienze Chimiche, Università di Padova, Padova, Italy
In this paper, the unusual reactivity of the complex Zn(II)-1,4,7-trimethyl-1, 4,7-triazacyclononane (2) in the transesterification of the RNA-model substrate, HPNP (3), is reported. The dependence of the reactivity (k2) with pH does not follow the characteristic bell-shape profile typical of complexes with penta-coordinated metal centers. By the contrary, two reactive species, featuring different deprotonation states, are present, with the tri-aqua complex being more reactive than the mono-hydroxy-diaqua one. Apparently, such a difference arises from the total complex charge which plays an important role in the stability of the transition state/s of the reactions. Relevant insight on the reaction mechanism were hence obtained.
Introduction
Phosphate diesters have a fundamental importance in the chemistry of life in particular because they constitute the backbone of essential biomolecules as DNA and RNA. Their hydrolytic stability is very high. When the sole P-O cleavage by water is considered, the half-life of DNA is estimated to be in the order of magnitude of millions of years, at 25°C and pH 7, and that of RNA is around a hundred years (Wolfenden et al., 1998; Schroeder et al., 2006). Still, the hydrolytic processing of nucleic acids occurs in living organism in few milliseconds, thanks to the enzymes devoted to this task, as nucleases and phosphatases (Westheimer, 1987; Kamerlin et al., 2013). Most of them contain metal ions, as Mg(II), Ca(II), and Zn(II), in their active sites.
In the attempt to reproduce the activity, and possibly the proficiency of enzymes, chemists have focused their attention on the creation of artificial nucleases (Morrow et al., 2008; Aiba et al., 2011; Lassila et al., 2011; Lönnberg, 2011; Mancin et al., 2012; Diez-Castellnou et al., 2017a). Despite the significant effort invested in understanding the mechanism of the enzyme catalyzed reaction (Korhonen et al., 2013; Erxleben, 2019) and, therefore, in creating efficient artificial hydrolytic agents, the enzyme reactivity is still unrivaled and several questions remain to be addressed.
A better understanding of the roles played by the metal ion in promoting the hydrolytic cleavage of phosphate esters could be achieved using simple mono- or bi-metallic complexes as models. In particular Zn(II), while not being Nature's first choice, is likely the metal ion most widely employed in artificial systems (Mancin and Tecilla, 2007). This is due to several reasons: (i) the possibility to produce well defined and relatively stable complexes with neutral ligands; (ii) the absence of a relevant redox chemistry; (iii) a good Lewis acid acidity; (iv) a modest field effect that allow the easy reorganization of the ligand shell to match the ligand or reaction requirements.
Design and investigation of bimetallic systems is often difficult since the possible formation of μ-hydroxo bridges may affect and even cancel their reactivity (Mancin and Tecilla, 2007). Mono-metallic Zn(II) complexes, on the other hand, produce usually modest rate accelerations when compared to the corresponding bimetallic ones. Still, they allow more detailed investigations of the cleavage reaction providing a simple and better defined model (Bonfá et al., 2003).
In this perspective, we report here a detailed investigation of the reactivity of the Zn(II) complexes of 1,4,7-triazacyclononane (1) and of its methyl derivative 1,4,7-trimethyl-1,4,7-triazacyclo-nonane (2) toward the hydrolysis of the RNA model 2-hydroxypropyl-p-nitrophenylphosphate (HPNP, 3) (see Scheme 1). We found that the reactivity of 2 follows an unprecedented behavior featuring three pH dependent reactivity breaks, providing new insight on the reactivity of these systems.
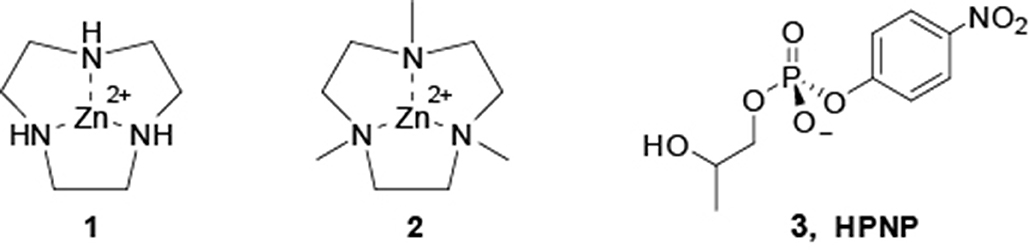
Scheme 1. Zn(II) metal complexes used in this work and RNA-model substrate, HPNP.
Results
HPNP cleavage rate in the presence of the two Zn(II) complexes was measured at 40°C. Kinetic experiments were performed in pseudo-first order conditions (i.e., in the presence of an excess of metal complex over the substrate, whose concentration was fixed at 20 μM) by monitoring the formation of 4-nitrophenolate by its UV-Vis absorption at 400 nm.
The complexes were prepared in situ by mixing a solution of Zn(NO3)2 with the corresponding ligand, in buffer. TACN and his derivatives have a high affinity for Zn(II). Indeed, the values reported in literature for the complex formation constants assure that, in the experimental condition here used, the complexes are fully formed (Yang and Zompa, 1976).
With both the complexes 1 and 2, the pseudo-firsts order rate constants (kobs) measured increase linearly with the Zn(II) complex concentration in the interval investigated (0–0.4 mM). Apparent second order rate constants (i.e., k2 = kcat/KM in the Michaelis-Menten formalism) can be obtained by linear regression fitting of the kobs vs. [complex] data.
Figure 1 reports the pH dependence of the apparent second order rate constants measured for the two complexes. As expected, the pH has a strong influence on the reaction rates suggesting, as generally observed with similar systems, that the deprotonation of metal bound-species has a relevant role in the reaction.
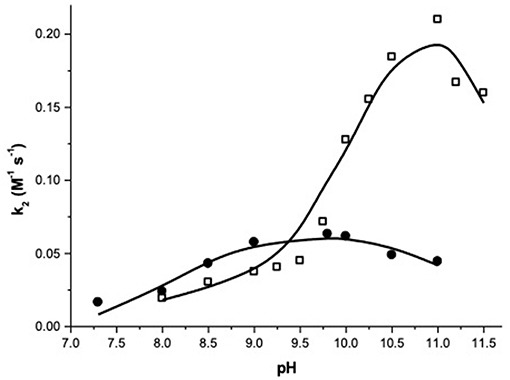
Figure 1. pH dependence of the second order rate constants (k2) for the transesterification of HPNP catalyzed by complex 1 (•) and 2 (□) at 40°C.
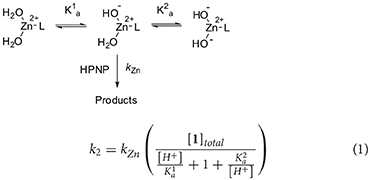
At a first sight, both the profiles show the bell-shape characteristic of metal complexes with two metal bound water molecules. Indeed, kinetic data for complex 1 were fitted with Equation (1), which accounts for the reactivity of a species with three protonation states where only the second one is active (Supplementary Material). The fit provides the pKa values of 8.13 and 11.28, respectively for the first and second deprotonation. The second order rate constant (kZn) for the reaction of the active mono-deprotonated catalyst's species with the substrate is 0.064 M−1 s−1.
The values of p and kZn obtained are in line with those reported in literature for complex 1 (Bonfá et al., 2003). The formation of the bis-deprotonated species and the consequent drop of the catalysts reactivity at higher pH have never been reported for 1, but a similar behavior is well-known for similar complexes of tridentate ligands, as 1,5,9-triazacyclododecane (Livieri et al., 2007).
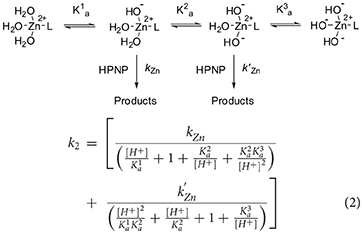
The fitting of the kinetic data for complex 2 with Equation (1) gives very poor results (see Supplementary Information). A closer inspection of the profile of complex 2 reveals that it does not follow exactly a bell-shape. Indeed, k2 smoothly increases from pH 8 to pH 9.5, with a reactivity similar to that of complex 1, suggesting that the first deprotonation is occurring in this range. When the pH reaches the value of 9.5, a stronger, reactivity increase is observed and k2 reaches values 4-fold larger than the maximum one reached by complex 1. Eventually, the reactivity starts to drop above pH 11.0. A good fit of the data was obtained by using Equation (2), which was written for a reactivity model that involves a species with four protonation states and with two of them, the second and the third, reactive (Supplementary Material). Note, one of the referees pointed out that highly correlated parameters are obtained in the case of 5 variables fitting. As discussed, reliability of the reactivity and acidity parameters obtained is however supported by the comparison with the pH reactivity profile of complex 1.
The results of the fittings are reported in Table 1. The three pKa values obtained (7.7, 10.2, and 11.6) are separated respectively by 2.5 and 1.4 pKa units, with the first pKa value quite close to the corresponding one measured for 1. Also the kZn value for the mono-deprotonated species is on the same order of magnitude to that measured for 1 (0.025 M−1 sec−1) but that of the bis-deprotonated species (k'Zn) is considerably larger (0.27 M−1 sec−1).
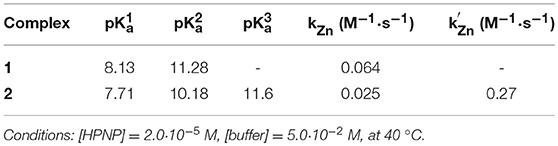
Table 1. Kinetic parameters from pH rate profiles.
Discussion
The presence of a third metal bound water molecule in complex 2 is confirmed by the crystal structure obtained by Trogler and coworkers (Silver et al., 1995) for the complex [Zn(Me3tacn)(H2O)3]2+ (2·3H2O)2+ where the Zn(II) ion adopts an octahedral environment, coordinated to three nitrogens of the macrocycle and also to three aqua ligands. In addition, Spiccia and coworkers (Fry et al., 2003) reported the crystal structure for the related complex [Zn2(Me3tacn)2(H2O)4(PhOPO3)]2+(22 ·4H2O·PhPO4)2+ where the Zn(II) ion is coordinated to three nitrogen atoms from N,N,N-trimethyl-1,4,7,-triazacyclononane, two water molecules and one oxygen from phenyl phosphate. These structures support our hypothesis that in complex 2, formed by Zn(II) with the ligand 1,4,7-trimethyl-1,4,7-triazacyclononane, the coordination number of the metal ion is 6, as the accessible surface of the metal ion is large enough to allow coordination of two water molecules and of the substrate simultaneously. Unfortunately, no crystal structures are available for complex 1 to confirm its preference for the coordination number 5 at the metal ion, as suggested by kinetic experiments. The flexibility of the Zn(II) ion, which does not have a defined coordination geometry, is well-known (Sigel and Bruce Martin, 1994).
Potentiometric titration data available are also quite scarce. In the case of complex 1, precipitation above pH 8 prevented the determination of the acidity of the metal bound water molecules. Kinetic experiments, as already mentioned, confirm a pKa value around 8 for the first water molecule (Bonfá et al., 2003). In the case of complex 2, Trogler and coworkers reported the values of 10.9 and 12.3 (Silver et al., 1995). Such values appear surprisingly high when compared with complex 1 and with other Zn(II) complexes of macrocyclic polyamides (Kimura et al., 1990; Koike et al., 1990, 1991, 1992; Kimura and Koike, 1991). Indeed, one would expect that electron-donating methyl group will lower the acidity of the substituted metallic complex with respect to the unsubstituted one (Canary et al., 1995). Interestingly, the similarity of these values with those here reported for the second and third pKa is clear (Fry et al., 2005).
Focusing the attention on the hydrolytic reactivity of the two complexes, it is relevant to note that the pH profiles obtained confirm the coordination of the substrate to the complex. No other possible mechanism, as general base catalysis or nucleophile catalysis, would account for the reactivity decrease observed at high pH values. There are three kinetically equivalent mechanisms that have been proposed to explain the pH dependence observed for the metal catalyzed HPNP transesterification (Korhonen et al., 2013; Diez-Castellnou et al., 2017a). In the first, the reaction involves the deprotonation of the substrate's hydroxyl group by a metal bound hydroxide, which acts as a general base, simultaneously to the nucleophilic attack on the phosphorus atom (Scheme 2A). In the second, the substrate alcoholic group is bound to the metal ion and deprotonates before the nucleophilic attack (Scheme 2B). This mechanism is indistinguishable from the others because the pKa values of metal bound water molecules and alcoholic hydroxyls are similar (Kimura et al., 1995; Livieri et al., 2007). In the third mechanism (Scheme 2C) the substrate deprotonation occurs before metal coordination and the deprotonation of a metal bound water molecules decreases (or hamper) the interaction of the metal with the substrate.
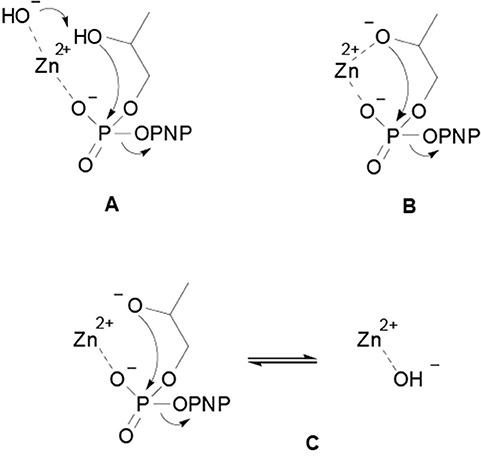
Scheme 2. Proposed mechanisms for the metal catalyzed HPNP transesterification reaction: (A) the substrate deprotonation by a metal bound hydroxide occurs simultaneously to the nucleophilic attack on the phosphorus atom; (B) the substrate alcoholic group is bound to the metal ion and deprotonates before the nucleophilic attack; (C) the substrate deprotonation occurs before metal coordination and the deprotonation of a metal bound water molecule decreases the interaction of metal with the substrate.
Several experimental evidences strongly support the last mechanism (2C) in the case of catalysts that have coordination sites available on the metal ion to accommodate only the substrate (Morrow et al., 2008; Korhonen et al., 2013; Diez-Castellnou et al., 2017a). On the other hand, it is quite likely that when the catalyst have more coordination sites, the substrate alkoxide will bind to the metal ion, turning the reaction mechanism into path 2B (Livieri et al., 2007). In this mechanism, the metal ion plays two opposite roles: (i) it increases the reactivity of the substrate toward the nucleophilic attack, acting as a Lewis acid, (ii) it decreases the reactivity of the nucleophile, by decreasing its pKa.
A few years ago, we calculated the kZn values for the HPNP cleavage promoted by several mononuclear Zn(II) complexes of polyamine ligands (Figure 2) (Bonfá et al., 2003; Bonomi et al., 2013). In the case of triamine complexes, we found a positive correlation of the reactivity with the pKa of the first deprotonation of a metal bound species. Such a behavior suggested that, among the two concomitant and counterbalancing effects active, the loss of reactivity of the nucleophile prevails over the Lewis acid activation of the substrate.
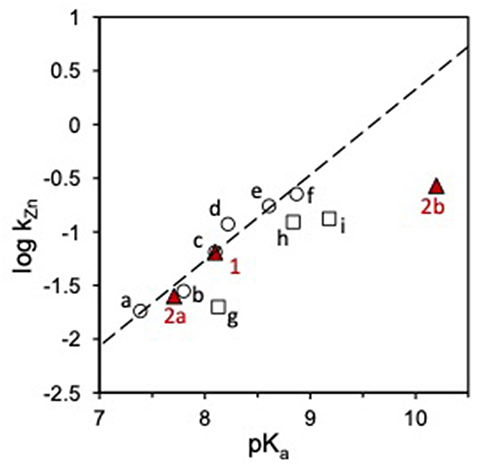
Figure 2. Plot of log kZn vs. pKa for the transesterification of HPNP catalyzed by Zn(II) complexes in water. Red triangles represent the reactivity of complexes 1 and 2, circles (○) are cyclic triamine ligands (a-f), squares (□) linear triamine ligands (g-i), see ref Bonfá et al. (2003) and Bonomi et al. (2013). The dashed line shows the linear fit of the reactivity data for the complexes of cyclic triamines (slope = 0.80). The reactivity of 1 and 2 was corrected to account for the temperature difference by matching the reactivity of 1 with the reported data (ref. Bonfá et al., 2003).
When the reactivity of complexes 1 and 2 is analyzed in this perspective, interesting insight is provided. The reactivity of complexes 1 and 2a (Scheme 3) is in line with that of other triamine complexes (Figure 2). On the other hand, the reactivity of mono-deprotonated complex 2b is about 10 times smaller than that expected for a complex of a triamine ligand with that pKa. This suggest that a further effect is at play in this case besides substrate Lewis acid activation and nucleophile reactivity modulation. Significant modifications of the coordination geometries in the two complexes 2a and 2b, which could justify their different reactivity, are unlikely as confirmed by DFT optimization of the complex geometries (see Supplementary Material). The other relevant differences between the reactive protonation states of complex 2 are essentially two. First, in complex 2a there is a metal bound water molecule, which could provide intramolecular H-bonds or acid catalysis. Since this water molecule is turned into an hydroxide in 2b, its positive effect on reactivity would be canceled. If this was the case (or if a general base catalysis mechanism was active), however, reactivity of 2a would be greater than that of 1 and of the other triamine complexes. The second difference is in the total charge of the reactive species, which is 0 in the ternary complex of 2a with HPNP and−1 in the case of 2b. Upon the nucleophilic attack of the substrate alkoxide, additional negative charge builds up in the phosphoryl group and this is electrostatically disfavored by the overall complex charge, resulting into a lower reactivity. Indeed, we earlier reported as the insertion of an additional positive charge in the ligand structure, located in close proximity to the reaction site, can increase the reactivity of a mononuclear Zn(II) complex up to one order of magnitude. This is in good agreement with the 10-fold rate decrease estimated for complex 2b (Bonomi et al., 2009).
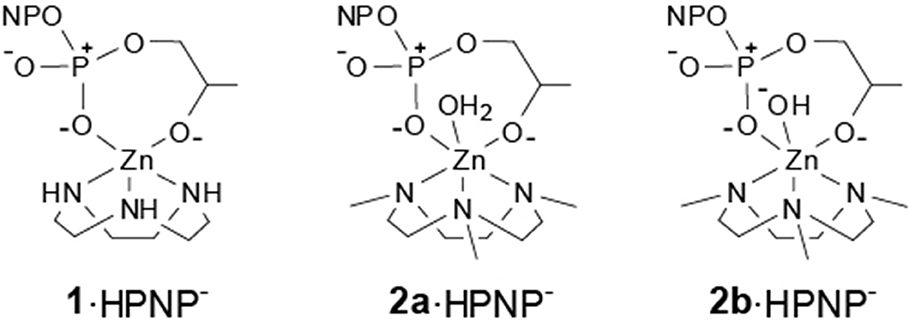
Scheme 3. Proposed structure of the pre-reactive complexes of 1 and 2 with HPNP.
Conclusions
In conclusion, in this paper we have demonstrated as even small modifications in the structure of the ligands, as the insertion of methyl groups in the nitrogen atoms of triazacyclononane, may have a relevant effect on the ability of the corresponding Zn(II) complex in promoting the phosphoryl transfer reaction. The complex 2 is, at the best of our knowledge, the only monometallic Zn(II) catalyst of HPNP cleavage that is active in two different protonation states. Such a reactivity is due to the peculiar ability of 1,4,7-trimethyl-1,4,7-triazacyclononane to increase the number of coordination sites to 6 on the metal ion with respect of other similar ligands. The most relevant result of this study is however the mechanistic information obtained. The reactivity of complex 2 further supports mechanism 2B as the most likely in the case of complexes where enough solvent-occupied coordination sites on the metal are available. It also confirms that hydrolytic reactivity of these systems is the result of a delicate counterbalance between Lewis acid and electrostatic activation of the substrate (and stabilization of the transition state) and the decrease of the nucleophile reactivity. Several evidences obtained with model systems, and confirmed by our results, suggest that optimum reactivity should be obtained when the nucleophile is not bound to the metal ion and the overall complex charge is as great as possible (Tirel et al., 2014; Tirel and Williams, 2015). This explains also the greater reactivity usually observed with trivalent cations, as well as the detrimental effect of negatively charged ligands (Mancin et al., 2012). Metal ion coordination of the nucleophile may however be necessary to increase the fraction of nucleophile available at physiological pH and to provide a better preorganization of the latter for the attack to the phosphorous atom. The need to consider of all these factors explains the difficulties in reproducing enzymes' proficiency with simple models. Indeed only a precise spatial organization of multiple active species can optimize the positive effects while minimizing the detrimental one. It is hence not surprising the fact that only by using sophisticated supramolecular architectures, where several beneficial factors can be implemented, remarkable reactivities were obtained (Manea et al., 2004; Feng et al., 2006; Bonomi et al., 2008; Gruber et al., 2011; Diez-Castellnou et al., 2014).
Equations
Data Availability
All datasets generated for this study are included in the manuscript and/or the Supplementary Files.
Author Contributions
MD-C contributed conception and design of the present work, collected and analyzed the kinetic data, and wrote the manuscript. GS performed the DFT calculations. FM and PS supervised this project. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
Funded by the EU-FP7 Marie Curie-ITN Project PhosChemRec (MD-C and PS Grant 238679), by the ERC Starting Grants Project MOSAIC (GS and FM Grant 259014) and by H2020 MSCA-ITN MMBio (PS and FM Grant 721613).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
MD-C thanks the PhosChemRec for a fellowship.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00469/full#supplementary-material
DFT calculations and additional kinetic experiments.
Abbreviations
HPNP, 2-hydroxypropyl-p-nitrophenylphosphate.
References
Aiba, Y., Sumaoka, J., and Komiyama, M. (2011). Artificial DNA cutters for DNA manipulation and genome engineering. Chem. Soc. Rev. 40, 5657–5668. doi: 10.1039/c1cs15039a
Bonfá, L., Gatos, M., Mancin, F., Tecilla, P., and Tonellato, U. (2003). The ligand effect on the hydrolytic reactivity of Zn(II) complexes toward phosphate diesters. Inorgan. Chem. 42, 3943–3949. doi: 10.1021/ic034139x
Bonomi, R., Saielli, G., Scrimin, P., and Mancin, F. (2013). An experimental and theoretical study of the mechanism of cleavage of an RNA-model phosphate diester by mononuclear Zn(II) complexes. Supramol. Chem. Taylor Francis. 25, 665–671. doi: 10.1080/10610278.2013.830724
Bonomi, R., Saielli, G., Tonellato, U., Scrimin, P., and Mancin, F. (2009). Insights on nuclease mechanism: the role of proximal ammonium group on phosphate esters cleavage. J. Am. Chem. Soc. 131, 11278–11279. doi: 10.1021/ja9033236
Bonomi, R., Selvestrel, F., Lombardo, V., Sissi, C., Polizzi, S., Mancin, F., et al. (2008). Phosphate diester and DNA hydrolysis by a multivalent, nanoparticle-based catalyst. J. Am. Chem. Soc. 130, 15744–15745. doi: 10.1021/ja801794t
Canary, J. W., Xu, J., Castagnetto, J. M., Rentzeperis, D, and Marky, L. A. (1995). Enthalpic control of zinc—water acidity in complexes of Tris(2-Aminoethyl)Amine and Tris(2-(Dimethylamino)Ethyl)Amine. J. Am. Chem. Soc. 117, 11545–11547. doi: 10.1021/ja00151a019
Diez-Castellnou, M., Mancin, F., and Scrimin, P. (2014). Efficient phosphodiester cleaving nanozymes resulting from multivalency and local medium polarity control. J. Am. Chem. Soc. 136, 1158–1161. doi: 10.1021/ja411969e
Diez-Castellnou, M., Martinez, A., and Mancin, F. (2017a). Phosphate ester hydrolysis: the path from mechanistic investigation to the realization of artificial enzymes. Adv. Phys. Org. Chem. 51. 129–186. doi: 10.1016/bs.apoc.2017.09.003
Erxleben, A. (2019). Mechanistic studies of homo- and heterodinuclear zinc phosphoesterase mimics: what has been learned? Front. Chem. 7, 1–22. doi: 10.3389/fchem.2019.00082
Feng, G., Natale, D., Prabaharan, R., Mareque-Rivas, J. C., and Williams, N. H. (2006). Efficient phosphodiester binding and cleavage by a ZnII complex combining hydrogen-bonding interactions and double lewis acid activation. Angew. Chem. Int. Ed. 45, 7056–7059. doi: 10.1002/anie.200602532
Fry, F. H., Fischmann, A. J., Belousoff, M. J., Spiccia, L., and Brügger, J. (2005). Kinetics and mechanism of hydrolysis of a model phosphate diester by [Cu(Me3tacn)(OH2)2]2+(Me3tacn = 1,4,7-Trimethyl-1,4,7-Triazacyclononane). Inorg. Chem. 44, 941–950. doi: 10.1021/ic049469b
Fry, F. H., Jensen, P., Kepert, C. M., and Spiccia, L. (2003). Macrocyclic copper(II) and zinc(II) complexes incorporating phosphate esters. Inorg. Chem. 42, 5637–5644. doi: 10.1021/ic026091d
Gruber, B., Kataev, E., Aschenbrenner, J., Stadlbauer, S., and König, B. (2011). Vesicles and micelles from amphiphilic zinc(II)-cyclen complexes as highly potent promoters of hydrolytic DNA cleavage. J. Am. Chem. Soc. 133, 20704–20707. doi: 10.1021/ja209247w
Kamerlin, S. C., Sharma, P. K., Prasad, R. B., and Warshel, A. (2013). Why nature really chose phosphate. Quart. Rev. Biophys. 46, 1–132. doi: 10.1017/S0033583512000157
Kimura, E., Kimura, E., Shiota, T., Koike, T., Shiro, M., and Kodama, M. (1990). A Zinc(II) Complex of 1,5,9-triazacyclododecane ([12]AneN3) as a model for carbonic anhydrase. J. Am. Chem. Soc. 112, 5805–5811. doi: 10.1021/ja00171a020
Kimura, E., Kodama, Y., Koike, T., and Shiro, M. (1995). Phosphodiester hydrolysis by a new zinc(II) macrocyclic tetraamine complex with an alcohol pendant: elucidation of the roles of Ser-102 and Zinc(II) in alkaline phosphatase. J. Am. Chem. Soc. 117, 8304–8311. doi: 10.1021/ja00137a002
Kimura, E., and Koike, T. (1991). Macrocyclic polyamines as a probe for equilibrium study of the acid functions of zinc(II) ion in hydrolysis enzymes. Inorg. Chem. 11, 285–301. doi: 10.1080/02603599108035829
Koike, T., Kimura, E., and Kimura, E. (1991). Roles of zinc(II) ion in phosphatases. A model study with zinc(II)-macrocyclic polyamine complexes. J. Am. Chem. Soc. 113, 8935–8941. doi: 10.1021/ja00023a048.
Koike, T., Kimura, E., Nakamura, I., Hashimoto, Y., and Shiro, M. (1992). The first anionic sulfonamide-binding zinc(II) complexes with a macrocyclic triamine: chemical verification of the sulfonamide inhibition of carbonic anhydrase. J. Am. Chem. Soc. 114, 7338–7345. doi: 10.1021/ja00045a002.
Koike, T., Shiota, T., Kimura, E., and Iitaka, Y. (1990). Acid properties of zinc(II) and cadmium(II) in complexation with macrocyclic oxo polyamine ligands. Inorg. Chem. 29, 4621–4629. doi: 10.1021/ic00348a009
Korhonen, H., Koivusalo, T., Toivola, S., and Mikkola, S. (2013). There is no universal mechanism for the cleavage of RNA model compounds in the presence of metal ion catalysts. Org. Biomol. Chem. 11, 8324–8339. doi: 10.1039/c3ob41554f
Lassila, J. K., Zalatan, J. G., and Herschlag, D. (2011). Biological phosphoryl-transfer reactions: understanding mechanism and catalysis. Annu. Rev. Biochem. 80, 669–702. doi: 10.1146/annurev-biochem-060409-092741
Livieri, M., Mancin, F., Saielli, G., Chin, J., and Tonellato, U. (2007). Mimicking enzymes: cooperation between organic functional groups and metal ions in the cleavage of phosphate diesters. Chem. A Eur. J. 13, 2246–2256. doi: 10.1002/chem.200600672
Lönnberg, H. (2011). Cleavage of RNA phosphodiester bonds by small molecular entities: a mechanistic insight. Org. Biomol. Chem. 9, 1687–1703. doi: 10.1039/c0ob00486c
Mancin, F., Scrimin, P., and Tecilla, P. (2012). Progress in artificial metallonucleases. Chem. Commun. 48, 5545–5559. doi: 10.1039/c2cc30952a
Mancin, F., and Tecilla, P. (2007). Zinc(II) complexes as hydrolytic catalysts of phosphate diester cleavage: from model substrates to nucleic acids. New J. Chem. 31, 800. doi: 10.1039/b703556j
Manea, F., Houillon, F. B., Pasquato, L., and Scrimin, P. (2004). Nanozymes: gold-nanoparticle-based transphosphorylation catalysts. Angew. Chem. Int. Ed. 43, 6165–6169. doi: 10.1002/anie.200460649
Morrow, J. R., Amyes, T. L., and Richard, J. P. (2008). Phosphate binding energy and catalysis by small and large molecules. Acc. Chem. Res. 41, 539–548. doi: 10.1021/ar7002013
Schroeder, G. K., Lad, C., Wyman, P., Williams, N. H., and Wolfenden, R. (2006). The time required for water attack at the phosphorus atom of simple phosphodiesters and of DNA. Proc. Nat. Acad. Sci. U.S.A.103, 4052–4055. doi: 10.1073/pnas.0510879103
Sigel, H., and Bruce Martin, R. (1994). The colourless ‘chameleon’ or the peculiar properties of Zn2+ in complexes in solution. Quantification of equilibria involving a change of the coordination number of the metal ion. Chem. Soc. Rev. 23, 83–91. doi: 10.1039/CS9942300083
Silver, G. C., Gantzel, P., and Trogler, W. C. (1995). Characterization of (Trimethyltriazacyclononane)triaquozinc(II) nitrate. A Nonbridged Isomer. Inorg. Chem. 34, 2487–2489. doi: 10.1021/ic00113a038
Tirel, E. Y., Bellamy, Z., Adams, H., Duarte, F., and Williams, N. H. (2014). Catalytic zinc complexes for phosphate diester hydrolysis. Angew. Chem. Int. Ed. 53, 8246–50. doi: 10.1002/anie.201400335
Tirel, E. Y., and Williams, N. H. (2015). Enhancing phosphate diester cleavage by a zinc complex through controlling nucleophile coordination. Chem. Eur. J. 21, 7053–7056. doi: 10.1002/chem.201500619
Wolfenden, R., Ridgway, C., and Young, G. Chapel Hill North Carolina. (1998). Spontaneous hydrolysis of ionized phosphate monoesters and diesters and the proficiencies of phosphatases and phosphodiesterases as catalysts. J. Am. Chem. Soc. 120, 833–834.
Keywords: RNA, monometallic Zn(II)-complexes, azacrown, phosphate cleavage, phosphoesterase models, kinetics
Citation: Diez-Castellnou M, Salassa G, Mancin F and Scrimin P (2019) The Zn(II)-1,4,7-Trimethyl-1,4,7-Triazacyclononane Complex: A Monometallic Catalyst Active in Two Protonation States. Front. Chem. 7:469. doi: 10.3389/fchem.2019.00469
Received: 07 April 2019; Accepted: 19 June 2019;
Published: 03 July 2019.
Edited by:
Soumyajit Roy, Indian Institute of Science Education and Research Kolkata, IndiaReviewed by:
Riccardo Salvio, Sapienza University of Rome, ItalyMuhammad Hanif, The University of Auckland, New Zealand
Copyright © 2019 Diez-Castellnou, Salassa, Mancin and Scrimin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marta Diez-Castellnou, bWRjN0BzdC1hbmRyZXdzLmFjLnVr