 Pantaleo Musci1
Pantaleo Musci1 Marco Colella1
Marco Colella1 Flavio Fanelli1,2Angela Altomare2
Flavio Fanelli1,2Angela Altomare2 Luisa Pisano3
Luisa Pisano3 Claudia Carlucci1
Claudia Carlucci1 Renzo Luisi1*
Renzo Luisi1* Leonardo Degennaro1*
Leonardo Degennaro1*- 1Flow Chemistry and Microreactor Technology FLAME-Lab, Department of Pharmacy–Drug Sciences, University of Bari “A. Moro”, Bari, Italy
- 2Crystallography Institute of the National Research Council (IC-CNR), Bari, Italy
- 3Department of Chemistry and Pharmacy, University of Sassari, Sassari, Italy
A new synthetic route to N-alkyl-2-acylazetidines was developed through a highly stereoselective addition of organolithiums to N-alkyl-2-oxazolinylazetidines followed by acidic hydrolysis of the resulting oxazolidine intermediates. This study revealed an unusual reactivity of the C=N bond of the oxazoline group when reacted with organolithiums in a non-polar solvent such as toluene. The observed reactivity has been explained considering the role of the nitrogen lone pair of the azetidine ring as well as of the oxazolinyl group in promoting a complexation of the organolithium, thus ending up with the addition to the C=N double bond. The high level of stereoselectivity in this addition is supported by DFT calculations and NMR investigations, and a model is proposed for the formation of the oxazolidine intermediates, that have been isolated and fully characterized. Upon acidic conditions, the oxazolidine moieties were readily converted into 2-acylazetidines. This synthetic approach has been applied for the preparation of highly enantioenriched 2-acylazetidines starting from chiral not racemic N-alkyl-2-oxazolinylazetidines.
Introduction
The four-membered saturated heterocycle azetidine is a valuable scaffold exploited in several active research areas (Singh et al., 2008; Couty and Evano, 2009; Antermite et al., 2017). The strain and dynamic phenomena associated to the azetidine ring allows exploring new chemical space for organic synthesis and drug discovery purposes (Degennaro et al., 2014a,b; De Ceglie et al., 2011). Recently, different approaches have been reported for the synthesis of azetidine derivatives for targeting lead compounds and bioisosteres for drug discovery (Couty et al., 2004; Ferraris et al., 2007; Pérez-Faginas et al., 2011). Despite the great interest for this small-sized heterocycle, some structural motifs, such as 2-ketoazetidines, seems to be poorly investigated. Biologically active compounds, incorporating the 2-acylazetidine moiety, include natural products such as alkaloids found in the genus Daphniphyllum (Kobayashi and Kubota, 2009) as well as 2-aroylazetidines known as potent inhibitors of dipeptidyl peptidase IV (DPP-IV) (Ferraris et al., 2004), a proline-specific serine protease used as target in several therapeutic areas such as diabetes (Weber, 2004) pain (Ronai et al., 1999), and cognition enhancement (During et al., 2003; Figure 1).
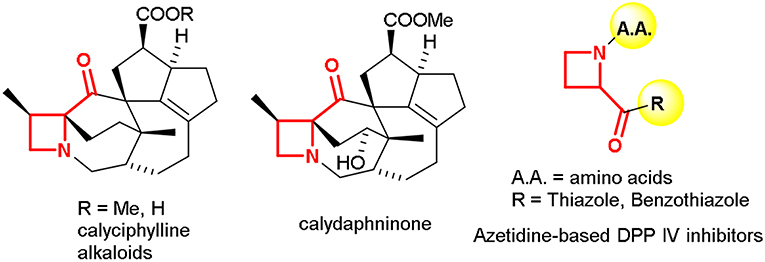
Figure 1. Examples of 2-acylazetidine motif in natural products and biologically active compounds.
Methods for the preparation of 2-acylazetidines could be traced back to 1969 with the first synthesis reported by Cromwell, by cyclization of 2,4-dibromoketones with primary amines (Scheme 1, A) or the addition of organolithiums to azetidines carboxylic acids (Rodebaugh and Cromwell, 1969, 1971; Kulkarni and Cromwell, 1977; Arnould et al., 1980; Scheme 1, B). Selected examples, for the stereoselective synthesis of 2-acylazetidines, include the nucleophilic addition of aryllithiums (Couty and Prim, 2002) and Grignard reagents (Couty et al., 2011) to optically active 2-cyanoazetidines (Scheme 1, E) or the addition of Grignard reagents to enantiopure Weinreb amides prepared from methyl 1-phenylethylazetidine-2-carboxylates (Ma et al., 2007; Scheme 1, C). Recently, the synthesis of 2-acylazetidines, by a one-pot tetramethylguanidine/I2-mediated formal [2+2] cycloaddition reaction of α-amidomalonate with enones, has been reported by Miao and Sun (Miao et al., 2013; Scheme 1, F). Yadav reported the ring expansion of aziridines, employing phenacyl bromide derivatives via in situ generated ammonium ylides in a silica gel-water reaction medium (Garima et al., 2010; Scheme 1, D).
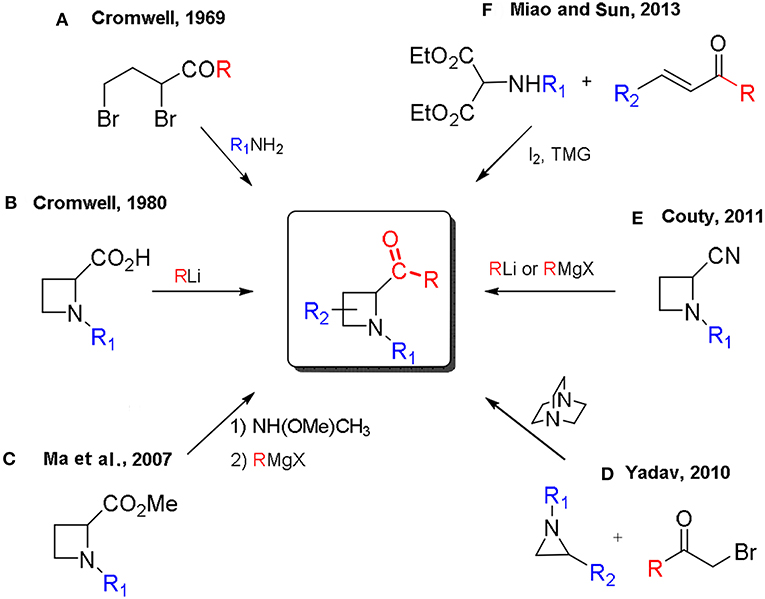
Scheme 1. Reported synthesis of 2-acylazetidines.
In 2011, our group reported an intriguing approach for the synthesis of 2-ketoaziridines exploiting an unprecedented reactivity profile of chiral 2-oxazolinylaziridines when subjected to reaction with organolithiums (Degennaro et al., 2011). This work focused on a regioselective α-lithiation of optically active N-phenylethyl-2-oxazolinylaziridines by using a strong base such as n-buthyllithium (n-BuLi) in a coordinating solvent (THF) at −78°C within 1 h. The resulting lithiated species proved to be chemically and configurationally stables, under the experimental conditions, and were trapped with electrophiles in highly stereoselective manner (Scheme 2, path A). In particular, this study demonstrated the presence of a mixture of two equilibrating invertomers for N-alkyl-2-oxazolinylaziridines as a result of the nitrogen inversion (Capriati et al., 2002; Luisi et al., 2005; Scheme 2). It was demonstrated by NMR and DFT calculations, that the dynamics at nitrogen was dependent on the nature of the substituent at the C2 of the azetidine ring. A dynamic model was proposed to account for a diverse and unexpected reactivity observed in a non-polar solvent such as toluene. In fact, as a consequence of a competing complexation of the organolithium reagent with the lone pair of the aziridine nitrogen led to an unusual attack to the C=N bond of the oxazoline ring with consequent formation of an oxazolidine intermediate, useful precursor of the corresponding 2-acylaziridine (Scheme 2, path B). Better results in term of yield and stereoselectivity were obtained by reacting C2-substituted oxazolinylaziridines with organolithiums in toluene and at higher temperature (0°C). Therefore, this model suggests that controlling the nitrogen dynamics, in these systems, could be possible to address the reactivity providing both α-substituted aziridines and 2-ketoaziridines.
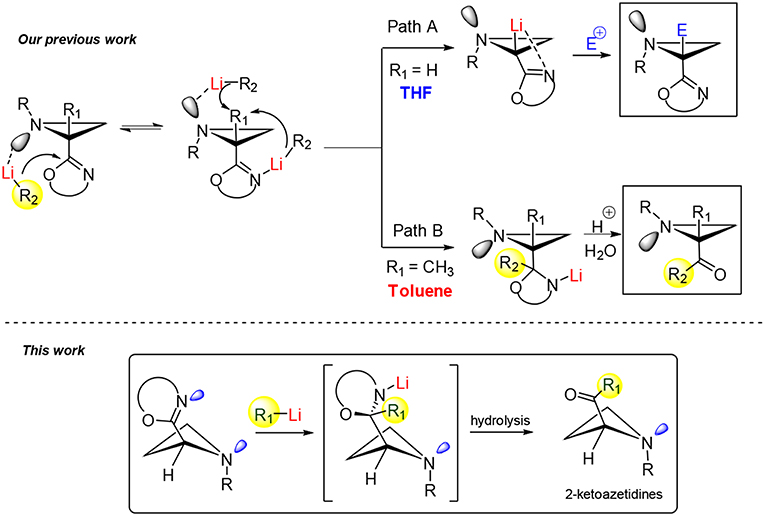
Scheme 2. Example of reactivity controlled by dynamics in small nitrogenated heterocycles.
Inspired by these preliminary results on 2-oxazolinylaziridines, highlighting the role of nitrogen dynamics on the reactivity of this three-membered heterocycles, we were keen to build on this ground a stereoselective synthesis of 2-ketoazetidines, likely starting from the corresponding 2-oxazolinyl azetidines (Scheme 2). We are glad to report herein the results obtained in this investigation.
Results and Discussion
Substrates Synthesis
The first step for the synthesis of 2-oxazolinylazetidines 4a (R, R)-4b and (R, S)-4b, reported in Scheme 3, involved the reaction of methyl 2,4-dibromobutanoate 1, commercially available, with a suitable primary amine. When ester 1 was reacted with benzylamine, azetidinylester 2a was obtained in high yield, and, by subsequent treatment with 2-amino-2-methylpropanol, n-hexyllithium, in presence of catalytic amount of LaCl3 afforded hydroxamide 3a (Jang et al., 2015; Scheme 3). The last step consisted in an intramolecular cyclization of hydroxamide 3a mediated by diethylaminosulfur trifluoride at low temperature, that provided the desired oxazoline 4a. A similar protocol was applied to the preparation of 2-methylcarboxylates (R, R)-2b, and (R, S)-2b, by using (R)-1-phenylethylamine as chiral not racemic amine, to yield orthogonally protected and chromatographically separable diastereomeric mixture of esters in 64% yield in 1:1 ratio. Hydroxamides (R, R)-3b and (R, S)-3b where separately synthesized as previously described for amide 3a and, in the case of amide (S, S)-3b, prepared from (S)-1-phenylethylamine, the structure has been solved by X-Ray analysis, confirming the chemical structure and the absolute stereochemistry of the compound (see Supplementary Material). Finally, the cyclization step yielded 2-oxazolinylazetidines (R, R)-4b and (R, S)-4b, as single diastereoisomers, each with excellent enantiomeric ratio (er = 98:2).
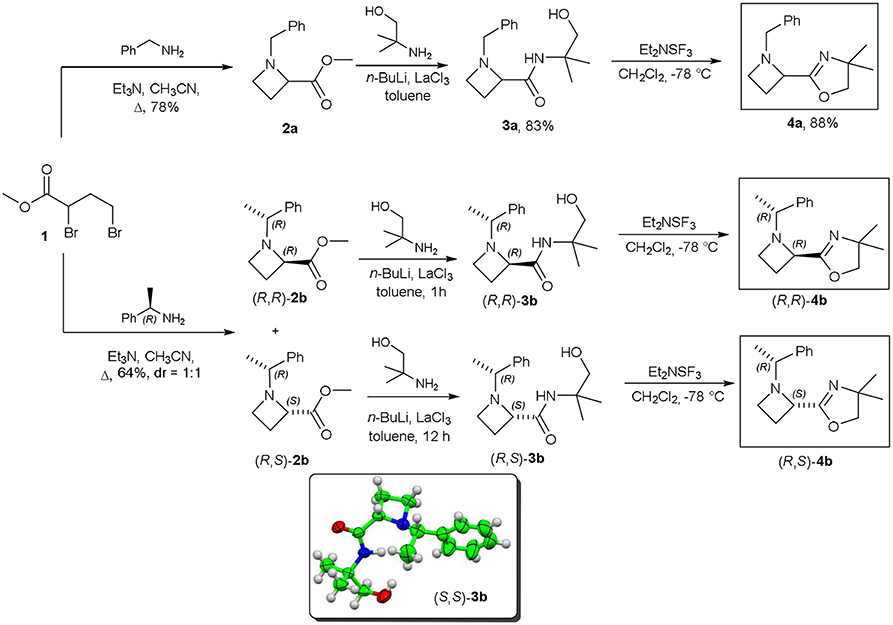
Scheme 3. Substrates synthesis.
Optimization Study and Scope
According to a recent report by Couty and coworkers, the addition of organolithiums or Grignard reagents to azetidine carboxylic esters (R, S)-2b, occurred with low chemoselectivity, providing the corresponding tertiary alcohol as the main product (Couty et al., 2011). This prompted us to consider the approach reported in Scheme 2 as an alternative route to 2-acylazetidines. We started our investigation considering the reaction of azetidine 4a with n-BuLi in toluene (Table 1). Pleasingly, oxazolidine 5a was the sole product observed in these experiments with no evidence for the α-deprotonation of the azetidine ring.
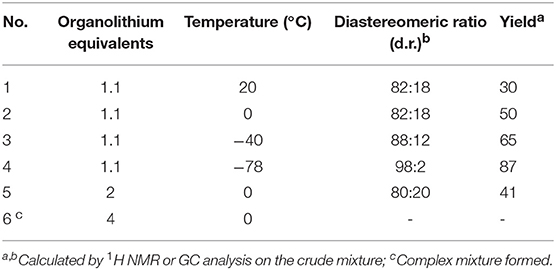
Table 1. Reaction of azetidine 4a with n-BuLi.
By adding n-BuLi at room temperature, within 20 min, azetidine 5a was obtained in 30% yield, after aqueous work up, as a 82:18 mixture of diastereomers (Table 1, No. 1). With the aim to improve both yield and diastereoselectivity, the reaction was conducted at lower temperature. To our delight, running the reaction at −78°C furnished the product 5a in high yield and as single diastereoisomer (Table 1, No. 4). The use of an excess n-BuLi, considerably lowered the yields, giving also a complex mixture of products (Table 1, No. 5-6). We considered the conditions in No. 4 as optimal for the examination of the reaction scope. Under optimized conditions, the nucleophilic addition of n-hexylLi, MeLi and EtLi proceeded in highly stereoselective manner affording compounds 5b-d as single diastereoisomer with satisfactory yields (Scheme 4). By using PhLi as nucleophile, the reaction required longer times (3 h) and product 5e was obtained with 82% yield and 98:2 dr. Surprisingly, the reaction with 2-thienyllithium did not afford any product, even keeping the reaction up to 6 h at −78 and 0°C, presumably as a consequence of the presence of ethereal solvents, namely THF, in 2-thienyllithium solution. Based on these results, the same protocol (i.e., toluene, −78°C), was tested using optically active oxazolinylazetidine (R, R)-4b. Unexpectedly, the addition reaction was not observed at −78°C, but full conversion was obtained running the reaction at 0°C in 20 min, obtaining oxazolidine 6a in 86% yield as a single diastereomer (dr > 98:2) and highly enantioenriched (er = 98:2). The modified conditions were also employed for preparing in good yield and diastereoselectivity oxazolidines 6b-d, and 7a-d starting from oxazolinylazetidine (R, R)-4b and (R, S)-4b, respectively (Scheme 4).
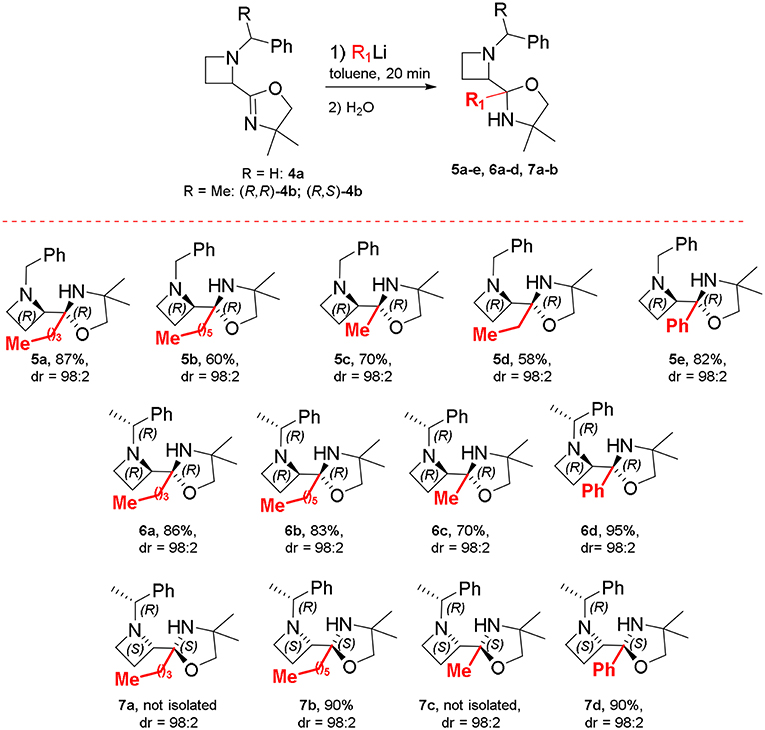
Scheme 4. Oxazolidine synthesis.
Proposed Model for the Stereoselective Addition
The high stereoselectivity observed for the C=N addition prompted us to consider the stereochemistry at the quaternary stereogenic center of the oxazolidine ring. All attempts to get suitable crystals for X-ray analysis were unsuccessful, and NOESY experiments were inconclusive. Based on our previous experience on the stereochemical assignments in three and four-membered heterocycles by using DFT calculations and NMR predictions, we decided to pursue this approach for solving this stereochemical puzzle (Azzena et al., 2018). The diastereoiomers (R, R, R)-6c and (R, R, S)-6c were considered. A relaxed potential energy surface (PES) scan of the dihedral angles C43N16C17C19 and O2C3C11C12, which define the relative position of the two substituents (oxazolidine ring and benzyl group) on the azetidinyl ring, was performed. All other parameters were allowed to vary freely during the PES scan; the single point energies were calculated at DFT/B3LYP/3-21G level in vacuo. After conformational minimization, the lowest energy conformers of the two diastereoisomers was subjected to fully unconstrained geometry optimization at SMD/DFT/B3LYP/6-311++G(d,p) level, followed by vibrational analysis. The free energy values provided by the vibrational analysis calculations indicated that diastereoisomer (R, R, R)-6c was 3.8 Kcal/mol more stable with respect to (R, R, S)-6c. At the end, optimized structures were used for prediction of nuclear shieldings using the gauge independent atomic orbital (GIAO) approach. GIAO NMR calculations were performed at SMD(CHCl3)/DFT/ MPW1PW91/gen level and the shielding tensors (σcalc) scaled to obtain the predicted chemical shifts (δscal, see Supplementary Material). The NMR chemical shifts (δ) were calculated as the differences of isotropic shielding constants (σ) with respect to the TMS (tetramethylsilane) reference, calculated at the same level of theory. The pcS-2 basis set, specifically designed for NMR shielding constant calculations (Jensen, 2008), was used for H and C atoms and 6-311++G(d,p) for N and O atoms. For indirect spin-spin coupling constants JHH calculation, we selected the SMD/DFT/B3LYP/6-311++G(d,p) level of theory, a good compromise between accuracy and computational cost. Indeed in our earlier studies, we noticed the performance of this method in predicting proton and carbon NMR shieldings as well as the spin–spin coupling constants JHH (Azzena et al., 2011; Carroccia et al., 2014; Zenzola et al., 2014; Degennaro et al., 2015a,b; Pisano et al., 2016). All calculations were performed with the Gaussian 09 program at DFT level and the solvent effect was modeled using the self-consistent reaction field (SCRF) calculations within the SMD model (Marenich et al., 2009; Supplementary Material). The statistical parameters CMAE and R2 of δscaled/δexpt were determined to establish the consistency between the theoretical and experimental magnetic parameters of the two possible diastereoisomers, and the best fit in all cases was found for the diastereoisomer (R, R, R)-6c (see Supplementary Material). In Figure 2 the comparison between experimental and calculated 1H NMR spectra of azetidine 6c is reported. A better match can be assessed between the real and calculated spectra of (R,R,R)-6c (Figures 2A,B).
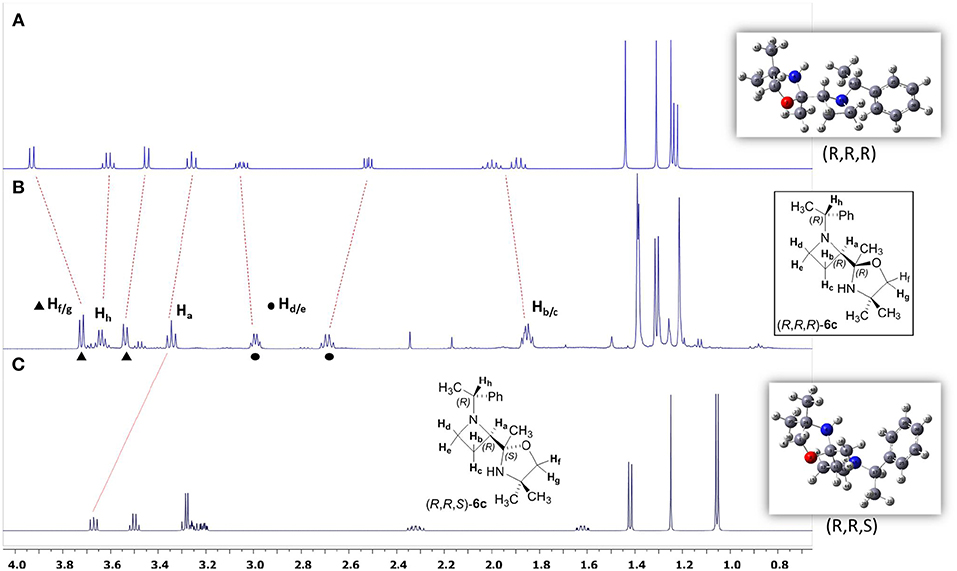
Figure 2. (A) Calculated spectrum of (R,R,R)-6c; (B) Real spectrum of (R,R,R)-6c; (C) Calculated spectrum of (R,R,S)-6c.
Based on the results obtained by DFT calculations, we assumed that the configuration at the new created stereocentre of 6c might be (R). With the aim to rationalize the stereochemical outcome of the nucleophilic addition of organolithiums to the C=N double bond of the oxazoline ring, we considered the stereodynamic model proposed for oxazolinylaziridines (Scheme 2). Assuming that oxazolinylazetidines could show dynamic phenomena associated with both nitrogen inversion and ring puckering (Parisi et al., 2016), the stereochemistry of substrates (R, R)-4b and (R, S)-4b was first assessed by NOESY experiments (see Supplementary Material). This stereochemical evaluation demonstrated a trans relationship between the nitrogen substituent and the oxazoline ring. Considering such stereochemical arrangement, the two lone pairs belonging to both azetidine and oxazoline nitrogens could likely be oriented in such a way to promote an easy formation of a complex with the organolithium reagent (Scheme 5). Under these stereochemical restrictions, it is reasonable to foresee the nucleophilic addition of the organolithium to the most accessible face of the planar C=N bond, resulting an (R) configuration at the new stereocenter starting from (R, R)-4b, and (S) configuration starting from (R, S)-4b (Scheme 5). On the basis of this model, the absolute configuration for compounds 6a-d and 7a-b have been supposed to be (R,R,R) and (R,S,S), respectively. Similar reasoning can be made for azetidine 4a leading to adducts 5a-e with (R,*R*) relative configuration. It is worth pointing out that, in striking contrast to what previously observed for aziridines, where a preferential α-lithiation took place under similar reaction conditions, with the nucleophilic addition occuring in 15–20% extent and low stereoselectivity, in the case of oxazolinylazetidines α-lithiation was never observed even at higher temperature. It is reasonable to assume that such peculiar stereoelectronic requirements realized with azetidines prevented the deprotonation event, leading to a stable complex prone to undergo exclusively nucleophilic addition to the C=N double bond.
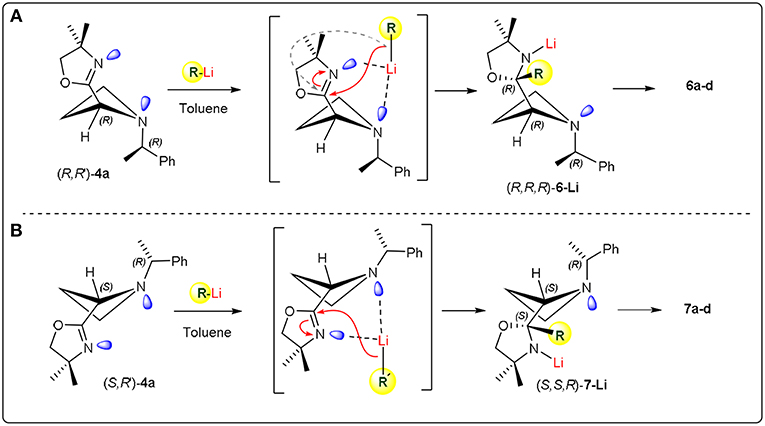
Scheme 5. Proposed model.
Synthesis of 2-acylazetidines
All the products 5a-e were isolated by flash chromatography, even though a partial and expected hydrolysis of the oxazolidine moiety in acidic media by silica gel occurred. This evidence prompted us to explore a mild and easy acidic hydrolysis by using silica gel in dichloromethane. Treatment of 5a-d with SiO2 in dichloromethane (DCM) as solvent for 3 h afforded N-alkyl-2-acylazetidines 8a-d in quantitative yields (Scheme 6). The oxazolidine 5e resulted unreactive under these conditions, and all the attempts to hydrolyze the cyclic aminal failed. Similarly, the hydrolytic protocol was applied to compounds 6a-c and 7b giving good yields of 2-acylazetidines 9a-c and 10b, respectively, in enantiopure form as confirmed by HPLC analysis (see Supplementary Material). A partial epimerization for azetidine 9c in acidic media by silica gel has been observed, leading to the formation of corresponding diastereoisomer 10c (dr = 50/50). Moreover, complete hydrolysis of oxazolidines 7a and 7c taken place already during purification by chromatography on silica gel giving satisfactory yields of 2-acylazetidines 10a and 10c. The absolute stereochemistry of all optically active compounds was assigned according to data reported in the literature (Couty et al., 2011).
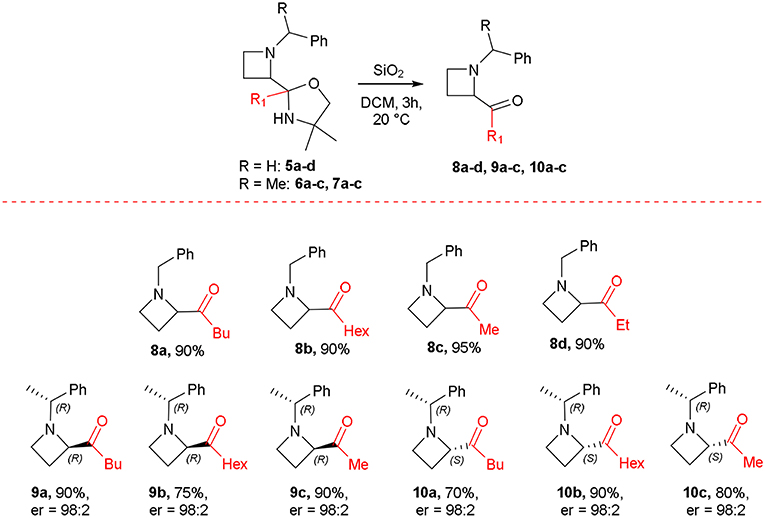
Scheme 6. Synthesis of 2-acylazetidines.
Conclusions
We have demonstrated that in an apolar solvent, such as toluene, different organolithiums were capable to give an unexpected regio- and stereoselective addition at the C=N bond of the oxazoline group of N-alkyloxazolinylazetidines. Different 1,3-oxazolidinyl azetidines formed in high yield and resulted useful precursors of 2-acylazetidines by acidic hydrolysis. The expected deprotonation event in α position with respect to oxazoline ring did not take place in this conditions. With the aim to rationalize the mechanism and the stereochemical outcome of the addition reaction, a stereodynamic model has been proposed, taking into consideration complexation and dynamic phenomena associated with the azetidine's nitrogen inversion. The configuration assignment, performed on oxazolidinyl azetidines as intermediates by NMR and DFT calculations, resulted mandatory for the validation of proposed model. Even though the stereochemical information generated in the addition reaction is lost in the hydrolysis of oxazolidine ring, this work furnishes an outstanding example of reactivity controlled by dynamics of small nitrogenated heterocycles. Work is in progress to further explore the reactivity of 2-oxazolinylazetidines with organolithium in coordinating solvents.
Materials and Methods
General Information
Flash chromatography was performed using 70–230 mesh Al2O3 (either neutral or basic activity II-IV), with the indicated solvent system according to standard techniques. Analytical thin layer chromatography (TLC) was carried out on precoated 0.25 mm thick plates of Kieselgel 60 F254; visualization was accomplished by UV light (254 nm) or by spraying a solution of 5 % (w/v) ammonium molybdate and 0.2 % (w/v) cerium(III) sulfate in 100 ml 17.6 % (w/v) aq. sulphuric acid and heating to 200°C for some time until blue spots appear. Infrared spectra (vmax, FT-IR) were recorded in reciprocal centimeters (cm−1). Nuclear magnetic resonance spectra were recorded on 300 or 500 MHz spectrometers. The frequency used to record the NMR spectra is given in each assignment and spectrum (1H NMR at 300 or 500 MHz; 13C NMR at 101 MHz or 176 MHz). Chemical shifts for 1H NMR spectra are recorded in parts per million with the residual protic solvent resonance as the internal standard (CDCl3: δ = 7.27 ppm). Data are reported as follows: chemical shift [multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet and bs = broad singlet), coupling constant (in Hz), integration and assignment]. 13C NMR spectra were recorded with complete proton decoupling. Chemical shifts are reported in parts per million with the residual protic solvent resonance as the internal standard (CDCl3: δ = 77.0 ppm). Assignments of 1H and 13C spectra were based upon the analysis of δ and J values, as well as DEPT, COZY and HSQC experiments where appropriate. All reactions involving air sensitive reagents were performed under nitrogen in oven-dried glassware using syringe-septum cap technique. All organolithiums are commercially available by Sigma Aldrich and were titrated before use. All other chemicals were commercially available and used without further purification. Enantiomeric excess was assessed by HPLC (Chiralcel ODH, ADH). Diastereomeric ratio was assessed by GC-MS or 1H NMR analysis on the reaction crude.
Synthesis of Substrates
Methyl 1-benzyl-2-azetidinecarboxylate 2a
According to procedure reported in literature (Nocquet et al., 2012).
1H NMR (500 MHz, CDCl3) δ 7.45–7.14 (m, 5H, Ar-H overlapping CHCl3), 3.80 (d, J = 12.6 Hz, 1H, CH2Ph), 3.74 (t, J = 8.4 Hz, 1H, CHCO), 3.63 (s, 3H, CH3), 3.59 (d, J = 12.6 Hz, 1H, CH2Ph), 3.34–3.29 (m, 1H, NCH2), 2.94 (ddd, J = 9.2, 7.8, 6.9 Hz, 1H, NCH2), 2.41–2.32 (m, 1H, NCH2CH2), 2.21 (dtd, J = 10.5, 8.1, 2.4 Hz, 1H, NCH2CH2).
Methyl 1-[(1R)-methyl]Benzylazetidine-(2R)-carboxylate (R,R)-2b
According to procedure reported in literature (Starmans et al., 1998).
1H NMR (300 MHz, CDCl3) δ 7.37–7.14 (m, 5H, Ar-H), 3.84–3.65 (m, 4H, CHCOOCH3), 3.45 (q, J = 6.6 Hz, 1H, CHCH3), 3.11 (ddd, J = 8.2, 7.7, 2.9 Hz, 1H, NCH2), 2.80 (td, J = 8.3, 7.1 Hz, 1H, NCH2), 2.36–2.10 (m, 2H, NCH2CH2), 1.22 (d, J = 6.6 Hz, 1H, CHCH3).
Methyl 1-[(1R)-methyl]Benzylazetidine-(2S)-carboxylate (R,S)-2b
According to procedure reported in literature (Starmans et al., 1998).
1H NMR (500 MHz, CDCl3) δ 7.33–7.14 (m, 5H, Ar-H), 3.63–3.52 (m, 2H, CHCO and NCHH), 3.40–3.29 (m, 4H, CHPh and OCH3), 3.04–2.96 (m, 1H, NCHH), 2.36–2.24 (m, 1H NCH2CH2), 2.13 (dtd, J = 10.4, 8.0, 2.3 Hz, 1H, NCH2CH2), 1.28 (d, J = 6.5 Hz, 1H, CHCH3).
1-benzyl-N-(1-hydroxy-2-methylpropan-2-yl)azetidine-2-carboxamide 3a
General procedure A: To a perfectly dry flask charged with LaCl3 (587 mg, 2.40 mmol) was dried by heating under reduced pressure. Then, toluene dry (50 mL) and 2-amino-2-methylpropan-1-ol (4.11 g, 46.13 mmol) were added and after cooling to 0°C, n-hexyllithium (2.0 M in hexane, 23.0 mL, 46.13 mmol) was added dropwise and the reaction was heated to 100°C for 15 min. The solution was cooled to room temperature and methyl 1-benzylazetidine-2-carboxylate 2a (3.78 g, 18.45 mmol) was added and the mixture was stirred over night. The reaction was quenched with water (1 mL) and filtered over celite. The organic phase was dried over sodium sulfate and the solvent was evaporated under reduced pressure to afford the product as a brown oil (4.50 g, yield 93%).
FT-IR (film) cm−1 3321, 3054, 2984, 2932, 2856, 2305, 1649, 1532, 1454, 1422, 1265, 1071, 909, 738, and 705.
1H NMR (500 MHz, CDCl3) δ 7.35–7.27 (m, 3H, Ar-H), 7.25–7.20 (m, 2H, Ar-H), 7.04 (bs, 1H, NH), 5.08 (bs, 1H, OH), 3.66 (d, J = 12.3 Hz, 1H, CH2Ph), 3.62 (t, J = 8.6 Hz, 1H, CH), 3.51 (d, J = 12.3 Hz, 1H, CH2Ph), 3.45–3.38 (m, 2H, CH2OH and NCH2), 3.31 (d, J = 11.6 Hz, 1H, CH2OH), 3.08 (dd, J = 16.2, 8.7 Hz, 1H, NCH2), 2.47–2.38 (m, 1H, NCH2CH2), 2.08–1.95 (m, 1H, NCH2CH2), 1.15 (s, 1H, CH3), 0.96 (s, 1H, CH3).
13C NMR (126 MHz, CDCl3) δ 173.9 (C = O), 137.4 (Ar-Cq), 129.1(2 x Ar-C), 128.9 (2 x Ar-C), 127.8 (Ar-C), 70.9 (CH2OH), 66.9 (CH), 62.7 (CH2Ph), 55.5 (Cq), 51.0 (NCH2), 24.7 (CH3), 24.4 (CH3), 23.2 (NCH2CH2).
HRMS (ESI-TOF) [M+Na]+ calculated for C15H22N2NaO2: 285.1579, found 285.1558.
(R)-N-(1-hydroxy-2-methylpropan-2-yl)-1-[(R)-1-phenylethyl]azetidine-2-carboxamide (R,R)-3b
According to the general procedure A, starting from (R,R)-2b (800 mg, 3.65 mmol) amide (R,R)-3b was isolated as white solid, 907 mg, yield 90%, mp 107–109°C, = + 69.49° (c = 1, CHCl3).
FT-IR (KBr, cm−1) ν 3334, 3234, 2969, 2852, 1645, 1538, 1451, 1377, 1282, 1072, 767, 702, and 565.
1H NMR (500 MHz, CDCl3): δ 7.70 (s, 1H, NH-OH), 7.34–7.21 (m, 5H, Ar-H), 5.24 (s, 1H, NH - OH), 3.75–3.64 (m, 3H, CH2OH and NCH), 3.45 (q, J = 6.4 Hz, 1H, CHCH3), 3.08 (t, J = 8.0 Hz, 1H, CH2), 2.78 (q, J = 8.0 Hz, 1H, CH2), 2.38–2.29 (m, 1H, CH2), 2.00–1.91 (m, 1H, CH2), 1.36 (s, 3H, CH3), 1.33 (s, 3H, CH3), 1.14 (d, J = 6.6 Hz, 3H, CHCH3).
13C NMR (125 MHz, CDCl3): δ 174.7 (C = O), 142.61 (Ar-Cq), 128.6 (2x Ar-C), 127.4 (Ar-C), 126.9 (2x Ar-C), 70.8 (CH2OH), 67.3 (CH-Ph), 65.3 (NCH), 55.6 (Cq), 49.6 (CH2N), 24.8 (CH3), 24.7 (CH3), 22.1 (CH2CH2CH), 21.2 (CH3CH).
HRMS (ESI-TOF) [M+H]+ calculated for C16H25N2O2: 277.1911, found 277.1917.
(S)-N-(1-hydroxy-2-methylpropan-2-yl)-1-[(R)-1-phenylethyl]azetidine-2-carboxamide (R,S)-3b
According to the general procedure A, starting from (S,R)-2b (860 mg, 3.94 mmol) amide (R,S)-3b was isolated as colorless oil, 979 mg, yield 90%, = −118.15° (c = 0.4, CHCl3).
FT-IR (film, cm−1) ν 3323, 2969, 2930, 2850, 1651, 1532, 1456, 1380, 1278, 1168, 1071, 709, and 656.
1H NMR (500 MHz, CDCl3) δ 7.29 (m, 5H, Ar-H), 6.71 (bs, 1H, NH), 4.99 (t, J = 6.1 Hz, 1H, OH), 3.57 (t, J = 8.6 Hz, 1H, CH-CO), 3.46 (t, J = 7.6 Hz, 1H, CH2N), 3.40–3.28 (m, 2H, CHPh and CHHN), 3.15–3.05 (m, 2H, CHHN and CHHOH), 2.44–2.27 (m, 1H, CH2), 1.99–1.86 (m, 1H, CH2), 1.32 (d, J = 6.6 Hz, 3H, CHCH3), 1.11 (s, 3H, CH3), 0.88 (s, 3H, CH3).
13C NMR (126 MHz, CDCl3): δ 173.9 (C = O), 141.5 (Ar-Cq), 128.9 (2 x Ar-C), 128.2 (Ar-C), 128.0 (2 x Ar-C), 71.0 (CH2OH), 67.0 (CH-Ph), 66.0 (CHCH2), 55.3 (Cq), 49.8 (CH2N), 24.4 (CH3), 24.1 (CH3), 22.47 (CH2CH2CH), 18.3 (CH3CH).
HRMS (ESI-TOF) [M+H]+ calculated for C16H25N2O2: 277.1911, found 277.1933.
General procedure B (cyclization to 4,5-dihydrooxazole): To a solution of hydroxamide (943 mg, 3.42 mmol) in dry dichloromethane (23 mL) at −78°C, diethylaminosulfur trifluoride (497 μL, 3.76 mmol) was added dropwise and the reaction was stirred for 1 h. Then the reaction was stirred at room temperature overnight. The solution was washed with NaHCO3 0.1 M (3 × 7 mL), the organic phase was dried over Na2SO4 and the solvent was evaporated under reduced pressure.
2-(1-benzylazetidin-2-yl)-4,4-dimethyl-4,5-dihydrooxazole 4a
According to the general procedure B, dihydrooxazole 4a was isolated as yellow oil by chromatography on alumina (20% AcOEt/hexane, Rf 0.4) (1.18 g, yield 88%).
FT-IR (film, cm−1) 3062, 3027, 2966, 2928, 2865, 1666, 1454, 1363, 1297, 1174, 1004, 935, 735, and 701.
1H NMR (700 MHz, CDCl3) δ 7.31–7.27 (m, 4H, Ar-H), 7.22 (t, J = 7.1 Hz, 1H, Ar-H), 3.86 (d, J = 8.0 Hz, 1H, OCH2), 3.82 (t, J = 8.2 Hz, 1H, azetidine-CH), 3.77 (d, J = 8.0 Hz, 1H, OCH2), 3.67 (AB system, d, J = 12.6 Hz, 2H, CH2Ph), 3.41–3.37 (m, 1H, azetidine-CH2), 3.00 (dt, J = 9.4, 7.4 Hz, 1H, azetidine-CH2), 2.45–2.37 (m, 1H, azetidine-CH2), 2.16 (m, 1H, azetidine-CH2), 1.18 (s, 3H, CH3), 1.07 (s, 3H, CH3).
13C NMR (126 MHz, CDCl3): δ 165.2 (C=N), 137.4 (Ar-Cq), 129.4 (2 × Ar-C), 128.3 (2 × Ar-C), 127.2 (Ar-C), 79.23 (oxazoline-CH2), 66.9 (oxazoline-Cq), 62.8 (CH2Ph), 61.5 (azetidine-CH), 51.8 (azetidine-CH2), 28.3 (CH3), 28.3 (CH3) 22.0 (azetidine-CH2).
HRMS (ESI-TOF) [M+H]+ calculated for C15H21N2O: 245.1654, found 245.1649.
4,4-dimethyl-2-{(R)-1-[(R)-1-phenylethyl]azetidin-2-yl}-4,5-dihydrooxazole (R,R)-4b
According to the general procedure B, dihydrooxazole (R,R)-4b was isolated as light brown oil by chromatography on silica (40% AcOEt/hexane, Rf 0.5) (536 mg, yield 92%), = + 81.20° (c = 1, CHCl3). (ADH, 99:1 Hex:iPrOH, 1 mL/min).
FT-IR (film, cm−1) ν 2967, 2929, 2869, 1737, 1660, 1493, 1453, 1364, 1175, 1072, 1028, 1004, 975, 764, and 701.
1H NMR (500 MHz, CDCl3): δ 7.35–7.26 (m, 4H, Ar-H), 7.24–7.20 (m, 1H, Ar-H), 4.04–3.98 (m, 2H, OCH2), 3.87 (t, J = 8.1 Hz, 1H, NCH), 3.49–3.40 (m, 1H, CH-Ph), 3.12–3.04 (m, 1H, CH2), 2.82–2.73 (m, 1H, CH2), 2.38–2.29 (m, 1H, CH2), 2.14–2.06 (m, 1H, CH2), 1.29 (s, 3H, CH3), 1.28 (s, 3H, CH3), 1.23 (d, J = 6.4 Hz, 3H, CHCH3).
13C NMR (126 MHz, CDCl3): δ 166.3 (C=N), 142.9 (Ar-Cq), 128.4 (2 × Ar-C), 127.6 (2 × Ar-C), 127.2 (Ar-C), 79.4 (OCH2), 68.1 (CH-Ph), 67.0 (Cq), 61.2 (NCH), 50.2 (CH2), 28.2 (CH3), 28.2 (CH3), 21.1 (CHCH3), 21.07 (CH2).
HRMS (ESI-TOF) [M+H]+ calculated for C16H23N2O: 259.1805, found 259.1805.
4,4-dimethyl-2-{(S)-1-[(R)-1-phenylethyl]azetidin-2-yl}-4,5-dihydrooxazole (R,S)-4b
According to the general procedure B, dihydrooxazole (R,S)-4b was isolated as light yellow oil by chromatography on silica (40% AcOEt/hexane, Rf 0.4) (747 mg, yield 94%), = −56.30° (c = 1, CHCl3). (ADH, 99:1 Hex:iPrOH, 0.5 mL/min).
FT-IR (film, cm−1) ν 2960, 2932, 2874, 1735, 1658, 1497, 1444, 1371, 1168, 1072, 1022, 1010, 985, 760, and 704.
1H NMR (500 MHz, CDCl3) δ 7.34–7.16 (m, 5H overlapping CHCl3, Ar-H), 3.76–3.68 (m, 2H, OCHH and CHCH2), 3.58–3.49 (m, 2H, OCHH and NCHH), 3.34 (q, J = 6.5 Hz, 1H, CHCH3), 2.99 (q, J = 7.7 Hz, 1H, OCHH), 2.38–2.27 (m, 1H, CH2CH), 2.13–2.04 (m, 1H, CH2CH), 1.26 (d, J = 6.6 Hz, 3H, CH3CH), 1.03 (s, 1H, CH3), 0.81 (s, 1H, CH3).
13C NMR (126 MHz, CDCl3): δ 164.8 (C=N), 142.6 (Ar-Cq), 128.3 (2 × Ar-C), 128.1 (2 × Ar-C), 127.4 (Ar-C), 79.0 (0CH2), 68.5 (CHPh), 66.5 (Cq), 61.4 (NCH), 50.9 (NCH2), 28.2 (CH3), 28.0 (CH3), 21.3 (CH2CH), 20.3 (CH3CH).
HRMS (ESI-TOF) [M+H]+ calculated for C16H23N2O: 259.1805, found 259.1805.
Addition TO 4,5-dihydrooxazoles
General procedure: To a solution of 2-(1-benzylazetidin-2-yl)-4,4-dimethyl-4,5-dihydrooxazole 4a (1 eq) in dry toluene cooled at −78°C, organolithium (R-Li, 1.1-1.5 eq) was added dropwise. The reaction was stirred for 20 min and quenched with water (1 mL). The crude was extracted with water/ethyl acetate and the collected organic phases were dried over sodium sulfate. The solvent was evaporated under reduced pressure and the alumina chromatography afforded the desire product.
(R*, R*)-2-(1-benzylazetidin-2-yl)-2-butyl-4,4-dimethyl-1,3-oxazolidine 5a
According to the General Procedure, the reaction was carried out using 2-(1-benzylazetidin-2-yl)-4,4-dimethyl-4,5-dihydrooxazole 4a (60 mg, 0.25 mmol) in dry toluene (5 mL) and butyllithium (1.15 M in hexane, 235 μL, 0.27 mmol) affording 5a as yellow oil (66 mg, yield 87%, dr = 98:2). Rf 0.9 (20% AcOEt/hexane).
FT-IR (film, cm−1) 3265, 2958, 2928, 2857, 1599, 1463, 1364, 1268, 1142, 1045, 935, 799, and 732.
1H NMR (500 MHz, CDCl3) δ 7.31–7.21 (m, 5H, Ar-H), 4.03 (d, J = 13.6 Hz, 1H, CH2Ph), 3.63 (d, J = 7.6 Hz, 1H, oxazolidine-CH2), 3.54 (t, J = 8.4 Hz, 1H, CH), 3.41 (d, J = 7.6 Hz, 1H, oxazolidine-CH2), 3.39 (d, J = 13.6 Hz, 1H, CH2Ph), 3.15 (m, 1H, azetidine-CH2), 2.71 (dd, J = 15.6, 8.7 Hz, 1H, azetidine-CH2), 1.99–1.91 (m, 2H, azetidine-CH2), 1.80–1.68 (m, 1H, butyl-H), 1.55–1.46 (m, 1H, butyl-H), 1.34 (s, 3H, oxazoline-CH3), 1.33 (s, 3H, oxazolidine-CH3), 1.31–1.23 (m, 5H, butyl-H), 0.90 (t, J = 7.0 Hz, 1H, CH2-CH3).
13C NMR (176 MHz, CDCl3) δ 138.8 (Ar-Cq), 128.4 (2 × Ar-C), 128.3 (2 × Ar-C), 126.9 (Ar-C), 100.6 (OCqNH), 77.4 (OCH2), 68.7 (CH), 62.9 (CH2Ph) 62.8 (butyl-CH2), 59.6 (butyl-CH2), 50.52 (NCH2), 36.9 (butyl-CH2), 29.9 (oxazolidine-CH3), 29.1 (oxazolidine-CH3), 28.0 (butyl-CH2), 26.51 (butyl-CH2), 23.55 (butyl-CH2), 20.9 (NCH2CH2), 14.17 (butyl-CH3).
HRMS (ESI-TOF) [M+H]+ calculated for C19H31N2O: 303.2436, found 303.2428.
(R*, R*)-2-(1-benzylazetidin-2-yl)-2-hexyl-4,4-dimethyl-1,3-oxazolidine 5b
According to the General Procedure, the reaction was carried out using 2-(1-benzylazetidin-2-yl)-4,4-dimethyl-4,5-dihydrooxazole 4a (70 mg, 0.29 mmol) in dry toluene (6 mL) and hexyllithium (1.25 M in hexane, 255 μL, 0.32 mmol) affording 5b as yellow oil (58 mg, yield 60%, dr = 98:2), Rf 0.9 (20% AcOEt/hexane).
1H NMR (500 MHz, CDCl3) δ 7.41–7.10 (m, 5H, Ar-H overlapping CHCl3 signal), 4.03 (d, J = 13.5 Hz, 1H, CH2Ph), 3.63 (d, J = 7.5 Hz, 1H, oxazolidine-H), 3.54 (t, J = 8.5 Hz, 1H, CH), 3.44 – 3.33 (m, 2H, CH2Ph and oxazolidine-H), 3.17–3.12 (m, 1H, azetidine-CH2), 2.70 (q, J = 8.0 Hz, 1H, azetdine-CH2), 1.94 (m, 2H, azetdine-CH2), 1.77–1.68 (m, 1H, hexyl-CH2), 1.49 (t, J = 10.3 Hz, 1H, hexyl-CH2), 1.34 (s, 3H, oxazoline- CH3), 1.32 (s, 3H, oxazoline-CH3), 1.31–1.23 (m, 9H, hexyl-CH2), 0.88 (t, J = 6.2 Hz, 3H, CH2-CH3).
13C NMR (126 MHz, CDCl3) δ 138.7 (Ar-Cq), 128.4 (2 × Ar-C), 128.3 (2 × Ar-C), 126.9 (Ar-C) 100.6 (OCqN), 77.4 (oxazolidine-CH2), 68.6 (azetidine-CH), 62.8 (CH2Ph), 59.6 (oxazolidine-Cq), 50.5 (azetidine-CH2), 37.2 (hexyl-CH2), 31.9 (hexyl-CH2), 30.15 (hexyl-CH2), 29.11 (oxazolidine-CH3), 28.05 (oxazolidine-CH3), 24.3 (hexyl-CH2), 22.7 (hexyl-CH2), 20.9 (azetidine-CH2), 14.25 (hexyl-CH3).
HRMS (ESI-TOF) [M+H]+ calculated for C21H35N2O: 331.2749, found 331.2743.
(R*, R*)-2-(1-benzylazetidin-2-yl)-2,4,4-trimethyl-1,3-oxazolidine 5c
According to the General Procedure, the reaction was carried out using 2-(1-benzylazetidin-2-yl)-4,4-dimethyl-4,5-dihydrooxazole 4a (50 mg, 0.21 mmol) in dry toluene (5 mL) and methyllithium (1.1 M in diethoxy methane, 210 μL, 0.23 mmol) affording 2-(1-benzylazetidin-2-yl)-2,4,4-trimethyloxazolidine 5c as yellow oil (37 mg, yield 70%, dr = 98:2). Rf 0.9 (5% AcOEt/hexane).
FT-IR (film, cm−1) 3029, 2963, 2928, 2853, 1637, 1454, 1371, 1261, 1044, 797, and 748.
1H NMR (300 MHz, CDCl3): δ 7.39–7.11 (m, 5H, Ar-H overlapping CHCl3 signal), 4.01 (d, J = 13.6 Hz, 1H, CH2Ph), 3.68 (d, J = 8.2 Hz, 1H, OCH2), 3.43–3.33 (m, 3H, CH2Ph, OCH2 and azetidine-CH), 3.15 (t, J = 6.5 Hz, 1H, azetidine-CH2), 2.73 (q, J = 7.9 Hz, 1H, azetidine-CH2), 2.02–1.86 (m, 2H, azetidine-CH2), 1.34 (s, 6H, oxazolidine-CH3), 1.25 (s, 1H, CH3).
13C NMR (126 MHz, CDCl3) δ 138.7 (Ar-Cq), 128.4 (2 × Ar-C), 128.4 (2 × Ar-C), 127.0 (Ar-C), 98.8 (OCqN), 77.7 (OCH2), 70.8 (azetidine-CH), 62.6 (CH2Ph), 59.9 (oxazoline-Cq), 50.2 (azetidine-CH2), 28.8 (oxazolidine-CH3), 27.5 (oxazolidine-CH3), 23.5 (CH3), 20.8 (azetidine-CH2).
HRMS (ESI-TOF) [M+H]+ calculated for C16H25N2O: 261.1967, found 261.1957.
(R*, R*)-2-(1-benzylazetidin-2-yl)-2-ethyl-4,4-dimethyl-1,3-oxazolidine 5d
According to the General Procedure, the reaction was carried out using 2-(1-benzylazetidin-2-yl)-4,4-dimethyl-4,5-dihydrooxazole 4a (120 mg, 0.49 mmol) in dry toluene (10.5 mL) and ethyllithium (0.1 M in benzene/cyclohexane, 5.4 mL, 0.54 mmol) affording 2-(1-benzylazetidin-2-yl)-2-ethyl-4,4-dimethyloxazolidine 5d as colorless oil (78 mg, yield 58%, dr = 98:2). Rf 0.9 (10% AcOEt/hexane).
FT-IR (film, cm−1) 3267, 2963, 2927, 2855, 1455, 1366, 1260, 1208, 1045, 916, 798, 734, and 697.
1H NMR (500 MHz, CDCl3) δ 7.34–7.27 (m, 4H, Ar-H), 7.25–7.20 (m, 1H, Ar-H), 4.03 (d, J = 13.6 Hz, 1H, oxazolidine-CH2), 3.62 (d, J = 7.7 Hz, 1H, CH2Ph), 3.56 (t, J = 8.5 Hz, 1H, azetidine-CH), 3.42 (d, J = 7.7 Hz, 1H, CH2Ph), 3.39 (d, J = 13.6 Hz, 1H, oxazolidine-CH2), 3.15 (td, J = 6.8, 3.8 Hz, 1H, azetidine-CH2), 2.76–2.68 (m, 1H, azetidine-CH2), 1.98–1.91 (m, 2H, azetidine-CH2), 1.84–1.74 (m, 1H, CH2CH3), 1.52 (tt, J = 14.4, 7.2 Hz, 1H, CH2CH3), 1.34 (s, 3H, oxazolidine-CH3), 1.32 (s, 3H, oxazolidine-CH3), 0.91 (t, J = 7.6 Hz, 3H, CH2CH3).
13C NMR (126 MHz, CDCl3) δ 138.7 (Ar-Cq), 128.4 (2 × Ar-C), 128.3 (2 × Ar-C), 126.9 (Ar-C), 100.7 (OCqN), 77.3 (OCH2), 68.1 (azetidine-CH), 62.8 (CH2Ph), 59.6 (Cq), 50.5 (azetidine-CH2), 29.5 (CH2CH3), 29.0 (oxazolidine-CH3), 28.1 (oxazoline-CH3), 20.8 (azetidine-CH2), 8.68 (CH2CH3).
HRMS (ESI-TOF) [M+H]+ calculated for C17H27N2O: 275.2123, found 275.2113.
(R*, R*)-2-(1-benzylazetidin-2-yl)-4,4-dimethyl-2-phenyl-1,3-oxazolidine 5e
According to the General Procedure, the reaction was carried out using 2-(1-benzylazetidin-2-yl)-4,4-dimethyl-4,5-dihydrooxazole 4a (73 mg, 0.30 mmol) in dry toluene (7 mL) and phenyllithium (1.0 M in dibutyl ether, 330 μL, 0.33 mmol) affording 2-(1-benzylazetidin-2-yl)-4,4-dimethyl-2-phenyloxazolidine 5e as colorless oil (79 mg, yield 82%, dr = 98:2). Rf 0.9 (5% AcOEt/hexane).
FT-IR (film, cm−1) 3262, 3084, 3061, 3028, 3001, 2963, 2928, 2857, 1953, 1887, 1812, 1652, 1494, 1453, 1366, 1235, 1038, 941, 738, and 700.
1H NMR (300 MHz, CDCl3) δ 7.62 (d, J = 8.1, 2H, Ar-H), 7.35–7.21 (m, 8H, Ar-H), 4.17 (d, J = 13.4 Hz, 1H, CH2Ph), 3.51–3.41 (m, 4H, HCHPh, oxazolidine-CH2 overlapping azetidine-CH), 3.18–3.09 (m, 1H, azetidine-CH2), 2.63 (dd, J = 16.2, 7.7 Hz, 1H, azetidine-CH2), 1.96 (dt, J = 17.9, 9.0 Hz, 1H, azetidine-CH2), 1.52 (dtd, J = 10.2, 7.9, 2.1 Hz, 1H, azetidine-CH2), 1.38 (s, 3H, CH3), 0.93 (s, 3H, CH3).
13C NMR (176 MHz, CDCl3) δ 143.1 (Ar-Cq), 138.9 (Ar-Cq), 128.5 (2 × Ar-C), 128.4 (2 × Ar-C), 127.8 (2 x Ar-C), 127.4 (Ar-C), 126.9 (Ar-C), 126.9 (2 × Ar-C), 101.2 (OCqN), 77.3 (OCH2), 70.8 (CH), 62.6 (CH2Ph), 60.1 (Cq), 49.7 (azetidine-CH2), 28.00 (CH3), 27.9 (CH3), 20.6 (azetidine-CH2).
HRMS (ESI-TOF) [M+H]+ calculated for C21H27N2O: 323.2123, found 323.2109.
(R)-2-butyl-4,4-dimethyl-2-{(R)-1-[(R)-1-phenylethyl]azetidin-2-yl}-1,3-oxazolidine 6a
According to the General Procedure, the reaction was carried out using dihydrooxazole (R,R)-4b (50 mg, 0.19 mmol) in dry toluene (3 mL) and butyllithium (1.15 M in hexane, 235 μL, 0.28 mmol) affording 6a as a pale yellow oil, yield 86%, 53 mg, dr = 95:5, = + 59.15° (c = 1, CHCl3).
FT-IR (film, cm−1) ν 3412, 3028, 2960, 2930, 2860, 1492, 1453, 1380, 1260, 1227, 1164, 1047, 968, 859, 761, and 701.
1H NMR (500 MHz, CDCl3) δ 7.35–7.30 (m, 2H, Ar-H), 7.29–7.22 (m, 3H, Ar-H), 3.65 (m, 2H, CHPh and OCH2), 3.58 (d, J = 7.8 Hz, 1H, OCH2), 3.49 (t, J = 8.5 Hz, 1H, NCH), 3.00–2.95 (m, 1H, NCH2), 2.67 (q, J = 8.8 Hz, 1H, NCH2), 1.92–1.70 (m, 3H, 2 x NCH2CH2 and butyl-H), 1.49–1.35 (m, 8H, 2 × butyl-H and 2 x oxazolidin-CH3), 1.30 (d, J = 6.9 Hz, 3H, CHCH3), 1.29–1.24 (m, 2H, butyl-H), 1.23–1.16 (m, 1H, butyl-H), 0.87 (t, J = 7.2 Hz, 3H, butyl-CH3).
13C NMR (126 MHz, CDCl3): δ 142.2 (Ar-C) 128.2 (2 × Ar-C), 128.2 (2 × Ar-C), 127.0 (Ar-C), 100.7 (NHCqO), 77.2 (OCH2), 65.4 (NCH), 64.2 (CHCH3), 59.3 (oxazolidine-Cq), 46.2 (NCH2), 35.9 (butyl-CH2), 30.0 (oxazolidine-CH3), 28.9 (oxazolidine-CH3), 26.3 (butyl-CH2), 23.5 (butyl-CH2), 20.9 (CHCH3), 20.1 (NCH2CH2), 14.2 (butyl-CH3).
HRMS (ESI-TOF) [M+H]+ calculated for C20H33N2O: 317.2587, found 317.2587.
(R)-2-hexyl-4,4-dimethyl-2-{(R)-1-[(R)-1-phenylethyl]azetidin-2-yl}-1,3-oxazolidine 6b
According to the General Procedure, the reaction was carried out using dihydrooxazole (R,R)-4b (60 mg, 0.23 mmol) in dry toluene (4 mL) and hexyllithium (2.0 M in hexane, 170 μL, 0.35 mmol) affording 6b as yellow oil, 66 mg, yield 83%, dr = 95:5, Rf 0.9 (20% AcOEt/hexane), = + 69.49° (c = 1, CHCl3).
FT-IR (film, cm−1) ν 3261, 2959, 2927, 2856, 1492, 1454, 1380, 1265, 1210, 1164, 1047, 928, 851, 761, 701, and 665.
1H NMR (500 MHz, CDCl3) δ 7.35–7.30 (m, 2H, Ar-H), 7.28–7.22 (m, 3H, Ar-H), 3.67–3.61 (m, 2H, CHPh and OCH2), 3.58 (d, J = 7.8 Hz, 1H, OCH2), 3.49 (t, J = 8.5 Hz, 1H, NCH), 3.00–2.95 (m, 1H, NCH2), 2.67 (q, J = 8.8 Hz, 1H, NCH2), 1.93–1.70 (m, 3H, NCH2CH2 and hexyl-H), 1.49–1.40 (m, 1H, hexyl-H), 1.39 (s, 3H, oxazolidine-CH3), 1.36 (s, 3H, oxazolidine-CH3), 1.32–1.21 (m, 9H, CHCH3 and 6 × hexyl-H), 0.87 (t, J = 7.0 Hz, 3H, hexyl-CH3).
13C NMR (126 MHz, CDCl3) δ 142.2 (Ar-Cq), 128.2 (2 × Ar-C), 128.2 (2 × Ar-C), 127.0 (Ar-C), 100.7 (NHCqO), 77.2 (OCH2), 65.4 (NCH), 64.2 (CHCH3), 59.3 (oxazolidine-Cq), 46.2 (NCH2), 36.2 (hexyl-CH2), 31.9 (hexyl-CH2), 30.1 (oxazolidine-CH3), 30.0 (hexyl-CH2), 28.9 (oxazolidine-CH3), 24.1 (hexyl-CH2), 22.7 (hexyl-CH2), 20.9 (CHCH3), 20.1 (NCH2CH2), 14.2 (hexyl-CH3).
HRMS (ESI-TOF) [M+H]+ calculated for C22H37N2O: 345.2900, found 345.2894.
(R)-2,4,4-trimethyl-2-{(R)-1-[(R)-1-phenylethyl]azetidin-2-yl}-1,3-oxazolidine 6c
According to the General Procedure, the reaction was carried out using dihydrooxazole (R,R)-4b (50 mg, 0.19 mmol) in dry toluene (3 mL) and methyllithium (1.0 M in diethoxymethane, 290 μL, 0.29 mmol) affording oxazolidine 6c as yellow oil, 36 mg, yield 70%, dr = 95:5). Rf 0.9 (5% AcOEt/hexane). = + 69.10° (c = 1, CHCl3).
FT-IR (film, cm−1) ν 3258, 2965, 2928, 2861, 1707, 1452, 1371, 1262, 1230, 1166, 1046, 846, and 701.
1H NMR (300 MHz, CDCl3) δ 7.37–7.20 (m, 5H, Ar-H overlapping CHCl3), 3.72 (d, J = 7.7 Hz, 1H, OCH2), 3.64 (q, J = 6.8 Hz, 1H, CHPh), 3.53 (d, J = 7.7 Hz, 1H, OCH2), 3.34 (t, J = 8.5 Hz, 1H, NCH), 2.99 (dd, J = 12.4, 6.1 Hz, 1H, NCH2), 2.69 (td, J = 8.9, 7.1 Hz, 1H, NCH2), 1.85 (td, J = 8.8, 6.0 Hz, 2H, NCH2CH2), 1.39 (s, 1H, oxazolidine-CH3), 1.38 (s, 1H, oxazolidine-CH3), 1.31 (d, J = 6.9 Hz, 1H, CHCH3), 1.21 (s, 1H, oxazolidine-CH3).
13C NMR (126 MHz, CDCl3) δ 142.1 (Ar-Cq), 128.3 (2 × Ar-C), 128.2 (2 × Ar-C), 127.1 (Ar-C), 99.0 (OCqNH), 77.5 (OCH2), 67.6 (NCH), 64.1 (CHPh), 59.6 (oxazolidine-Cq), 46.1 (NCH2), 29.7 (oxazolidine-CH3), 28.4 (oxazolidine-CH3), 23.0 (oxazolidine-CH3), 20.8 (CHCH3), 20.2 (NCH2).
HRMS (ESI-TOF) [M+Na]+ calculated for C17H26N2NaO: 297.1943, found 297.1946.
(R)-4,4-dimethyl-2-phenyl-2-{(R)-1-[(R)-1-phenylethyl]azetidin-2-yl}-1,3-oxazolidine 6d
According to the General Procedure, the reaction was carried out using dihydrooxazole (R,R)-4b (50 mg, 0.19 mmol) in dry toluene (3 mL) and phenyllithium (1.0 M in dibutyl ether, 290 μL, 0.29 mmol) affording oxazolidine 6d as colorless oil, 62 mg, yield 95%, dr = 95:5). Rf 0.8 (10% AcOEt/hexane). = + 40.10° (c = 1, CHCl3). (ADH, 99.5:0.5 Hex:iPrOH + 0.2% DEA, 0.3 mL/min).
FT-IR (film, cm−1) ν 3257, 3026, 2927, 2865, 1450, 1382, 1268, 1200, 1164, 1039, 961, 851, 752, 720, 701, and 647.
1H NMR (500 MHz, CDCl3) δ 7.62–7.58 (m, 2H, Ar-H), 7.37–7.20 (m, 8H, Ar-H), 3.75–3.68 (m, 1H, CHPh), 3.55 (d, J = 7.5 Hz, 1H, OCH2), 3.47–3.38 (m, 2H, NCH and OCH2), 3.01–2.90 (m, 1H, NCH2), 2.60–2.52 (m, 1H, NCH2), 1.87–1.77 (m, 1H, NCH2CH2), 1.44 (s, 3H, oxazolidine-CH3), 1.41 (d, J = 6.7 Hz, 1H, CHCH3), 1.33–1.23 (m, 1H, NCH2CH2), 0.99 (s, 1H, oxazolidine-CH3).
13C NMR (126 MHz, CDCl3) δ 142.9 (Ar-Cq), 142.5 (Ar-Cq), 128.2 (2 × Ar-C), 128.2 (2 × Ar-C), 127.6 (2 × Ar-C), 127.4 (Ar-C), 127.3 (2 × Ar-C), 125.8 (Ar-C), 101.2 (NHCqO), 77.1 (OCH), 68.0 (NCH), 64.4 (CHPh), 59.9 (oxazolidine-Cq), 45.9 (NCH2), 28.6 (2 × oxazolidine-CH3), 21.2 (CHCH3), 19.8 (NCH2CH2).
HRMS (ESI-TOF) [M+H]+ calculated for C22H29N2O: 337.2274, found 337.2277.
(S)-2-hexyl-4,4-dimethyl-2-{(S)-1-[(R)-1-phenylethyl]azetidin-2-yl}-1,3-oxazolidine 7a
According to the General Procedure, the reaction was carried out using dihydrooxazole (R,S)-4b (80 mg, 0.31 mmol) in dry toluene (3 mL) and hexyllithium (1.6 M in hexane, 290 μL, 0.46 mmol) affording 7a as yellow oil, 96 mg, yield 90%, dr = 95:5, Rf 0.8 (30% AcOEt/hexane = −8.30° (c = 1, CHCl3).
FT-IR (film, cm−1) ν 3259, 2960, 2928, 2857, 1453, 1380, 1365, 1196, 1173, 1047, 930, 773, and 699.
1H NMR (500 MHz, CDCl3) δ 7.34–7.27 (m, 4H, Ar-H), 7.23–7.18 (m, 1H, Ar-H), 3.85 (q, J = 6.8 Hz, 1H, CHPh), 3.79 (t, J = 8.3 Hz, 1H, NCH), 3.53 (d, J = 7.6 Hz, 1H, OCH), 3.25 (d, J = 7.6 Hz, 1H, OCH), 3.19 (dd, J = 15.3, 8.6 Hz, 1H, NCH2), 2.93 (td, J = 7.9, 3.0 Hz, 1H, NCH2), 1.95–1.88 (m, 2H, NCH2CH2), 1.74–1.64 (m, 1H, hexyl-H), 1.51–1.42 (m, J = 13.0, 8.4 Hz, 1H, hexyl-H), 1.41–1.20 (m, 14H, CHCH3, oxazolidine-CH3 and 8 × hexyl-H), 1.19 (s, J = 8.0 Hz, 3H, oxazolidine-CH3), 0.88 (t, J = 6.9 Hz, 3H, hexyl-CH3).
13C NMR (126 MHz, CDCl3) δ 144.3 (Ar-Cq), 128.2 (2 × Ar-C), 127.2 (2 × Ar-C), 126.6 (Ar-C), 100.7 (OCqNH), 77.1 (OCH2), 64.3 (NCH), 59.4 (oxazolidine-Cq), 58.9 (CHPh), 42.9 (NCH2), 36.9 (hexyl-CH2), 31.9 (hexyl-CH2), 30.2 (hexyl-CH2), 29.6 (oxazolidine-CH3), 28.6 (oxazolidine-CH3), 24.2 (hexyl-CH2), 22.8 (hexyl-CH2), 20.6 (NCH2CH2), 14.2 (hexyl-CH3), 13.8 (CHCH3).
HRMS (ESI-TOF) [M+Na]+ calculated for C22H36N2NaO: 367.2725, found 367.2718.
(S)-4,4-dimethyl-2-phenyl-2-{(S)-1-[(R)-1-phenylethyl]azetidin-2-yl}-1,3-oxazolidine 7d
According to the General Procedure, the reaction was carried out using dihydrooxazole (R,S)-4b (50 mg, 0.19 mmol) in dry toluene (3 mL) and phenyllithium (1.0 M in dibutyl ether, 290 μL, 0.29 mmol) affording oxazolidine 7b as yellow oil, 59 mg, yield 90%, dr = 95:5). Rf 0.7 (30% AcOEt/hexane). = −3.09 (c = 0.5, CHCl3). (LUX-1, 99.5:0.5 Hex:iPrOH + 0.2% DEA, 0.5 mL/min).
FT-IR (film, cm−1) ν 3060, 3027, 2966, 2929, 2866, 1687, 1493, 1451, 1366, 1273, 1233, 1036, 742, and 700.
1H NMR (500 MHz, CDCl3) δ 7.59–7.55 (m, 2H, Ar-H), 7.39–7.21 (m, 8H, Ar-H overlapping CHCl3), 3.96 (q, J = 6.8 Hz, 1H, CHPh), 3.64 (t, J = 8.3 Hz, 1H, NCH), 3.32 (d, J = 7.3 Hz, 1H, OCH2), 3.16 (d, J = 7.3 Hz, 1H, OCH2), 3.06 (dt, J = 15.1, 7.5 Hz, 1H, NCH2), 2.97–2.90 (m, 1H, NCH2), 1.94 (tt, J = 17.4, 8.7 Hz, 1H, NCH2CH2), 1.47–1.36 (m, 4H, CHCH3 and NCH2CH2), 1.24 (s, 3H, oxazolidine-CH3), 0.86 (s, 3H, oxazolidine-CH3).
13C NMR (126 MHz, CDCl3) δ 144.4 (Ar-Cq), 143.2 (Ar-Cq), 128.9 (2 × Ar-C), 127.7 (2 × Ar-C), 127.5 (2 × Ar-C), 127.4 (Ar-C), 127.1 (2 × Ar-C), 126.6 (Ar-Cq), 101.2 (OCqNH), 76.9 (OCH2) 67.3 (NCH), 59.9 (oxazolidine-Cq), 59.8 (CHPh), 42.7 (NCH2), 28.5 (oxazolidine-CH3), 28.0 (oxazolidine-CH3), 20.2 (NCH2CH2), 14.6 (CHCH3).
HRMS (ESI-TOF) [M+Na]+ calculated for C22H28N2NaO: 359.2099, found 359.2092.
Hydrolysis of 1,3-oxazolidines
General Procedure: To a solution of oxazolidines 5a-d, 6a-c and 7b (0.16 mmol, 1 eq) in dichloromethane, silica (150 mg) was added. The reaction was stirred for 3 h at room temperature. The crude was filtered and the solvent was evaporated under reduced pressure to obtain the desire product.
1,3-Oxazolidines 7a and 7c undergo quantitative hydrolysis to corresponding acyl derivatives 10a and c during the chromatography on silica gel.
1-(1-benzylazetidin-2-yl)pentan-1-one 8a
According to the General Procedure, the reaction was carried out using oxazolidine 5a (50 mg, 0.16 mmol), ketoazetidine 8a was obtained as colorless oil (33 mg, yield 90%).
FT-IR (film, cm−1) 2957, 2926, 2854, 1711, 1455, 1360, 1260, 1029, 913, 801, 747, and 700.
1H NMR (500 MHz, CDCl3): δ 7.33–7.23 (m, 5H, Ar-H overlapping CHCl3 signal), 3.73 (d, J = 12.5 Hz, 1H, CH2Ph), 3.68 (t, J = 8.6 Hz, 1H, azetidine-CH), 3.58 (d, J = 12.5 Hz, 1H, CH2Ph), 3.35–3.30 (m, 1H, azetidine-CH2), 2.94 (dd, J = 16.2, 8.0 Hz, 1H, azetidine-CH2), 2.53 (ddd, J = 17.5, 8.6, 6.3 Hz, 1H, butyl-CH2), 2.28-2.12 (m, 3H, azetidine-CH2 and butyl-CH2), 1.50–1.36 (m, 2H, butyl-CH2), 1.25-1.19 (m, 2H, butyl-CH2), 0.86 (t, J = 7.4 Hz, 3H, CH3).
13C NMR (126 MHz, CDCl3): δ 212.0 (C = O), 137.2 (Ar-Cq), 129.0 (2 x Ar-C), 128.4 (2 x Ar-C), 127.3 (Ar-C), 71.33 (azetidine-CH), 62.9 (CH2Ph), 50.8 (azetidine-CH2), 37.7 (butyl-CH2), 25.1 (butyl-CH2), 22.3 (butyl-CH2), 21.8 (azetidine-CH2), 13.9 (CH3).
HRMS (ESI-TOF) [M+H]+ calculated for C15H22NONa: 254.1521, found 254.1510.
1-(1-benzylazetidin-2-yl)heptan-1-one 8b
According to the General Procedure, the reaction was carried out using from oxazolidine 5b (50 mg, 0.15 mmol), ketoazetidine 8b was obtained as colorless oil (35 mg, yield 90%).
FT-IR (film, cm−1) 2926, 2853, 1707, 1454, 1364, 1460, 1028, 912, 802, 735, and 699. 1H NMR (500 MHz, CDCl3): δ 7.36–7.22 (m, 5H, Ar-H overlapping CHCl3 signal), 3.73 (d, J = 12.6 Hz, 1H, CH2Ph), 3.68 (t, J = 8.7 Hz, 1H, azetidine-CH), 3.58 (d, J = 12.5 Hz, 1H, CH2Ph), 3.34–3.29 (m, 1H, azetidine-CH2), 2.94 (dd, J = 16.2, 8.0 Hz, 1H, azetidine-CH2), 2.53 (ddd, J = 17.5, 8.5, 6.3 Hz, 1H, hexyl-CH2), 2.28–2.11 (m, 3H, azetidine-CH2 and hexyl-CH2), 1.50–1.40 (m, 2H, hexyl-CH2), 1.30–1.17 (m, 6H, hexyl-CH2), 0.87 (t, J = 7.0 Hz, 3H, CH3).
13C NMR (126 MHz, CDCl3): δ 212.2 (C = O), 137.5 (Ar-Cq), 129.2 (2 x Ar-C), 128.5 (2 x Ar-C), 127.5 (Ar-C), 71.5 (azetidine-CH), 63.1 (CH2Ph), 51.0 (azetidine-CH2), 31.8 (hexyl-CH2), 29.9 (hexyl-CH2), 29.1 (hexyl-CH2), 23.2 (hexyl-CH2), 22.7 (hexyl-CH2), 21.9 (azetidine-CH2), 14.2 (CH3).
HRMS (ESI-TOF) m/z: calcd for C17H25NONa [M+Na]+ 282.1834; found 282.1828.
1-(1-benzylazetidin-2-yl)ethanone 8c
According to the General Procedure, the reaction was carried out using oxazolidine 5c (50 mg, 0.19 mmol) in dichloromethane (1 mL) affording 2-acylazetidine 8c as yellow oil, yield 95%, 32 mg, dr = 95:5).
FT-IR (film, cm−1) 3086, 3062, 3028, 2965, 2930, 2867, 1955, 1881, 1734, 1658, 1652, 1531, 1454, 1162, 1069, and 914.
1H NMR (700 MHz, CDCl3) δ 7.33–7.23 (m, 5H, Ar-H overlapping CHCl3 signal), 3.73 (d, J = 12.6 Hz, 1H, CH2Ph), 3.66 (dd, J = 14.5, 5.8 Hz, 1H, azetidine-CH), 3.59 (d, J = 12.6 Hz, 1H, CH2Ph), 3.34–3.31 (m, 1H, azetidine-CH2), 2.94 (ddd, J = 9.3, 8.0, 6.9 Hz, 1H, azetidine-CH2), 2.23 (dtd, J = 10.8, 8.3, 2.5 Hz, 1H, azetidine-CH2), 2.18 (ddd, J = 19.4, 10.5, 8.9 Hz, 1H, azetidine-CH2), 2.05 (s, 3H, CH3).
13C NMR (176 MHz, CDCl3): δ 210.5 (C = O), 137.4 (Ar-Cq), 129.1 (2 × Ar-C), 128.5 (2 × Ar-C), 127.5 (Ar-C), 71.8 (azetidine-CH), 63.2 (CH2Ph), 51.0 (azetidine-CH2), 25.7 (CH3), 21.8 (azetidine-CH2).
HRMS (ESI-TOF) [M+H]+ calculated for C12H16NO: 190.1232, found 190.1226.
1-(1-benzylazetidin-2-yl)propan-1-one 8d
According to the General Procedure, the reaction was carried out using oxazolidine 5d (50 mg, 0.18 mmol), ketoazetidine 8d was obtained as yellow oil (33 mg, yield 90%).
FT-IR (film, cm−1) 3063, 3029, 2964, 2928, 2875, 1955, 1709, 1634, 1495, 1454, 1404, 1241, 1077, 1028, 735, and 700.
1H NMR (500 MHz, CDCl3): δ 7.36–7.22 (m, 5H, Ar-H overlapping CHCl3 signal), 3.75 (d, J = 12.6 Hz, 1H, CH2Ph), 3.69 (t, J = 8.6 Hz, 1H, azetidine-CH), 3.57 (d, J = 12.6 Hz, 1H, CH2Ph), 3.33–3.28 (m, 1H, azetidine-CH2), 2.93 (q, J = 7.5 Hz, 1H, azetidine-CH2), 2.60 (dq, J = 18.4, 7.3 Hz, 1H, COCH2), 2.35–2.11 (m, 3H, azetidine-CH2 and COCH2), 0.95 (t, J = 7.3 Hz, 3H, CH3).
13C NMR (75 MHz, CDCl3): δ 212.6 (C = O), 137.6 (Ar-Cq) 129.1 (2 x Ar-C), 128.5 (2 x Ar-C), 127.5 (Ar-C), 71.4 (azetidine-CH), 63.1 (CH2Ph), 51.1 (azetidine-CH2), 31.4 (COCH2), 22.0 (azetidine-CH2), 7.33 (CH3).
HRMS (ESI-TOF) [M+H]+ calculated for C13H18NO: 204.1388, found 204.1381.
1-{(R)-1-[(R)-1-phenylethyl]azetidin-2-yl}pentan-1-one 9a
Starting from oxazolidine 6a (50 mg, 0.16 mmol), ketoazetidine 9a was obtained as pale yellow oil. (35 mg, yield 90%), er = 98:2 (LUX-1, 99:1 Hex:iPrOH + 0.5% DEA, 0.5mL/min). = + 149.46° (c = 1, CHCl3).
FT-IR (KBr, cm−1) ν 3391, 3028, 2960, 2930, 2871, 1705, 1493, 1453, 1371, 1282, 1029, 760, and 701.
1H NMR (500 MHz, CDCl3): δ 7.36–7.29 (m, 4H, Ar-H), 7.25–7.21 (m, 1H, Ar-H), 3.72 (t, J = 8.7 Hz, 1H, CHCO), 3.39 (q, J = 6.6 Hz, 1H, CHPh), 3.14–3.05 (m, 1H, NCH2), 2.80–2.60 (m, 3H, NCHH and COCH2), 2.21–2.12 (m, 1H, NCH2CH2 overlapping acetone), 2.10–2.00 (m, 1H, NCH2CH2), 1.66–1.54 (m, 2H, COCH2CH2), 1.40–1.31 (m, 2H, COCH2CH2CH2), 1.12 (d, J = 6.6 Hz, 3H, CHCH3), 0.94 (t, J = 7.4 Hz, 1H, CH2CH3).
13C NMR (126 MHz, CDCl3): δ 212.6 (CO), 143.1 (Ar-Cq), 128.5 (2 x Ar-C), 127.4 (2 x Ar-C), 127.3 (Ar-C), 71.3 (CHCO), 67.7 (CHPh), 50.0 (NCH2), 37.4 (COCH2), 25.7 (COCH2CH2), 22.6 (CH2CH3), 21.4 (CH3CH), 21.1 (NCH2CH2), 14.1 (CH2CH3).
HRMS (ESI-TOF) [M+H]+ calculated for C16H24NO: 246.1858, found 246.1852.
1-{(R)-1-[(R)-1-phenylethyl]azetidin-2-yl}heptan-1-one 9b
Starting from oxazolidine 6b (50 mg, 0.145mmol), ketoazetidine 9b was obtained as pale yellow oil (30 mg, yield 75%). = +102.22° (c = 0.7, CHCl3) (ADH, 99:1 Hex:iPrOH, 0.5 mL/min).
FT-IR (KBr, cm−1) ν 2958, 2928, 2854, 1706, 1493, 1452, 1370, 1282, 1068, 1029, 60, and 700.
1H NMR (500 MHz, CDCl3): δ 7.39–7.29 (m, 4H, Ar-H), 7.27–7.22 (m, 1H, Ar-H overlapping CHCl3), 3.72 (t, J = 8.6 Hz, 1H, NCH), 3.39 (q, J = 6.5 Hz, 1H, CHPh), 3.10 (dd, J = 11.0, 4.3 Hz, 1H, NCH), 2.86–2.58 (m, 3H, NCHH and COCH2), 2.22–2.11 (m, 1H, NCH2CH2), 2.10–2.01 (m, 1H, NCH2CH2), 1.64–1.54 (m, 2H, hexyl-CH2), 1.37–1.28 (m, 6H, hexyl-CH2), 1.12 (d, J = 6.6 Hz, 3H, CHCH3), 0.90 (t, J = 6.7 Hz, 3H, hexyl-CH3).
13C NMR (126 MHz, CDCl3): δ 212.7 (C = O), 143.1 (Ar-Cq), 128.5 (2 × Ar-C), 127.5 (2 × Ar-C), 127.3 (Ar-C), 71.4 (NCH), 67.7 (CHPh), 50.0 (NCH2), 37.8 (hexyl-CH2), 31.9 (hexyl-CH2), 29.2 (hexyl-CH2), 23.5 (hexyl-CH2), 22.7 (hexyl-CH2), 21.5 (CHCH3), 21.1(NCH2CH2), 14.2 (hexyl-CH3).
HRMS calcd. for C18H27NNaO [M+Na]+ 296.1990; found 296.1978.
1-{(R)-1-[(R)-1-phenylethyl]azetidin-2-yl}ethanone 9c
Starting from oxazolidine 6c (50 mg, 0.18 mmol), ketoazetidine 9c was obtained as pale yellow oil (37 mg, yield 90%).
FT-IR (KBr, cm−1) ν 2968, 2929, 2850, 1705, 1493, 1453, 1354, 1282, 1245, 1172, 1072, 758, and 701.
1H NMR (500 MHz, CDCl3): δ 7.35–7.29 (m, 4H, Ar-H), 7.27–7.22 (m, 1H, Ar-H), 3.69 (t, J = 8.7 Hz, 1H, CHCO), 3.40 (q, J = 6.6 Hz, 1H, CHPh), 3.13–3.08 (m, 1H, NCH2), 2.78 (dd, J = 16.4, 8.0 Hz, 1H, NCH2), 2.34 (s, 3H, CH3CO), 2.22–2.02 (m, 2H, CH2CH), 1.13 (d, J = 6.6 Hz, 3H, CHCH3). 13C NMR (126 MHz, CDCl3): δ 211.2 (CO), 143.1 (Ar-Cq), 128.5 (2 × Ar-C), 127.4 (2 × Ar-C), 127.3 (Ar-C), 71.6 (CHCO), 67.7 (CHPh), 50.0 (NCH2), 25.4 (CH3CO), 21.4 (CH3CH), 20.9 (NCH2CH2). HRMS (ESI-TOF) [M+Na]+ calculated for C13H17NNaO: 226.1208, found 226.1200.
1-{(S)-1-[(R)-1-phenylethyl]azetidin-2-yl}pentan-1-one 10a
Pale yellow oil. er = 98:2 (LUX-1, 99:1 Hex:iPrOH + 0.5% DEA, 0.5 mL/min). = −26.61° (c = 1, CHCl3).
FT-IR (KBr, cm−1) ν 2959, 2930, 2871, 1705, 1493, 1453, 1373, 1303, 1276, 1159, 1028, 760, and 701.
1H NMR (500 MHz, CDCl3): δ 7.26–7.17 (m, 5H, Ar-H), 3.63 (t, J = 8.6 Hz, 1H, CHCO), 3.52 (td, J = 7.3, 2.8 Hz, 1H, NCH2), 3.31 (q, J = 6.5 Hz, 1H, CHPh), 2.98 (dd, J = 16.0, 8.4 Hz, 1H, NCH2), 2.12–2.06 (m, 2H, NCH2CH2), 2.05–1.97 (m, 1H, COCH2), 1.92–1.83 (m, 1H, COCH2), 1.29 (d, J = 6.6 Hz, 3H, CHCH3), 1.22–0.98 (m, 4H, CH2CH2CH3), 0.76 (t, J = 7.2 Hz, 3H, CH2CH3).
13C NMR (126 MHz, CDCl3): δ 211.1 (CO), 142.0 (Ar-Cq), 128.4 (2 × Ar-C), 128.4 (2 × Ar-C), 127.8 (Ar-C), 70.9 (CHCO), 68.5 (CHPh), 50.6 (NCH2), 38.3 (COCH2), 25.3 (COCH2CH2), 22.3 (COCH2CH2CH2), 21.1 (NCH2CH2), 19.4 (CHCH3), 14.0 (CH2CH3).
HRMS (ESI-TOF) [M+H]+ calculated for C16H24NO: 246.1858, found 246.1852.
1-((S)-1-((R)-1-phenylethyl)azetidin-2-yl)heptan-1-one 10b
Starting from oxazolidine 7b (50 mg, 0.145 mmol), ketoazetidine 10b was obtained as pale yellow oil (36 mg, yield 90%), = −10.81° (c = 0.3, CHCl3). (LUX-1, 99:1 Hex:iPrOH +0.5% DEA, 0.5 mL/min).
FT-IR (KBr, cm−1) ν 2957, 2928, 2855, 1705, 1453, 1372, 1302, 1276, 1159, 1082, 1029, and 701.
1H NMR (500 MHz, CDCl3): δ 7.38–7.29 (m, 1H, Ar-H), 7.27–7.10 (m, 4H, 4 × Ar-H overlapping CDCl3), 3.63 (t, J = 8.5 Hz, 1H, NCH), 3.53 (td, J = 7.3, 2.7 Hz, 1H, NCH2), 3.32 (q, J = 6.6 Hz, 1H, CHPh), 2.99 (dd, J = 15.9, 8.6 Hz, 1H, NCH2), 2.13–1.97 (m, 3H, hexyl-H and NCH2), 1.88 (ddd, J = 17.5, 8.8, 5.9 Hz, 1H, hexyl-H), 1.33–1.08 (m, 9H, 6 × hexyl-H and CHCH3), 1.06–0.99 (m, 2H, hexyl-H), 0.85 (t, J = 7.3 Hz, 3H, hexyl-H).
13C NMR (126 MHz, CDCl3): δ 211.1 (C = O), 142.0 (Ar-Cq), 128.4 (2 × Ar-C), 128.4 (2 × Ar-C), 127.8 (Ar-C), 71.0 (NCH), 68.5 (CHPh), 50.6 (NCH2), 38.6 (hexyl-C), 31.7 (hexyl-C), 28.9 (hexyl-C), 23.1 (hexyl-C), 22.6 (hexyl-C), 21.1 (NCH2CH2), 19.4 (CHCH3), 14.2 (hexyl-C).
HRMS (ESI-TOF) [M+H]+ calculated for C18H27NNaO: 296.1990, found 296.1985.
1-{(S)-1-[(R)-1-phenylethyl]azetidin-2-yl}ethanone 10c
Pale yellow oil. Yield 80%. = −3.52° (c = 0.3, CHCl3).
FT-IR (KBr, cm−1) ν 2965, 2928, 2851, 1706, 1493, 1453, 1352, 1244, 1167, 1030, and 702.
1H NMR (500 MHz, CDCl3): δ 7.35–7.30 (m, 1H, Ar-H), 7.28–7.20 (m, 4H, Ar-H overlapping CHCl3), 3.59 (t, J = 8.6 Hz, 1H, NCH), 3.52 (td, J = 7.4, 2.7 Hz, 1H, NCH2), 3.32 (q, J = 6.6 Hz, 1H, CHPh), 3.04–2.97 (m, 1H, NCH2), 2.15–2.02 (m, 2H, NCH2CH2), 1.69 (s, 3H, COCH3), 1.30 (d, J = 6.5 Hz, 3H, CHCH3).
13C NMR (126 MHz, CDCl3): δ 207.1 (C = O), 142.7 (Ar-Cq), 128.5 (2 × Ar-C), 128.4 (2 × Ar-C), 128.0 (Ar-C), 71.5 (NCH), 68.3 (CHPh), 50.6 (NCH2), 25.5 (COCH3), 20.8 (NCH2CH2), 19.2 (CHCH3).
HRMS (ESI-TOF) [M+Na]+ calculated for C13H17NNaO: 226.1208, found 226.1202.
X-ray Structural Study of Compound (S,S)-3b
Colorless needles of compound (S,S)-3b were obtained by slow evaporation of solvent (Methanol) at room temperature. A single crystal (dimensions 0.500 × 0.350 × 0.220 mm) was selected and mounted on a glass fiber for the X-ray diffraction measurements. The X-ray diffraction experiment was carried out at room temperature by a Bruker-Nonius KappaCCD single crystal diffractometer, equipped with a charge-coupled device (CCD detector), using monochromatized MoKα radiation (λ 0.71073 Å). The automatic data collection was performed by the COLLECT software, cell determination and refinement by DIRAX and data reduction by EVAL. Absorption effects were corrected by SADABS program via a semi-empirical approach. Additional software used: WinGX21 for preparing the material for publication. The structure was solved by direct methods by using SIR2014 program and refined via full-matrix least squares on F2 by SHELXL2014/7. Non-hydrogen atoms were refined anisotropically. The hydrogen atoms were placed at calculated positions and refined isotropically using a riding model approximation with the displacement parameters set to Uiso(H) = 1.5·Ueq(C) in the case of methyl carbon and to Uiso(H) = 1.2·Ueq(C) for all other carbon atoms where Ueq is the equivalent isotropic displacement parameter of carbon. The compound (S,S)-3b belongs to the monoclinic crystal system with cell lengths (a 7.422(1); b 11.655(3); c 10.382(7); alpha 90; beta 110.40(1); gamma 90). Cell volume 841.8(2) Å3; Z 2. ρcalc 1.090 g/cm3; space group p21. A total of 3,806 reflections were collected at 298 K in the θ range from 3.410 to 27.489°, of which 2254 were observed (I > 2σ(I)). The final values of agreement factors were R 7.34 and wR 17.02% with a number of refined parameters was 187 and the maximum and minimum residual densities were Δρmax 0.52 and Δρmin −0.60. The complete crystallographic information on compound (S,S)-3b has been deposited at the Cambridge Crystallographic Data Center (deposit CCDC 1947700).
Data Availability
All datasets generated for this study are included in the manuscript/Supplementary Files.
Author Contributions
PM, MC, and FF have been involved in the synthesis of all compounds with the help of CC. AA and FF performed X-Ray analysis for compound (S,S)-3b. LP performed DFT and NMR calculations. RL and LD supervised this work and wrote the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by the project MISE, Horizon 2020 – PON 2014/2020 FARMIDIAB code 338 and Support for research Funds Es. Fin. 2017-18, University of Bari.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00614/full#supplementary-material
References
Antermite, D., Degennaro, L., and Luisi, R. (2017). Recent advances in the chemistry of metallated azetidines Org. Biomol. Chem. 15, 34–50. doi: 10.1039/C6OB01665K
Arnould, J. C., Cossy, J., and Pete, J. P. (1980). Reactivite photochimique des α-aminoenones: reactions de cyclisation et nouveau type de reaction dans les α-sulfonamido-cyclohexenones. Tetrahedron 36, 1585–1592. doi: 10.1016/S0040-4020(01)83126-4
Azzena, U., Carraro, M., and Pisano, L. (2018). Addressing stereochemistry of heterocyclic compounds by DFT NMR calculations. Chem. Hetrocycl. Comp. 54, 380–388. doi: 10.1007/s10593-018-2279-x
Azzena, U., Dettori, G., Pisano, L., Musio, B., and Luisi, R. (2011). BH3 -promoted stereoselective β-lithiation of N-alkyl-2-phenylaziridines. J. Org. Chem. 76, 2291–2295 doi: 10.1021/jo102474u
Capriati, V., Degennaro, L., Florio, S., Flavia, R., and Luisi, R. (2002). Stereospecific β-lithiation of oxazolinyloxiranes: synthesis of α,β-epoxy-γ-butyrolactones. Org. Lett. 4, 1551–1554. doi: 10.1021/ol025781i
Carroccia, L., Degennaro, L., Romanazzi, G., Cuocci, C., Pisano, L., and Luisi, R. (2014). Straightforward access to 4-membered sulfurated heterocycles: introducing a strategy for the single and double functionalization of thietane 1-oxide. Org. Biomol. Chem. 12, 2180–2184. doi: 10.1039/c4ob00173g
Couty, F., Drouillat, B., and Lemée, F. (2011). Ring Expansion of 2-(1-Hydroxyalkyl)azetidines to 4-(2-Chloroethyl)oxazolidinones. Eur. J. Org. Chem. 794–801. doi: 10.1002/ejoc.201001375
Couty, F., and Evano, G. (2009). Azetidines: new tools for the synthesis of nitrogen heterocycles Synlett, 19, 3053–3064. doi: 10.1055/s-0029-1218299
Couty, F., Evano, G., and Prim, D. (2004). Synthesis of chiral not racemic azetidines. Mini ReV. Org. Chem. 1, 133–148. doi: 10.2174/1570193043488845
Couty, F., and Prim, D. (2002). Synthesis of enantiopure 2-acyl azetidines and the application of amino alcohols derived therefrom in enantioselective catalysis. Tetrahedron Asymmet. 13, 2619–2624. doi: 10.1016/S0957-4166(02)00710-3
De Ceglie, M. C., Musio, B., Affortunato, F., Moliterni, A., Altomare, A., Florio, S., et al. (2011). Solvent- and temperature-dependent functionalisation of enantioenriched aziridines. Chem. A Eur. J. 17, 286–296. doi: 10.1002/chem.201002172
Degennaro, L., Carroccia, L., Parisi, G., Zenzola, M., Romanazzi, G., Fanelli, F., et al. (2015a). Regio- and stereoselective synthesis of sulfur-bearing four-membered heterocycles: direct access to 2,4-disubstituted thietane 1-oxides. J. Org. Chem. 80, 12201–12211. doi: 10.1021/acs.joc.5b02126
Degennaro, L., Giovine, A., Carroccia, L., and Luisi, R. (2014a). “Lithium compounds in organic synthesis: from fundamentals to applications,”in Practical Aspects of Organolithium Chemistry, eds R. Luisi and V. Capriati. (Weinheim: VCH Verlag & Co. KGaA, 513–538.
Degennaro, L., Mansueto, R., Carenza, E., Rizzi, R., Florio, S., Pratt, L. M., et al. (2011). Nitrogen dynamics and reactivity of chiral aziridines: generation of configurationally stable aziridinyllithium compounds. Chem. – Eur. J. 17, 4992–5003. doi: 10.1002/chem.201003424
Degennaro, L., Pisano, L., Parisi, G., Mansueto, R., Clarkson, G. J., Shipman, M., et al. (2015b). Nitrogen stereodynamics and complexation phenomena as key factors in the deprotonative dynamic resolution of alkylideneaziridines: a spectroscopic and computational study. J. Org. Chem. 80, 6411–6418. doi: 10.1021/acs.joc.5b00848
Degennaro, L., Zenzola, M., Trinchera, P., Carroccia, L., Giovine, A., Romanazzi, G., et al. (2014b). Regioselective functionalization of 2-arylazetidines: Evaluating the ortho-directing ability of the azetidinyl ring and the α-directing ability of the N-substituent. Chem. Commun. 50, 1698–1700. doi: 10.1039/c3cc48555b
During, M. J., Cao, L., Zuzga, D. S., Francis, J. S., Fitzsimons, H. L., Jiao, X., et al. (2003). Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nat. Med. 9, 1173–1179. doi: 10.1038/nm919
Ferraris, D., Belyakov, S., Li, W., Oliver, E., Ko, Y.-S., Calvin, D., et al. (2007). Azetidine-based inhibitors of dipeptidyl peptidase IV (DPP IV). Curr. Top. Med. Chem. 7, 597–608. doi: 10.2174/156802607780090993
Ferraris, D., Ko, Y.-,Sen Calvin, D., Chiou, T., Lautar, S., Thomas, B., Wozniak, K., et al. (2004). Ketopyrrolidines and ketoazetidines as potent dipeptidyl peptidase IV (DPP IV) inhibitors. Bioorg. Med. Chem. Lett. 14, 5579–5583. doi: 10.1016/j.bmcl.2004.08.057
Garima, P., Srivastava, V. P., and Yadav, L. D. S. (2010). The first example of ring expansion of N-tosylaziridines to 2-aroyl-N-tosylazetidines with nitrogen ylides in an aqueous medium. Green Chem. 12, 1460–1465. doi: 10.1039/C004736H
Jang, H. J., Lee, J. T., and Yoon, H. J. (2015). Aziridine in polymers: a strategy to functionalize polymers by ring-opening reaction of aziridine. Polym. Chem. 6, 3387–3391. doi: 10.1039/C5PY00266D
Jensen, F. (2008). Basis set convergence of nuclear magnetic shielding constants calculated by density functional methods. J. Chem. Theor. Comp. 4, 719–727. doi: 10.1021/ct800013z
Kobayashi, J., and Kubota, T. (2009). The Daphniphyllum alkaloids. Nat. Prod. Rep. 26, 936–962. doi: 10.1039/B813006J
Kulkarni, S. B., and Cromwell, N. H. (1977). The synthesis of l-alkyl-2-benzoyl-4-methylazetidine. Configurational and conformational studies. J. Heterocycl. Chem. 14, 981–983. doi: 10.1002/jhet.5570140606
Luisi, R., Capriati, V., Florio, S., Di Cunto, P., and Musio, B. (2005). Synthesis and lithiation of oxazolinylaziridines: the N-substituent effect. Tetrahedron 61, 3251–3260. doi: 10.1016/j.tet.2005.01.045
Ma, S. H., Yoon, D. H., Ha, H. J., and Lee, K. W. (2007). Preparation of enantiopure 2-acylazetidines and their reactions with chloroformates. Tetrahedron Lett. 48, 269–271. doi: 10.1016/j.tetlet.2006.11.033
Marenich, A. V., Cramer, C. J., and Truhlar, D. G. (2009). Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phy. Chem. B 113, 6378–6396. doi: 10.1021/jp810292n
Miao, C. B., Dong, C. P., Zhang, M., Ren, W. L., Meng, Q., Sun, X. Q., et al. (2013). Highly Stereoselective, One-Pot Synthesis of Azetidines and 2,4-Dioxo-1,3-diazabicyclo[3.2.0] Compounds Mediated by I2. J. Org. Chem. 78, 4329–4340. doi: 10.1021/jo400181r
Nocquet, P.-A., Hazelard, D., and Compain, P. (2012). Synthesis of spirocyclopropyl γ-lactams by tandem intramolecular azetidine ring-opening/closing cascade reaction: synthetic and mechanistic aspects. Tetrahedron 68, 4117–4128. doi: 10.1016/j.tet.2012.03.111
Parisi, G., Zenzola, M., Capitanelli, E., Carlucci, C., Romanazzi, G., Pisano, L., et al. (2016). Exploiting structural and conformational effects for a site-selective lithiation of azetidines. Pure Appl. Chem. 88, 631–648. doi: 10.1515/pac-2016-0602
Pérez-Faginas, P., Aranda, M. T., García-López, M. T., Snoeck, R., Andrei, G., Balzarini, J., et al. (2011). Synthesis and SAR studies on azetidine-containing dipeptides as HCMV inhibitors. Bioorg. Med. Chem. 19, 1155–1161. doi: 10.1016/j.bmc.2010.12.052
Pisano, L., Degennaro, L., Carraro, M., Fanelli, F., Azzena, U., Mastrorilli, P., et al. (2016). Computational NMR as useful tool for predicting structure and stereochemistry of four-membered sulfur heterocycles. Eur. J. Org. Chem. 2016, 3252–3258. doi: 10.1002/ejoc.201600372
Rodebaugh, R. M., and Cromwell, N. H. (1969). The synthesis of 1-(t-butyl)-2-benzoylazetidine. J. Heterocycl. Chem. 6, 439–440. doi: 10.1002/jhet.5570060332
Rodebaugh, R. M., and Cromwell, N. H. (1971). Conformational isomerism of 2-carboazetidines. J. Heterocycl. Chem. 8, 421–430. doi: 10.1002/jhet.5570080312
Ronai, A. Z., Timar, J., Mako, E., Erdo, F., Gyarmati, Z., Toth, G., et al. (1999). Diprotin, an inhibitor of dipeptidyl aminopeptidase IV(EC 3.4.14.5) produces naloxone — reversible analgesia in rats. Life Sci. 64, 145–152. doi: 10.1016/S0024-3205(98)00544-X
Singh, G. S., D'hooghe, M., and De Kimpe, N. (2008). Azetidines, Azetines and Azetes: Monocyclic Comprehensive Heterocyclic Chemistry III. Vol. 2. Available online at: http://www.sciencedirect.com/science/referenceworks/9780080449920
Starmans, W. A. J., Starmans, W. A. J., Walgers, R. W. A., Thijs, L., de Gelder, R., Smits, J. M. M., et al. (1998). Azetidine based Ligands in Boron catalyzed Asymmetric Dies-Alder Reactions. Tetrahedron 54, 4991–5004. doi: 10.1016/S0040-4020(98)00204-X
Weber, A. E. (2004). Dipeptidyl peptidase IV inhibitors for the treatment of diabetes. J. Med. Chem. 47, 4135–4141. doi: 10.1021/jm030628v
Keywords: azetidine, lithiation, oxazoline, stereoselectivity, NMR calculations, oxazolidine
Citation: Musci P, Colella M, Fanelli F, Altomare A, Pisano L, Carlucci C, Luisi R and Degennaro L (2019) Stereo- and Enantioselective Addition of Organolithiums to 2-Oxazolinylazetidines as a Synthetic Route to 2-Acylazetidines. Front. Chem. 7:614. doi: 10.3389/fchem.2019.00614
Received: 27 May 2019; Accepted: 23 August 2019;
Published: 10 September 2019.
Edited by:
Hyun-Joon Ha, Hankuk University of Foreign Studies, South KoreaReviewed by:
Albert Moyano, University of Barcelona, SpainEun Jin Cho, Chung-Ang University, South Korea
Copyright © 2019 Musci, Colella, Fanelli, Altomare, Pisano, Carlucci, Luisi and Degennaro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Renzo Luisi, cmVuem8ubHVpc2lAdW5pYmEuaXQ=; Leonardo Degennaro, bGVvbmFyZG8uZGVnZW5uYXJvQHVuaWJhLml0