 Xiangfei Zhang
Xiangfei Zhang Ping Li1
Ping Li1 Binju Wang
Binju Wang Zexing Cao
Zexing Cao- 1State Key Laboratory of Physical Chemistry of Solid Surfaces, Fujian Provincial Key Laboratory of Theoretical and Computational Chemistry, and College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, China
- 2School of Chemistry and Pharmaceutical Engineering, Huanghuai University, Zhumadian, China
The oxidative addition of a range of robust aryl C–F bonds to a single Al(I) center supported by a (NacNac)− bidentate ligand ((NacNac)− = [ArNC(Me)CHC(Me)NAr]− and Ar = 2,6–C6H3) have been explored by density functional theory calculations. Our calculations demonstrate that the Al(I) center-mediated C–F insertion generally proceeds via the concerted mechanism that involve both the donation () and back-donation () interactions. In addition, the predicted free energy barriers for the C–F bond activation show good agreement with the experimental information available. Finally, the comparative studies show that B(I) is the most active among group III metals (B, Al, Ga), thus supplying a testable prediction for experiments.
Introduction
In natural organic halides, the fluorinated compounds have relatively low abundance (Harper et al., 2003), but their importance has been increasingly recognized in pharmaceuticals, advanced materials, agrochemicals, and polymer chemistry (Hiyama and Yamamoto, 2000; Müller et al., 2007; Purser et al., 2008; O'Hagan, 2010). This is mainly because the introduction of fluorine can significantly modify the electron-density distribution in a molecule or a building block, resulting in a dramatic change in their reactivity and properties, but inducing little effects in their steric hindrance.
The C–F bond is one of the strongest σ covalent single bonds, and its activation typically requires the transition metal catalysis (TMs) (Huang et al., 2018; Lin et al., 2019). In last decades, the functionalization of C–F bonds has received considerable interest, and extensive efforts have been made to develop various strategies for the C–F activation (Mazurek and Schwarz, 2003; Panetier et al., 2011; Johnson et al., 2012; Klahn and Rosenthal, 2012; Nova et al., 2012; Kuehnel et al., 2013). In the transition metal-mediated C–F activations, the strong repulsion interactions between the d-occupied orbital of TM and the electron-rich F atom would raise the activation energies for the concerted oxidative addition reactions. As such, the C–F activation would have a preference for the stepwise manner (Choi et al., 2011), which usually involves σ-bond metathesis or insertion–elimination reactions during these processes (Watson et al., 2001; Kraft et al., 2010; Nova et al., 2010).
In addition to the transition metals, the compounds of the some main-group elements may have the transition-metal-like reactivity and catalyze the oxidative addition of the C–F bond (Jana et al., 2010; Stahl et al., 2013; Swamy et al., 2017; Chu and Nikonov, 2018). In particular, recent experimental studies demonstrated that a monomeric Al(I) center supported by the NacNac− ligand (NacNac− = [ArNC(Me)CHC(Me)NAr]− and Ar = 2,6–C6H3) could catalyze the oxidative addition of the C–F bond (Chu et al., 2015; Crimmin et al., 2015). As aluminum is non-toxic and the most abundant metal in nature, Al(I) compound represents a promising strategy for the C–F activation. In NacNacAl (1), the conjugate bidentate ligand NacNac− can combine with Al+ through the N→ Al dative bond, in which HOMO is basically composed of an sp2-like lone pair occupied orbital at Al, while LUMO+1 is mainly contributed by the p-type orbital of Al (Scheme 1) (Cui et al., 2000; Schoeller and Frey, 2013).
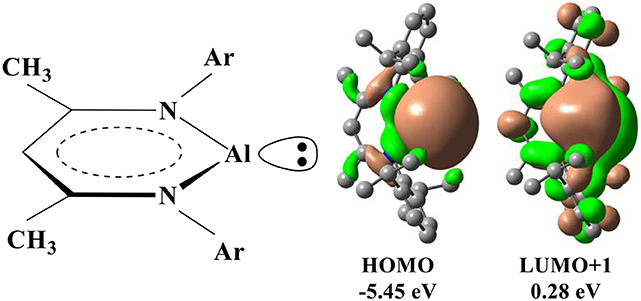
Scheme 1. Schematic drawings for the structure of NacNacAl and its representative molecular orbitals.
Our previous studies show that the aluminum center of NacNacAl (1), as the N-heterocyclic carbene (NHC) analog, has similar electronic characters with the transition metal (Zhang and Cao, 2016). Although the C–F bond activation by the main-group element centers has been investigated, both experimentally (Meier and Braun, 2009; Caputo et al., 2013; Stahl et al., 2013; Chen et al., 2017; Bayne and Stephan, 2019; Pait et al., 2019) and theoretically (Mondal et al., 2017), the detailed mechanisms for the oxidative addition of C–F bonds at the Al(I) of NacNacAl (1) are still largely unknown. Herein, we have performed extensive density functional theory (DFT) calculations on the C–F bond activation, and the possible mechanisms for the diversity of oxidative addition reactions and dependence of the ease of C–F oxidative addition on the fluorination and position have been explored.
Computational Details
For convenience, the aryl C(sp2)–F substrates are labeled as 4, 7, 9, 11, 14, and 18 [Scheme 2 and Figure SI1], while the intermediates and transition states are labeled as IM1−Y (IM1−Yiso) and TS1−X (TS1−Yiso), respectively, where Y is the substrate label (Scheme 2 and Figures 3, 4). The reactant complex is labeled as R1−Y, and the corresponding products are labeled with Y+1, Y+2, etc. for multiple reaction channels.
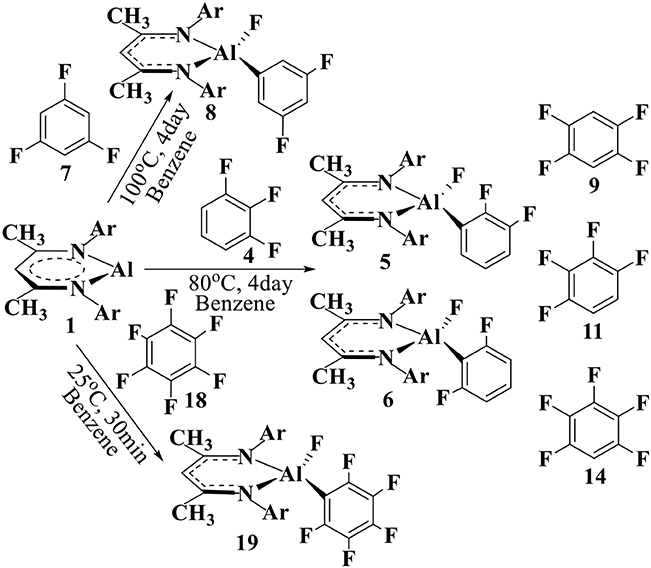
Scheme 2. C–F addition reactions of NacNacAl (1) complex with the fluorinated benzenes. 4, 7, and 18 are from experiments, while the additional fluorobenzenes 9, 11, and 14 are also considered in this study.
The geometries of reactants, intermediates, transition states, and products have been fully optimized by the DFT calculations with the functionals M06-2X (Zhao and Truhlar, 2008), and a comparison of structures by experiment and theory with different functionals is shown in Figure SI2. In particular, based on previous computational investigations and the evaluation of functional performance (Zhang and Cao, 2016), several selected functionals have been used for the aryl C–F bond systems (Figure SI3).
Frequency calculations at the same level of theory have been carried out to confirm if the optimized structures are local minima without any imaginary frequency, or transition states (TS) with only one imaginary frequency on the potential-energy surface (PES). The intrinsic reaction coordinate (IRC) (Fukui, 1970, 1981) analysis is used to track the minimum energy path correlating the transition state with the corresponding reactant and product. The natural bond orbital (NBO) analysis (Reed et al., 1988) has been also carried out to further examine the electronic and bonding properties of the optimized structures.
Here, two different basis sets are considered for all atoms. The relatively small basis set 6-31G(d) (Hehre et al., 1972; Francl et al., 1982) (BS1) is used for all of the geometry optimizations and frequency calculations, and the larger 6-311+G(d,p) basis set (BS2) is used for the single-point calculations with the SMD solvation model for estimation of the solvent effect of benzene (Marenich et al., 2009). The corrections of Gibbs free energy and zero-point energy (ZPE) from the gas-phase frequency calculations are used to determine the relative reaction energetics. In consideration of overestimation of the entropic effect from the gas-phase calculation, a correction of −2.6 (2.6) kcal/mol (Benson, 1982) (at T = 298.15 K) was applied to calibrate the relative free energies for the reaction step with the molecular number ratio of reactant to product of 2:1 (or 1:2) according to the free volume theory and previous theoretical calculations (Schoenebeck and Houk, 2010; Ariafard et al., 2011; Liu et al., 2012; Wang and Cao, 2013). Here, all calculations have been performed by the Gaussian 09 program (Frisch et al., 2009).
Results and Discussion
Herein, NacNacAl (1) catalyzed oxidative addition of the aryl C–F bonds from various fluorobenzene derivatives has been investigated (see Scheme 2). In particular, substrates 4, 7, and 18 have been studied in experiments. For comparison, we also considered additional fluorobenzene derivatives 9, 11, and 14 in this study. It is seen from Scheme 2 that the reaction conditions for the C–F activation in substrate 7 is much harsher than that in substrate 18, suggesting that substrate 18 is much more reactive than substrate 7. Based on DFT calculations, the predicted relative energies are collected in Table SI1. The Cartesian coordinates of all molecules, intermediates, and transition states for their optimized structures are compiled into Table SI2.
Addition of the Aryl σ C–F Bond
NacNacAl (1)-mediated oxidative additions of the aryl C–F bonds in 4, 7, 9, 11, 14, and 18 have been explored.
Figure 1 shows the optimized structures of complexes of NacNacAl (1) with substrates 4 and 7; the optimized structures for other substrates are summarized in the Supporting Information (Table SI2). Note that F atoms from the substrates maintain a long distance with the Al center, but a relatively short distance with methyl H atoms. All these suggest that interactions between NacNacAl (1) and substrates are dominated by C–H···F-type H-bond interactions (Saha et al., 2018). For substrate 4, two binding conformations (IM1−4 and IM1−4iso) are located in calculation, which would lead to two distinct products as observed in experiments (Scheme 2).
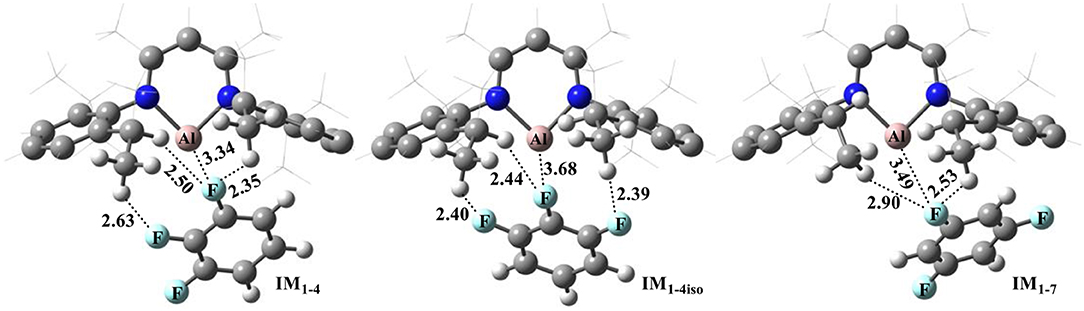
Figure 1. Optimized structures of reactant complexes of NacNacAl (1) with the trifluorobenzene derivatives (4 and 7). For clarity, only the key atoms are highlighted with the ball-and-stick model.
Considering that the electronic structure of the Al(I) center in NacNacAl (1) resembles that of a transition metal (Cui et al., 2000; Schoeller and Frey, 2013), the NacNacAl (1)-mediated oxidative addition reactions may proceed via the concerted insertion mechanism. Indeed, the concerted insertion transition states have been located for all fluorobenzene substrates in this study (Figure SI4). Figure 2 presents the mechanism and the representative transition state structures for the C–F activation of substrate 4.
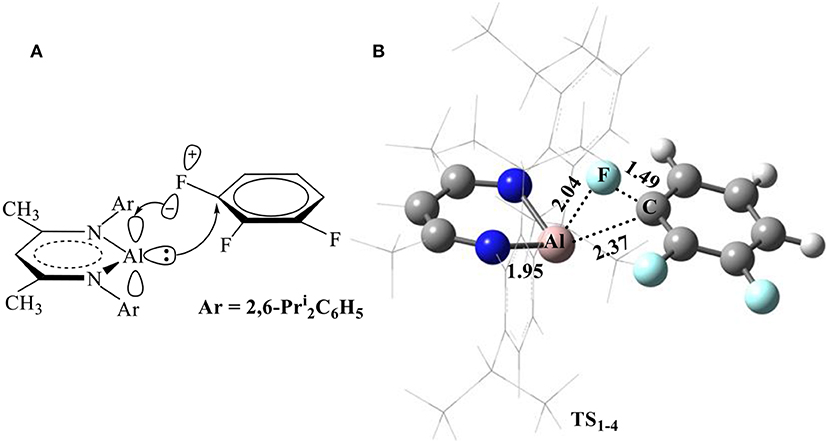
Figure 2. Proposed mechanism for the C–F bond activation (A) and the optimized transition-state structure for the C–F activation of substrate 4 (B, bond lengths in Å) in reactions R1−4.
It is seen from Figure 2B that NacNacAl (1)-mediated oxidative C–F addition involves a transition state of the three-membered ring structure TS1−4, in which the Al–F and C–F distances are 2.04 and 1.49 Å, respectively. It was found that the C–F bond can be efficiently activated by the donation () and back-donation () interactions (Figure 2A) (Schoeller and Frey, 2013). In contrast to transition metals (Clot et al., 2011), the Al center in NacNacAl (1) has the additional empty p-orbital, which renders the donation () and back-donation () interactions simultaneously, thus resulting in the concerted mechanism for the C–F bond insertion reactions. The predicted free energy barrier for substrate 4 is 26.1 kcal/mol relative to 1 + 4, and this reaction is remarkably exothermic, with the Gibbs free energies of the reaction ΔG of −91.9 kcal/mol (298.15 K). The C–F bond insertion reaction leads to the formation of the fluorinated species NacNacAlF(C6H3F2) (O'Hagan, 2010), in which the Al center has been converted to sp3 hybridization, from the initial sp2 hybridization.
Figure 3 compares the calculated relative energy profiles for all the fluorinated benzene substrates. We note that substrate 18 has the lowest overall free energy barrier (ΔG≠ = 19.5 kcal/mol) among these substrates, indicating that it is the most reactive. This is in accordance with the experimental findings (Scheme 2). For substrates 4 and 7, the calculated lowest free energy barriers are 24.1 and 26.6 kcal/mol, respectively. The predicted reactivity order 18 > 4 > 7 is indeed in good agreement with experiments. In addition, our calculations predict that substrates 9 and 11 have similar reactivity with 4 and 7, as all these substrates have similar free energy barriers for C–F activation. For comparison, we also considered the C–F activations of other isomers of 4 and 11 (20 and 24 in Figure SI5), and the calculated lowest barriers are 26.8 kcal/mol for 20 and 24.5 kcal/mol for 24 (Figure SI6). It was found that the predicted reactivities for the C–F activation are correlated with the C–F bond strength of different substrates. Among all these substrates, 18 has the lowest bond dissociation energy (BDE = 122.4 kcal/mol, Figure SI5), while 7 has the highest bond dissociation energy (BDE = 126.5 kcal/mol). These BDE values show good correlations with the predicted reactivity of 18 and 7.
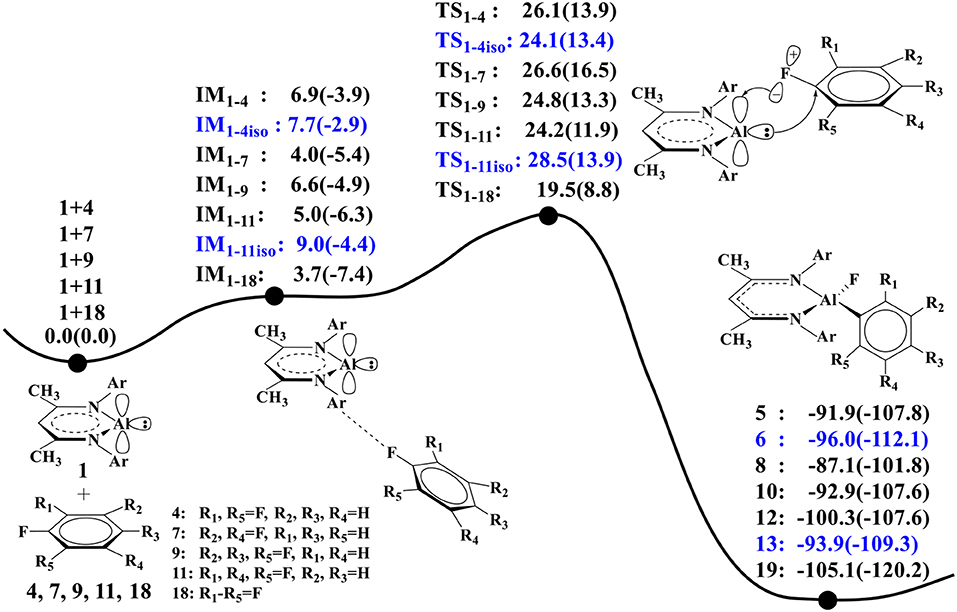
Figure 3. The predicted relative free energies and energies [ΔG(ΔE) in kcal/mol, the same below] here (the ZPE correction was included in relative energy in parentheses) for these oxidative additions are calculated by the M06-2X approach. The reactions of 1 with 4 and 11 produce two isomers 5, 6, and 12, 13.
As B and Ga atoms in Group III have similar electronic configurations with Al, we also compared their performance in C–F activation. Figure 4 summarizes the calculated energy profiles for C–F activation of 14 by NacNacAl (1), NacNacB (1B), and NacNacGa (1Ga) (Asay et al., 2011). For NacNacAl (1), we have compared C–F activations at three different positions: para-, meta-, and ortho-positions, and the calculated barriers are 20.6 (TS1−14), 23.7 (TS1−14ortho), and 23.1 kcal/mol (TS1−14meta), respectively. All these indicate that the C–F bond at the para-position is the most active. As such, all the NacNacB (1B)- and NacNacGa (1Ga)-mediated C–F activations occur at the para-position. It is seen from Figure 4 that the calculated barriers for the para C–F activation are 20.6 kcal/mol for NacNacAl (1), 10.8 kcal/mol for triangle singlet transition state NacNacB (1B), and 24.7 kcal/mol for NacNacGa (1Ga), indicating that the electron-deficient compound 1B has the highest reactivity toward C–F activation. Among these three elements, the B atom has the smallest atomic radius (Kutzelnigg, 1984), which results in stronger σ donor interactions than Al and Ga atoms. Meanwhile, we found that the ground state of NacNacB (1B) is actually the triplet state, instead of the singlet state as in NacNacAl (1) or NacNacB (1Ga). In the triplet state, we located both the linear and the triangle transition states that are characterized as “diradical” TS; the calculated barriers are 33 and 19.9 kcal/mol, respectively (Figures SI7, SI8). Clearly, the triangle transition state in the closed shell singlet state, with a barrier of 10.8 kcal/mol (Figure 4), is preferred over all other pathways (Table SI1). As such, we located a two-state reactivity for the NacNacB (1B)-mediated C–F activations.
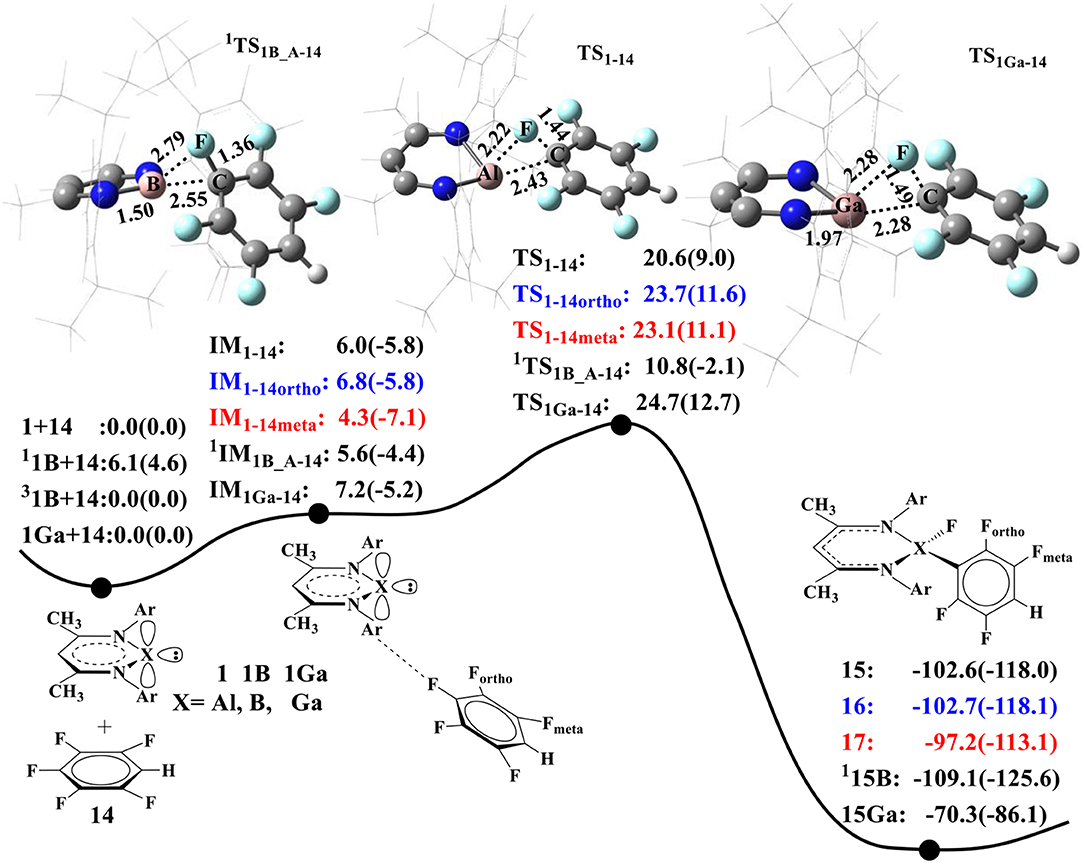
Figure 4. The predicted relative free energies and energies [ΔG(ΔE) in kcal/mol]. Optimized transition-state structures involved in the oxidative additions of 1,2,4,5-tetrafluorobenzene (Choi et al., 2011) to the NacNacAl (1), NacNacB (1B), and NacNacGa (1Ga) by M06-2X.
Conclusions
Extensive density functional calculations have been used to explore the oxidative additions of robust σ C(sp2)–F bonds to the Al(I) center, and plausible reaction mechanisms and optimized structures of reactants, intermediates, transition states, and products have been predicted. Our calculations demonstrate that all Al(I) center-mediated C–F insertions proceed via the concerted mechanism, which is governed by both the donation () and back-donation () interactions. All the calculated C–F insertion mechanisms resemble that of the conventional transition-metal-like catalysis, and the predicted free energy barriers show good agreement with experiments.
Data Availability
The datasets generated for this study are available on request to the corresponding author.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by the National Natural Science Foundation of China (21673185 and 21873078).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00596/full#supplementary-material
References
Ariafard, A., Brookes, N. J., Stranger, R., and Yates, B. F. (2011). DFT study on the mechanism of the activation and cleavage of CO2 by (NHC)CuEPh3 (E = Si, Ge, Sn). Organometallics 30, 1340–1349. doi: 10.1021/om100730h
Asay, M., Jones, C., and Driess, M. (2011). N-heterocyclic carbene analogues with low-valent group 13 and group 14 elements: syntheses, structures, and reactivities of a new generation of multitalented ligands. Chem. Rev. 111, 354–396. doi: 10.1021/cr100216y
Bayne, J. M., and Stephan, D. W. (2019). C–F bond activation mediated by phosphorus compounds. Chem. Eur. J. 25, 9350–9357. doi: 10.1002/chem.201900542
Caputo, C. B., Hounjet, L. J., Dobrovetsky, R., and Stephan, D. W. (2013). Lewis acidity of organofluorophosphonium salts: Hydrodefluorination by a saturated acceptor. Science 341, 1374–1377. doi: 10.1126/science.1241764
Chen, W., Bakewell, C., and Crimmin, M. R. (2017). Functionalisation of carbon–fluorine bonds with main group reagents. Synthesis 49, 810–821. doi: 10.1055/s-0036–1588663
Choi, J., Wang, D. Y., Kundu, S., Choliy, Y., Emge, T. J., Krogh-Jespersen, K., et al. (2011). Net oxidative addition of C(sp3)–F bonds to iridium via initial C–H bond activation. Science 332, 1545–1548. doi: 10.1126/science.1200514
Chu, T., Boyko, Y., Korobkov, I., and Nikonov, G. I. (2015). Transition metal–like oxidative addition of C–F and C–O bonds to an aluminum(I) center. Organometallics 34, 5363–5365. doi: 10.1021/acs.organomet.5b00793
Chu, T., and Nikonov, G. I. (2018). Oxidative addition and reductive elimination at main-group element centers. Chem. Rev. 118, 3608–3680. doi: 10.1021/acs.chemrev.7b00572
Clot, E., Eisenstein, O., Jasim, N., Macgregor, S. A., Mcgrady, J. E., and Perutz, R. N. (2011). C–F and C–H bond activation of fluorobenzenes and fluoropyridines at transition metal centers: How fluorine tips the scales. Acc. Chem. Res. 44, 333–348. doi: 10.1021/ar100136x
Crimmin, M. R., Butler, M. J., and White, A. J. (2015). Oxidative addition of carbon–fluorine and carbon–oxygen bonds to Al(I). Chem. Commun. 51, 15994–15966. doi: 10.1039/c5cc07140b
Cui, C. M., Roesky, H. W., Schmidt, H.–G., Noltemeyer, M., Hao, H. J., and Cimpoesu, F. (2000). Synthesis and structure of a monomeric aluminum(I) compound [{HC(CMeNAr)2}Al](Ar = 2,6-iPr2C6H3): a stable aluminum analogue of a carbene. Angew. Chem. Int. Ed. 112, 4444–4446. doi: 10.1002/1521-3773(20001201)39:23<4274::AID-ANIE4274>3.0.CO;2-K
Francl, M. M., Pietro, W. J., Hehre, W. J., Binkley, J. S., Gordon, M. S., DeFrees, D. J., et al. (1982). Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 77, 3654–3665. doi: 10.1063/1.444267
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scueria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2009). Gaussian 09, Revision B.01. Wallingford, CT: Gaussian, Inc.
Fukui, K. (1970). Formulation of the reaction coordinate. J. Phys. Chem. 74, 4161–4163. doi: 10.1021/j100717a029
Fukui, K. (1981). The path of chemical reactions—the IRC approach. Acc. Chem. Res. 14, 363–368. doi: 10.1021/ar00072a001
Harper, D. B., O'Hagan, D., and Murphy, C. D. (2003). “Fluorinated natural products: occurrence and biosynthesis,” in The Handbook of Environmental Chemistry, Vol. 3, part P, ed. G. W. Gribble (Berlin: Springer-Verlag), 141–169. doi: 10.1007/b10454
Hehre, W. J., Ditchfield, R., and Pople, J. A. (1972). Self-consistent molecular orbital methods. XII. further extensions of Gaussian-type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 56, 2257–2261. doi: 10.1063/1.1677527
Hiyama, T., and Yamamoto, H. (2000). “Chapter 5: Biologically active organofluorine compounds,” in Organofluorine Compounds, ed. H. Yamamoto (Berlin: Springer-Verlag), 137–182. doi: 10.1007/978-3-662-04164-2
Huang, D. H., Vera, G. A. D., Chu, C. H., Zhu, Q. H., Stavitski, E., Mao, J., et al. (2018). Single-atom Pt catalyst for effective C–F bond activation via hydrodefluorination. ACS Catal. 8, 9353–9358. doi: 10.1021/acscatal.8b02660
Jana, A., Samuel, P. P., TavcAr, G., Roesky, H. W., and Schulzke, C. (2010). Selective aromatic C–F and C–H bond activation with silylenes of different coordinate silicon. J. Am. Chem. Soc. 132, 10164–10170. doi: 10.1021/ja103988d
Johnson, S. A., Hatnean, J. A., and Doster, M. E. (2012). “Chapter 5: Functionalization of fluorinated aromatics by nickelmediated C–H and C–F bond oxidative addition: prospects for the synthesis of fluorine-containing pharmaceuticals,” in Progress in Inorganic Chemistry, Vol. 57, ed. K. D. Karlin (New York, NY: Wiley & Sons), 255–352. doi: 10.1002/9781118148235.ch5
Klahn, M., and Rosenthal, U. (2012). ChemInform abstract: an update on recent stoichiometric and catalytic C–F bond cleavage reactions by lanthanide and group 4 transition metal complexes. Organometallics 31, 1235–1244. doi: 10.1002/chin.201217253
Kraft, B. M., Clot, E., Eisenstein, O., Brennessel, W. W., and Jones, W. D. (2010). Mechanistic investigation of vinylic carbon–fluorine bond activation of perfluorinated cycloalkenes using ZrH2 and ZrHF. J. Fluorine Chem. 131, 1122–1132. doi: 10.1016/j.jfluchem.2010.05.003
Kuehnel, M. F., Lentz, D., and Braun, T. (2013). Synthesis of fluorinated building blocks by transitionmetal–mediated hydrodefluorination reactions. Angew. Chem. Int. Ed. 52, 3328–3348. doi: 10.1002/anie.201205260
Kutzelnigg, W. (1984). Chemical bonding in higher main group elements. Angew. Chem. Int. Ed. 23, 272–295. doi: 10.1002/anie.198402721
Lin, Z. Y., Lan, Y., and Wang, C. (2019). Synthesis of gem-difluoroalkenes via nickel-catalyzed reductive C–F and C–O bond cleavage. ACS Catal. 9, 775–780. doi: 10.1021/acscatal.8b04348
Liu, B., Gao, M., Dang, L., Zhao, H. T., Marder, T. B., and Lin, Z. Y. (2012). DFT studies on the mechanisms of the platinum–catalyzed diboration of acyclic α, β-unsaturated carbonyl compounds. Organometallics 31, 3410–3425. doi: 10.1021/om3002153
Marenich, A. V., Cramer, C. J., and Truhlar, D. G. (2009). Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B. 113, 6378–6396. doi: 10.1021/jp810292n
Mazurek, U., and Schwarz, H. (2003). Carbon–fluorine bond activation—Looking at and learning from unsolvated systems. Chem. Commun. 12, 1321–1326. doi: 10.1039/B211850E
Meier, G., and Braun, T. (2009). Catalytic CF activation and hydrodefluorination of fluoroalkyl groups. Angew. Chem. Int. Ed. 48, 1546–1548. doi: 10.1002/anie.200805237
Mondal, T., De, S., and Koley, D. (2017). DFT Study on C–F bond activation by group 14 dialkylamino metalylenes: a competition between oxidative additions versus substitution reactions. Inorg. Chem. 56, 10633–10643. doi: 10.1021/acs.inorgchem.7b01615
Müller, K., Faeh, C., and Diederich, F. (2007). Fluorine in pharmaceuticals: Looking beyond intuition. Science 317, 1881–1886. doi: 10.1126/science.1131943
Nova, A., Mas–Ballest, É. R., and Lledòs, A. (2012). Breaking C–F bonds via nucleophilic attack of coordinated ligands: Transformations from C–F to C–X Bonds (X = H, N, O, S). Organometallics 31, 1245–1256. doi: 10.1021/om2010386
Nova, A., Reinhold, M., Perutz, R. N., Macgregor, S. A., and McGrady, J. E. (2010). Selective activation of the ortho C–F bond in pentafluoropyridine by zerovalent nickel: reaction via a metallophosphorane intermediate stabilized by neighboring group assistance from the pyridyl nitrogen. Organometallics 29, 1824–1831. doi: 10.1021/om100064z
O'Hagan, D. (2010). ChemInform abstract: fluorine in health care: Organofluorine containing blockbuster drugs. J. Fluorine Chem. 131, 1071–1081. doi: 10.1016/j.jfluchem.2010.03.003
Pait, M., Kundu, G., Tothadi, S., Karak, S., Jain, S., Vanka, K., et al. (2019). C–F bond activation by a saturated N-heterocyclic carbene: mesoionic compound formation and adduct formation with B(C6F5)3. Angew. Chem. Int. Ed. 58, 2804–2808. doi: 10.1002/anie.201814616
Panetier, J. A., Macgregor, S. A., and Whittlesey, M. K. (2011). Catalytic hydrodefluorination of pentafluorobenzene by [Ru(NHC)-(PPh3)2(CO)H2]: A nucleophilic attack by a metal-bound hydride ligand explains an unusual ortho-regioselectivity. Angew. Chem. Int. Ed. 123, 2835–2838. doi: 10.1002/ange.201006789
Purser, S., Moore, P. R., Swallow, S., and Gouverneur, V. (2008). Fluorine in medicinal chemistry. Chem. Soc. Rev. 37, 320–330. doi: 10.1039/b610213c
Reed, A. E., Curtiss, L. A., and Weinhold, F. (1988). Intermolecular interactions from a natural bond orbital, donor–acceptor viewpoint. Chem. Rev. 88, 899–926. doi: 10.1021/cr00088a005
Saha, B. K., Saha, A., Sharada, D., and Rather, S. A. (2018). F or O, which one is the better hydrogen bond (is it)? acceptor in the C–H···X–C (X– = F–, O =) interactions? Cryst Growth Des. 18, 1–16. doi: 10.1021/acs.cgd.7b01164
Schoeller, W. W., and Frey, G. D. (2013). On the acceptor properties of a Al–Nacnac carbene analogue, a density functional investigation. J. Org. Chem. 744, 172–177. doi: 10.1016/j.jorganchem.2013.06.031
Schoenebeck, F., and Houk, K. N. (2010). Ligand-controlled regioselectivity in palladium-catalyzed cross coupling reactions. J. Am. Chem. Soc. 132, 2496–2497. doi: 10.1021/ja9077528
Stahl, T., Klare, H. F. T., and Oestreich, M. (2013). Main-group Lewis acids for C–F bond activation. ACS Catal. 3, 1578–1587. doi: 10.1021/cs4003244
Swamy, V. S. V. S. N., Bisai, M. K., Das, T., and Sen, S. S. (2017). Metal free mild and selective aldehyde cyanosilylation by a neutral penta-coordinate silicon compound. Chem. Commun. 53, 9850–9853. doi: 10.1039/C7CC03948D
Wang, B. J., and Cao, Z. X. (2013). Sequential covalent bonding activation and general base catalysis: insight into N-heterocyclic carbene catalyzed formylation of N–H bonds using carbon dioxide and silane. RSC Adv. 3, 14007–14015. doi: 10.1039/C3RA41464G
Watson, L. A., Yandulov, D. V., and Caulton, K. G. (2001). C-D0 (D0 = π-donor, F) Cleavage in H2CdCH(D0) by (Cp2ZrHCl)n: Mechanism, agostic fluorines, and a carbene of Zr(IV). J. Am. Chem. Soc. 123, 603–611. doi: 10.1021/ja0024340
Zhang, X., and Cao, Z. (2016). Insight into reaction mechanisms for oxidative addition of strong σ bonds to an Al(I) center. Dalton Trans. 45, 10355–10365. doi: 10.1039/c6dt01154c
Zhao, Y., and Truhlar, D. G. (2008). The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 120, 215–241. doi: 10.1007/s00214-007-0310-x
Keywords: fluorobenzene, NacNacAl, density functional theory, C–F bond, reaction mechanism
Citation: Zhang X, Li P, Wang B and Cao Z (2019) Mechanistic Features in Al(I)-Mediated Oxidative Addition of Aryl C–F Bonds: Insights From Density Functional Theory Calculations. Front. Chem. 7:596. doi: 10.3389/fchem.2019.00596
Received: 26 June 2019; Accepted: 12 August 2019;
Published: 03 September 2019.
Edited by:
Nino Russo, University of Calabria, ItalyReviewed by:
Ming Lei, Beijing University of Chemical Technology, ChinaLung Wa Chung, Southern University of Science and Technology, China
Copyright © 2019 Zhang, Li, Wang and Cao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Binju Wang, d2FuZ2Jpbmp1MjAxOEB4bXUuZWR1LmNu; Zexing Cao, enhjYW9AeG11LmVkdS5jbg==