 Manyi Yang
Manyi Yang Guoqiang Wang
Guoqiang Wang Jingxiang Zou
Jingxiang Zou Shuhua Li
Shuhua Li- Key Laboratory of Mesoscopic Chemistry of Ministry of Education, School of Chemistry and Chemical Engineering, Institute of Theoretical and Computational Chemistry, Nanjing University, Nanjing, China
The detailed mechanism of the AuCN-catalyzed annulation of salicylaldehyde (SA) and phenyl acetylene leading to isoflavanone-type complexes has been investigated via density functional theory (DFT) calculations. Reaction pathways and possible stationary points are obtained with the combined molecular dynamics and coordinate driving (MD/CD) method. Our calculations reveal that the cyanide ion promoted umpolung hydroacylation/intramolecular oxa-Michael addition mechanism is more favorable than the Au(I)/Au(III) redox mechanism proposed previously. In the umpolung mechanism, the hydroxyl of SA is found to strongly stabilize the cyanide ion involved intermediates and transition states via hydrogen bond interactions, while the Au(I) ion always acts as a counter cation. The overall reaction is exergonic by 41.8 kcal/mol. The hydroacylation of phenyl acetylene is the rate-determining step and responsible for the regioselectivity with a free energy barrier of 27.3 kcal/mol. These calculated results are in qualitative accord with the experimental findings.
Introduction
The catalytic annulation of chelating aldehydes and unsaturated hydrocarbons provides a general, highly efficient, and atom-economical strategy to synthesize complex carbocyclic and heterocyclic frameworks (Willis, 2010). Salicylaldehyde (SA) is a highly favored chelating aldehyde because of its ready availability as well as common ortho-hydroxy or alkoxy arylcarbonyl motifs in natural products and bioactive molecules. In recent years, starting from SA, various transition-metal-catalyzed Csp2-H activation and heterocyclization reactions have been developed to efficiently construct various important heterocycles of biologically active compounds and drug molecules (Shimizu et al., 2008; Zeng and Li, 2014; Baruah et al., 2016; Yang and Yoshikai, 2016). For example, the Rh(III)-catalyzed (Shimizu et al., 2008), Ru(II)-catalyzed (Baruah et al., 2016), Co(I)-diphosphine-catalyzed (Yang and Yoshikai, 2016) Csp2-H activation and annulation reactions of SA with monosubstituted and disubstituted alkyne have been reported for the synthesis of chromone derivatives.
Over the last two decades, homogeneous gold catalysis has developed very rapidly and broadly in organic synthesis due to its rich chemistry and fascinating reactivity. It has emerged as a very efficient method for rapid construction of complex and highly functionalized molecules (Fukuda and Utimoto, 1991; Teles et al., 1998; Hashmi et al., 2000a,b; Yao and Li, 2004; Nguyen et al., 2006; Boorman and Larrosa, 2011; Xie et al., 2014; Dorel and Echavarren, 2015; Joost et al., 2015; Qian and Zhang, 2015; Kumar and Nevado, 2017; Akram et al., 2018; Mandal and Datta, 2018; Matsumoto et al., 2018; Wu et al., 2018; Kreuzahler et al., 2019; Mascareñas et al., 2019). Very recently, starting with SA and aryl acetylene, Li and co-workers developed an AuCN-catalyzed system (Skouta and Li, 2007) to afford the isoflavanone-type frameworks, which have many possible applications in the synthesis of isoflavanone natural products. Although the AuCN-catalyzed annulation of SA and aryl acetylene can effectively construct isoflavanone-type frameworks, the reaction conditions are very harsh. It takes place at high temperature (150°C), and over-stoichiometric amounts of alkynes (3 equivalent) are required, as shown in Scheme 1A. Moreover, the detailed mechanism of this reaction is still elusive. We believe that the elucidation of the mechanism of this reaction is helpful for designing new and milder catalytic reaction systems.
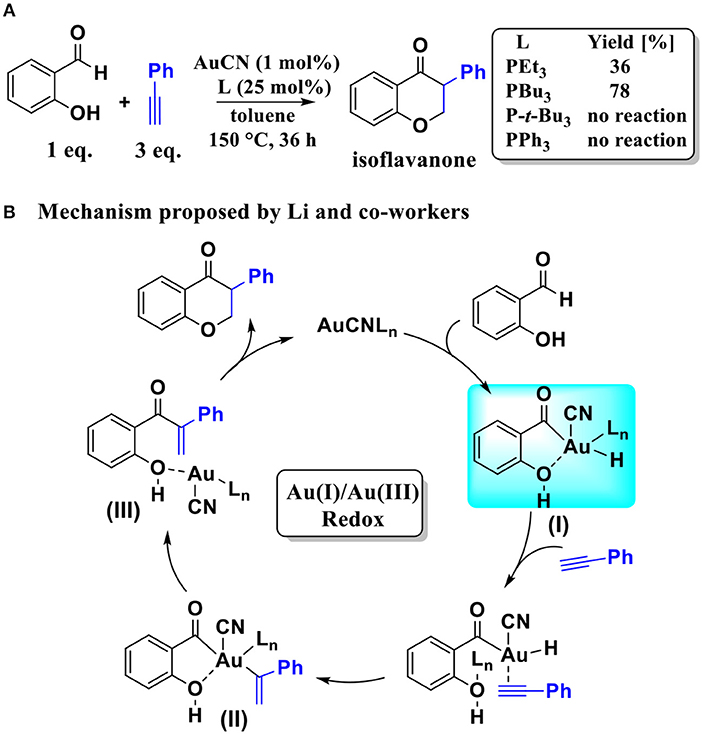
Scheme 1. (A) AuCN-catalyzed annulation of salicylaldehyde with aryl acetylene; (B) The Au(I)/Au(III) redox mechanism proposed by Li and co-workers.
Based on the regioselectivity of the AuCN-catalyzed annulation reaction, Li and co-workers suggested an Au(I)/Au(III) redox mechanism via Csp2-H activation of aldehyde, as shown in Scheme 1B. This mechanism starts with an oxidative addition of the aldehyde Csp2-H bond leading to an acyl Au(III) hydride I. Then a hydrometalation step occurs, followed by a reductive elimination process and then an intramolecular conjugate addition, affording the final isoflavanone derivative and regenerating the Au(I) catalyst. Experiments indicated that the phosphine ligands had significant influence on the yield of product. As shown in Scheme 1A, triethylphosphane (PEt3) and tributylphosphane (PBu3) exhibited moderate to good efficiency (36 and 78% yield, respectively), while other alternative ligands, such as triphenyphosphane (PPh3) and tri-tert-butylphosphane (P-t-Bu3), providing only trace amounts of product.
In the present work, we carry out density functional theory (DFT) calculations to investigate the mechanism of the AuCN-catalyzed annulation reaction between SA and phenyl acetylene using AuCN as the catalyst and PEt3 as the model ligand. The combined molecular dynamics and coordinate driving (MD/CD) method (Yang et al., 2017b, 2018) developed by us is applied to explore reaction pathways and determine the structures of all possible stationary points along reaction pathways. This method is a cost-effective theoretical tool for automatically searching low-energy reaction pathways for relatively large chemical reactions in both gas phase and solution. Our calculations indicate that the pathway initiated by the oxidative addition of aldehyde Csp2-H bond toward Au (I) center could be excluded, due to its high activation barrier. Instead, our calculations suggest a cyanide ion promoted umpolung hydroacylation/intramolecular oxa-Michael addition mechanism for this reaction.
Computational Details
Geometries of all stationary points, including minimum structures (reactants, intermediates, and products) and transition states (TSs), were fully optimized by the DFT calculations using the B3PW91 functional (Lee et al., 1988) with Grimme's dispersion correction (GD3BJ) (Grimme et al., 2010). The SDD (Figgen et al., 2005) basis set and associated effective core potentials were used for Au, 6-31+G(2d) basis set for P and 6-31+G(d) for other atoms (BS1). To obtain more accurate electronic energies, single-point energies of the optimized stationary points were recalculated (with the same functional) by employing the SDD basis set for Au and the 6-311+G(2d,p) basis set for all other atoms (BS2). The C-PCM Solvation Model (Cossi et al., 2003) was used as the implicit solvation model with the toluene as the solvent. For each TS, the intrinsic reaction coordinate (IRC) (Gonzalez and Schlegel, 1989) analysis was performed to verify whether the TS truly connects the reactant and the product. The Gibbs free energies were calculated at T = 423 K and 1.0 atm in toluene (corresponding to the experimental conditions) by the way described previously (Zeng and Li, 2011), in which the entropy of translational movement was evaluated with the method developed by Mammen et al. (1998). All calculations were performed with the Gaussian16 (Frisch et al., 2016) software package.
The MD/CD method implemented in the automated design of chemical reaction (ADCR) program (Yang et al., 2017a) is applied to locate all the stationary points along the most probable reaction pathways. Since the system is very large and the resulting lower-energy pathways will be further verified with more accurate calculations, we performed MD/CD calculations with the B3PW91 functional (also with GD3BJ) at a smaller basis set. In this basis set, the effective core potential LanL2DZ was used for Au and the 6-31G basis set was used for other atoms (BS3). The C-PCM model was used as the implicit solvation model with toluene as the solvent. In the MD/CD calculation, the MD simulations were skipped for all intermediate structures (due to their relatively rigid structures) and the energy cutoff 45 kcal/mol was used.
Activation free energy barriers (ΔG≠) used in this work are defined as the free energy difference between the TS and the most stable points (initial reactants and intermediates) along the energy profile. Given that the formation of the PEt3AuCN complex (from the initial AuCN catalyst and the PEt3 ligand) is highly exergonic, free energies discussed in this work are with respect to PEt3AuCN.
Results and Discussions
Au(I)/Au(III) Redox Mechanism
Initially, we investigated the “intuitive” Au(I)/Au(III) redox mechanism, which involves the direct oxidative addition of aldehyde Csp2-H bond to PEt3AuCN complex 1. As shown in Figure 1, the formation of a hydrogen bond between cyano group of PEt3AuCN and the hydroxyl of SA in the complex 5 is endergonic by 0.9 kcal/mol. However, the direct oxidative addition of the formyl Csp2-H bond of SA toward the Au(I) center via transition state TS5/Ia to form the Au(III) intermediate Ia is required to overcome a high activation barrier of 44.4 kcal/mol (relative to reactants), which is inconsistent with the experimental conditions that the reaction occurs at 150°C. Therefore, instead of Au(I)/Au(III) redox catalytic cycle proposed previously, there might be an energetically more favorable pathway.
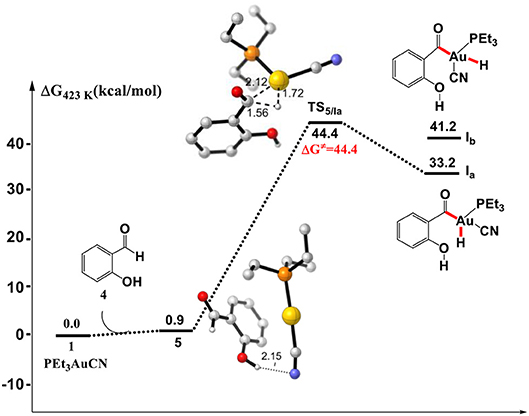
Figure 1. Gibbs free energy profile of the oxidative addition process. Relative free energies calculated at 423 K and 1 atm (with respect to separated reactants, in kcal/mol). All listed distances are in Å.
The Cyanide Ion Promoted Umpolung Pathway
Determination of the Active Catalyst
According to the previous experimental studies (Hormann et al., 1986), AuCN can readily react with a molecular phosphine ligand to generate a linear PEt3AuCN species 1. It is generally accepted that linear Au(I) could not associate with a second or third ligand because this is an entropy decreasing process (Schwerdtfeger et al., 2003; Carvajal et al., 2004). However, our calculations show that the association of PEt3AuCN with two additional PEt3 ligands to form a tetracoordinated Au(I) species 3 is exergonic by 9.9 kcal/mol (relative to complex 1, see Scheme 2. This result indicates the formation of the tetracoordinated Au (I) complex 3 is possible. In fact, in the related experimental studies (Skouta and Li, 2007), 25-fold amounts of phosphine ligand (with respect to AuCN) is required to achieve a good yield of the isoflavanone derivatives. These theoretical and experimental results suggest that the tetracoordinated Au (I) complex 3 could be formed under the experimental conditions. Although the reaction of Au (I) complex 3 with another PEt3 molecule could generate a zwitterionic intermediate (PEt3)4PAu+…CN−, this process is thermodynamically unfavorable (highly endergonic by 15.1 kcal/mol). Thus, we assume that the tetracoordinated complex 3 might be involved in the catalytic cycle. The elongation of Au-CN bond length in (PEt3)3PAuCN by 0.14 Å (compared to PEt3AuCN) indicates the Au-CN bond strength in (PEt3)3PAuCN is weakened, increasing the nucleophilic reactivity of cyanide ion.
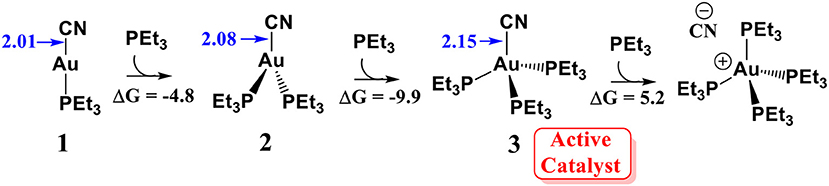
Scheme 2. Reaction energies (ΔG) for the association of PEt3AuCN with PEt3 ligand. All energies are in kcal/mol; All bond lengths are in Å.
HCN Promoted Formation of α-Cyano Carbanion Intermediate
With (PEt3)3AuCN 3 as the active catalyst, we found that the cyano group of catalyst 3 could react with the phenolic hydroxyl group of SA to liberate the HCN molecule, which then occurs nucleophilic addition toward the carbonyl of SA to generate a α-cyano carbanion intermediate 13. The Gibbs free energy profiles for these processes are presented in Figures 2, 3, respectively. The optimized geometries of the involved species along these reaction pathways are displayed in Figure 4.
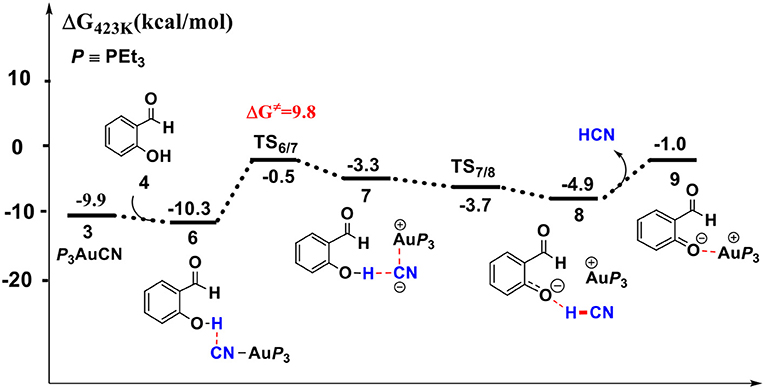
Figure 2. Gibbs free energy profile for the formation of free HCN molecule in toluene solution. Relative free energies (with respect to separated reactants) are given in kcal/mol.
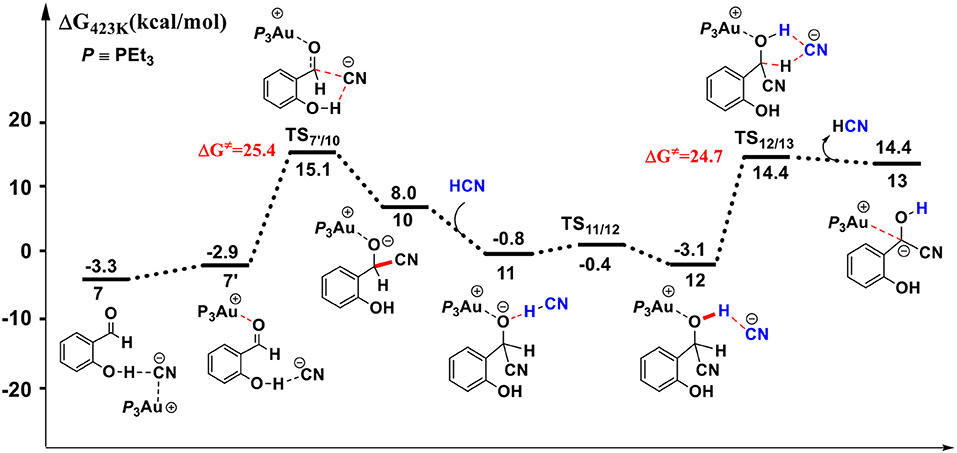
Figure 3. Gibbs free energy profile for the formation of the α-cyano carbanion intermediate 13 in toluene solution. Relative free energies (with respect to separated reactants) are given in kcal/mol.
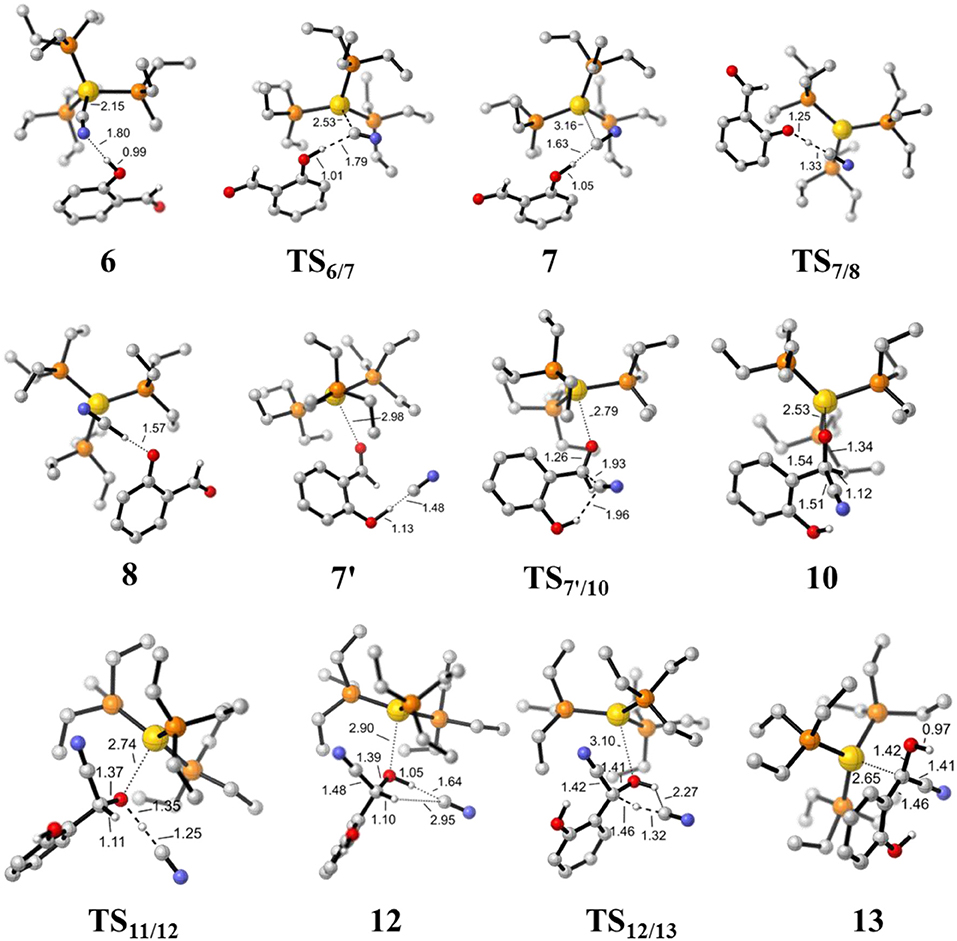
Figure 4. Optimized geometries of some species involved in the formation of the HCN and α-cyano carbanion intermediate 13. Hydrogen atoms except for those involved in the reaction are omitted for clarity. All bond lengths are in Å.
First, complexation of the catalyst 3 and SA forms the complex 6, which is stabilized by a O-H…N hydrogen bond interaction (dO−H…N = 1.80 Å) and exergonic by 0.4 kcal/mol, as shown in Figure 2. Then, the dissociation of cyanide ion from the Au(I) center in complex 6 leads to an [Au(I)]+…CN− ion-pair complex 7 (via the transition state TS6/7). Both the transition state TS6/7 and ion-pair complex 7 could be stabilized by strong hydrogen bond interaction with the phenolic hydroxyl of SA, in which the distances of O-H…CN− are 1.79 and 1.63 Å, respectively. This process is slightly endergonic by 7.0 kcal/mol with a barrier of 9.3 kcal/mol. Subsequently, complex 7 undergoes a proton transfer process from the acidic phenolic hydroxyl to the cyanide ion to generate a new ternary ion-pair complex 8, HCN-SA−-(PEt3)3Au+. The liberation of HCN from the complex 8 forms the complex 9, SA– – (PEt3)3Au+. Overall, the formation of the separated HCN and complex 9 is endergonic by 9.3 kcal/mol with a free energy barrier of only 9.8 kcal/mol (with respect to complex 6). These results indicate that a free HCN molecule may be generated under the experimental conditions (150°C).
For the species 7, it may convert into a new complex 7′, in which the cationic gold center of (PEt3)3Au+ is associated with the oxygen atom of carbonyl group (dAu−O = 2.86 Å) (see Figure 3). Cyanide ion in complex 7′ subsequently attacks the carbonyl carbon atom of SA (with the assistance of the phenolic hydroxy), leading to a gold-ligated alkoxide intermediate 10 via the transition state TS7′/10 (with a barrier of 25.4 kcal/mol). Then, the association of the basic alkoxide intermediate 10 and a free HCN (generated through the pathway described above) forms a hydrogen bond stabilized complex 11 (dO…H−CN = 1.39 Å). The following intramolecular proton transfer leads to an Au+-cyanohydrin-CN− complex 12 (via transition state TS11/12). The cyanide ion in complex 12, stabilized by hydrogen bond interaction with cyanohydrin, could abstract the α-H of cyanohydrin to form α-cyano carbanion intermediate 13 and regenerate the HCN simultaneously. The free energy barrier for the CN− assisted proton abstraction (via transition state T212/13) is 24.7 kcal/mol. In addition, the pathway involving SA-catalyzed intramolecular 1,2 H-shift of the gold-ligated alkoxide intermediate 10 could also form α-cyano carbanion intermediate 13 (via transition state TS12/13−SA, see Figure S1 for details). In summary, the HCN promoted carbonyl umpolung process is endergonic by 24.7 kcal/mol (relative to intermediate 6). The rate-limiting step for this stage is the nucleophilic attack of cyanide ion at the carbonyl carbon atom with an activation barrier of 25.4 kcal/mol.
Other reaction channels to form the alkoxide intermediate 10, starting from complex 6 or 9, could also be located with the MD/CD method (see Figure S2 for details). However, the related free energy barriers for these pathways are higher by 3~5 kcal/mol than the pathway listed in Figure 3. Therefore, the pathway, starting from complexes 7 or 7′ is responsible for the formation of alkoxide intermediate 10, which then converts into α-cyano carbanion intermediate 13.
Hydroacylation of Phenylacetylene
Addition of cyanide ion to SA leads to an umpolung of the carbonyl SA, and the corresponding α-cyano carbanion intermediate 13 could act as an acyl anion equivalent to react with phenylacetylene 14, forming a branched α-β unsaturated ketone complex 17 and regenerating the cyanide ion. The Gibbs free energy profile for this process is presented in Figure 5. The optimized geometries of some stationary points are displayed in Figure 6.
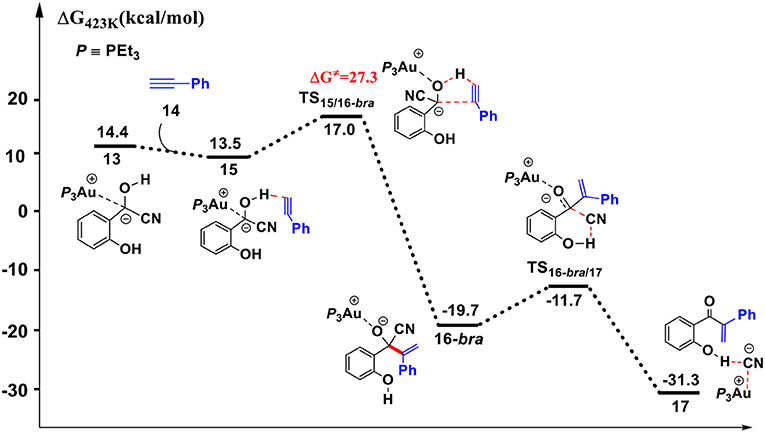
Figure 5. Gibbs free energy profile for the hydroacylation of phenylacetylene process. Relative free energies (with respect to separated reactants) are given in kcal/mol.
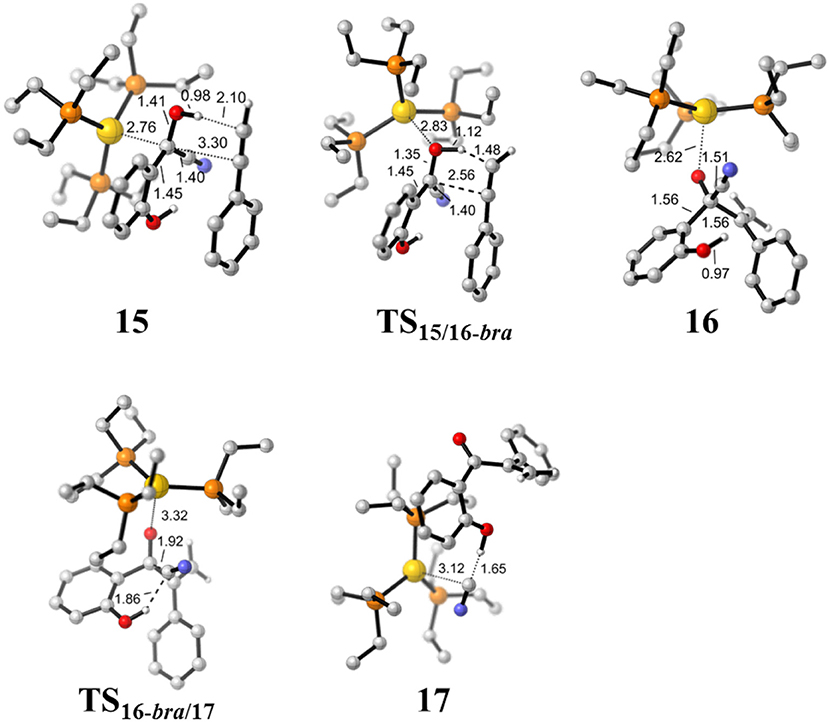
Figure 6. Optimized geometries of some species involved in the hydroacylation of phenylacetylene. All bond lengths are in Å.
First, complexation of the phenyl acetylene 14 with the α-cyano carbanion intermediate 13 forms an intermolecular complex 15. Then, a branched hydroacylation intermediate 16-bra could be generated via a concerted alkyne-Ene transition state (Piel et al., 2011; Schedler et al., 2013) (transition state TS15/16−bra), in which, the proton transfer from the α-cyano carbanion intermediate to the terminal carbon (C1) of phenylacetylene is accomplished with an nucleophilic attack of the acetyl anion center at the C2 position of phenyl acetylene. The formation of intermediate 16-bra is exergonic by 33.2 kcal/mol with a barrier of 27.3 kcal/mol. Subsequently, the elimination of the cyanide ion from intermediate 16, with the assistance of the phenolic hydroxyl (via transition state TS16-bra/17), forms a branched α-β unsaturated ketone complex 17 (Other possible reaction pathways starting from intermediate 16-bra are presented in Figure S3). This ternary intermediate 17 was stabilized by multiple non-covalent interactions, including the electrostatic interaction between the Au cation and cyanide ion (dAu…CN = 3.12 Å) and a strong hydrogen bond interaction (dO−H…CN− = 1.65 Å) formed between the cyanide ion and phenolic hydroxyl group. Along the energy profile described above, we can find that this hydroacylation process is exergonic by 45.7 kcal/mol, relative to the intermediates 13 and 14. The concerted alkyne-Ene process is the rate-determining step with a free energy barrier of 27.3 kcal/mol.
Intramolecular Oxa-Michael Addition
With the assistance of a basic cyanide ion, the branched α,β-unsaturated ketone proceeds through an intramolecular oxa-Michael addition to form the isoflavanone 20 and regenerate the active catalyst 3. The Gibbs free energy profile and the optimized geometries of involved stationary points along the reaction pathway are displayed in Figures 7, 8, respectively.
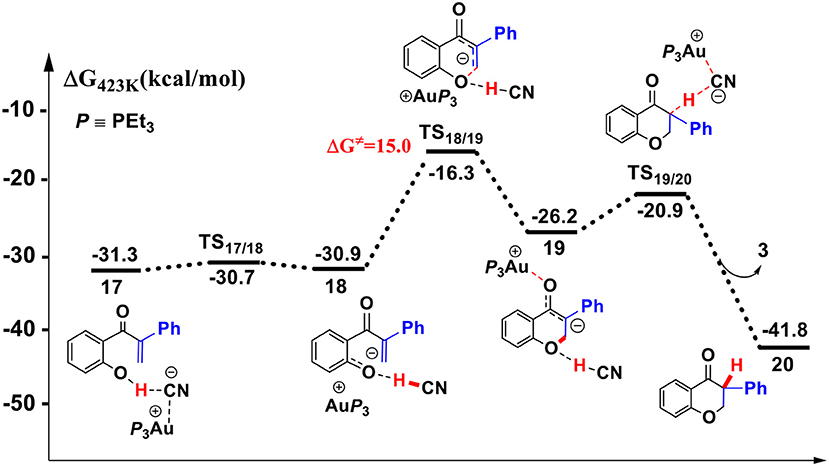
Figure 7. Gibbs free energy profile of the intramolecular oxa-Michael process in toluene solution. Relative free energies (with respect to separated reactants) are given in kcal/mol.
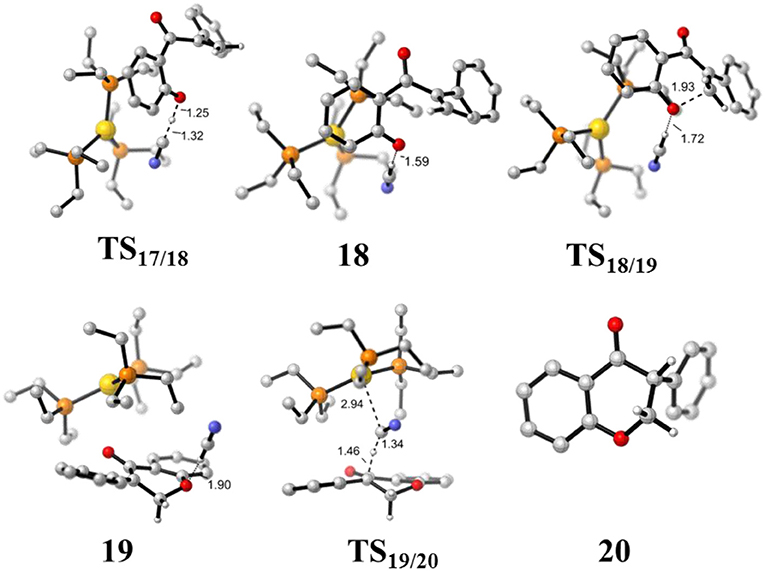
Figure 8. Optimized geometries of some species involved in the intramolecular oxa-Michael process. All bond lengths are in Å.
First, the deprotonation of the phenolic hydroxyl by the cyanide ion forms a complex 18 (via transition state TS17/18) containing phenol anion, HCN, and (PEt3)3Au cation. The formation of the complex 18 is slightly endergonic, in which the nucleophilicity of the oxygen atom of phenol is increased. Then, the complex 18 could convert into the complex 19 via an intramolecular oxa-Michael addition reaction (via transition state TS18/19). Finally, protonation of the enolate anion center with HCN forms the isoflavanone product 20 and regenerates the active catalyst (PEt3)3AuCN 3. The process described above is exergonic by 10.5 kcal/mol (relative to intermediate 17), and the rate-limiting step is the intramolecular nucleophilic addition reaction of complex 18 with a free energy barrier of 15.0 kcal/mol.
In summary, our calculations reveal that a cyanide ion promoted umpolung hydroacylation/intramolecular oxa-Michael addition mechanism, as shown in Scheme 3, is more favorable than the Au(I)/Au(III) redox mechanism proposed previously. The new pathway contains four stages described above. The overall reaction is exergonic by 41.8 kcal/mol. The hydroacylation of phenyl acetylene is the rate-determining step with a free energy barrier of 27.3 kcal/mol. The free energy barrier is in accord with the experimental fact that the studied reaction takes place at 150°C.
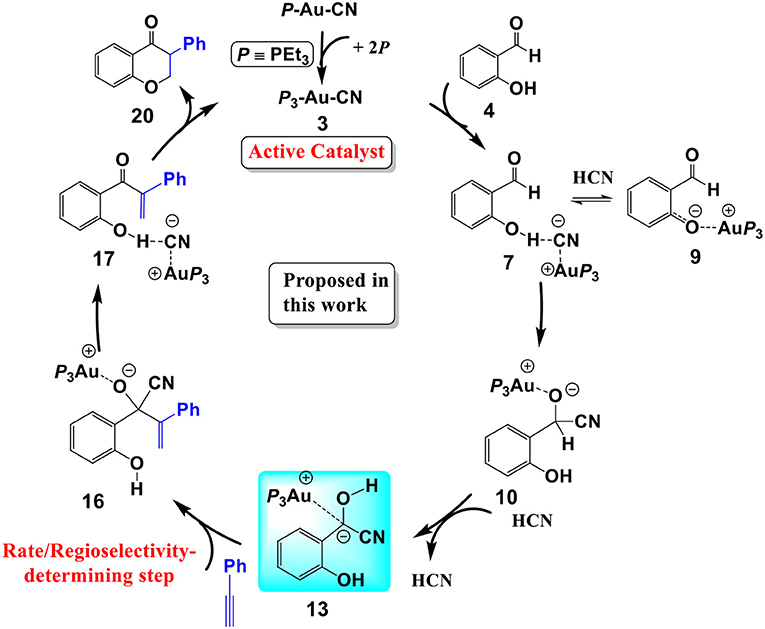
Scheme 3. The cyanide ion promoted umpolung hydroacylation/intramolecular oxa-Michael addition mechanism proposed in this work.
Possible Theoretical Explanations of Experimental Findings
To understand the experimental fact that only the isoflavanone product is observed, we also explored the pathway involving the formation of linear enone intermediate 16-lin (related to the formation of flavanone product) via TS15/16-lin (see Scheme 4). The free energy barrier of this transition state is 31.0 kcal/mol, which is higher than TS15/16-bra by 3.7 kcal/mol. These results indicate that the formation of isoflavanone product is dynamically favorable, which is in accord with the experimental observed regioselectivity.
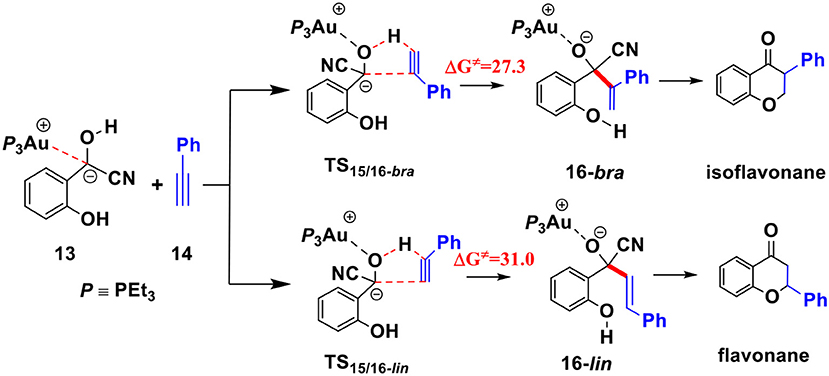
Scheme 4. Two possible pathways for the hydroacylation of phenylacetylene; Activation free energy barriers (ΔG≠) are given in kcal/mol.
In addition to PEt3, we also investigated the impact of the ligand on the reactivity of this annulation reaction, including PBu3, and PPh3. Key transition states with relative higher free energies (TS7′/10, TS12/13, TS15/16-bra), involved in the pathway, were determined to evaluate the ligand effect. The free energy barriers of these different TSs with different phosphines are listed in Table 1 and the related optimized geometries are presented in Figure S4. The calculated results show that the rate-determining step for the studied reaction is the hydroacylation of phenyl acetylene (TS15/16-bra) with free energy barriers of 27.3 and 32.0 kcal/mol, respectively, when the ligands are PEt3, and PPh3, respectively. However, for the PBu3 ligand, both the HCN-assisted 1,2 H-shift (TS12/13) and hydroacylation of phenyl acetylene (TS15/16-bra) may be the rate-determining steps, with comparable barriers (25.0 and 24.9 kcal/mol, respectively). The calculated energy barriers are in qualitative accord with the experimental fact that ligands PEt3 and PBu3 provided moderate to good yield (36 and 78%, respectively), while for the ligand PPh3, only trace amounts of products were observed.
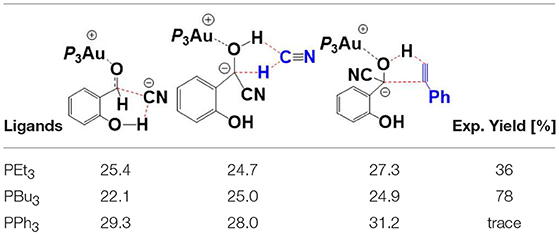
Table 1. Comparison of the free energy barriers (kcal/mol) of different TSs with various ligands (PEt3, PBu3, and PPh3).
The competing benzoin condensation (Wöhler, 1832; Zinin, 1839, 1840) pathway of SA via α-cyano carbanion intermediate 13 (see Scheme 5) is also possible. The free energy barrier of the rate-limiting step in this competing pathway is 24.7 kcal/mol (see Figures S5, S6 for more details). However, this process is almost thermally neutral (slightly exergonic by 0.6 kcal/mol, relative to separated reactants), which is incomparable with the pathway of the annulation of SA and phenyl acetylene (described above, exergonic by 41.8 kcal/mol). This result indicates that the generation of the isoflavanone-type product is thermodynamically much more favorable. Our calculations are in accord with the experimental finding that excessive amount of phenyl acetylene (3-fold amounts of SA) are required to improve the yield of the isoflavanone product.
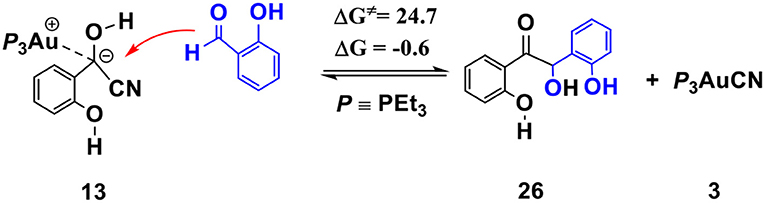
Scheme 5. Activation free energy barrier (ΔG≠) and reaction energy (ΔG) of the competitive benzoin condensation pathway of SA via α-cyano carbanion intermediate 13 (all energies are given in kcal/mol).
Conclusions
We have performed DFT calculations assisted by the MD/CD method to explore the detailed mechanism of the AuCN-catalyzed annulation of salicylaldehyde (SA) and phenyl acetylene. Our calculations reveal that a cyanide ion promoted umpolung hydroacylation/intramolecular oxa-Michael addition mechanism is more favorable than the Au(I)/Au(III) redox mechanism proposed previously. The new proposed pathway contains four stages, as shown in Scheme 3: (1) the association of AuCN with three PEt3 molecule generates an active catalyst (PEt3)3AuCN 3; (2) The nucleophilic attack of the cyanide ion on the carbonyl in SA and subsequent HCN-assisted 1,2 H-shift provide the α-cyano carbanion intermediate 13, which is a key intermediate involved in this mechanism; (3) the hydroacylation of phenylacetylene, followed by the elimination of the cyanide ion, leads to a branched α-β unsaturated ketone 17; (4) the intramolecular conjugate addition of the α,β unsaturated ketone forms the isoflavanone product 20 and regenerates the active catalyst 3. The overall reaction is exergonic by 41.8 kcal/mol. The hydroacylation of phenyl acetylene is the rate-determining step and responsible for the regioselectivity with a free energy barrier of 27.3 kcal/mol at T = 423 K and 1.0 atm in toluene. In the umpolung mechanism, the hydroxyl of SA is found to strongly stabilize the cyanide ion involved intermediates and transition states via hydrogen bond interactions, while the Au(I) ion always acts as a counter cation. Our results are in qualitative accord with the experimental findings. The results provide important insight into Au(I)-catalyzed annulation of SA and aryl acetylenes, which may be useful in designing more effective catalysts for synthesis of heterocyclic frameworks.
Data Availability
All datasets generated for this study are included in the manuscript/Supplementary Files.
Author Contributions
The work was completed by cooperation of all authors. MY, GW, JZ, and SL were responsible for the study of concept and design of the project. MY performed corresponding calculations. MY, GW, JZ, and SL drafted and revised the manuscript.
Funding
This project was supported by the National Natural Science Foundation of China (Grant Nos. 21833002, 21673110, and 21873046) and the program B for outstanding PhD candidate of Nanjing University. All calculations in this work have been done on the IBM Blade cluster system in the High-Performance Computing Center of Nanjing University.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00557/full#supplementary-material
References
Akram, M. O., Banerjee, S., Saswade, S. S., Bedi, V., and Patil, N. T. (2018). Oxidant-free oxidative gold catalysis: the new paradigm in cross-coupling reactions. Chem. Comm. 54, 11069–11083. doi: 10.1039/C8CC05601C
Baruah, S., Kaishap, P. P., and Gogoi, S. (2016). Ru(Ii)-Catalyzed C-H Activation and Annulation of Salicylaldehydes with Monosubstituted and Disubstituted Alkynes. Chem. Comm. 52, 13004–13007. doi: 10.1039/C6CC07204F
Boorman, T. C., and Larrosa, I. (2011). Gold-Mediated C-H Bond Functionalisation. Chem. Soc. Rev. 40, 1910–1925. doi: 10.1039/C0CS00098A
Carvajal, M. A., Novoa, J. J., and Alvarez, S. (2004). Choice of coordination number in D10 complexes of group 11 metals. J. Am. Chem. Soc. 126, 1465–1477. doi: 10.1021/ja038416a
Cossi, M., Rega, N., Scalmani, G., and Barone, V. (2003). Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comp. Chem. 24, 669–681. doi: 10.1002/jcc.10189
Dorel, R., and Echavarren, A. M. (2015). Gold(I)-catalyzed activation of alkynes for the construction of molecular complexity. Chem. Rev. 115, 9028–9072. doi: 10.1021/cr500691k
Figgen, D., Rauhut, G., Dolg, M., and Stoll, H. (2005). Energy-consistent pseudopotentials for group 11 and 12 atoms: adjustment to multi-configuration Dirac–Hartree–Fock Data. Chem. Phys. 311, 227–244. doi: 10.1016/j.chemphys.2004.10.005
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2016). Gaussian 16,Revision A.03. Wallingford, CT: Gaussian, Inc.
Fukuda, Y., and Utimoto, K. (1991). Effective transformation of unactivated alkynes into ketones or acetals with a Gold(Iii) Catalyst. J. Org. Chem. 56, 3729–3731. doi: 10.1021/jo00011a058
Gonzalez, C., and Schlegel, H. B. (1989). An improved algorithm for reaction path following. J. Chem. Phys. 90, 2154–2161. doi: 10.1063/1.456010
Grimme, S., Antony, J., Ehrlich, S., and Krieg, H. (2010). A consistent and accurate ab initio parametrization of density functional dispersion correction (Dft-D) for the 94 elements H-Pu. J. Chem. Phys. 132:154104. doi: 10.1063/1.3382344
Hashmi, A. S. K., Frost, T. M., and Bats, J. W. (2000a). Highly selective gold-catalyzed arene synthesis. J. Am. Chem. Soc. 122, 11553–11554. doi: 10.1021/ja005570d
Hashmi, A. S. K., Schwarz, L., Choi, J.-H., and Frost, T. M. (2000b). A new gold-catalyzed C–C bond formation. Angew. Chem. Int. Ed. 39, 2285–2288. doi: 10.1002/1521-3773(20000703)39:13<2285::AID-ANIE2285>3.0.CO;2-F
Hormann, A. L., Shaw, C. F., Bennett, D. W., and Reiff, W. M. (1986). Solid-state structure and solution equilibria of cyano(Triethylphosphine)Gold(I), Inorg. Chem. 25, 3953–3957. doi: 10.1021/ic00242a025
Joost, M., Amgoune, A., and Bourissou, D. (2015). Reactivity of gold complexes towards elementary organometallic reactions. Angew. Chem. Int. Ed. 54, 15022–15045. doi: 10.1002/anie.201506271
Kreuzahler, M., Daniels, A., Wolper, C., and Haberhauer, G. (2019). 1,3-Chlorine Shift to a vinyl cation: a combined experimental and theoretical investigation of the E-selective gold(I)-catalyzed dimerization of chloroacetylenes. J. Am. Chem. Soc. 141, 1337–1348. doi: 10.1021/jacs.8b11501
Kumar, R., and Nevado, C. (2017). Cyclometalated Gold(Iii) complexes: synthesis, reactivity, and physicochemical properties. Angew. Chem. Int. Ed. 56, 1994–2015. doi: 10.1002/anie.201607225
Lee, C., Yang, W., and Parr, R. G. (1988). Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 37, 785–789. doi: 10.1103/PhysRevB.37.785
Mammen, M., Shakhnovich, E. I., Deutch, J. M., and Whitesides, G. M. (1998). Estimating the entropic cost of self-assembly of multiparticle hydrogen-bonded aggregates based on the cyanuric acid·melamine lattice. J. Org. Chem. 63, 3821–3830. doi: 10.1021/jo970944f
Mandal, N., and Datta, A. (2018). Gold(I)-catalyzed intramolecular diels–alder reaction: evolution of trappable intermediates via asynchronous transition states. J. Org. Chem. 83, 11167–11177. doi: 10.1021/acs.joc.8b01752
Mascareñas, J. L., Varela, I., and López, F. (2019). Allenes and derivatives in gold(I)- and platinum(Ii)-catalyzed formal cycloadditions. Acc. Chem. Res. 52, 465–479. doi: 10.1021/acs.accounts.8b00567
Matsumoto, C., Yamada, M., Dong, X., Mukai, C., and Inagaki, F. (2018). The gold-catalyzed formal hydration, decarboxylation, and [4+2] cycloaddition of alkyne derivatives featuring L2/Z-Type diphosphinoborane ligands. Chem. Lett. 47, 1321–1323. doi: 10.1246/cl.180610
Nguyen, R.-V., Yao, X., and Li, C.-J. (2006). Highly efficient gold-catalyzed atom-economical annulation of phenols with dienes. Org. Lett. 8, 2397–2399. doi: 10.1021/ol0607692
Piel, I., Steinmetz, M., Hirano, K., Fröhlich, R., Grimme, S., and Glorius, F. (2011). Hoch asymmetrische nhc-katalysierte hydroacylierung nichtaktivierter alkene. Angew. Chem. Int. Ed. 123, 5087–5091. doi: 10.1002/ange.201008081
Qian, D., and Zhang, J. (2015). Gold-catalyzed cyclopropanation reactions using a carbenoid precursor toolbox. Chem. Soc. Rev. 44, 677–698. doi: 10.1039/C4CS00304G
Schedler, M., Wang, D. S., and Glorius, F. (2013). Nhc-catalyzed hydroacylation of styrenes. Angew. Chem. Int. Ed. 52, 2585–2589. doi: 10.1002/anie.201209291
Schwerdtfeger, P., Hermann, H. L., and Schmidbaur, H. (2003). Stability of the Gold(I)–phosphine bond. a comparison with other group 11 elements. Inorg. Chem. 42, 1334–1342. doi: 10.1021/ic026098v
Shimizu, M., Tsurugi, H., Satoh, T., and Miura, M. (2008). Rhodium-catalyzed oxidative coupling between salicylaldehydes and internal alkynes with C?H bond cleavage to produce 2,3-disubstituted chromones. Chem. Asian J. 3, 881–886. doi: 10.1002/asia.200800037
Skouta, R., and Li, C. J. (2007). Gold(I)-catalyzed annulation of salicylaldehydes and aryl acetylenes as an expedient route to isoflavanones. Angew. Chem. Int. Ed. 46, 1117–1119. doi: 10.1002/anie.200603495
Teles, J. H., Brode, S., and Chabanas, M. (1998). Cationic Gold(I) complexes: highly efficient catalysts for the addition of alcohols to alkynes. Angew. Chem. Int. Ed. 37, 1415–1418. doi: 10.1002/(SICI)1521-3773(19980605)37:10<1415::AID-ANIE1415>3.0.CO;2-N
Willis, M. C. (2010). Transition Metal Catalyzed Alkene and Alkyne Hydroacylation. Chem. Rev. 110, 725–748. doi: 10.1021/cr900096x
Wöhler, L. (1832). Untersuchungen ber das radikal der benzoesäure. Annalen Pharmacie 3, 249–282. doi: 10.1002/jlac.18320030302
Wu, H., Zhao, T., and Hu, X. (2018). Friedel-crafts reaction of N,N-dimethylaniline with alkenes catalyzed by cyclic diaminocarbene-Gold(I) complex. Sci Rep. 8:11449. doi: 10.1038/s41598-018-29854-0
Xie, J., Pan, C., Abdukader, A., and Zhu, C. (2014). Gold-Catalyzed C(Sp3)-H Bond Functionalization. Chem. Soc. Rev. 43, 5245–5256. doi: 10.1039/C4CS00004H
Yang, J., and Yoshikai, N. (2016). Cobalt-catalyzed annulation of salicylaldehydes and alkynes to form chromones and 4-chromanones. Angew. Chem. Int. Ed. 55, 2870–2874. doi: 10.1002/anie.201510999
Yang, M., Yang, L., Wang, G., Zhou, Y., Xie, D., and Li, S. (2018). Combined molecular dynamics and coordinate driving method for automatic reaction pathway search of reactions in solution. J. Chem. Theory Comput. 14, 5787–5796. doi: 10.1021/acs.jctc.8b00799
Yang, M., Zou, J., Wang, G., and Li, S. (2017a). Adcr Program,version1.0. Nanjing: Nanjing University.
Yang, M., Zou, J., Wang, G., and Li, S. (2017b). Automatic reaction pathway search via combined molecular dynamics and coordinate driving method. J. Phys. Chem. A. 121, 1351–1361. doi: 10.1021/acs.jpca.6b12195
Yao, X., and Li, C.-J. (2004). Highly efficient addition of activated methylene compounds to alkenes catalyzed by gold and silver. J. Am. Chem. Soc. 126, 6884–6885. doi: 10.1021/ja0482637
Zeng, G., and Li, S. (2011). Insights into dehydrogenative coupling of alcohols and amines catalyzed by a (Pnn)-Ru(Ii) hydride complex: unusual metal-ligand cooperation. Inorg Chem. 50, 10572–10580. doi: 10.1021/ic200205e
Zeng, H., and Li, C. J. (2014). A Complete Switch of the Directional Selectivity in the Annulation of 2-Hydroxybenzaldehydes with Alkynes. Angew. Chem. Int. Ed. 53, 13862–13865. doi: 10.1002/anie.201407589
Zinin, N. (1839). Beiträge zur kenntniss einiger verbindungen aus der benzoylreihe. Annalen Pharmacie 31, 329–332. doi: 10.1002/jlac.18390310312
Keywords: DFT, MD/CD, cyanide ion, umpolung, hydroacylation
Citation: Yang M, Wang G, Zou J and Li S (2019) Mechanistic Insight Into the AuCN Catalyzed Annulation Reaction of Salicylaldehyde and Aryl Acetylene: Cyanide Ion Promoted Umpolung Hydroacylation/Intramolecular Oxa-Michael Addition Mechanism. Front. Chem. 7:557. doi: 10.3389/fchem.2019.00557
Received: 31 May 2019; Accepted: 22 July 2019;
Published: 06 August 2019.
Edited by:
Zexing Cao, Xiamen University, ChinaReviewed by:
Ming Lei, Beijing University of Chemical Technology, ChinaZhixiang Wang, University of Chinese Academy of Sciences, China
Copyright © 2019 Yang, Wang, Zou and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuhua Li, c2h1aHVhQG5qdS5lZHUuY24=
†These authors have contributed equally to this work