 Christian A. Juillerat
Christian A. Juillerat Vancho Kocevski2,3
Vancho Kocevski2,3 Hans-Conrad zur Loye
Hans-Conrad zur Loye- 1Department of Chemistry and Biochemistry, University of South Carolina, Columbia, SC, United States
- 2Center for Hierarchical Wasteform Materials (CHWM), University of South Carolina, Columbia, SC, United States
- 3Nuclear Engineering Program, University of South Carolina, Columbia, SC, United States
Single crystals of four new layered uranyl phosphates, including three oxyfluoride-phosphates, were synthesized by molten flux methods using alkali chloride melts, and their structures were determined by single-crystal X-ray diffraction. Cs11[(UO2)12(PO4)3O13] (1) and Rb11[UO2)12(PO4)3O12F2] (2) contain uranyl phosphate layers exhibiting a new sheet topology that can be related to that of β-U3O8, while Cs4.4K0.6[(UO2)6O4F(PO4)4(UO2)] (3) and Rb4.4K0.6[(UO2)6O4F(PO4)4(UO2)] (4) contain layers of a known isomer of the prominent phosphuranylite topology. The location of the fluorine in structures 2-4 is discussed using bond valence sums. First principles calculations were used to explore why a pure oxide structure is obtained for the Cs containing phase (1) and in contrast an oxyfluoride phase for the Rb containing phase (2). Ion exchange experiments were performed on 1 and 2 and demonstrate the ability of these structures to exchange approximately half of the parent alkali cation with a target alkali cation in an aqueous concentrated salt solution. Optical measurements were performed on 1 and 2 and the UV-vis and fluorescence spectra show features characteristic of the uranyl group.
Introduction
Nuclear power has been well-established for several decades and, nonetheless, studies continue to develop a deeper understanding of the nuclear fuel cycle, including exploring improved methods of both long-term and short-term waste storage (zur Loye et al., 2018), and continuing to investigate the processes of radionuclide leaching into surrounding ecosystems. For these reasons, it is advantageous to further expand our understanding of uranium coordination chemistry, specifically in extended structures, as this can give us insights in understanding intermediate phases in the nuclear waste cycle, identifying potential structures useful in nuclear waste processing or storage, and possible pathways within the environment for the migration of U6+ and other actinides.
Nature often gives a good indication of what chemical compounds can be made synthetically. For example, uranium containing minerals present a few prominent sheet anion topologies that can be observed both in minerals and synthetic compounds. In Lussier et al. (2016) most recent review of hexavalent uranium compounds, autunite, phosphuranylite, and uranophane are significant minerals classes with 40 proposed autunite minerals, 16 phosphuranylite, and 10 uranophane minerals. Phosphuranylite and autunite minerals are the primary classes of phosphorus containing minerals, as phosphorus bearing uranium minerals make up nearly a quarter of all identified uranium minerals. The uranophane topology is specifically prominent among silicates, but as in the phosphuranylite topology the tetrahedrally coordinated Si or P sites can be replaced by other tetrahedrally—or even trigonal pyramidal or trigonal planar—coordinating elements. These sheet anion-topologies common among minerals have also been observed in numerous synthetic compounds including 38 belonging to the autunite, 18 belonging to the uranophane, and 16 belonging to the phosphuranylite classes (Lussier et al., 2016; Wang et al., 2017; Juillerat et al., 2018a;Juillerat and zur Loye, 2019).
While nature certainly gives a good indication of what we might be able to synthesize in the laboratory, many additional sheet topologies outside of those found in minerals are also reported [62 reported in Lussier et al. (2016)]. All of the discussed sheet topologies so far have been for uranium oxide compounds and one way to expand the number of known sheet topologies, and thus our understanding of uranium chemistry, is to partially exchange oxygen within these sheets with other anions such as a halides or sulfides, although in this paper we will only discuss halides. While one may expect to make radically different sheet structures using Cl− due to the commonly terminal nature and large size of this anion, causing it to stick out of the plane of the sheet, as seen in K4U5O16Cl2 and Cs5U7O22Cl3 (Read et al., 2014), the inclusion of F− could lead to new sheet structures or to those already observed in oxides. This arises partially due to the similarity in size of O and F, and examples can be seen in the existence of both rare earth oxides and oxyfluorides that adopt the prominent apatite structure (Latshaw et al., 2014, 2015). In rare earth silicates, the coordination of the rare earth to F limits the available connectivity to the silicate tetrahedra, as SiO3F tetrahedra are unreported in crystalline structures (Leinenweber et al., 2005; Morrison et al., 2016). Although PO3F tetrahedra exist, i.e., Sr(PO3F), they have yet to be reported in uranium extended structures, although it is unclear whether this is due to chemical principles or whether the proper conditions for this structure motif have yet to be explored.
Herein we present the synthesis and structural characterization of two examples of uranium oxyfluorides that adopt the phosphuranylite topology, Cs4.4K0.6[(UO2)6O4F(PO4)4(UO2)] (3) and Rb4.4K0.6[(UO2)6O4F(PO4)4(UO2)] (4), and a new sheet anion topology that is observed for both a pure oxide, Cs11[(UO2)12(PO4)3O13] (1), and an oxyfluoride, Rb11[UO2)12(PO4)3O12F2] (2).
Experimental
Synthesis
Compounds 1-4 were synthesized via molten flux methods using alkali chloride fluxes (Bugaris and zur Loye, 2012; Juillerat et al., 2019a). For all reactions UF4 (International Bio-Analytical Industries, powder, ACS grade) was used as the uranium starting material, AlPO4 (Alfa Aesar, powder, 99.99%) was used as the phosphate source, and an alkali halide, CsCl (Alfa Aesar, powder, 99.99%), KCl (Mallinckrodt Chemicals, powder, 99.6%), or RbCl (Alfa Aesar, powder, 99.8%), or a mix thereof was used as a flux. Caution! Although the uranium precursor used contained depleted uranium, standard safety measures for handling radioactive substances must be followed. Generally, all solid reactants were loaded into either an alumina or platinum crucible and heated to 875°C in 1.5 h, held at this temperature for 12 h, then cooled at 6°C/h to 550 or 450°C depending on the melting point of the flux. After slow cooling, the furnace was shut off and allowed to rapidly cool to room temperature before sonicating the reaction mixtures in water to remove the flux and isolate the crystalline products by vacuum filtration.
Cs11[(UO2)12(PO4)3O13] (1) was synthesized by loading 1 mmol UF4, 0.25 mmol AlPO4, and 20 mmol of CsCl in a platinum crucible with a loose-fitting platinum lid and was slow cooled to 550°C. The reaction produced red tablets (Figure 1) in a nearly quantitative yield with no identifiable by-products. Rb11[UO2)12(PO4)3O12F2] (2) was obtained by loading 0.5 mmol UF4, 0.125 mmol AlPO4, and 20 mmol of RbCl into a small alumina crucible in a concrete holder with a larger inverted crucible covering it. This mixture was heated as mentioned above and slow cooled to 550°C and produced similar looking orange-red tablets as in 1 in a nearly quantitative yield with no identifiable byproducts. Cs4.4K0.6[(UO2)6O4F(PO4)4(UO2)] (3) and Rb4.4K0.6[(UO2)6O4F(PO4)4(UO2)] (4) were obtained from reactions of 0.5 mmol UF4, 0.2 mmol AlPO4, and 5 mmol CsCl or RbCl and 5 mmol KCl loaded into an alumina crucible covered with an alumina plate held in place by rubber cement. These were heated and slow cooled to 450°C and produced a yellow crystalline product identified as a mixture of the newly reported F containing phases and either CsxK4−x[(UO2)3(PO4)2O2] orRb1.4K2.6[(UO2)3(PO4)2O2] that are visibly indistinguishable. The purity and identity of the products were determined by powder X-ray diffraction (PXRD) using a Bruker D2 Phaser equipped with a LYNXEYE silicon strip detector using a Cu Kα (λ = 1.54056 Å) source.

Figure 1. Optical images of orange-red tablet shaped single crystals of (A) Cs11[(UO2)12(PO4)3O13] (1) and (B) Rb11[UO2)12(PO4)3O12F2] (2).
Structure
The reported structure solutions were obtained from single crystal X-ray diffraction (SXRD) data collected on a Bruker D8 QUEST diffractometer equipped with an Incoatec IμS 3.0 microfocus radiation source (Mo Kα, λ = 0.71073 Å) and a PHOTON II area detector. The reduction absorption correction was applied to the raw data using SAINT+ and SADABS within the APEX3 software (Bruker, 2015). The SHELXL suite was used within the OLEX2 GUI to solve the structure using SHELXT and refine the solution using SHELXL (Sheldrick, 2015a,b). The TWINROTMAP functionality in PLATON was used to check for missed symmetry elements and twin laws, where 3 and 4 were both refined as two component inversion twins with a significant volume fraction of 0.414(6) for 3 and a minor twin component of 0.084(8) in 4 (Spek, 2009). Full crystallographic information can be found in Table 1.

Table 1. Crystallographic details of structures 1-4.
In all structures the refinement of the U sites is straightforward, while in all structures there is disorder among the alkali cation sites, and in structures 1 and 2 there is disorder in one of the phosphate tetrahedra. Generally, the disorder in the alkali sites was treated by freely refining the sites as Cs or Rb as appropriate, and if less than one, then it was assumed that either the sites is shared by a smaller alkali cation, K+ in 3 and 4, or a disordered site across multiple positions. The presence of significant nearby electron density peaks suggests a disordered site, while the absence of these suggests sharing of the site between Cs/K or Rb/K. Mixed sites, and multiple disordered sites were constrained to occupancies of one using free variables or SUMP commands in cases of more complicated disorder, and the use of ISOR and EADP commands were implemented to constrain thermal parameters. The full details of the structure refinements are located in Supporting Information and the checkoff report is contained in Supplementary Data Sheet 1.
In structures 1 and 2 the P3 site is half occupied because it is disordered across a mirror plane and the two disordered sites are too close to both be fully occupied sites. This is also true for O23, O27, O28 which are coordinated to P3 and O29 coordinated to P1 in 1, and O28A and F28B in 2. Labeling all sites within the coordination sphere of the U sites as O in structures 2-4, does not result in charge balance, as there is an excess of negative charge. This could not be resolved by reasonable models of the alkali cation disorder and this observation, along with the identification of F in all three structures by EDS in both powdered and singly crystalline forms, confirms the presence of F. While the fluorine site could be easily located in structure 2 by using bond valence sums (BVS) and knowledge of U coordination chemistry (discussed in structure description), it was not easily identified in 3 and 4. In 3 and 4 the O3 site was fixed as a half occupied O/F shared site to maintain charge balance in the crystallographic solution, and this arbitrary assignment will be discussed in later sections.
EDS was used to verify the presence of F in 2-4 and all other elements present in each single crystal used for structure determination as well as in bulk powder samples of 1 and 2. Data were collected on a TESCAN Vega-3 SBU equipped with an EDS detector.
Optical Spectroscopy
UV-vis and fluorescence measurements were performed on bulk powder samples of 1 and 2 using a PerkinElmer Lambda 35 UV-vis scanning spectrophotometer equipped with an integrating sphere and a PerkinElmer LS55 Luminescence spectrometer. The UV-vis diffuse reflectance data were internally converted using the Kulbelka-Munk equation and then normalized (Kubelka and Munk, 1931). Fluorescence excitation spectra were collected at emission wavelengths of 574 and 564 nm for 1 and 2, respectively, and emission spectra were collected at an excitation wavelength of 437 nm for both 1 and 2.
Ion Exchange
Ion exchange experiments were performed on powder and single crystalline samples of Cs11[(UO2)12(PO4)3O13] (1) and Rb11[UO2)12(PO4)3O12F2] (2) where 20 mg of sample was soaked in ~4 mL of concentrated salt solution in a drying oven set to 90°C for 3 days. The Rb analog, 2, was soaked in 11 m CsCl solutions while the Cs analog, 1, was soaked in 7 m RbCl or 4 m KCl solutions. Products were examined by EDS and PXRD as described above.
First Principles Calculations
We used first-principles calculations using the density functional theory (DFT) code VASP (Vienna Ab-initio Simulation Package) (Kresse and Furthmuller, 1996a,b) employing the projector augmented wave (PAW) method (Blöchl, 1994; Kresse and Joubert, 1999) and generalized gradient approximation of Perdew, Burke and Ernzerhof (PBE) (Perdew et al., 1996) to model the systems. These were spin-polarized calculations, using a plane wave basis set with an energy cut-off of 520 eV to expand the electronic wave functions, and 10−6 eV energy convergence criteria. A 2 × 2 × 2 k-point mesh was used for sampling the Brillouin zone. The ground state geometries at 0 K were obtained by relaxing the cell volume, atomic positions, and cell symmetry until the maximum forces on each atom were < 0.01 eV/Å. To better represent the correlated nature of the U f-electrons, we employed the DFT+U method (Anismov et al., 1993; Liechtenstein et al., 1995), with a Ueff for the U atoms of 4.0 eV (Ueff = U – J, with U = 4.0 eV, and J = 0.0 eV). The Ueff value was chosen to be close to that obtained from related experimental results for UO2 (Schoenes, 1987; Kotani and Takao, 1992). The valence electron configurations were [U] 6s26p65f36d17s2, [Cs] 5s25p66s1, [P] 3s23p3, [O] 2s22p4, and [F] 2s22p5, respectively.
In an effort to understand why Rb forms an oxyfluoride while Cs only an oxide, we also considered the two opposite cases, Rb11(UO2)12(PO4)3O13 and Cs11(UO2)12(PO4)3O13F2. Because the oxide and oxyfluoride compounds have different composition, we cannot directly compare their calculated total energies, i.e., thermodynamic stability. Therefore, we need to investigate their relative stability by analyzing the reaction enthalpies, ΔrH, considering the two reactions:
The ΔrH, values were calculated using:
where ΔfH are the formation energies per formula unit of the products, i, and reactants, j, and the sum is over all products and reactants. ci and cj are the stoichiometric coefficients of the products and reactants, respectively. For each of the reactants and products we calculated their ΔfH using the same VASP calculations input parameters listed above.
DFT gives only the reaction enthalpy at 0 K, and to include the temperature effect on the reactions, we calculate the finite temperature quasi-Gibbs formation energies, , using the equation:
Sconf is the configurational entropy, defined as:
where kb is the Boltzmann constant, xi is the mole fraction of the constituent i, and the sum is over each constituent i in the compound. Note that in our calculations we do not consider the vibrational contribution to the entropy because due to their very large size, calculating this term for the title compounds is outside the current capabilities of DFT, and hence the term quasi-Gibbs energy. In the case of the gases O2, F2, and Cl2, we use tabulated values for the standard entropies (Chase, 1998).
Discussion
Synthesis
There have been numerous reported uranium phosphate containing structures prepared by similar synthetic methods using UF4, AlPO4, and alkali chloride fluxes, predominantly loaded into alumina crucibles and heated at a temperature of 875°C (Juillerat et al., 2018a,b; Juillerat and zur Loye, 2019; Juillerat et al., 2019b). Cs11[(UO2)12(PO4)3O13] (1) and Rb11[UO2)12(PO4)3O12F2] (2) were first discovered as the minor product, previously unidentified red tablets, in the synthesis of Cs6[(UO2)7O4(PO4)4] and Rb6[(UO2)7O4(PO4)4], respectively (Juillerat et al., 2018b). The optimization of the Cs containing analog lead to the use of platinum crucibles, and was successful, but when using analogous synthetic techniques for the Rb analog, only simple rubidium oxides, such as Rb2U2O7, were obtained in platinum crucibles. The use of alumina crucibles for the synthesis of Rb11[UO2)12(PO4)3O12F2] (2) proved necessary, although the reason is not well-understood. Structures 3 and 4, related to the phosphuranylite topology, were discovered when trying to optimize synthetic conditions for Cs1.4K2.6[(UO2)3O2(PO4)2], Cs1.7K4.3[(UO2)5O5(PO4)2], Rb1.4K2.6[(UO2)3O2(PO4)2] and Rb1.6K4.4[(UO2)5O5(PO4)2]. The synthesis of these phases is described and discussed in a recent publication which concluded that higher flux to reactant ratios (40 mmol flux to 0.5 mmol UF4) favored the formation of the phases Cs1.4K2.6[(UO2)3O2(PO4)2] and Rb1.4K2.6[(UO2)3O2(PO4)2], while 20 mmol of flux and 0.5 mmol UF4 lead to synthesis of Cs1.4K2.6[(UO2)3O2(PO4)2] and Cs1.7K4.3[(UO2)5O5(PO4)2] or Rb1.4K2.6[(UO2)3O2(PO4)2] and Rb1.6K4.4[(UO2)5O5(PO4)2], which could not be successfully separated (Juillerat et al., 2018a). The title phases, Cs4.4K0.6[(UO2)6O4F(PO4)4(UO2)] (3) and Rb4.4K0.6[(UO2)6O4F(PO4)4(UO2)] (4), were obtained simultaneously with Cs1.4K2.6[(UO2)3O2(PO4)2] and Rb1.4K2.6[(UO2)3O2(PO4)2] by further reducing the flux to reactants ratio to 10 mmol and 5 mmol of flux with 0.5 mmol UF4; unfortunately, the title compounds could not be separated manually from Cs1.4K2.6[(UO2)3O2(PO4)2] and Rb1.4K2.6[(UO2)3O2(PO4)2], as all crystallize as yellow plates.
Cs4.4K0.6[(UO2)6O4F(PO4)4(UO2)] (3) and Rb4.4K0.6[(UO2)6O4F(PO4)4(UO2)] (4) contain relatively small amounts of potassium, and attempts were made to synthesize potassium-free Cs and Rb analogs of this uranyl oxyfluoride based on the phosphuranylite sheet topology; however, none were successful as pure CsCl or RbCl fluxes lead to the synthesis of 1, 2, Cs6[(UO2)7O4(PO4)4], Rb6[(UO2)7O4(PO4)4], Cs3[Al2O(PO4)3][(UO2)3O2], Rb3[Al2O(PO4)3][(UO2)3O2], or a mixture of these products.
Structure
Cs11[(UO2)12(PO4)3O13] (1) and Rb11[UO2)12(PO4)3O12F2] (2) are two dimensional sheet structures (Figure 2C) and crystallize in the orthorhombic space group Pnma with lattice parameters a = 14.9561(3), b = 17.9663(4), c = 20.8520(4), and a = 14.1258(3), b = 18.0121(4), c = 20.6241(4), respectively. To our knowledge, the sheet topology present in both structures is unreported in the literature and can be deconstructed into units of the β-U3O8 topology as shown in Figure 2A. The β-U3O8 topology can be deconstructed into U3O16 and U2O14 units, where the U3O16 unit is a square bipyramid, edge-sharing with two pentagonal bipyramids on either side, and the U2O14 unit is two corner-sharing pentagonal bipyramids. These two units alternate to form the β-U3O8 topology as shown in Figure 2A. Structure 1 is also built-up of alternating U3O16 and U2O14 units and 2 contains the U3O16, U2O14, and U2O13F units where the pentagonal bipyramids corner share through the F; however, every third U3O16 unit is missing in both structures (Figure 2B).
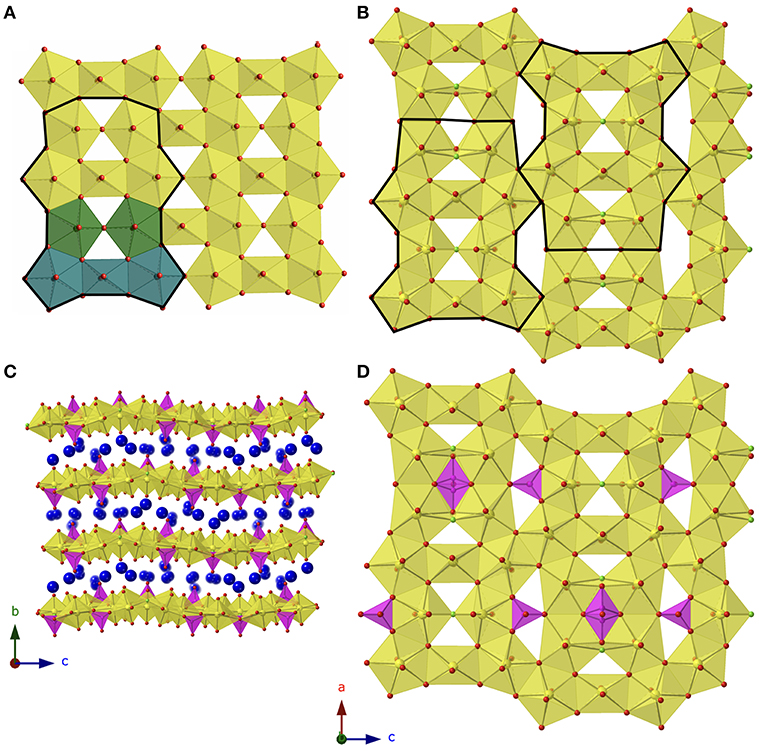
Figure 2. (A) β-U3O8 topology with building unit outlined in black and U3O16 and U2O14 units shown in blue and green, respectively. (B) Topology of 1 and 2 with the building unit boldened, and the phosphate tetrahedra omitted. (C) View of 1 in the a direction. (D) The sheets found in 2 with phosphate tetrahedra included. Uranium, phosphorus, oxygen, and alkali cations are shown in yellow, magenta, red, and blue, respectively.
Between the group of four pentagonal bipyramids (two edge sharing U2O14 or U2O13F units) is a disordered phosphate tetrahedra with two possible orientations as shown in Figure 2D. Additional phosphate tetrahedra edge share with the U2O14/U2O13F units that are located between two U3O16 units and corner share to the adjacent group of four pentagonal bipyramids. The disorder in the phosphate tetrahedron, P3 in both structures 1 and 2, is slightly different between the two structures as shown in Figure 3. In Figure 3A, depicting Rb11[UO2)12(PO4)3O12F2] (2), the phosphate tetrahedron is half occupied and accompanied by a split oxygen/fluorine site whose occupancies sum to 1. Therefore, either the phosphate tetrahedron points up or down (with respect to the plane of the sheet), where the oxygen site corresponds to the orientation of the phosphate tetrahedron, and the fluorine site corresponds to the absence of the phosphate tetrahedron. This is supported by the P3-O28A and P3-F28B bond distances which are 1.555(8) and 2.170(8) Å, respectively where the P-F bond is much too long for the tetrahedral coordination environment of P5+. Tables of bond valences and bond distances for U and P for all structures are collected in Tables S1–S4.
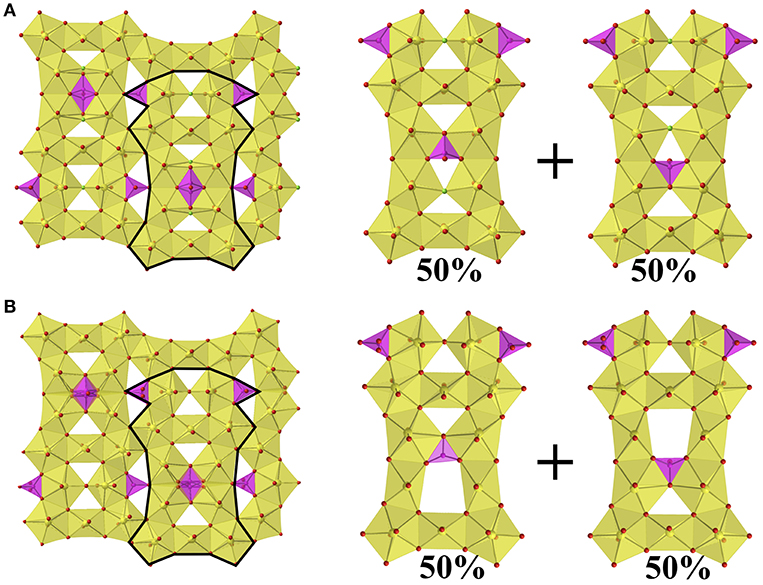
Figure 3. (A) Sheets found in Rb11[UO2)12(PO4)3O12F2] (2) showing the two possible orientations of the phosphate tetrahedron, where F in presence in the absence of the phosphate tetrahedron. (B) The sheets found in Cs11[(UO2)12(PO4)3O13] (1) showing the two possible orientations of the phosphate tetrahedron.
The disorder in the P3 tetrahedron in the Cs analog, structure 1, is similar by virtue of the phosphate tetrahedron pointing up or down within the plane of the sheet; however, the electron density near the O28 site freely refines to a half occupied oxygen site (rather than unity as in 2), and therefore in the absence of the phosphate tetrahedron there are two square bipyramids (Figure 3B) as opposed to two corner-sharing pentagonal bipyramids as in 2. The bond valence sums of U6 and U7, the corner-sharing pentagonal bipyramids in 2 and the two square bipyramids created by the absence of P3 in 1, are slightly lower at values of 5.749 and 5.767, respectively, for 1, as compared to 5.844 and 5.833, respectively for 2; however, all values are within the accepted range for U6+ (~5.6–6.1) (Burns et al., 1997). Tables of bond valences and bond distances for U and P for all structures are located in Tables S1–S4.
While EDS identifies the presence (or absence) of fluorine in structures 1 and 2, it does not identify the positions of the fluorine sites within structure 2. In order to locate the F sites in 2 we calculated BVS for all possible F sites, which includes all oxygen sites coordinated to the uranium sites except for the axial uranyl oxygens, as F on a uranyl oxygen site would be extremely unexpected given the bond order of the ~1.8 Å U-O “yl” bond (3) and multiple bonds are not possible for F. All of the uranyl U-O bond lengths in 2 are between 1.802(4) and 1.830(6) Å and show no significant deviation from the expected ~1.8 Å bond length. Bond valence sums of the remaining O atoms are between 1.82 and 2.30 using ro = 2.051 and B = 0.519 for U-O (Burns et al., 1997), ro = 1.617 and B = 0.370 for P-O (Brown and Altermatt, 1985), and ro = 2.081 and B = 0.515 for Rb-O (Brown and Altermatt, 1985), except for “O5” and “O28B” which have low values of 1.42 and 1.28, and which are significantly lower than the expected value of 2. If ro = 1.98 and B = 0.40 for U-F bonds (Zachariasen, 1978), these bond valence sums come out to 1.02 and 0.94, respectively, and match well with the expected value of 1 for F; therefore, these sites have been identified as F5 and F28B and are necessary for achieving charge balance in the structure. For comparison, the bond valence sums for O5 and O28 sites in structure 1, that contains no fluorine, are 1.86 and 1.90, respectively, and therefore these results also support our decision to assign the fluorine sites in 2 as F5 and F28B.
Cs4.4K0.6[(UO2)6O4F(PO4)4(UO2)] (3) and Rb4.4K0.6[(UO2)6O4F(PO4)4(UO2)] (4) also contain small amounts of fluorine as identified by EDS and by the need for charge balance in the structures. Similar methods as described above were used to identify the fluorine sites(s), again excluding uranyl oxygen sites as possibilities; however, the results are less definitive than the bond valence sums of 2. In 3 all O bond valence sums are between 1.92 and 2.22, none of which signal good candidates for fluorine doping, and similarly in 4, the O BVS are between 1.78 and 2.25. This suggests that there is no preferred site for F and for this reason we have arbitrarily set the occupancy of O3 to be half occupied by both F and O, as this site has the lowest BVS in both structures 3 and 4, and the rarity of PO3F tetrahedra leaves O3 and O6 as the most plausible options. [(UO2)5(HPO4)3(PO4)F4](H9C10N2)3 synthesized hydrothermally using as the F− source also contains phosphuranylite related layers and fluorine is present on sites similar to the ones found in 3 and 4 (Deifel et al., 2008).
The Cs4.4K0.6[(UO2)6O4F(PO4)4(UO2)] (3) and Rb4.4K0.6[(UO2)6O4F(PO4)4(UO2)] (4) structures are similar to the phosphuranylite mineral, KCa(H3O)3[(UO2)6O4(PO4)4(UO2)(H2O)8] (Demartin et al., 1991), as they contain the same phosphuranylite-type layers constructed of chains of alternating UO8 and U2O12 units connected to adjacent chains through corner- and edge- sharing phosphate tetrahedra (Figure 4A). There is also an additional uranium site, square bipyramid, corner sharing with four phosphate tetrahedra to link adjacent sheets, as there is in phosphuranylite (Figure 4B). The alkali sites fill the voids between the uranyl phosphate layers (Figures 4D,E). Several recent papers have further characterized phosphuranylite type layers by the direction in which the phosphate tetrahedra point (up or down orthogonal to the plane of the sheet) and there are seven known isomers (Locock and Burns, 2003; Juillerat et al., 2018a; Juillerat and zur Loye, 2019). Both structures 3 and 4 are the uudduuO isomer, observed in the mineral phurcalite (Atencio et al., 1991), while phosphuranylite is the uudduuS isomer. In addition to the presence of fluorine in the uranyl phosphate sheets, the stacking of the uranyl phosphate sheets in 3 and 4 is also different from those observed in other phosphuranylite based structures. In phosphuranylite and recently reported phosphuranylite type structures, the chains of UO8 and U2O12 units in adjacent sheets run parallel to each other, while in 3 and 4 there are two orientations of these chains that alternate between layers, where the torsion angle between chains in two adjacent layers is 37.8°. This is illustrated in Figure 4C where parallel layers are shown in yellow, and the layer between these is shown in orange.
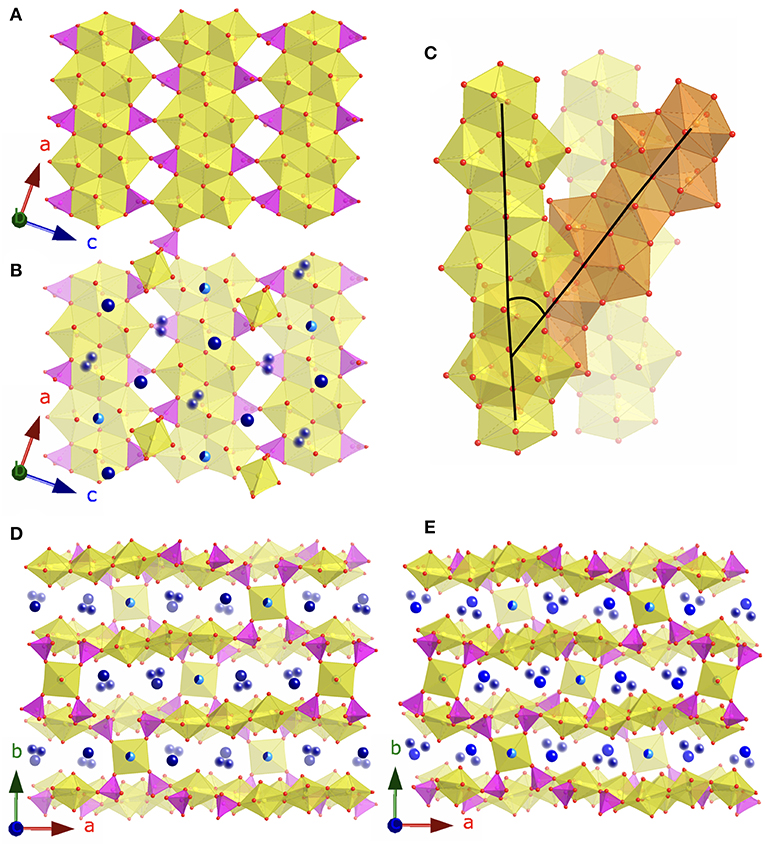
Figure 4. (A) The phosphuranylite layers found in 3 and 4. (B) The phosphuranylite layers plus the square bipyramid uranyl sites and alkali cations. (C) Depiction of the 37.8° torsion angle between chains in adjacent layers. (D) View of 3 in the c direction and (E) view of 4 in the c direction. Uranium, phosphate, and oxygen are shown in yellow, magenta, and red, respectively. Alkali cations are shown in blues, where blurred spheres represent partially occupied disordered sites, and the half light/dark blue spheres represent the Cs/K and Rb/K shared sites.
First-Principles Calculations
As was mentioned previously, Cs prefers to form the oxide structure, while Rb prefers the oxyfluoride structure, and to understand the cause of this different behavior, we studied their reaction enthalpies using DFT. Shown in Table 2 are the calculated ΔrH values which indicate reactions (1) and (2) are thermodynamically unfavorable, i.e., their ΔrH values are positive. Also, the [A, O12F2] compounds have more negative ΔrH compared to the respective [A, O13] compound, indicating that forming the [A, O12F2] is preferred over the [A, O13]. Experimental results confirm that this is the case for the Rb containing compound, it disagrees for that containing Cs. This discrepancy between experiment and calculations may come from the fact that the calculations are performed at 0 K, and thus, finite temperature enthalpy values could provide a different conclusion.
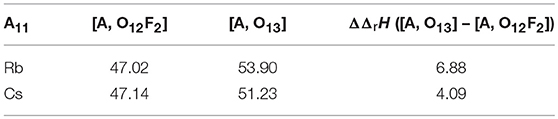
Table 2. Reaction enthalpies (ΔrH) and difference in reaction enthalpies (ΔΔrH), in kJ/mol/atom, of the A11(UO2)12(PO4)3O12F2 ([A, O12F2]) and A11(UO2)12(PO4)3O13 ([A, O13]) compounds (A = Rb, Cs).
To consider the temperature effect, we calculated the using Equation (4), and substituted them in Equation (3) to obtain the quasi-Gibbs reaction energies, . Shown in Figure 5 is the calculated as a function of the temperature. With increasing temperature the becomes more negative, eventually becoming < 0 at T > 2,200 K, implying that above that temperature the reactions are thermodynamically favorable. It is also important to note that the of the [Cs, O13] compound becomes more negative than the of the [Cs, O12F2] compound for T > 1,900 K, the temperature at which a phase change occurs. Moreover, above the temperature at which the becomes negative, the [Cs, O13] is more negative compared to the [Cs, O12F2] , indicating that above 2,200 K the formation of [Cs, O13] is thermodynamically preferred over the formation of [Cs, O12F2]. The results suggest that the formation of the oxide is driven by the entropy, and that at high enough temperatures, above 2,925 K from our calculations, the [Rb, O13] can also be formed over the [Rb, O12F2]. Also, note that the difference between the phase change temperatures of the Cs and Rb compounds is 1,025 K, which is big enough so that a phase change is observed in one case but not the other. However, the calculated reaction temperature, ~2,200 K is much higher than the experimental one, 1,148 K. The difference arises from the absence of the other entropic contributions in our calculations, except for the gases. Because of the large size of the systems we are not able to calculate the phonon spectra, whereas introducing the vibrational contribution might lower the calculated reaction temperature to better match the experimental results.
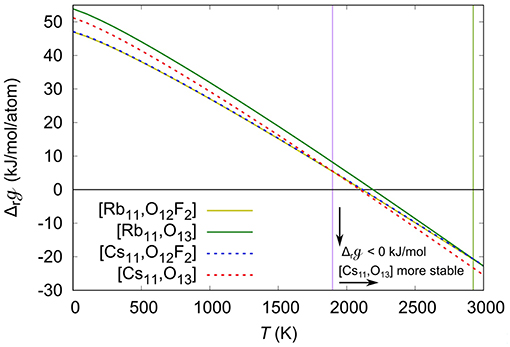
Figure 5. Quasi-Gibbs reaction energies (), in kJ/mol/atom, as a function of the temperature (T), in K. The ΔrG of the Rb11(UO2)12(PO4)3O12F2 ([Rb,O12F2]), Rb11(UO2)12(PO4)3O13 ([Rb,O13]) Cs11(UO2)12(PO4)3O12F2 ([Cs,O12F2]), and Cs11(UO2)12(PO4)3O13 ([Cs,O13]) are shown in yellow, green, blue and red lines, respectively. The purple and green vertical lines respectively show the temperature at which the CsO13 and RbO13 become more stable than the CsO12F2 and RbO12F2.
Ion Exchange
The ion exchange products of 1 soaked in RbCl and KCl and 2 soaked in CsCl show small changes in the PXRD patterns shown in Figures S1, S2, indicating the layered structures are maintained throughout the ion exchange process. EDS was used to qualitatively analyze the alkali contents of each sample before and after ion exchange. The results showed that after 3 days of soaking in concentrated salt solution approximately half of the alkali species are exchanged. In the exchange of Cs in 1 for Rb and K, the approximate contents of the post ion-exchange products are 5.3 Cs, 5.7 Rb, and 5.4 Cs, 5.6 K, respectively. While in the exchange of Rb for Cs in 2, results in 6.3 Rb, 4.7 Cs.
Optical Properties
The UV-vis absorption spectra (Figure S3) and fluorescence emission spectra (Figure S4) of Cs11[(UO2)12(PO4)3O13] (1) and Rb11[UO2)12(PO4)3O12F2] (2) are typical of U6+ species in uranium oxide extended structures. 1 and 2 absorb broadly from 200 to 575 nm and can be classified as semiconductors. A careful look at the DFT calculated density of states (Figure S5) showed that these compounds are actually Mott insulators. The ligand to metal charge transfer transitions are at 377 and 379 nm, respectively and the transitions from the core are at 473 and 463 nm, respectively. The maximum fluorescence emission occurs at an excitation wavelength of 437 nm where the emission peak is centered on 574 and 564 nm, respectively for 1 and 2.
Conclusions
Three new uranyl phosphate oxyfluorides, and one uranyl phosphate, have been synthesized by molten flux methods using alumina crucible reaction vessels and alkali chloride fluxes. Their structures were determined by SXRD and the presence of fluorine was confirmed by qualitative EDS. The location of the fluorine sites was deduced using bond valence sums, although they were inconclusive for determining the location of F in 3 and 4. Structures 1 and 2 were further characterized by PXRD, DFT calculations, ion-exchange experiments, and optical spectroscopy. The DFT calculations support the observation of the Rb, oxyfluoride structures (2) in contrast to the pure Cs, oxide structure of (1), indicating that the formation of pure oxide structure may be driven by entropy, and it might be obtained for both Rb and Cs, given high enough temperatures. The temperature difference in the temperatures at which the pure oxide structures can be obtained is 1,025 K between the Rb and Cs, which hints at the reason why we see an oxyfluoride in the Rb containing (2) and the pure oxide in the Cs (1) containing phases. Structures 1 and 2 are capable of alkali ion exchange, where approximately half of the alkali cations in the parent structure can be replaced by a target alkali species in concentrated salt solutions, although the Cs containing structure, 1, undergoes more extensive ion exchange than the Rb analog, 2, perhaps due to the larger interlayer spacing in 1. Alkali chloride fluxes continue to be a viable synthetic technique for crystallizing new uranium phosphate structures containing new structure types and further exploration should continue, in addition to expanding to alkaline fluxes in order to incorporate divalent cations and hopefully obtain new novel structure types.
Data Availability
CCDC deposition numbers 1898442–1898445 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailingZGF0YV9yZXF1ZXN0QGNjZGMuY2FtLmFjLnVrLA== or by contacting The Cambridge Crystallographic Data Center, 12 Union Road, Cambridge CB21EZ, UK; fax: +44 1223 336033.
Author Contributions
CJ and H-CzL were responsible for the synthesis and structural characterization of the four new compounds and most of the written manuscript. VK and TB contributed the DFT calculations, methods, and discussion.
Funding
Research was conducted by the Center for Hierarchical Wasteform Materials (CHWM), an Energy Frontier Research Center (EFRC). Research was supported by the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Materials Sciences and Engineering under Award DE-SC0016574. CJ is additionally supported by an NSF IGERT Graduate Fellowship under grant number 1250052.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
VK and TB acknowledge the use of computational resources provided by the National Energy Research Scientific Computing Center (NERSC) and the HPC cluster Hyperion, supported by The Division of Information Technology at University of South Carolina.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00583/full#supplementary-material
An extended discussion of the crystallographic refinements of 1 and 2, additional details on the DFT calculations, bond valence sums and selected bond distances, powder X-ray diffraction data, optical spectra of 1 and 2, and DFT calculated density of states can be found in the Supporting Information.
References
Anismov, V. I., Solovyev, I. V., Korotin, M. A., Czyzyk, M. T., and Sawatzky, G. A. (1993). Density-functional theory and NiO photoemission spectra. Phys. Rev. B 48, 16929–16934. doi: 10.1103/PhysRevB.48.16929
Atencio, D., Neumann, R., Silva, A. J. G. C., and Mascarenhas, Y. P. (1991). Phurcalite from Perus, São Paulo, Brazil, and redetermination of its crystal structure. Can. Mineral. 29, 95–105.
Blöchl, P. E. (1994). Projector augmented-wave method. Phys. Rev. B 50, 17953–17979. doi: 10.1103/PhysRevB.50.17953
Brown, I. D., and Altermatt, D. (1985). Bond-valence parameters obtained from a systematic analyses of the inorganic cyrstal structure database. Acta Cryst. B41, 244–247. doi: 10.1107/S0108768185002063
Bugaris, D. E., and zur Loye, H.-C. (2012). Materials discovery by flux crystal growth: quartenary and higher oxides. Angew. Chem. Int. Ed. 51, 3780–3811. doi: 10.1002/anie.201102676
Burns, P. C., Ewing, R. C., and Hawthorne, F. C. (1997). The crystal chemistry of hexavalent uranium: polyhedron geometries, bond-valence parameters, and polymerization of polyhedral. Can. Mineral. 35, 1551–1570.
Chase, M. W. Jr. (1998). NIST-JANAF thermochemical tables, Fourth Edition, Part I, Al-Co. J. Phys. Chem. Ref. Data Monogr. 9:1745.
Deifel, N. P., Holman, K. T., and Cahill, C. L. (2008). hydrolysis as a route to unique uranium phosphate materials. Chem. Commun. 45, 6037–6039. doi: 10.1039/b813819b
Demartin, F., Diella, V., Donzelli, S., Gramaccioli, C. M., and Pilati, T. (1991). The importance of accurate crystal structure determination of uranium minerals. I. Phosphuranylite KCa(H3O)3(UO2)7(PO4)4O4[8H2O]. Acta Cryst. B47, 439–446. doi: 10.1107/S010876819100099X
Juillerat, C. A., Klepov, V. V., Alekseev, E. V., and zur Loye, H.-C. (2019b). Overstepping Löwenstein's rule—a route to unique aluminophosphate frameworks with 3D salt-inclusion and ion exchange properties. Inorg. Chem. 58, 724–736. doi: 10.1021/acs.inorgchem.8b02906
Juillerat, C. A., Klepov, V. V., Morrison, G., Pace, K. A., and zur Loye, H.-C. (2019a). Flux crystal growth: a versatile technique to reveal the crystal chemistry of complex uranium oxides. Dalton Trans. 48, 3162–3181. doi: 10.1039/C8DT04675A
Juillerat, C. A., Moore, E. E., Besmann, T. B., and zur Loye, H.-C. (2018b). Observation of an unusual uranyl cation-cation interaction in the strongly fluorescent layered uranyl phosphates Rb6[(UO2)7O4(PO4)4] and Cs6[(UO2)7O4(PO4)4]. Inorg. Chem. 57, 3675–3678. doi: 10.1021/acs.inorgchem.8b00302
Juillerat, C. A., Moore, E. E., Kocevski, V., Besmann, T. M., and zur Loye, H.-C. (2018a). A family of layered phosphates crystallizing in a rare geometrical isomer of the phosphuranylite topology: synthesis, characterization, and computational modeling of A4[(UO2)3O2(PO4)2] (A = alkali metals) exhibiting intra-layer ion exchange. Inorg. Chem. 57, 4726–4738. doi: 10.1021/acs.inorgchem.8b00434
Juillerat, C. A., and zur Loye, H.-C. (2019). Crystal growth and structure characterization of three layered uranyl phosphates and their relation to the phosphuranylite family. Cryst. Growth Des. 19, 1183–1189. doi: 10.1021/acs.cgd.8b01643
Kotani, A., and Takao, Y. (1992). Systematic analysis of core photoemission spectra for actinide di-oxides and rare-earth sesqui-oxides. Prog. Theor. Phys. Suppl. 108, 117–131. doi: 10.1143/PTPS.108.117
Kresse, G., and Furthmuller, J. (1996a). Efficient iterative schemes for ab-initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186. doi: 10.1103/PhysRevB.54.11169
Kresse, G., and Furthmuller, J. (1996b). Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50. doi: 10.1016/0927-0256(96)00008-0
Kresse, G., and Joubert, D. (1999). From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775. doi: 10.1103/PhysRevB.59.1758
Kubelka, P., and Munk, F. Z. (1931). Ein beitrag zur optik der farbanstriche. Z. Techn. Phys. 12, 593–601.
Latshaw, A. M., Smith, M. D., and zur Loye, H.-C. (2014). Crystal growth and structure of three new neodymium containing silicates: Na0.50Nd4.50(SiO4)3O, Na0.63Nd4.37(SiO4)3O0.74F0.26 and Na4.74Nd4.26(O0.52F0.48)[SiO4]4. Solid State Sci. 35, 28–32. doi: 10.1016/j.solidstatesciences.2014.06.005
Latshaw, A. M., Wilkins, B. O., Hughey, K. D., Yeon, J., Williams, D. E., Tran, T. T., et al. (2015). A5RE4X[TO4]4 crystal growth and photoluminescence. Fluoride flux synthesis of sodium and potassium rare earth silicate oxyfluorides. CrystEngComm 17, 4654–4661. doi: 10.1039/C5CE00671F
Leinenweber, K., Johnson Hennifer, E., and Groy Thomas, L. (2005). American mineralogist. J. Earth Planet. Mater. 90:115. doi: 10.2138/am.2005.1528
Liechtenstein, A. I., Anisimov, V. I., and Zaanen, J. (1995). Density-functional theory and strong interactions: orbital ordering in Mott-Hubbard insulators, Phys. Rev. B 52, R5467–R5470. doi: 10.1103/PhysRevB.52.R5467
Locock, A. J., and Burns, P. C. (2003). The crystal structure of bergenite, a new geometrical isomer of the phosphuranylite group. Can. Mineral. 41, 91–101. doi: 10.2113/gscanmin.41.1.91
Lussier, A. J., Lopez, R. A. K., and Burns, P. C. (2016). A revised and expanded structure hierarchy of natural and synthetic hexavalent uranium compounds. Can. Mineral. 54, 177–283. doi: 10.3749/canmin.1500078
Morrison, G., Smith, M. D., and zur Loye, H.-C. (2016). Understanding the formation of salt-inclusion phases: an enhanced flux growth method for the targeted synthesis of salt-inclusion cesium halide uranyl silicates. J. Am. Chem. Soc. 138, 7121–7129. doi: 10.1021/jacs.6b03205
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868. doi: 10.1103/PhysRevLett.77.3865
Read, C. M., Yeon, J., Smith, M. D., and zur Loye, H.-C. (2014). Crystal growth, structural characterization, cation–cation interaction classification, and optical properties of uranium (vi) containing oxychlorides, A4U5O16Cl2 (A = K, Rb), Cs5U7O22Cl3, and AUO3Cl (A = Rb, Cs). CrystEngComm 16:7259. doi: 10.1039/C4CE00281D
Schoenes, J. (1987). Recent specroscopic studies of UO2. J. Chem. Soc. Faraday Trans. 83, 1205–1213. doi: 10.1039/f29878301205
Sheldrick, G. M. (2015a). Crystal structure refinement with SHELXL. Acta Cryst. C71, 3–8. doi: 10.1107/S2053229614024218
Sheldrick, G. M. (2015b). SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. A71, 3–8. doi: 10.1107/S2053273314026370
Spek, A. L. (2009). Structure validation in chemical crystallography. Acta Cryst. D65, 148–155. doi: 10.1107/S090744490804362X
Wang, Y., Duan, T., Weng, Z., Ling, J., Yin, X., Chen, L., et al. (2017). Mild periodic acid flux and hydrothermal methods for the synthesis of crystalline f-element-bearing iodate compounds. Inorg. Chem. 56, 13041–13050. doi: 10.1021/acs.inorgchem.7b01855
Zachariasen, W. H. (1978). Bond lengths in oxygen and halogen compounds of d and f elements, J. Less-Common Met. 62, 1–7. doi: 10.1016/0022-5088(78)90010-3
Keywords: flux crystal growth, uranyl, phosphuranylite, single crystal, DFT
Citation: Juillerat CA, Kocevski V, Besmann TM and zur Loye H-C (2019) Observation of the Same New Sheet Topology in Both the Layered Uranyl Oxide-Phosphate Cs11[(UO2)12(PO4)3O13] and the Layered Uranyl Oxyfluoride-Phosphate Rb11[(UO2)12(PO4)3O12F2] Prepared by Flux Crystal Growth. Front. Chem. 7:583. doi: 10.3389/fchem.2019.00583
Received: 26 June 2019; Accepted: 02 August 2019;
Published: 21 August 2019.
Edited by:
Federico Cesano, University of Turin, ItalyCopyright © 2019 Juillerat, Kocevski, Besmann and zur Loye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hans-Conrad zur Loye, enVybG95ZUBtYWlsYm94LnNjLmVkdQ==