 Mohsen Ahmadi
Mohsen Ahmadi Christian Fischer1
Christian Fischer1 Carola Schulzke
Carola Schulzke- 1Institut für Biochemie, Universität Greifswald, Greifswald, Germany
- 2Departement de Chimie Moléculaire, Université Grenoble Alpes, UMR CNRS 5250, Grenoble, France
A MoIV mono-oxido bis-dithiolene complex, [MoO(mohdt)2]2− (mohdt = 1-methoxy-1-oxo-4-hydroxy-but-2-ene-2,3-bis-thiolate) was synthesized as a structural and functional model for molybdenum oxidoreductase enzymes of the DMSO reductase family. It was comprehensively characterized by inter alia various spectroscopic methods and employed as an oxygen atom transfer (OAT) catalyst. The ligand precursor of mohdt was readily prepared by a three-step synthesis starting from dimethyl-but-2-ynedioate. Crystallographic and 13C-NMR data support the rationale that by asymmetric substitution the electronic structure of the ene-dithio moiety can be fine-tuned. The MoIVO bis-dithiolene complex was obtained by in situ reaction of the de-protected ligand with the metal precursor complex trans-[MoO2(CN)4]4−. The catalytic oxygen atom transfer mediated by the complex was investigated by the model OAT reaction from DMSO to triphenylphosphine with the substrate transformation being monitored by 31P NMR spectroscopy. [MoO(mohdt)2]2− was found to be catalytically active reaching 93% conversion, albeit with a rather low reaction rate (reaction time 56 h). The observed overall catalytic activity is comparable to those of related complexes with aromatic dithiolene ligands despite the novel ligand being aliphatic in nature and originally perceived to perform more swiftly. The respective results are rationalized with respect to a potential intermolecular interaction between the hydroxyl and ester functions together with the electron-withdrawing functional groups of the dithiolene ligands of the molybdenum mono-oxido complex and equilibrium between the active monomeric MoIVO and MoVIO2 and the unreactive dimeric MO3 species.
Introduction
Molybdenum dependent enzymes are essential contributors to the life of nearly every known organism on earth being it an ancient archaeon, a plant or a mammal which includes the modern human being (Mendel, 2007; Edwards et al., 2015). To date, four such molybdenum dependent enzymes have been discovered to be part of the human organism, which are sulfite oxidase (SO), xanthine dehydrogenase (XDH), aldehyde oxidase (AO), and mARC (Garner and Bristow, 1985; Hille, 2013; Hille et al., 2014; Schulzke and Ghosh, 2014). Defects in the maturation of the molybdenum cofactors (Figure 1), which can occur at different stages of the respective multistep biosynthesis, cause diseases (e.g., isolated sulfite oxidase deficiency: iSOD) due to the non-functioning of the molybdenum enzymes. This has consequences such as brain damage, motor retardation, convulsions etc. beginning right after birth and typically leading to infancy or early childhood death (Reiss, 2000). The extreme instability of the molybdenum cofactor prevents it from being biotechnologically produced and applied as treatment. Understanding exactly what makes Moco unstable and what makes it catalytically active is therefore of great interest for those aiming at developing an artificial cofactor which might be used as a respective drug in the future. This constitutes the motivation for our group and specifically for the study discussed in the following as one of many approaches. A moiety including molybdenum and one or two dithiolene ligands (representing molybdopterin—MPT; see Figure 1) is one of the most common motives in molybdenum cofactor bio-inorganic chemistry (Rajagopalan, 1997; Schulzke and Samuel, 2011).
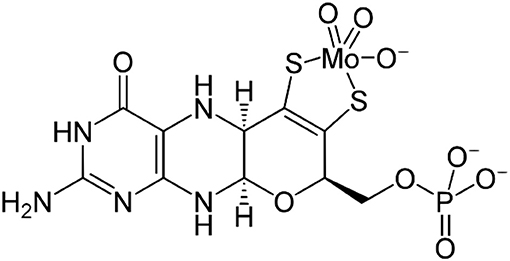
Figure 1. The chemical structure of the “free” biosynthesized molybdenum cofactor (Moco) prior to insertion into the apoproteins including the tricyclic molybdopterin ligand (MPT). Note: for XO and AO one oxido ligand will be replaced by a sulfido ligand before the active site is formed; in SO a cysteine binds molybdenum in the active site pocket.
During the last 20 years, various bis-dithiolene mono-oxido molybdenum complexes have been developed and investigated (Donahue et al., 1998; Lim and Holm, 2001; Enemark et al., 2004; Döring et al., 2010; Schulzke, 2016; Ghosh et al., 2017). Such model compounds have helped understanding the roles of the dithiolene type ligands in the active sites of the DMSOR family enzymes, e.g., how they affect the electron and atom transfer reactivity during catalysis. Still, a comprehensive understanding of the roles of the different substituents is yet to be accomplished. During the catalytic reactions of these enzymes, molybdenum cycles between the oxidation states MoIV (d2) and MoVI (d0) constituting the fully reduced and fully oxidized active species. The oxidation state MoV (d1) is part of the regeneration of the active site by two proton coupled electron transfer steps (PCET). Dithiolenes are non-innocent ligands which can affect the electronic structure of their molybdenum (and tungsten) complexes by providing the central metal with electron density shifted from a sulfur p-orbital bearing a lone pair to an empty metal d-orbital by respective orbital overlap or even by full ligand to metal charge transfer (LMCT) (Kirk et al., 2004; Sugimoto et al., 2009). Although the role of molybdenum in the DMSOR enzymes for the catalysis of the oxygen atom transfer reactions (OAT) is quite well-understood, the role of the molybdopterin ligand (MPT) remains to be comprehensively deciphered. The synthesis of MPT or any artificial close relative of it represents a major chemical challenge and the respective attempts are still ongoing in a small number of research groups, although some significant advances have already been reported (Bradshaw et al., 1998, 2001a; Sugimoto et al., 2005; Williams et al., 2012, 2015; Basu and Burgmayer, 2015; Gisewhite et al., 2018). Holm and coworkers have not only developed OAT model reactions relevant for the molybdenum enzymes' interconversion but have also extensively reviewed them already in the 1980's (Berg and Holm, 1985; Holm, 1987). In many model reactivity studies dimethyl sulfoxide (DMSO) was employed for the oxidation of MoIVO complexes, which is a natural substrate of DMSO reductase, and organic phosphines (PR3, as easy to handle non-natural co-substrates) were used for the reduction of MoVIO2, Scheme 1.
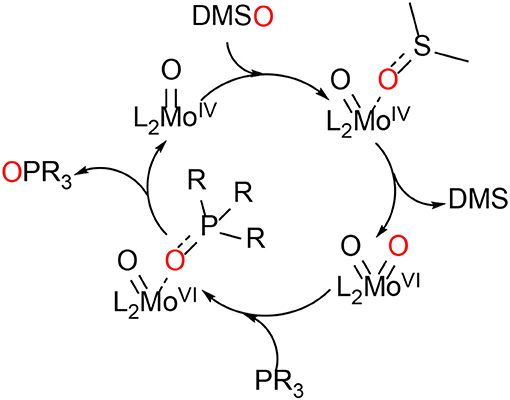
Scheme 1. The model OAT reaction between DMSO and a phosphine catalyzed by a molybdenum bis-dithiolene complex (L = ene-dithiolate ligand; R = alkyl or aryl).
Both, [MoIVO(dt)2] as well as [MoVIO2(dt)2] complexes (dt = dithiolene ligand) employing distinct dithiolenes were reported by us before and shown to be active catalysts for the OAT reaction with varied capabilities (Ghosh et al., 2017). What became apparent from many studies from others as well as our own, was the detrimental influence of aromatic dithiolenes on the catalytic performance, in particular of those in which the ene of the dithiolene is actually part of the aromatic moiety, e.g., in benzenedithiolate (Fischer and Fischer, 2017). Aliphatic dithiolene ligands, in contrast, have proven to be much more instable species and consequently also much better catalysts due to the higher activity. Introduced here are now a new aliphatic dithiolene ligand and its MoIVO bis-dithiolene complex. Both were characterized comprehensively as were all ligand precursors. The IR and UV-vis spectroscopic data of the complex were compared to known data of related compounds and the complex' ability to catalyze OAT reactions was investigated. The observed surprisingly poor performance is discussed referring to (i) the presence of specific substituents (ester and alcohol groups), (ii) crystallographic and spectroscopic data revealing inter alia information about bond lengths and strengths, (iii) substrate formation monitoring, and (iv) probable intermolecular interactions.
Experimental
Synthetic Procedures
All reactions and manipulations were carried out using standard Schlenk and glove box techniques under an atmosphere of high purity nitrogen (Schlenk) or argon (glove box). All solvents were dried, distilled and either degassed or purged with dinitrogen or argon prior to use. Ethylene trithiocarbonate (Kim et al., 2008) and the molybdenum precursor K3Na[MoO2(CN)4]·6H2O (Smit et al., 1993) were synthesized according to previously reported procedures.
Dimethyl 2-Thio-1,3-Dithiole-4,5-Dicarboxylate (1)
In a modification of a literature procedure (Easton and Leaver, 1965) dimethyl but-2-ynedioate (18.3 mmol, 2.25 mL) and ethylene trithiocarbonate (18.3 mmol, 2.52 g) were heated to reflux for 10 h under N2 in anhydrous toluene. The solution was left to cool to r.t. and filtered. The remaining solution was kept at −20°C and adding n-hexane to the solution led to precipitation of yellowish crystalline compound 1. Yield: 3.8 g, 85%. 1H NMR (CDCl3, 300 MHz): δ (ppm): 3.90 (s, 6H, CH3). 13C NMR (CDCl3, 75 MHz): δ (ppm): 207.2 (C=S), 157.9 (C=O), 138.1 (C=C), 53.85 (CH3). FT–IR bands (KBr pallet, cm−1): 3446 (br), 2954 (s), 2918 (w), 1745 (s), 1720 (s), 1552 (s), 1257 (br), 1101 (s), 1087 (m), 1060 (s), 1008(s), 993(s), 921(s), 837 (w), 777(w), 761(w), 744(w), 698(w), 511 (m). APCI-MS (EI): m/z calculated for C7H6O4S3: 249.94; Found: 250.71 [M+H+]. Elemental analysis for C7H6O4S3: calc. (%): C, 33.59; H, 2.42; S, 38.43. Found: C, 34.65; H, 2.46; S, 37.38.
Methyl 5-(Hydroxymethyl)-2-Thioxo-1,3-Dithio-4-Carboxylate (2)
To a well-stirred solution of 1 (3.62 g, 14.5 mmol) and dry LiCl (1.22 g, 29 mmol) in anhydrous THF (40 mL) and EtOH (15 mL) at −15 to −10°C powdered sodium borohydride (NaBH4, 1.15 g, 30.5 mmol) was slowly added in small portions over a duration of 20 min. An exothermic reaction took place and the temperature was kept under −10°C at all times and for further 30 min. Then H2O (150 mL, 0°C) was added followed by concentrated aqueous HCl (4 N, carefully and portion-wise) until the evolution of H2 gas ceased. The mixture was extracted with EtOAc (3 × 100 mL), and the extract was dried over Na2SO4. Evaporation of the solvent gave a yellow oily residue which was re-dissolved in CH2Cl2/EtOAc (2:1, 25 mL) and purified by column chromatography. The first yellow fraction contained trace amounts of the starting material and the second fraction contained the mono-alcohol. The second fraction was concentrated in vacuo to give brownish-yellow crystalline compound 2 (see Scheme 2). Yield: 1.6 g, 54%. 1H NMR (CDCl3, 300 MHz): δ (ppm): 4,94 (s, 2H, CH2), 3.88 (s, 3H, CH3). 13C NMR (CDCl3, 75 MHz): δ (ppm): 210.7 (C=S), 163.6 (C=O), 158.6 (CO–C=C), 124.82 (CH2–C=C), 60.5 (CH2), 52.9 (CH3). FT–IR bands (KBr pallet, cm−1): 3446 (br), 3012 (w), 2951 (s), 2924 (w), 2017 (br), 1994 (br), 1745 (s), 1718 (m), 1627 (m), 1618 (m), 1550 (m), 1435 (s), 1261 (br), 1070 (s), 758 (s), 599 (w), 514 (w), 460 (m). APCI-MS (EI): m/z calculated for C6H6O3S3: 221.95; Found: 222.8 [M+H+]. Elemental analysis for C8H10O4S3 (1/2 × EtOAc as co-crystallized lattice solvent) calc. (%): C, 36.07; H, 3.71; S, 36.11. Found: C, 36.31; H, 3.32; S, 36.16. The side product (4,5-bis(hydroxymethyl)-1,3-ene-dithio-2-thione (3, di-alcohol) was collected from the third fraction by column chromatography as yellow needle shaped microcrystalline solid (see Scheme 2). 1H NMR (CD3OD, 300 MHz): δ (ppm): 4.52 (s, 2H, CH2). 13C NMR (CD3OD, 75 MHz): δ (ppm): 214.4 (C=S), 143.5 (C=C), 57.8 (CH2). FT–IR bands (KBr pallet, cm−1): 3421 (br), 2953 (s), 1982 (br), 1718 (br), 1436 (m), 1361 (w), 1327 (w), 1247 (m), 1201 (m), 1180 (m), 1074 (m), 1053 (s), 1035 (m), 991 (s), 635 (m), 518 (m). APCI-MS (EI): m/z calculated for C5H6O2S3: 193.95; Found: 194.80 [M+H+].
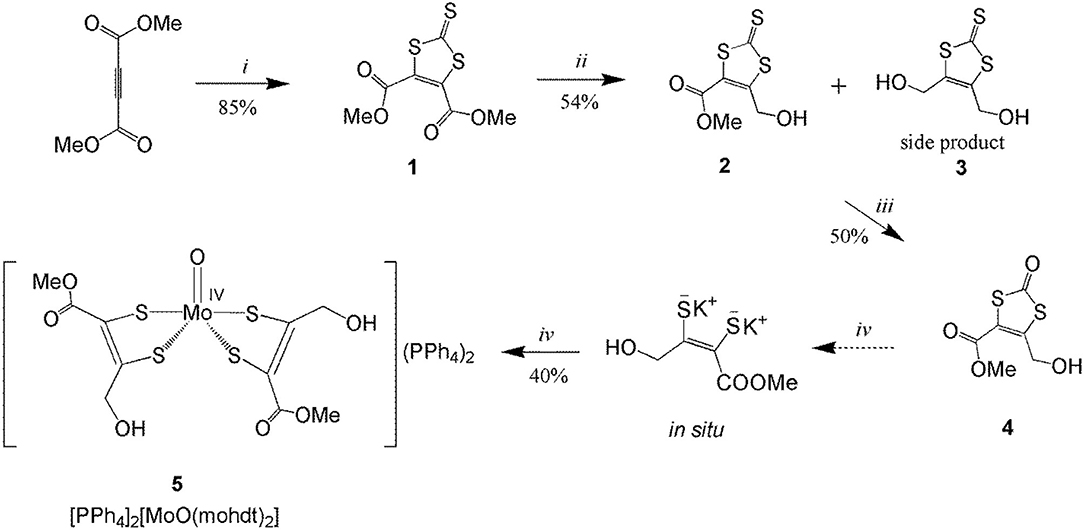
Scheme 2. Synthesis of compounds 1–4 and molybdenum complex 5. (i) ethylene trithiocarbonate, toluene, reflux, 10 h; (ii) 2 eq. NaBH4, dry LiCl, (anhydrous THF:EtOH), −15°C to 0°C, 1 h; (iii) Hg(OAc)2, AcOH/CHCl3, 6 h; (iv) KOH, degassed MeOH, [MoO2(CN)4]K3Na, degassed H2O, Ph4PCl salt, under N2, 50°C, 3 h.
4-Methyl-Carboxylate-5-Hydroxymethyl-1,3-ene-Dithio-2-One (4, mohdtC=O)
Four equivalents of mercury acetate, Hg(OAc)2 (7 g, 21.8 mmol) were added to a stirred solution of 2 (1 g, 5 mmol) in 100 mL AcOH/CHCl3 (2:1) for 6 h. The reaction was followed by TLC (silica, DCM). The resulting pale green mixture was filtered through a Celite pad to remove the mercury salts (mainly HgS). The resulting solution was washed first with water and then with aqueous NaHCO3 and dried over Na2SO4. The final light yellowish powder was collected after short silica column chromatography (DCM/EtOAc; 3:1). Yield: 0.5 g, 50%. 1H NMR (CDCl3, 300 MHz): δ (ppm): 4.93 (s, 2H, CH2), 3.86 (s, 3H, CH3). 13C NMR (CDCl3, 75 MHz): δ (ppm) = 188.7 (C=Ooxo), 160.2 (C=OCOOMe), 151.6 (CO–C=C), 117.6 (CH2–C=C), 60.1 (CH2), 53.1 (CH3). FT–IR bands (KBr pallet, cm−1): 3483 (br), 2956 (w), 1701(s), 1654 (s), 1618 (m), 1544 (m), 1435 (s), 1352 (m), 1286 (br), 1220 (m), 1068 (m), 1029 (w), 970 (w), 952 (w), 894 (w), 813 (w), 759 (w), 607 (w), 470 (w). APCI-MS (EI): m/z calculated for C6H6O4S2: 205.97; Found: 206.80 [M+H+]. Elemental analysis for C6H6O4S2: calc. (%): C, 34.94; H, 2.93; S, 31.09. Found: C, 35.01; H, 2.95; S, 30.52. Electronic absorption spectral data in CH3CN (λmax, nm (ε/M−1 cm−1)): 211 (2682), 285 (br, 2340).
[Ph4P]2[MoO(mohdt)2] (5)
The ligand precursor 4 (0.12 g, 0.6 mmol) was added to a Schlenk flask containing 16 mL of 0.1 M KOH solution in anhydrous methanol under N2 atmosphere and stirred for 2 h. The solution turned light yellow and to this a blue solution of K3Na[MoO2(CN)4]·6H2O (0.15 g, 0.3 mmol) dissolved in 8 mL degassed water was added by cannula under N2. The reaction mixture was stirred at 50°C for 3 h. Then 0.21 g of tetraphenylphosphine chloride, Ph4PCl dissolved in 8 mL degassed water was added to the reaction mixture. The final red solution was concentrated in vacuum to dryness. It was then dissolved in 40 ml of CH3CN and the residue was filtered off. The organic solution was transferred to another Schlenk flask and anhydrous diethyl ether was added slowly. The brownish-red precipitate was collected and dried under reduced pressure. Yield: 0.3 g, 40%. 1H NMR (CD3CN, 300 MHz): δ (ppm): 7.80-8.93 (m, 4H, Ph4P+), 7.51-7.75 (m, 16H, Ph4P+), 4.57 (s, 4H, CH2), 3.66 (s, 3H, CH3). 13C NMR (CD3CN, 75 MHz): δ (ppm): 165.2 (CO), 152.8 (CO–C=C), 136.35, 135.7, 135.5, 131.3, 131.2, 119.4 (CH2–C=C), 63.7 (CH2), 54.7 (CH3). FT–IR bands (KBr pallet, cm−1): 3431 (br), 3055 (w), 3022 (w), 2924 (s), 1718 (br), 1585 (m), 1541(s), 1483 (s), 1436 (s), 1330 (br), 1228 (s), 1188 (w), 1165, 1109 (s), 1026 (w), 997 (s), 977 (s), 925 (s), 885 (s), 758 (s), 723 (s), 688 (s), 615 (w), 526 (s), 459 (w). MALDI-TOF-MS (Negative ion linear mode using 2,5-dihydroxybenzoic acid, DHB as matrix): m/z calculated for C10H12MoO7S4: 469.85, Found: 469.26. Elemental analysis for C58H54MoO7P2S4: calc. (%): C, 60.62; H, 4.74; S, 11.16. Found: C, 60.70; H, 4.37; S, 11.10. Electronic absorption data in CH3CN (λmax, nm (ε = M−1 cm−1)): 225 (10653), 256 (sh, 2563), 265 (2758), 277 (2340), 323 (1023).
Physical Measurements
NMR measurements were recorded on a Bruker Avance II-300 MHz instrument. All samples were dissolved in deuterated solvents and chemical shifts (δ) are given in parts per million (ppm) using solvent signals as reference (CDCl3 1H: δ = 7.24 ppm; 13C: δ = 77.0 ppm; CD3OD 1H: δ = 3.31, 4.87 ppm; 13C: δ = 49.15 ppm, CD3CN 1H: δ = 1.94 ppm; 13C: δ = 1.3 ppm) related to external tetramethylsilane (δ = 0 ppm). Spectra were obtained at 25°C unless otherwise noted. Coupling constants (J) are reported in Hertz (Hz) and splitting patterns are designated as s (singlet), d (doublet), t (triplet), q (quartet), quint (quintet), sext (sextet), m (multiplet), dd (doublet of doublet). Infrared spectra were recorded as KBr disks in the range 4000–400 cm−1 on a PerkinElmer Fourier-Transform Infrared (FT–IR) spectrophotometer. The assignment of the bands was done with subjective appreciation: w, weak; m, medium; s, strong; vs, very strong; br, broad. UV/Vis spectra were recorded on a Shimadzu UV-3600 spectrophotometer. Elemental analyses (C, H, N and S) were carried out with an Elementar Vario Micro Cube elemental analyzer. Mass spectra of organic molecules (APCI) were recorded with the high performance compact mass spectrometer Advion Expression CMS. Resolution: 0.5–0.7 m/z units (FWHM) at 1,000 m/z units sec−1 over the entire acquisition range. For compound 5 the mass spectra were measured on a Bruker microflex matrix assisted laser desorption/ionization (MALDI-TOF) spectrometer and the Advion Expression CMS spectrometer.
Electrochemical measurements were carried out with an AUTOLAB PGSTAT12 potentiostat/galvanostat using a glassy carbon working electrode with a reaction surface of 1 mm2 in acetonitrile solution with 0.1 M of [nBu4N][PF6] as supporting electrolyte. A platinum knob electrode (together with internal referencing vs. ferrocene/ferrocenium; Fc/) was used as reference electrode and a platinum rod electrode as auxiliary electrode. All measurements were controlled with the NOVA software and carried out inside a glove box under argon atmosphere.
X-Ray Crystallography
Suitable single crystals of compounds 1, 2, 3, and 4 were mounted on a thin glass fiber coated with paraffin oil. X-ray single-crystal structural data were collected at low temperature (170 K) using a STOE-IPDS II diffractometer equipped with a normal-focus, 2.4 kW, sealed-tube X-ray source with graphite-mono-chromated MoKα radiation (λ = 0.71073 Å). The program XArea was used for integration of diffraction profiles; numerical absorption correction was made with the programs X-shape and X-red32; all from STOE. The structure was solved by SIR92 (A. Altomare et al., 1993) or SHELXL-2013 (Sheldrick, 2008) and refined by full-matrix least-squares methods using SHELXL-2013 or SHELXL-2016 (Sheldrick, 2008, 2015). The non-hydrogen atoms were refined anisotropically. The oxygen bound alcohol hydrogen atoms in 2, 3 and 4 were refined freely. All other hydrogen atoms were refined isotropically on calculated positions using a riding model with their Uiso values constrained to 1.5Ueq of their pivot atoms for methyl and hydroxyl groups and to 1.2Ueq for all other C-H bonds. All calculations were carried out using SHELXL-2013/16 and the WinGX system, Ver2014.01 (Farrugia, 2012). Crystallographic data were deposited with the Cambridge Crystallographic Data Centre, CCDC, 12 Union Road, Cambridge CB21EZ, UK. These data can be obtained free of charge on quoting the depository numbers CCDC (1) 1858446, (2) 1858448, (3) 1858449, and (4) 1858447 by FAX (+44-1223-336-033), email (deposit@ccdc.cam.ac.uk) or their web interface (at http://www.ccdc.cam.ac.uk). Crystal and refinement data are summarized in Table 1. Details of crystal structural data are tabulated in the electronic supporting information (Tables S1–S18).
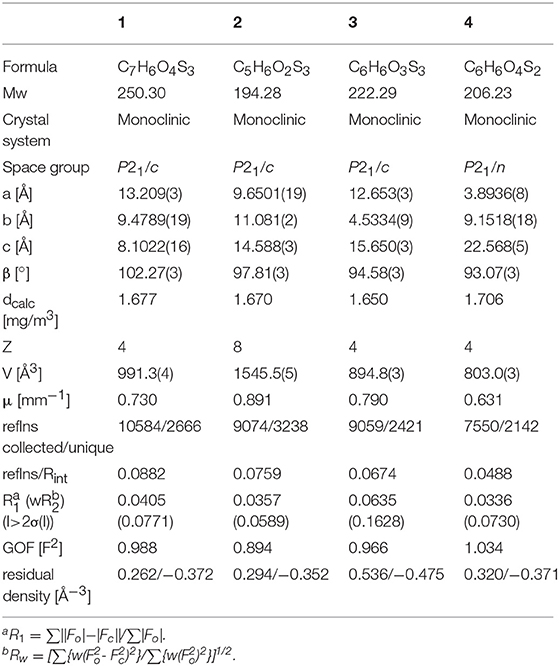
Table 1. Crystal and refinement data for 1, 2, 3, and 4 at 170 K.
Results and Discusion
Syntheses
The synthetic route to dithiolene ligand precursors 1–4 along with the complexation reaction are displayed in Scheme 2. The ligand mohdt is readily obtained by a three-step procedure starting from dimethyl but-2-ynedioate. Trithiocarbonate 1 was synthetized by reaction of the symmetrical alkyne with ethylene trithiocarbonate in anhydrous toluene under reflux conditions in a modified literature procedure (Easton and Leaver, 1965). Compound 1 was then reduced by little more than two equivalents of sodium borohydride (NaBH4) in the presence of LiCl and dry THF/EtOH similar to procedures applied previously (Jeppesen et al., 1999; Bellanger et al., 2012) but in distinct stoichiometry as a different, asymmetric compound was targeted here. The reduced unsymmetrical trithiocarbonate 2 was collected as major compound after column chromatography. Also a small amount of symmetrically substituted compound 3 (which was the target in the two reports cited above) as side product could be isolated and identified suggesting that the stoichiometric amount of NaBH4 needs to be controlled very carefully in this reduction reaction.
An often applied procedure for the synthesis of oxo-dithiocarbonate compounds is oxidizing the thione moiety in trithiocarbonate backbones with mercury acetate in the presence of acetic acid utilizing mercury's thiophilicity (Nguyen et al., 2010). Compound 2 was oxidized accordingly and then purified by column chromatography yielding the final dithiolene precursor 4, mohdtC=O (see Scheme 2).
A detailed comparison of infrared, 1H and 13C NMR spectroscopic data of the trithiocarbonates (1, 2, and 3) and the dithiocarbonate 4 reveals some interesting aspects of the C=S, C=O and C=C functional groups. The C=S signals in the 13C NMR spectra of 1, 2 and 3 were observed at δ 207.2, 210.8, and 214.4 ppm, respectively, which differs as expected from the C=O signal of 4 at δ 188.7 ppm (see Supplementary Material). The differences in the 13C NMR spectra for the C=C bonds in 1 (138.1 ppm; ester only), 2 (158.6 and 124.8 ppm; mixed) and 3 (143.5 ppm, hydroxyl only) are predominantly due to the presence (or absence) of the electron-donating and -withdrawing ester and hydroxyl functions, respectively. Most notably, in the unsymmetrically substituted 2 the comparable downfield and upfield shifts are more pronounced than in the symmetric compounds indicating a considerable push-pull effect induced by the asymmetry (see also the discussion in the structural characterization part below). Changing the C=S (2) to a C=O (4) function results in an upfield shift by ca. 7 ppm for both carbon atoms of the C=C moiety (151.6 and 117.6 ppm, mixed). The 1H NMR spectrum of 4 in CDCl3 displays two singlets at δ = 3.86 and 4.93 ppm, assigned to the methyl and methylene protons, respectively, which is almost identical to the values obtained for 2. This indicates that an exchange of C=O for C=S has no effect on these protons. Compared to the symmetric species the methylene protons are shifted downfield by ca. 0.4 ppm and the methyl protons are shifted upfield by an average of 0.03 ppm. The C=S stretching frequencies in the IR spectra for 1, 2, and 3 were observed at 1067, 1070, and 1053 cm−1 (Liu et al., 2010), respectively, while the C=Ooxo frequency in 4 is found at 1,654 cm−1 in accordance with the generally stronger bond between C and O (see Supplementary Material).
Complex 5 was synthesized according to a modified method already reported in the literature (Bradshaw et al., 2001b). The elemental analysis, the infrared, electronic absorption and NMR spectroscopic data, and the MALDI-TOF mass spectrometric data of 5 unambiguously support the formation of the mono-oxido MoIV center coordinated by two {S2C2(CO2Me)(CH2OH)}2− ligands. A comparison with known data of a closely related compound from the literature (Coucouvanis et al., 1991) and the similarity of the respective analyses further validates the proposed chemical structure of the complex. The molecular ion peak of 5 was detected at m/z 469.3 by MALDI-TOF-MS in the negative ion linear mode using 2,5-dihydroxy benzoic acid (DHB, 10 mg/mL in acetonitrile/water mixture (1/1, v/v) containing 0.1% TFA) (see Supplementary Material). The molecular ion peak of complex 5 was also detected by ESI-MS (-) analysis with a fitting isotopic pattern at m/z 460.2 to 469.1. The tetraphenylphosphonium counter cations (PP) were observed at m/z 339.0 in the positive ESI-MS mode (see Supplementary Material).
The 13C-NMR signals for the C=C bond in 4 are slightly shifted to the downfield/deshielded/higher frequency region in complex 5, which is a characteristic difference between a free dithiolene ligand precursor and the de-protected ene-dithiolate ligand coordinated to a MoIVO center. The π-delocalization within the dithiolene and the charge donation to the metal can be assessed considering the frequency of the C=C stretching mode in the FT-IR spectrum typically found in a range of 1,400–1,600 cm−1 as the C=C bond weakens with increased donation to the metal (Garton et al., 1997). A tentative assignment of the band at 1,541 cm−1 to this vibration, which is only marginally shifted from 1,544 cm−1, supports the presence of the ene-dithiolate rather than reduction of the metal with concomitant oxidation of the ligand to a radical species (partial thione character). The M=O stretching frequency of 5 at 925 cm−1 exhibits a substantial shift from the Mo precursor 728 cm−1 (Figure 2) (Ghosh et al., 2017). This is comparable to reported related MoIVO complexes such as [MoO{S2C2(COOMe)2}2]2− (Coucouvanis et al., 1991), [MoO{S2C2(CN)2}2]2− (Donahue et al., 1998) and [MoO{S2C2(CONH2)2}2]2− (Oku et al., 1997). The IR spectrum of mohdtC=O (4) further shows two sharp bands at 1,710 cm−1, at 1,654 cm−1 and one medium signal at 1,618 cm−1 belonging to (C=O)ester and (C=O)oxo dithiolene stretching frequencies, respectively. The (C=O)ester vibration is shifted to higher frequency in complex 5 (ν(C=O)ester: 1,718 cm−1) and (C=O)oxo has disappeared after complexation as expected (see Figure 2). Further C–O and C–S frequencies are difficult to identify/assign as they are masked by the dominating C–H stretching bands of the Ph4P+ counter-cation in the region 688–758 cm−1 (Tchouka et al., 2011).
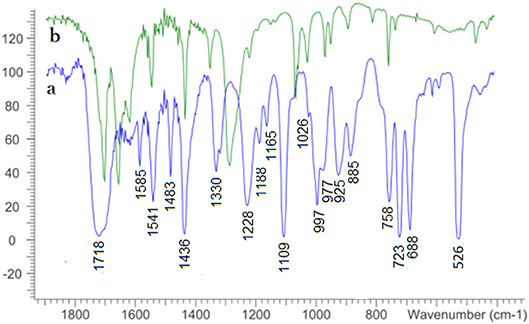
Figure 2. Solid state FT-IR spectra of complex 5 (a, bottom) and ligand precursor 4 (b, top) measured as KBr pellets.
Electronic spectra of the molybdenum precursor, ligand precursor 4 and the resulting MoIVO complex (5) were recorded in CH3CN solution (Figure 3). The UV-vis spectra display absorption bands in the region 256–323 nm, characteristic of ligand to metal charge transfer (LMCT) and of intra-ligand charge transfer as also strongly suggested by comparison with the spectrum of the protected ligand. The two broad bands of rather similar shape for the ligand (ca. 250–310 nm) and complex 5 at λmax 323 nm (ε = 1,023 M−1 cm−1) are most likely due to the same transition albeit shifted as would be expected after coordination. The corresponding bands and extinction coefficients of molybdenum complexes bearing two different dithiolene ligands, representing another type of non-symmetry, such as (Et4N)2[MoIVO(S2C2(CO2Me)2)(bdtCl2)] (λmax 531 nm; ε = 340 M−1 cm−1) (Sugimoto et al., 2009) and (Ph4P)2[MoIVO(edt)(mnt)] (λmax 433 nm; ε = 1,110 M−1 cm−1) (Donahue et al., 1998) are comparable to those of complex 5 reported here. The intensities (extinction coefficients) of the reported bands are well in accordance, while the observed band energy for complex 5 is higher (band at lower wavelength). In the complex we therefore tentatively assign the band at 323 nm to an LLCT transition. The single very broad absorption signal belonging to the dithiolene ligand precursor 4 is narrowed and exhibits a bathochromic shift in complex 5 indicative of a significant change in the electronic structure with more distinct LLCT transitions at slightly lower energy, as expected upon loss of the protecting C=O function (potentially engaged in resonance structures), coordination to a metal center and formation of new mixed metal-ligand molecular orbitals. Similar observations were made previously with a series of moderately related strictly aliphatic complexes (cyclohexane, pyrane, and thiopyrane derived ligands) with signals in the region 260–476 nm (Sugimoto et al., 2005). Coucouvanis and co-workers reported UV-vis data of the complex (Et4N)2[MoO(S2C2COOMe)2], which can be considered the closest relative of the new complex at λmax: 360, 460(sh) and 550 nm (Coucouvanis et al., 1991) albeit without extinction coefficients. Replacing the strongly electron withdrawing ester function by the ethyl alcohol substituent apparently shifts the transitions to higher energy; possibly also due to increasing ligand field strength when more electron density can be pushed toward the central metal. The recorded values for 5 are further comparable to related (Et4N)2[MoO(S2C2(COPh)2)2] with phenyl-keto substituents (λmax: 310 (sh), 338 (sh), 400 nm), the bands of which fall in between those of complex 5 and the Coucouvanis complex (Ansari et al., 1987).
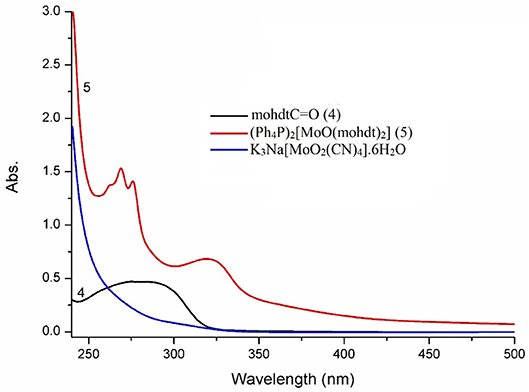
Figure 3. Comparison of UV-vis spectra of mohdtC=O (compound 4; λmax, nm (ε/M−1 cm−1): 211 (2682), 285 (br, 2340)), molybdenum precursor K3Na[MoO2(CN)4]·6H2O and the molybdenum complex 5 (λmax, nm (ε = M−1 cm−1): 225 (10653), 256 (sh, 2563), 265 (2758), 277 (2340), 323 (1023)) in acetonitrile solution.
The observed shift in the UV-vis data from the Cocouvanis complex (all ester) to complex 5 (mixed; ester and alcohol) corresponds also to the electrochemical properties of both. The cyclic voltammogram of 5 (see Figure S26) exhibits a reversible redox process for the MoIV↔MoV transition ([MoO(mohdt)2]2−/[MoO(mohdt)2]−) at −0.62 V vs. [Fc]/[Fc]+ which constitutes a decrease in potential compared to the all ester complex at −0.074 V vs. [Fc]/[Fc]+ (value given in the report: −0.03 V vs. SCE) (Coucouvanis et al., 1991); i.e., the electron pushing alcohol substituent facilitates oxidation of the complex whereas the complex with two electron withdrawing ester substituents is more easily reduced.
Structural Characterization
The molecular structures of 1, 2, 3, and 4 are shown in Figure 4 and the selected comparable bond distances and angles are listed in Table 2. All ligands were (re-)crystallized by the slow diffusion method. The structure of 1 was published previously in a databank without any accompanying discussion (Neil Bricklebank et al., 2003). In the X-ray structure of compound 3 two independent molecules are present in the unit cell, which differ slightly with respect to the angles of the -CH2-OH substituents to the ene moiety (rmsd 0.305; max. distance 0.5464 Å). All three ene-trithiocarbonate compounds (1–3) crystallized in the monoclinic P21/c space group whereas compound 4 (ene-dithiocarbonate) crystallized in the monoclinic P21/n space group. A description of a crystal structure of 3 in a different space group (C2/c) and with only one molecule in the asymmetric unit is available in Acta Cryst (Pløger et al., 2006). The ene-trithiocarbonate rings are structurally all similar and exhibit C=S, C–S and C=C distances in ranges of 1.632(4)−1.659(3), 1.718(3)−1.748(3) and 1.339(4)−1.347(4) Å, respectively.
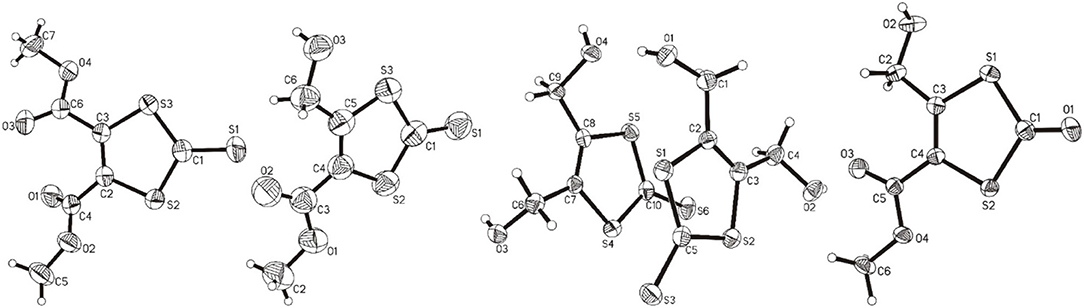
Figure 4. From left to right: the molecular structures of 1, 2, 3, and 4, respectively, shown at the 50% probability level.
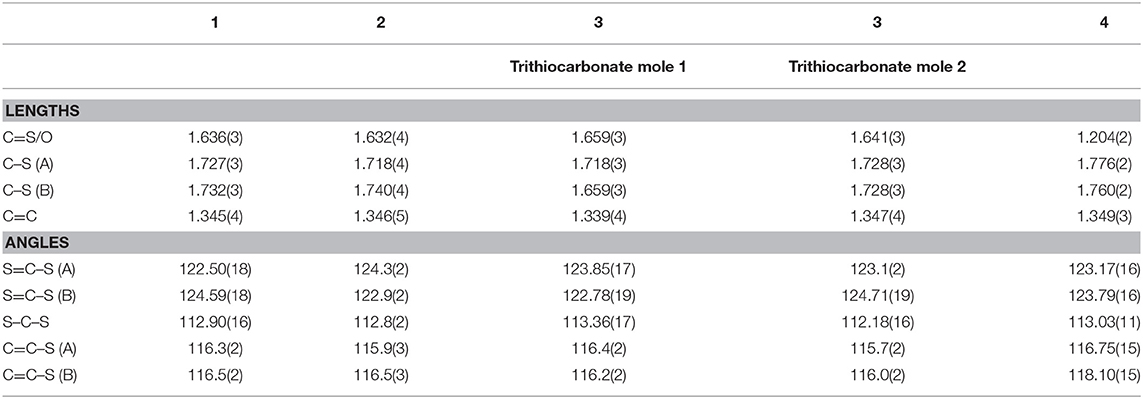
Table 2. Selected bond lengths [Å] and angles [°] for 1, 2, 3, and 4.
However, the ene carbon atoms' C–S bond distances in the four ligand precursor molecules upon close inspection are rather noteworthy. The intention of utilizing distinct substituents and an asymmetrically substituted dithiolene ligand for the complex synthesis was to fine-tune the electronic properties of the ligand (and consequently of the complex) hypothesizing that with a push-pull-effect the ligand's non-innocence (i.e., its ability to donate electron density/electrons to the metal center) should be raised. While one half of the ene-dithiolate moiety has a stronger preference for donating electron density toward the metal center than the other, then such donation should be facilitated compared to a system in which both substituents compete for the exact same effect and having the exact same properties. The resonance of such system, however, is expected to be decreased, translating into lower stability which is typically concomitant to higher reactivity (secondary effect; see Figure S27). That such primary effect was indeed realized at least in the ligand precursors is strongly supported by the distances between the ene-carbon atoms and the sulfur atoms as well as by the 13C-NMR data as already discussed above (Figure 5). The two (or four in case of 3 with the two independent molecules in the asymmetric unit) ene-C-S bond distances in the symmetric molecules of 1 and 3 are much more similar to each other than to those in the unsymmetric molecules of 2 and 4. Most notably, the C-S bond lengths involving the ester substituted ene carbon are significantly longer in 2 and 4 than in the case of 1 (ester only) and those involving the alcohol substituted ene carbon atoms are much shorter than in the case of 3 (alcohol only). The unsymmetric substitution apparently increases the C-S single bond character of the ester side of the ene-dithio moiety and the C-S double bond character of the alcohol side. The latter will facilitate electron density donation toward the coordinated metal upon complex formation from this side of the molecule, as there is apparently already more density available in the respective bonds compared to the ester sides of the molecules. These metrical observations coincide with 13C-NMR data of the ene functional group discussed above. In fact, the chemical shifts of the symmetric molecules (1 and 3) are even closer to each other than they are to the shifts of either of the ene carbon atoms in the unsymmetric molecules. This means, that exchanging just one of two substituents results in a stronger modulation of the electronic structure compared to replacing both substituents.
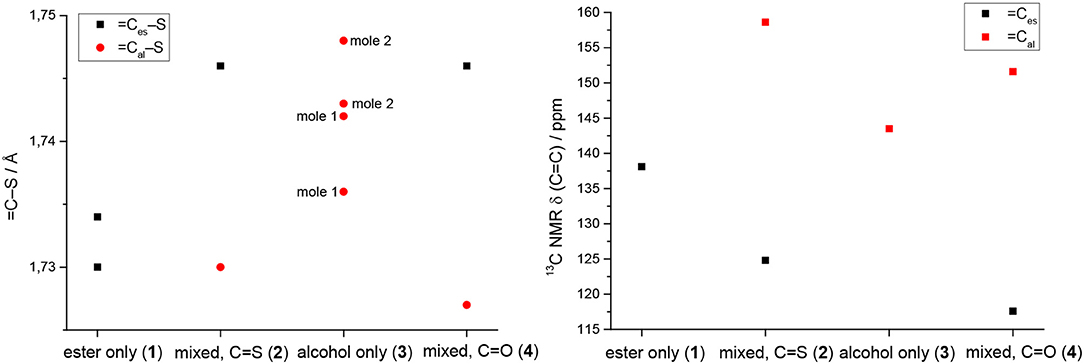
Figure 5. Ene-carbon to sulfur (=C–S) distances in Å (left) and 13C NMR chemical shifts of the ene moiety in ppm (right) visualizing the substituents' influence in compound 1 (ester only), compound 2 (mixed ester/alcohol), compound 3 (alcohol only; the individual molecules of the structure are indicated by mole 1 and mole 2 in the graph) and compound 4 (mixed ester/alcohol) strongly supporting the proposed push-pull effect in the asymmetric molecules. =Ces-S and =Cal-S indicate the ene-carbon atoms which are substituted by the ester and alcohol functions, respectively.
With respect to the influence of the protecting group, the C=O oxo distance in 4 is 1.204(2) Å, which is necessarily shorter than the C=S distances in ene-trithiocarbonates due to the smaller size of oxygen atoms compared to sulfur. The other bond distances of the ene-dithiocarbonate moiety are slightly longer than the observed ranges for the ene-tritiocarbonates (OC–S: 1.760/1.776 Å; C=C 1.349 Å) (see Table 2). This indicates somehow stronger donation of electron density toward the C=O functional group of the ene-dithiocarbonate than to the respective C=S of the ene-trithiocarbonates.
Figure 6 shows projections of the crystal packing in the structures of 1, 2, 3, and 4 along the a or b axes. The hydrogen bonding/short contacts present in all structures are depicted in blue. Only for 1 all ene-trithiocarbonate moieties are coplanar within the crystal lattice whereas for the other three compounds the planar parts of the molecular structures are arranged in angles up to nearly perpendicular (87.63°; 3) to each other. X-ray suitable single crystals of complex 5 remained elusive, unfortunately, despite considerable and repeated efforts of recrystallization.
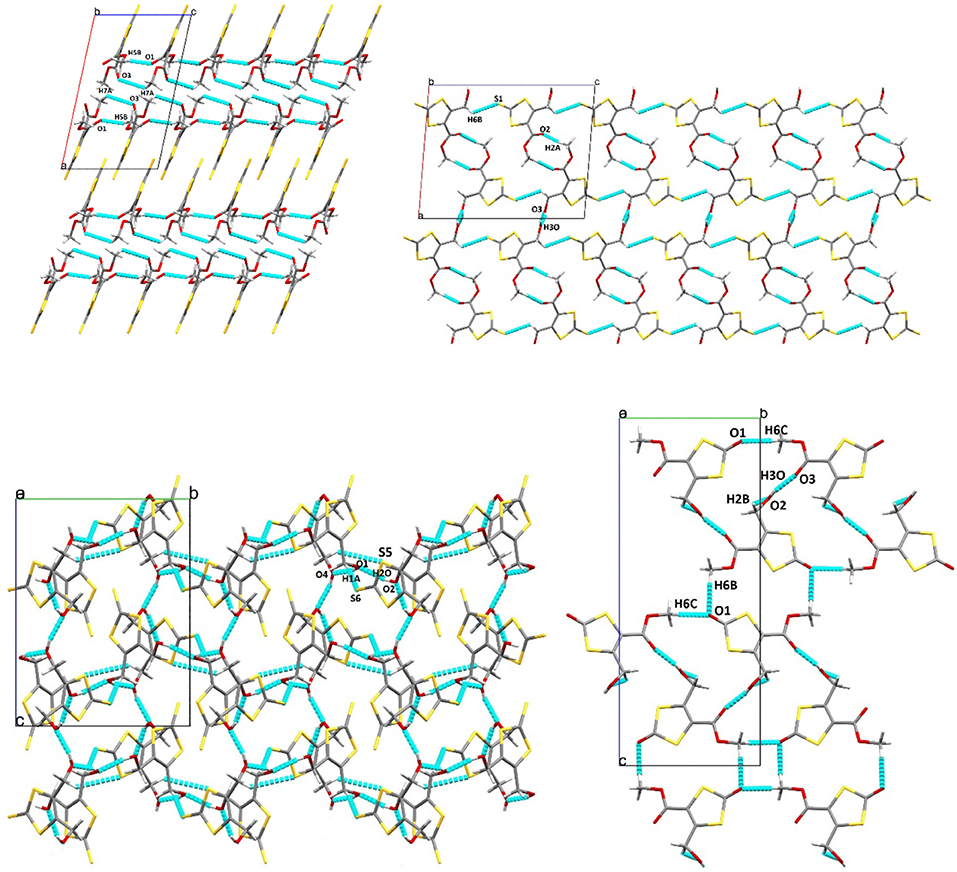
Figure 6. Crystal packing projection views along the b axis of 1 (top left), and 2 (top right), and along the a axis of 3 (bottom left) and 4 (bottom right). Selected hydrogen bonding/short contacts (Å): (1): C(5)–H(5B)···O(1): 2.522, C(7)–H(7A) ···O(3): 2.708, C(4)–O(3): 3.001; (2): O(3)–H(3O)···O(3): 1.89(6), C(2)–H(2A)···O(2): 2.59, S(1)–H(6B): 2.960; (3): O(1)–H(1O)···O(4): 1.95(4), CO(2)–H(2O)···O(1): 1.90(4), O(3)–H(3O)···O(2): 2.10(4), O(4)–H(4O)···O(3): 1.89(4), H(1A)–S(6): 2.946, S(5)–H(9B): 2.979; (4): O(1)–H(6C): 2.503, O(2)–H(2B): 2.441, O(3)–H(3O): 1.999, O(1)–H(6B): 2.594.
OAT Catalysis
The OAT activity of MoIVO bis-dithiolene complex 5 was investigated with the model oxygen atom transfer reaction between DMSO and PPh3 (see Scheme 3; based on Ref. Berg and Holm, 1985). The reaction progress was monitored by 31P-NMR spectroscopy. The reaction typically proceeds via oxygen atom transfer from DMSO to the MoIVO moiety resulting in dimethyl sulfide and a MoVIO2 species which then oxidizes the acceptor substrate PPh3 yielding OPPh3 and concurrently completing the catalytic cycle (Lorber et al., 1997; Tucci et al., 1998). In this mechanism, the phosphorous atom of the alkyl-phosphines is performing a nucleophilic attack on one of the two oxido ligands on the MoVI center by donation into the empty Mo=O π* orbital generating the phosphine oxide intermediate, while a free electron pair of the oxygen atom is simultaneously attacking the P–C σ* orbital (Holm, 1987; Smith et al., 2000). I.e., an electron pair on phosphorous establishes an initial single bond with oxygen and for the respective P=O double bond a lone pair on oxygen is used. At the same time one electron pair of the Mo=O double bond becomes the new second lone pair on oxygen and the other electron pair of the former metal oxygen double bond remains entirely at the metal center (severing the bond between Mo and O), so that the two-electron reduction of the metal/oxidation of phosphorous proceeds smoothly together with the transfer of oxygen from metal to substrate.
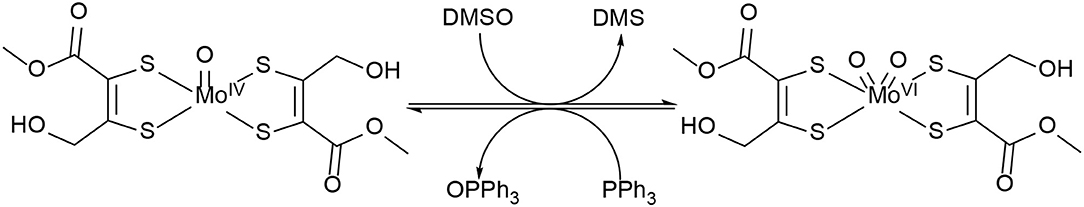
Scheme 3. Proposed reactions for the oxygen atom transfer catalyzed by 5.
DMSO is used as oxygen donor source and simultaneously employed as solvent and substrate with consequentially very high excess to the catalyst. PPh3 was chosen as expedient model substrate for its high solubility in organic solvents and its suitable affinity toward oxygen. A 3 mM catalyst loading was employed together with 3 eq. of PPh3 and 0.5 mL of deoxygenated DMSO in an airtight NMR tube at room temperature. 31P-NMR spectroscopy is the most convenient method to monitor the reaction progress since substrate (PPh3) and product (PPh3O) demonstrate well-separated resonance signals (PPh3: s, −5.8 ppm and PPh3O: s, 26.6 ppm in DMSO-d6). Reaction monitoring by NMR started immediately after preparation of the reaction mixture under N2 atmosphere. The concentration of PPh3 (at −5.8 ppm) decreased gradually with the reaction time and at the end of the reaction PPh3O was the dominating species (Figure 7).
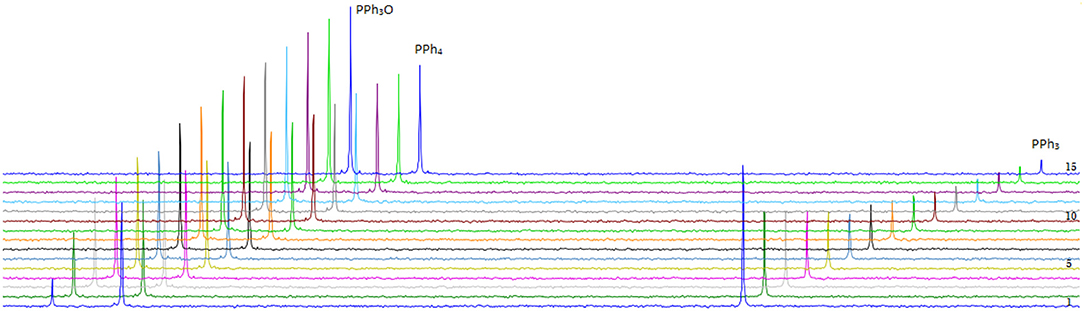
Figure 7. Transformation of PPh3 to OPPh3 catalyzed by 5 in the presence of PPh3 and DMSO-d6 as oxidizing agent monitored by 31P-NMR spectroscopy at r.t (unregulated temperature). Fifteen spectra are shown taken with a frequency of 1 every 4 h.
As the central metal is involved in a two electron redox-process, electron density buffering by a non-innocent ligand is considered beneficial for such reactions. The aim of this study was to optimize this electron density buffering by the asymmetric ligand substitution and a respective push-pull effect (present in the ligand precursor as evidenced by structural and spectroscopic data). A one-sided preference for electron donation induced by the introduction of one electron donating alcohol substituent was supposed to better support the involved redox processes and increase the complex' reactivity. However, the reaction proceeds very slowly with the maximum conversion (93%) of applied PPh3 (9 mM) reached after ~2.5 days with a not entirely steady progress under the applied reaction conditions (rather hot summer days, cooler nights, no temperature control; see Figure S22). We therefore abstained from trying to extract specific kinetic parameters for this transformation.
The disappointingly low reaction velocity can be attributed not entirely to the low activity of 5 but also to the comparably mild oxidizing substrate. Although well-established by the Holm group, the DMSO/PPh3 system has its disadvantages with respect to the known very slow conversion of MoIV to MoVI with DMSO. When adding Me3NO as a stronger oxidizing agent to a freshly prepared solution of 5 and PPh3 in DMSO-d6 we observed 36.52% conversion overnight (within 15 h), which constitutes a slight acceleration in comparison but still not the anticipated rapid catalytic process.
The most frequently investigated molybdenum centers in oxidoreductase model chemistry are MoIVO and MoVIO2 species which are comparable to the native co-factors regarding the oxidation states and they bear transferable oxido ligands (McMaster et al., 2004b). These complexes, however, in particular when mixed in a reaction medium while circling through catalysis, can also form dimeric or oligomeric assemblies transforming terminal oxido ligands into μ-oxido functions, e.g., dimeric and chemically inert MO3 moieties, which are catalytically inactive (McMaster et al., 2004a; Mitra and Sarkar, 2013; Hille et al., 2016). Confirming their chemical structures, a number of X-ray diffraction studies of such dimeric species are reported in the literature (Tatsumisago et al., 1982; Ratnani et al., 1990; Sellmann et al., 1992; Thompson et al., 1993; Awwal et al., 2007; Pal et al., 2007; Mitra and Sarkar, 2013) albeit not with bis-dithiolene molybdenum centers. In fact Subramanian et al. have stated that the reduction of MoVIO2L2 species necessarily leads to μ-oxo-bridged dimers (Subramanian et al., 1984). The formation of such species constitutes a general problem associated with catalytic/kinetic investigations of OAT, although in the best cases monomeric MoIVO plus MoVIO species and dimeric MO3 are in equilibrium with considerable amounts left of the former pair so that catalytic activity can still persist (Holm, 1987, 1990).
In order to verify whether the slowness of the OAT reaction was indeed due to dimer formation, a solution of 3 mM complex 5 in the presence of oxidizing agent trimethylaminoxid (TMAO) in acetonitrile was prepared and monitored by UV-Vis spectroscopy under anaerobic condition. It was observed that after the initial formation of the transient MoVIO2 species (here very broad signal at λmax: 540 nm), it swiftly decomposed again while the signal for the catalytically inert MO3 species (typical signal at λmax: 375 nm) (Villata et al., 2000; Sugimoto et al., 2003; Pal et al., 2007) exhibited a steady rise (see Figure S22). The change in the UV-vis with the progress of the reaction exhibits clean isosbestic points indicating the simultaneous presence of only two (not three) species. This can be explained by the fact that the transient di-oxo species is of particularly low concentration, hence, nearly invisible in the UV-vis as the dimer formation is essentially instant as soon as the di-oxo species is available.
The typical dimerization is particularly problematic for those systems with bis-dithiolene co-ligands bearing aliphatic backbones without steric or electronic protection. When aromatic dithiolene ligands are used (and this refers to both, an aromatic substituent on the ene- moiety as well as the ene moiety being part of an aromatic ring as in benzene-dithiolate) molybdenum dithiolene complexes are typically much more stable and, hence, much less reactive than those with aliphatic dithiolene systems (Fischer and Fischer, 2017). Dithiolene ligands bearing electron withdrawing groups as substituents typically exhibit weak Mo–S bonds. This does promote the Mo=O bond and concomitantly stabilizes the monomeric species by electronic tuning but it also decreases the catalytic activity (Hille et al., 2016). In contrast, dithiolene ligands with electron donating groups push electron density toward the metal center and by that decrease the Lewis acidity of molybdenum. It was shown previously that the Mo=O bond is weakened in dithiolene complexes with aliphatic backbones in particular with electron donating substituents (Hille et al., 2016). Taking all this into consideration we attribute the slowness of the catalyzed OAT reaction observed for complex 5 predominantly to the formation of the dimeric and chemically inert MO3 species as the aliphatic substituents on the used dithiolene ligand have mixed electron donating and electron withdrawing character, intended to fine tune the electronic structure of the complex and balance stability and reactivity. The dimerization may benefit from the presence of ester and hydroxyl functions on the dithiolene which constitute excellent functionalities for hydrogen bonding, which potentially results in close proximity of the catalytic centers even in solution. Previously, the presence of hydrogen bonding potential, in particular for intramolecular hydrogen bonding was perceived to be beneficial for catalysis (Oku et al., 1997; Okamura et al., 2016a,b). However, in this case it appears to be rather detrimental. The respective potential interactions between two complexes might facilitate formation of the catalytically inactive dimer by intermolecular hydrogen bonds (see Figure S23 for a proposed interaction). An accelerated dimer formation would slow down the catalytic OAT reaction significantly by inactivation of the present catalyst species and in addition by preventing the catalytically active species to diffuse freely into the solution. The observed actual reactivity, in fact, is more comparable to a system with aromatic backbone, e.g., to one previously reported by our group ((Bu4N)2[MoIVO(ntdt)2] and (Ph4P)2[MoVIO2(ntdt)2]; ntdt = 2-naphthyl-1,4-dithiolate) (Ghosh et al., 2017) than to other aliphatic systems. Although the strategy of combining electron pushing and electron withdrawing substituents generally appears to achieve the anticipated fine-tuning of the electronic structure (evident at least for the ligand precursor) it did not translate into the targeted increase in reactivity. Apparently, further consideration needs to go into the exact nature of the utilized substituents. For respective next generation compounds the ability to engage in hydrogen bonding should be assessed from the very beginning.
Conclusions
The aliphatic dithiolene ligand, 1-methoxy-1-oxo-4-hydroxy-but-2-ene-2,3-bis-thiolate (mohdt) and its Mo bis-dithiolene complex were synthetized and comprehensively characterized. The unsymmetrically substituted dithiolene ligand is subject to a push-pull effect modulating its electronic structure. A comparison of structural-metrical and 13C-NMR data of four related ligand precursor compounds reveals that substituent effects are indeed much more pronounced in unsymmetric than in symmetric molecules. 13C-NMR data in particular turned out to be rather sensitive to such effects and should be considered a valuable tool for respective assessments. Since the synthesized complex can be considered a structural model for the molybdopterin bearing DMSO reductases with respect to the immediate coordination sphere, it was also tested for its catalytic oxygen atom transfer ability in DMSO by mixing the catalyst with PPh3 at room temperature. The molybdenum complex catalyzes the OAT reaction from DMSO to PPh3 up to a 93% conversion within 56 h. In contrast to the expectations based on the evidenced push-pull effect, the catalytic performance of complex 5 is unexpectedly slow most likely due to the formation of dimeric MoV species after initial oxidative transformation of MoIVO to MoVIO2 species. This is possibly supported by hydrogen bonding effects of the ligands' substituents and certainly not hindered by any steric bulk.
Author Contributions
MA: syntheses, experiments, and drafting the manuscript. AG: syntheses, experiments, and supporting manuscript drafting. CF: scientific support of the project, catalysis, and kinetic evaluation. CS: study design, in charge of overall direction, managing the project, and finalizing the report.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Generous financial support from the European Research Council (project MocoModels, grant 281257) and the DFG funded SPP 1927 (project SCHU 1480/4-1) is gratefully acknowledged. We are also indebted to the reviewers of this manuscript for very helpful comments and their constructive criticism.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00486/full#supplementary-material
References
Altomare, A., Cascarano, G., Giacovazzo, C., Guagliardi, A., Burla, M. C., Polidori, G., et al. (1993). SIR92—a program for automatic solution of crystal structures by direct methods. J. Appl. Crystallogr. 26:343.
Ansari, M. A., Chandrasekaran, J., and Sarkar, S. (1987). Synthesis and characterization of a mononuclear molybdenum(IV) oxo complex (Et4N)2[MoO(S2C2(COPh)2)2]. Inorg. Chim. Acta 133, 133–136. doi: 10.1016/s0020-1693(00)84384-4
Awwal, L., Drake, J. E., Hursthouse, M. B., Kumar, R., Light, M. E., Ratnani, R., et al. (2007). Synthesis and characterization of binuclear oxomolybdenum(V) and dioxomolybdenum(VI) O,O′-ditolyl dithiophosphate complexes: crystal structures of Mo2O3[S2P(OC6H4Me-o)2]4, Mo2O3[S2P(OC6H4Me-m)2]4 and MoO2[S2P(OC6H4Me-p)2]2. Polyhedron 26, 3973–3979. doi: 10.1016/j.poly.2007.04.034
Basu, P., and Burgmayer, S. J. (2015). Recent developments in the study of molybdoenzyme models. J. Biol. Inorg. Chem. 20, 373–383. doi: 10.1007/s00775-014-1228-0
Bellanger, H., Darmanin, T., Taffin de Givenchy, E., and Guittard, F. (2012). Superhydrophobic hollow spheres by electrodeposition of fluorinated poly(3,4-ethylenedithiopyrrole). RSC Adv. 2, 10899–10906. doi: 10.1039/C2RA21665E
Berg, J. M., and Holm, R. H. (1985). Model for the active site of oxo-transfer molybdoenzymes: synthesis, structure, and properties. J. Am. Chem. Soc. 107, 917–925. doi: 10.1021/ja00290a029
Bradshaw, B., Collison, D., Garner, C. D., and Joule, J. A. (2001a). Stable pyrano[2,3-b]quinoxalines and pyrano[2,3-g]pteridines related to molybdopterin. Chem. Commun. 1, 123–124. doi: 10.1039/B008544H
Bradshaw, B., Dinsmore, A., Collison, D., Garner, C. D., and Joule, J. A. (2001b). The synthesis of pyrano[2,3-b]quinoxalines related to molybdopterin. J. Chem. Soc. 24, 3232–3238. doi: 10.1039/b108576j
Bradshaw, B., Dinsmore, A., Garner, C. D., and Joule, J. A. (1998). Synthesis of a cobalt complex of a pyrano[2,3-b]quinoxaline-3,4-dithiolate related to molybdopterin. Chem. Commun. 3, 417–418. doi: 10.1039/a708366a
Coucouvanis, D., Hadjikyriacou, A., Toupadakis, A., Koo, S. M., Ileperuma, O., Draganjac, M., et al. (1991). Studies of the reactivity of binary thio- and tertiary oxothiomolybdates toward electrophiles. Reactions with dicarbomethoxyacetylene and the synthesis and structures of the [Et4N2[MoO(L)2], anti-[Et4N]2[Mo2O2S2(L)2]], syn-[Ph4P]2[Mo2O2S2(L)2].cntdot.2DMF, Ph4P]2[Mo(L)3]DMF.cntdot.C6H6, and [Ph4P]2[Mo2S2(L)4]2CH2Cl2 complexes (L = 1,2-dicarbomethoxy-1,2-ethylenedithiolate). Inorg. Chem. 30, 754–767. doi: 10.1021/ic00004a028
Donahue, J. P., Goldsmith, C. R., Nadiminti, U., and Holm, R. H. (1998). Synthesis, structures, and reactivity of bis(dithiolene)molybdenum(IV,VI) complexes related to the active sites of molybdoenzymes. J. Am. Chem. Soc. 120, 12869–12881. doi: 10.1021/ja982914f
Döring, A., Schulzke, C., and Zhang, Q. (2010). Synthesis, characterization and structural analysis of isostructural dinuclear molybdenum and tungsten oxo-bis-mu-sulfido-benzenedithiolene complexes. Inorg. Chim. Acta 363, 4140–4144. doi: 10.1016/j.ica.2010.05.036
Easton, D. B. J., and Leaver, D. (1965). 1,3-Dipolar reactivity of 1,2-dithiole-3-thiones and 1,3-dithiolan-2-thiones. Chem. Commun. 22, 585–586. doi: 10.1039/C19650000585
Edwards, M., Roeper, J., Allgood, C., Chin, R., Santamaria, J., Wong, F., et al. (2015). Investigation of molybdenum cofactor deficiency due to MOCS2 deficiency in a newborn baby. Meta Gene 3, 43–49. doi: 10.1016/j.mgene.2014.12.003
Enemark, J. H., Cooney, J. J., Wang, J. J., and Holm, R. H. (2004). Synthetic analogues and reaction systems relevant to the molybdenum and tungsten oxotransferases. Chem. Rev. 104, 1175–1200. doi: 10.1021/cr020609d
Farrugia, L. (2012). WinGX and ORTEP for windows: an update. J. Appl. Crystallogr. 45, 849–854. doi: 10.1107/S0021889812029111
Fischer, C., and Fischer, L. (2017). Molybdenum and tungsten enzymes: bioinorganic chemistry. R. Soc. Chem. 20, 94–129. doi: 10.1039/9781782628828-00094
Garner, C. D., and Bristow, S. (1985). “Molybdenum enzymes,” in Metal Ions in Biology, Vol. 7, ed T. G. Spiro (New York, NY: John Wiley and Sons, Inc.), 343.
Garton, S. D., Hilton, J., Oku, H., Crouse, B. R., Rajagopalan, K. V., and Johnson, M. K. (1997). Active site structures and catalytic mechanism of rhodobacter sphaeroides dimethyl sulfoxide reductase as revealed by resonance Raman spectroscopy. J. Am. Chem. Soc. 119, 12906–12916. doi: 10.1021/ja972109l
Ghosh, A. C., Samuel, P. P., and Schulzke, C. (2017). Synthesis, characterization and oxygen atom transfer reactivity of a pair of Mo(iv)O- and Mo(vi)O2-enedithiolate complexes—a look at both ends of the catalytic transformation. Dalton Trans. 46, 7523–7533. doi: 10.1039/C7DT01470H
Gisewhite, D. R., Yang, J., Williams, B. R., Esmail, A., Stein, B., Kirk, M. L., et al. (2018). Implications of pyran cyclization and pterin conformation on oxidized forms of the molybdenum cofactor. J. Am. Chem. Soc. 140, 12808–12818. doi: 10.1021/jacs.8b05777
Hille, R. (2013). The molybdenum oxotransferases and related enzymes. Dalton Trans. 42, 3029–3042. doi: 10.1039/c2dt32376a
Hille, R., Hall, J., and Basu, P. (2014). The mononuclear molybdenum enzymes. Chem. Rev. 114, 3963–4038. doi: 10.1021/cr400443z
Hille, R., Schulzke, C., and Kirk, M. L. (2016). Molybdenum and Tungsten Enzymes: Bioinorganic Chemistry, eds R. Hille, C. Schulzke, and M. L. Kirk. Cambridge, UK: The Royal Society of Chemistry.
Holm, R. H. (1987). Metal-centered oxygen atom transfer reactions. Chem. Rev. 87, 1401–1449. doi: 10.1021/cr00082a005
Holm, R. H. (1990). The biologically relevant oxygen atom transfer chemistry of molybdenum—from synthetic analog systems to enzymes. Coordinat. Chem. Rev. 100, 183–221. doi: 10.1016/0010-8545(90)85010-P
Jeppesen, J. O., Takimiya, K., Thorup, N., and Becher, J. (1999). A novel tetrathiafulvalene building block. Synthesis 5, 803–810.
Kim, H. S., Nguyen, D. Q., Cheong, M., Kim, H., Lee, H., Ko, N. H., et al. (2008). One-pot synthesis of ethylene trithiocarbonate from ethylene carbonate. Appl. Catalysis A General 337, 168–172. doi: 10.1016/j.apcata.2007.12.012
Kirk, M. L., McNaughton, R. L., and Helton, M. E. (2004). “The electronic structure and spectroscopy of metallo-dithiolene complexes,” In Dithiolene Chemistry, Vol. 52, ed E. I. Stiefel (New York, NY: John Wiley and Sons, Inc., 111–212.
Lim, B. S., and Holm, R. H. (2001). Bis(dithiolene)molybdenum analogues relevant to the DMSO reductase enzyme family: synthesis, structures, and oxygen atom transfer reactions and kinetics. J. Am. Chem. Soc. 123, 1920–1930. doi: 10.1021/ja003546u
Liu, B., Wu, B., Xu, J., Wu, Z., Zhao, Y., Zheng, X., et al. (2010). Resonance Raman intensity analysisof the excited-state photochemical dynamicsof dimethyl 1,3-dithiole-2-thione-4,5-dicarboxylate in the A-band absorption. J. Raman Spectroscopy 41, 1185–1193. doi: 10.1002/jrs.2580
Lorber, C., Rosaria Plutino, M. I., Elding, L., and Nordlander, E. (1997). Kinetics of oxygen-atom transfer reactions involving molybdenum dithiolene complexes. J. Chem. Soc. Dalton Trans. 21, 3997–4004.
McMaster, J., Tunney, J. M., and Garner, C. D. (2004a). “Chemical analogues of the catalytic centers of molybdenum and tungsten dithiolene-containing enzymes,” in Dithiolene Chemistry, ed E. I. Stiefel (New York, NY: John Wiley and Sons, Inc., 539–583.
McMaster, J., Tunney, J. M., and Garner, C. D. (2004b). “Chemical analogues of the catalytic centers of molybdenum and tungsten ditholene-containing enzymes,” in Dithiolene Chemistry, Vol. 52, ed E. I. Stiefel (New York, NY: John Wiley and Sons, Inc., 539–584.
Mendel, R. R. (2007). Biology of the molybdenum cofactor. J. Exp. Bot. 58, 2289–2296. doi: 10.1093/jxb/erm024
Mitra, J., and Sarkar, S. (2013). Oxo–Mo(IV)(dithiolene)thiolato complexes: analogue of reduced sulfite oxidase. Inorg. Chem. 52, 3032–3042. doi: 10.1021/ic302485c
Neil Bricklebank, S. J. C., and Michael, B., Hursthouse. (2003). Crystal Structure Report Archive. U.S. National Library of Medicine National Center for Biotechnology Information, Bethesda, MD, United States. doi: 10.5258/ecrystals/902
Nguyen, Tle A., Demir-Cakan, R., Devic, T., Morcrette, M., Ahnfeldt, T., Auban-Senzier, P., et al. (2010). 3-D coordination polymers based on the tetrathiafulvalenetetracarboxylate (TTF-TC) derivative: synthesis, characterization, and oxidation issues. Inorg. Chem. 49, 7135–7143. doi: 10.1021/ic100950n
Okamura, T., Omi, Y., Hirano, Y., and Onitsuka, K. (2016a). Comparative studies on the contribution of NH···S hydrogen bonds in tungsten and molybdenum benzenedithiolate complexes. Dalton Trans. 45, 15651–15659. doi: 10.1039/C6DT02250B
Okamura, T.-A., Yamada, T., Hasenaka, Y., Yamashita, S., and Onitsuka, K. (2016b). Unexpected reaction promoted by NH+···O=Mo hydrogen bonds in nonpolar solvents. Eur. J. Inorg. Chem. 2016, 2952–2961. doi: 10.1002/ejic.201600081
Oku, H., Ueyama, N., and Nakamura, A. (1997). Association of oxo–molybdenum dithiolene complexes with a multiamide additive and its influence on the ease of O-atom transfer. Inorg. Chem. 36, 1504–1516. doi: 10.1021/ic951277g
Pal, K., Maiti, R., Chaudhury, P. K., and Sarkar, S. (2007). Synthesis, structure and reactions of a series of 1,2-dicyanoethylenedithiolate coordinated dimeric Mo(V) complexes. Inorg. Chim. Acta 360, 2721–2733. doi: 10.1016/j.ica.2007.01.018
Pløger, J. M., Petersen, B. M., Bond, A. D., and Jeppesen, J. O. (2006). 4,5-Bis(hydroxymethyl)-1,3-dithiole-2-thione. Acta Crystallogr. E62, o2066–o2068. doi: 10.1107/S1600536806014140
Rajagopalan, K. V. (1997). The molybdenum cofactors—perspective from crystal structure. J. Biol. Inorg. Chem. 2, 786–789. doi: 10.1007/pl00010648
Ratnani, R., Bohra, R., Srivastava, G., and Mehrotra, R. C. (1990). Crystal and molecular structure of μ-oxobis[bis(dipropyl dithiophosphato) oxomolybdenum(V)]. J. Crystallogr. Spectroscopic Res. 20, 541–544. doi: 10.1007/bf01221894
Reiss, J. (2000). Genetics of molybdenum cofactor deficiency. Hum. Genet. 106, 157–163. doi: 10.1007/s004390051023
Schulzke, C. (2016). “An overview of the synthetic strategies, reaction mechanisms and kinetics of model compounds relevant to molybdenum- and tungsten-containing enzymes,” in Molybdenum and Tungsten Enzymes: Bioinorganic Chemistry, Vol. 6, eds R. Hille, C. Schulzke, and M. L. Kirk (Cambridge: Royal Soc Chemistry, 1–7.
Schulzke, C., and Ghosh, A. C. (2014). “Molybdenum and tungsten oxidoreductase models,” in Bioinspired Catalysis, eds W. Weigand and P. Schollhammer (Weinheim: Wiley-VCH Verlag GmbH and Co. KGaA, 349–382.
Schulzke, C., and Samuel, P. P. (2011). Molybdenum and Tungsten Cofactor Model Chemistry. New York, NY: Nova Science Publishers Inc.
Sellmann, D., Grasser, F., Knoch, F., and Moll, M. (1992). Übergangsmetallkomplexe mit Schwefelliganden, LXXX. Synthese, Struktur, Oxotransfer- und redoxgekoppelte Kondensationsreaktionen von Molybdän-Oxo-Komplexen mit dem sterisch anspruchsvollen vierzähnigen Thioether-Thiolat-Liganden ‘buS4'2− (‘buS4'2- = 1,2-Bis(2-mercapto-3,5-di-t-butylphenylthio)ethan(2−)). Z. Naturforschung B 47:61. doi: 10.1515/znb-1992-0114
Sheldrick, G. (2015). SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. 71, 3–8. doi: 10.1107/S2053273314026370
Sheldrick, G. M. (2008). A short history of SHELX. Acta Cryst. A 64, 112–122. doi: 10.1107/S0108767307043930
Smit, J. P., Purcell, W., Roodt, A., and Leipoldt, J. G. (1993). Kinetics of the substitution reaction between aquaoxotetracyanomolybdate(IV) and cyanide/hydrogen cyanide. Polyhedron 12, 2271–2277. doi: 10.1016/S0277-5387(00)88267-4
Smith, P. D., Millar, A. J., Young, C. G., Ghosh, A., and Basu, P. (2000). Detection, isolation, and characterization of intermediates in oxygen atom transfer reactions in molybdoenzyme model systems. J. Am. Chem. Soc. 122, 9298–9299. doi: 10.1021/ja0008362
Subramanian, P., Kaul, B., and Spence, J. T. (1984). Modelling the molybdenum centers of the molybdenum hydroxylases. J. Mol. Catal. 23, 163–177. doi: 10.1016/0304-5102(84)80004-8
Sugimoto, H., Harihara, M., Shiro, M., Sugimoto, K., Tanaka, K., Miyake, H., et al. (2005). Dioxo-molybdenum(VI) and mono-oxo-molybdenum(IV) complexes supported by new aliphatic dithiolene ligands: new models with weakened MoO bond characters for the arsenite oxidase active site. Inorg. Chem. 44, 6386–6392. doi: 10.1021/ic050234p
Sugimoto, H., Siren, K., Tsukube, H., and Tanaka, K. (2003). Mono-dithiolene molybdenum(IV) complexes of cis-1,2-dicyano-1,2-ethylenedithiolate (mnt2−): new models for molybdenum enzymes. Eur. J. Inorg. Chem. 2003, 2633–2638. doi: 10.1002/ejic.200200638
Sugimoto, H., Tatemoto, S., Suyama, K., Miyake, H., Itoh, S., Dong, C., et al. (2009). Dioxomolybdenum(VI) complexes with Ene-1,2-dithiolate ligands: synthesis, spectroscopy, and oxygen atom transfer reactivity. Inorg. Chem. 48, 10581–10590. doi: 10.1021/ic901112s
Tatsumisago, M., Matsubayashi, G. E., Tanaka, T., Nishigaki, S., and Nakatsu, K. (1982). Synthesis, spectroscopy, and X-ray crystallographic analysis of (η3-dithiobenzoato-SCS′)oxo(trithioperoxybenzoato-S,S′S″)molybdenum(IV) and μ-oxo-bis[bis(dithiobenzoato-SS′)oxomolybdenum(V)]. J. Chem. Soc. Dalton Trans. 1982, 121–127. doi: 10.1039/DT9820000121
Tchouka, H., Meetsma, A., Molnár, G., Rechignat, L., and Browne, W. R. (2011). Solution and single crystal spectroscopic characterization of (PPh4)2[Fe(CN)5(imidazole)]·2H2O. J. Mol. Struct. 999, 39–48. doi: 10.1016/j.molstruc.2011.05.027
Thompson, R. L., Lee, S., Geib, S. J., and Cooper, N. J. (1993). Intramolecular bridge/terminal oxo exchange within oxomolybdenum [MO3]4+ complexes containing linear oxo bridges. Inorg. Chem. 32, 6067–6075. doi: 10.1021/ic00078a026
Tucci, G. C., Donahue, J. P., and Holm, R. H. (1998). Comparative kinetics of oxo transfer to substrate mediated by bis(dithiolene)dioxomolybdenum and -tungsten complexes. Inorg. Chem. 37, 1602–1608. doi: 10.1021/Ic971426q
Villata, L. S. A., Féliz, M. R., and Capparelli, A. L. (2000). Photochemical and catalytic properties of dimeric species of molybdenum(V). Coordinat. Chem. Rev. 196, 65–84. doi: 10.1016/S0010-8545(99)00186-1
Williams, B. R., Fu, Y., Yap, G. P., and Burgmayer, S. J. (2012). Structure and reversible pyran formation in molybdenum pyranopterin dithiolene models of the molybdenum cofactor. J. Am. Chem. Soc. 134, 19584–19587. doi: 10.1021/ja310018e
Keywords: artificial molybdenum active site, aliphatic dithiolene, oxygen atom transfer, Moco model, MoIV oxo complex
Citation: Ahmadi M, Fischer C, Ghosh AC and Schulzke C (2019) An Asymmetrically Substituted Aliphatic Bis-Dithiolene Mono-Oxido Molybdenum(IV) Complex With Ester and Alcohol Functions as Structural and Functional Active Site Model of Molybdoenzymes. Front. Chem. 7:486. doi: 10.3389/fchem.2019.00486
Received: 07 September 2018; Accepted: 24 June 2019;
Published: 11 July 2019.
Edited by:
Andrea Erxleben, National University of Ireland Galway, IrelandReviewed by:
Wolfgang Weigand, Friedrich Schiller University Jena, GermanyThomas Werner, Leibniz Institut für Katalyse (LG), Germany
Wolfram Willy Seidel, Institut für Chemie, Universität Rostock, Germany
Copyright © 2019 Ahmadi, Fischer, Ghosh and Schulzke. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carola Schulzke, Y2Fyb2xhLnNjaHVsemtlQHVuaS1ncmVpZnN3YWxkLmRl