 Meaghan McKinnon
Meaghan McKinnon Veronika Belkina1
Veronika Belkina1 Mehmed Z. Ertem
Mehmed Z. Ertem David C. Grills
David C. Grills Jonathan Rochford
Jonathan Rochford- 1Department of Chemistry, University of Massachusetts Boston, Boston, MA, United States
- 2Chemistry Division, Brookhaven National Laboratory, Upton, NY, United States
The subject of this study [fac-Mn(bqn)(CO)3(CH3CN)]+ (bqn = 2,2′-biquinoline), is of particular interest because the bqn ligand exhibits both steric and electronic influence over the fundamental redox properties of the complex and, consequently, its related catalytic properties with respect to the activation of CO2. While not a particularly efficient catalyst for CO2 to CO conversion, in-situ generation and activity measurements of the [fac-Mn(bqn)(CO)3]− active catalyst allows for a better understanding of ligand design at the Mn center. By making direct comparisons to the related 2,2′-bipyridyl (bpy), 1,10-phenanthroline (phen), and 2,9-dimethyl-1,10-phenanthroline (dmphen) ligands via a combination of voltammetry, infrared spectroelectrochemistry, controlled potential electrolysis and computational analysis, the role of steric vs. electronic influences on the nucleophilicity of Mn-based CO2 reduction electrocatalysts is discussed.
Introduction
The catalytic reduction of CO2 into useful C-1 chemical feedstocks offers one potential strategy to develop a carbon-neutral alternative to our current dependence on fossil fuels. A major challenge for the catalysis community is to develop molecular catalysts capable of coupling electrochemical reduction with protonation of the CO2 substrate toward useful C-1 products. One such example is the proton-coupled two-electron reduction of CO2 to CO, which comes at a cost of just −0.52 V vs. SHE in water at pH 7 (Equation 1) (Arakawa et al., 2001), while in dry acetonitrile at pH 0 (Equation 2), the standard electrode potential for this reaction has been estimated, using two different thermodynamic cycles, as −0.13 V vs. Fc+/0 (Matsubara et al., 2015) or −0.12 V vs. Fc+/0 (Pegis et al., 2015).
This seemingly simple reaction is limited by slow kinetic parameters, however, which demands an appropriate catalyst to access a kinetically efficient pathway for CO2 utilization – albeit at the cost of an electrochemical overpotential (η) (Appel and Helm, 2014). Of the homogeneous transition metal-based molecular catalysts used in this field (Francke et al., 2018; Sinopoli et al., 2018; Stanbury et al., 2018), manganese(I) polypyridyl tricarbonyl catalysts of the type [fac-MnI(N∧N)(CO)3X]n (where N∧N = polypyridyl ligand, X = Br− (n = 0) or CH3CN (n = +1)), and their analogs, have been of keen interest due to their high selectivity, low cost, and low overpotential (Grills et al., 2018). Recently we have reported on tuning both the inner coordination sphere (McKinnon et al., 2019) and second coordination sphere (Ngo et al., 2017) of the ligand to optimize catalytic efficiency and selectivity for CO formation, notably against the competitive two-electron two-proton coupled redox transformations of CO2 to HCO2H and H+ to H2. More subtle modification on the periphery of the bpy ligand is also known to strongly influence the reduction potentials observed for these catalysts. This is evident, for example, with the inductive electron donating influence of the dtbpy (dtbpy = 4,4′-tBu2-bpy) ligand, which shifts the reduction potential 0.11 V more negative relative to the simple bpy analog (Smieja et al., 2013). Similarly, while maintaining the same inner coordination sphere of [fac-Mn(N∧N)(CO)3L]n catalysts but introducing redox non-innocence on the ligand backbone, a recent report on [fac-MnBr(phen-dione)(CO)3] (phen-dione = 1,10-phenanthroline-5,6-dione) demonstrated vastly different electrochemistry, maintaining a high selectivity for CO evolution, compared to the analogous bpy and phen (phen = 1,10-phenanthroline) complexes, notwithstanding significant ligand-based redox activity at the dione functional group to generate a bis-carboxylate phenanthroline intermediate species (Stanbury et al., 2017).
In the current study we have focused on a rather simple modification of the polypyridyl ligand, but one which allows us to directly investigate both steric and electronic influences in the [fac-Mn(bqn)(CO)3(CH3CN)]+([4-CH3CN]+) pre-catalyst, where bqn = 2,2′-biquinoline (Figure 1). While fundamental in approach, a simple systematic extension of the π-conjugated system of the polypyridyl ligand has not yet been reported for Mn(I) CO2 reduction electrocatalysts. Four pre-catalysts of the general structure [fac-Mn(N∧N)(CO)3(CH3CN)]+ are here investigated using the benchmark 2,2′-bipyridyl (bpy) ligand ([1-CH3CN]+) alongside the 1,10-phenanthroline (phen) ([2-CH3CN]+), 2,9-dimethyl-1,10-phenanthroline (dmphen) ([3-CH3CN]+), and aforementioned bqn ligands. Although the phen ligand does not have as extensive a π-conjugation as the bqn ligand, it serves a critical role in this study by bridging the gap between the sterically related dmphen and bqn ligands. Indeed, a direct comparison of the sterically and electronically expanded bqn ligand to the sterically related dmphen ligand probes the question of whether steric or electronic effects dominate in determining the redox, and ultimately catalytic, properties of this class of CO2 reduction electrocatalyst. As demonstrated below using a combination of IR-SEC and computational studies, the combined electronic and steric influences of the π-extended bqn ligand hinder formation of a [4–4]0 dimer, facilitating a concerted two-electron ECE mechanism for the generation of [4]−. However, this comes at the cost of a change in product selectivity for the [4-CH3CN]+ pre-catalyst to favor H2 evolution in the presence of excess TFE. Selective CO2 activation is observed for all catalysts here studied upon in-situ generation of a three-electron reduced [fac-Mn(−1)(N∧N•−)(CO)3]2− active catalyst.
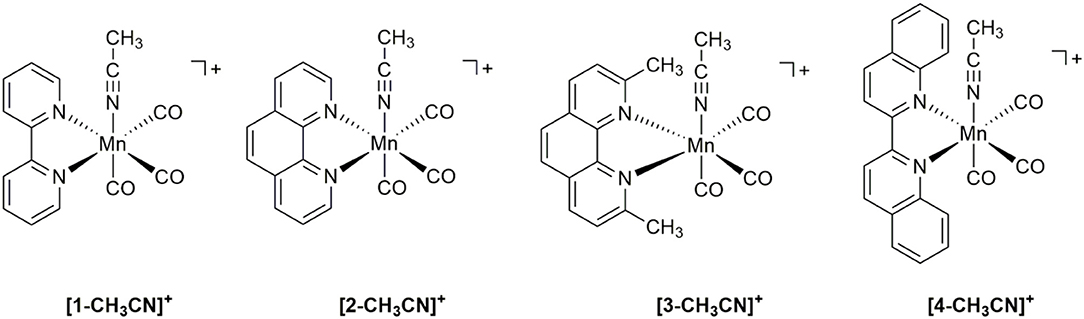
Figure 1. Molecular structures of complexes investigated.
Results and Discussion
Synthesis and Structural Characterization
Initial metathesis of [MnBr(CO)5] with silver triflate was completed as a first step to produce the [Mn(CO)5(OTf)] intermediate (Scheiring et al., 2000), allowing for the subsequent straightforward isolation of pure [fac-MnI(N∧N)(CO)3(OTf)] products. Microwave reflux of [Mn(CO)5(OTf)] with one equivalent of the appropriate ligand in tetrahydrofuran afforded the pale yellow [fac-Mn(bpy)(CO)3(OTf)] (1-OTf), [fac-Mn(phen)(CO)3(OTf)] (2-OTf), and [fac-Mn(dmphen)(CO)3(OTf)] (3-OTf) products, and the red-orange [fac-Mn(bqn)(CO)3(OTf)] (4-OTf) solid, in quantitative yield following precipitation in excess diethyl ether. Each product was satisfactorily characterized by 1H NMR and FTIR spectroscopies as well as elemental analysis. The facial (fac) arrangement of the Mn(CO)3 core structure in each complex was confirmed by FTIR spectroscopy, where characteristic ν(CO) vibrational stretching modes for each of the solvated [1-CH3CN]+, [2-CH3CN]+, [3-CH3CN]+ and [4-CH3CN]+ complexes in neat acetonitrile provides a very useful comparison of structural and electronic properties of all four complexes using a basic knowledge of point-group symmetry and Mn(dπ)-CO(π*) back-bonding, respectively (Table 2). The bpy and phen complexes, [1-CH3CN]+ and [2-CH3CN]+, each exhibit pseudo-C3v point group symmetry with identical FTIR spectra composed of a sharp, symmetric ν(CO) stretching mode at 2,050 cm−1 and a second broad, lower frequency asymmetric stretching mode at 1,958 cm−1. Consistent with voltammetry and computational analysis presented below, the identical ν(CO) stretching modes illustrates the negligible electronic influence of the π-extended phen ligand in comparison to the bpy system. This observation is consistent with related reports of the analogous bromide complexes (Kurtz et al., 2015; Stanbury et al., 2017; Tignor et al., 2018). In contrast, the dmphen complex [3-CH3CN]+ exhibits a descent in symmetry to pseudo-Cs, evident in a breaking of degeneracy for its lower frequency asymmetric ν(CO) stretching modes, which occur at 1,959 and 1,944(sh) cm−1. This suggests a sterically induced distortion of the fac-Mn(CO)3 core, likely by the 2,10-dimethyl substituents of the dmphen ligand. Furthermore, the higher frequency symmetric ν(CO) stretching mode of [3-CH3CN]+ exhibits a 4 cm−1 shift to lower frequency at 2,046 cm−1, consistent with an inductive electron-donating influence of the two methyl substituents at dmphen, likely due to increased Mn(dπ) → CO(π*) back-bonding. Interestingly, the bqn complex, [4-CH3CN]+ exhibits an FTIR spectral profile with ν(CO) = 2,047 and 1,959 cm−1, indicative again of pseudo-C3v symmetry, similar to [1-CH3CN]+ and [2-CH3CN]+. However, with the high frequency symmetric ν(CO) stretching mode occurring at 2,047 cm−1, this suggests that the inductive electron-donating influence of the bqn ligand is similar to that of the dmphen ligand in [3-CH3CN]+. Experimental FTIR spectral profiles are consistent with calculated spectra, as illustrated in Figure 2.
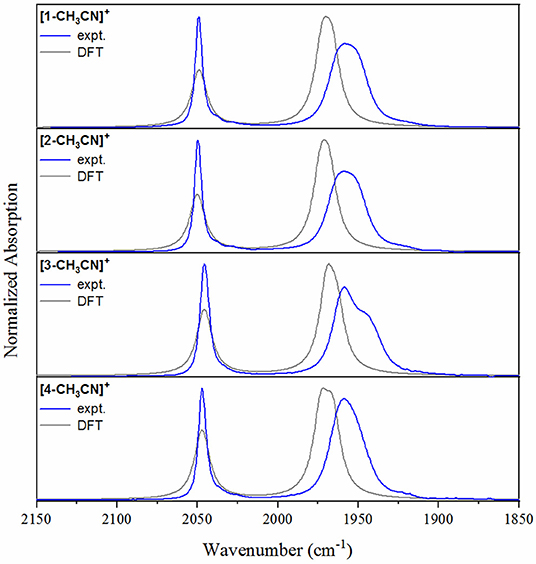
Figure 2. Experimental FTIR spectra (blue) of [1-CH3CN]+, [2-CH3CN]+, [3-CH3CN]+, and [4-CH3CN]+ recorded in acetonitrile displaying characteristic ν(CO) stretching modes. Density functional theory-computed IR spectra (gray) are included for comparison (empirically-derived frequency scaling factor = 0.9633, FWHM = 12 cm−1).
Cyclic Voltammetry Under Non-catalytic Conditions
Electrochemical characterization of each complex was carried out under inert conditions (1 atmosphere of argon), in the absence of auxiliary Brønsted acid, prior to screening for catalytic activity. Each complex displays an irreversible Mn(II/I) oxidation event within 20 mV of each other in the range of +1.02 to +1.04 V vs. the ferricenium/ferrocence (Fc+/0) pseudo-reference (Figure 3, Table 1). This suggests that there is little difference in electron density between each Mn-center upon varying the polypyridyl ligand across the four complexes. The very subtle differences observed in the v(CO) stretching modes by FTIR spectroscopy are unlikely to be reproduced by cyclic voltammetry. Furthermore, voltammetry data is more complex due to the in-situ generation of the Mn(II) oxidation state. We will focus on the reduction properties of these complexes forthwith due to their greater relevance toward our subsequent catalytic studies. The electrochemical properties of [1-CH3CN]+ have been recently reported (Grills et al., 2018; McKinnon et al., 2019). Three sequential one-electron cathodic peaks are observed at Epc = −1.48 V, −1.83 V and −2.94 V with only the third reduction exhibiting quasi-reversible behavior (E1/2 = −2.90 V, ΔEp = 81 mV at υ = 0.1 V s−1) vs. Fc+/0. The phen complex, [2-CH3CN]+ exhibits a first one-electron irreversible reduction at Epc = −1.48 V vs. Fc+/0, identical to that observed for [1-CH3CN]+, further supporting the electronic similarity of bpy and phen at least in the [fac-Mn(N∧N)(CO)3L]n class of complexes (Tignor et al., 2018). This first reduction has been previously established (Grills et al., 2018) as ligand-based according to an electrochemical-chemical (EC) reaction scheme whereby, upon one-electron reduction of the bpy π* orbital, rapid CH3CN dissociation occurs with a concurrent shift in radical character from the ligand to form the neutral five-coordinate 17-valence electron intermediate, [fac-Mn0(bpy)(CO)3] ([1]0). This metastable Mn(0) complex, in the absence of any steric hindrance (Sampson et al., 2014), rapidly forms the Mn0-Mn0 bound 18-valence electron [fac-Mn0(bpy)(CO)3]2 dimer, [1–1]0. While unequivocal evidence of [fac-Mn0(N∧N)(CO)3] dimer formation is presented below via infrared spectroelectrochemical (IR-SEC) studies, its oxidation is often evident in the reverse anodic scan in cyclic voltammetry, observed here at −0.61 V and −0.58 V for [1–1]0 and [2–2]0, respectively. Thus, the second reduction event for [fac-Mn(N∧N)(CO)3L]n complexes is often attributed to an irreversible two-electron EC event whereby the dimer is reductively cleaved to generate two equivalents of the two-electron reduced [fac-Mn0(N∧N•−)(CO)3]− anion, now established as the principal active catalyst for CO2 activation by this class of complex via the most common reduction-first pathway (Riplinger et al., 2014). Reduction of the phen based dimer, [2–2]0 is observed at Epc = −1.86 V, suggesting that [2]− is more nucleophilic than the benchmark bpy analog, [1]− generated at the slightly more positive potential of Epc = −1.83 V. Similar to its bpy analog, the native [2-CH3CN]+ precursor exhibits a quasi-reversible third reduction event at Epc = −2.72 V (E1/2 = −2.66 V, ΔEp = 114 mV at υ = 0.1 V s−1). This third quasi-reversible reduction has rarely been discussed in the literature but computational analysis here suggests that it is a predominantly ligand based reduction giving rise to a 19-electron [fac-Mn0(N∧N2−)(CO)3]2− dianion. The related dmphen complex, [3-CH3CN]+ exhibits unexceptionally similar voltammetry in comparison to both its bpy and phen analogs. Its first reduction again involves a ligand based EC pathway leading to formation of the [3–3]0 dimer. This EC reaction is shifted negatively by 10 mV to Epc = −1.49 V, relative to formation of both [1–1]0 and [2–2]0, consistent with the inductive donating character of the 2,9-dimethyl substituents observed by FTIR studies. Although there is weak evidence of dimer oxidation in the full scan voltammogram presented in Figure 3, reversing the scan after just one-electron reduction clearly demonstrates a significant dimer oxidation peak at Epa = −0.65 V (Figure S5). Reductive cleavage of the [3–3]0 dimer to form the two-electron reduced five-coordinate [fac-Mn0(dmbpy•−)(CO)3]− anion ([3]−), however, occurs at a more positive potential of −1.78 V. This observation suggests that the steric influence of dmphen slightly hinders electronic coupling between the Mn(0) and dmphen•− radical centers in [3]−, rendering it slightly less nucleophilic as a result. The third reduction wave for [3]−/2− is observed at −2.86 V and, in contrast to its bpy and phen analogs, is completely irreversible.
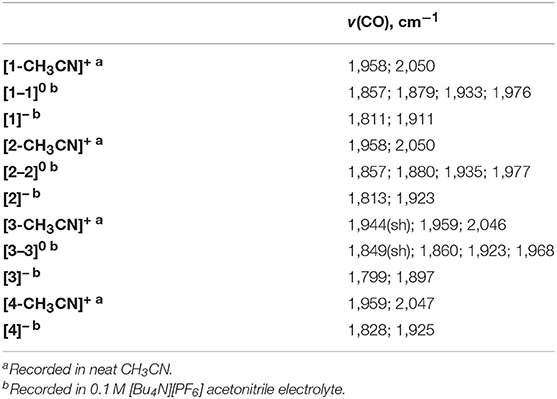
Table 1. Redox potentials recorded by cyclic voltammetry for [1-CH3CN]+, [2-CH3CN]+, [3-CH3CN]+, and [4-CH3CN]+ reported vs. the ferricenium/ferrocene (Fc+/0) pseudo reference.
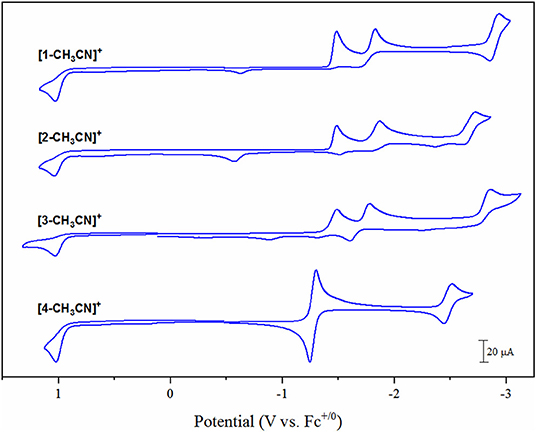
Figure 3. Cyclic voltammograms of [1-CH3CN]+, [2-CH3CN]+, [3-CH3CN]+ and [4-CH3CN]+ recorded in 0.1 M [Bu4N][PF6] acetonitrile supporting electrolyte at a glassy carbon working electrode with a scan rate of υ = 0.1 V s−1 under 1 atmosphere of argon.
Noticeably in Figure 3, the bqn complex, [4-CH3CN]+ exhibits a single, reversible, concerted two-electron reduction event at Epc = −1.30 V (E1/2 = −1.28 V, ΔEp = 53 mV at υ = 0.1 V s−1) followed by a single additional quasi-reversible one-electron reduction event at Epc = −2.51 V (E1/2 = −2.48 V, ΔEp = 71 mV at υ = 0.1 V s−1) vs. Fc+/0. Equally important here is the lack of any evidence for dimer oxidation in the reverse anodic scan of [4-CH3CN]+. This electrochemical behavior, at least in its peak profile, is uncannily similar to previously reported bulky 6,6′-substituted bpy ligands investigated at the identical [fac-Mn0(N∧N)(CO)3(CH3CN)]+ center (Sampson et al., 2014; Ngo et al., 2017). Mn0-Mn0 formation is known to favor a staggered structure of both bpy ligands in [1–1]0 (Machan et al., 2014), and thus far only the inclusion of steric bulk orthogonal to the plane of the polypyridyl ligand has been demonstrated to prevent dimer formation (Sampson et al., 2014; Ngo et al., 2017). Thus, it is appropriate here to question whether the redox behavior exhibited by [4-CH3CN]+ is in fact due to an electronic influence of this relatively electron deficient ligand, or rather, alternatively is the result of a steric influence of the bqn ligand. The bqn ligand is, after all, π-extended from the same 6,6′-positions as previously reported 6,6′-orthoganol-sterically bulky bpy stystems, albeit here non-orthogonal to, but in the same plane as, the core bpy structure. This question is addressed explicitly via computational analysis presented below.
Infrared Spectroelectrochemistry (IR-SEC)
To gain structural insight into electrochemical activation of these pre-catalysts, IR-SEC was carried out in 0.1 M [Bu4N][PF6] acetonitrile supporting electrolyte under 1 atmosphere of argon (Table 2). IR-SEC is a powerful investigative tool that takes advantage of the structural specificity of IR spectroscopy for transition metal carbonyl complexes and the ability to prepare in-situ catalytic intermediates simply by gradually stepping the potential of the working electrode (Kaim and Fiedler, 2009; Machan et al., 2014). At open-circuit potential, i.e., resting potential of the native pre-catalyst, [1-CH3CN]+ and [2-CH3CN]+ both display identical ν(CO) stretching modes as presented earlier (Figure 2), confirming negligible influence of the 0.1 M [Bu4N][PF6] electrolyte on their FTIR spectra. When the potential was biased beyond the first-reduction at −1.60 V vs. Fc+/0 for both [1-CH3CN]+ and [2-CH3CN]+, a progression was observed where the native ν(CO) stretching modes diminished while four unique but related ν(CO) stretching modes grew in concurrently at 1,857, 1,879, 1,933, and 1,976 cm−1 from [1-CH3CN]+ (Figure S10) and at 1,857, 1,880, 1,935, and 1,977 cm−1 from [2-CH3CN]+ (Figure S11). These data are consistent with an earlier report of the [1–1]0 dimer (Hartl et al., 1995), and the peak profile associated with related Mn0-Mn0 dimers previously observed via IR-SEC (Grills et al., 2018), and they are also consistent with our computational frequency analysis. A similar assignment is made for [2–2]0 which exhibits almost identical ν(CO) stretching frequencies (vide supra). Upon biasing the working electrode potential further negative (−2.00 V vs. Fc+/0) beyond the second Mn0-Mn0/Mn− based reduction for both [1-CH3CN]+ and [2-CH3CN]+, loss of the four dimer stretches was observed (Figures S10, S11) with concurrent growth of two new ν(CO) stretching modes at lower wavenumber, corresponding to the five-coordinate two-electron reduced [fac-Mn0(N∧N•−)(CO)3]− active catalysts, [1]− [ν(CO) = 1,811 and 1,911 cm−1] and [2]− [ν(CO) = 1,813 and 1,923 cm−1]. Consistent with its comparable voltammetry behavior, the dmphen based complex, [3-CH3CN]+ exhibits a similar IR-SEC transition to the two-electron reduced active catalyst, [3]− [ν(CO) = 1,799 and 1,897 cm−1] via the [3–3]0 dimer intermediate [ν(CO) = 1,849(sh), 1,860, 1,923, and 1,968 cm−1] as illustrated in Figure 4. Although clean transformation of [3-CH3CN]+ to [3]− was observed upon direct electrolysis at −2.00 V, it is worth noting that selective electrolysis to the intermediate [3–3]0 dimer species did exhibit evidence of minor decomposition to an unidentified side-product [ν(CO) = 1,919 and 2,026 cm−1] with concurrent growth of a broad weak infrared absorption consistent with free CO (Figure S13). This is consistent with an earlier report of poor stability of the simpler phen-based dimer, [2–2]0 (Stanbury et al., 2017).
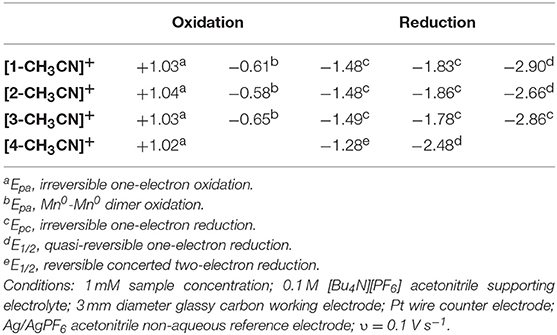
Table 2. ν(CO) infrared stretches for [1-CH3CN]+, [2-CH3CN]+, [3-CH3CN]+, and [4-CH3CN]+ precatalysts, and their one-electron reduced, dimeric, and two-electron reduced derivatives, obtained by solution phase FTIR and IR-SEC spectroscopy.
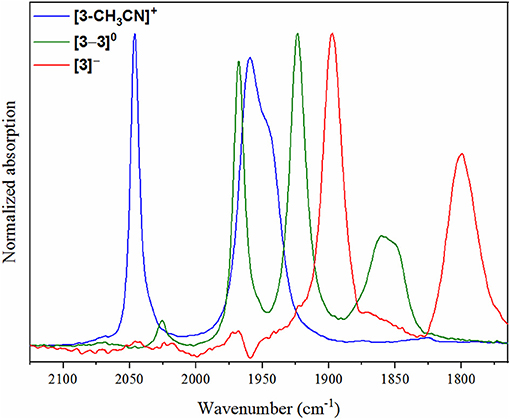
Figure 4. IR-SEC spectra recorded on [3-CH3CN]+ at the resting potential (blue), upon one-electron reduction (green) and upon two-electron reduction (red).
As a concerted two-electron reduction event occurs for [4-CH3CN]+, the potential was set at −1.65 V vs. Fc+/0 for quantitative in-situ formation of the two-electron reduced product. Indeed, no evidence for an intermediate one-electron reduced monomer or the Mn0-Mn0 dimer was observed. Instead, consistent with the observed voltammetry (vide supra) only a transition to two lower-frequency stretching modes at ν(CO) = 1,828 and 1,925 cm−1 was observed, which are attributed to the two-electron reduced [fac-Mn0(bqn•−)(CO)3]− species, [4]− (Figure 5).
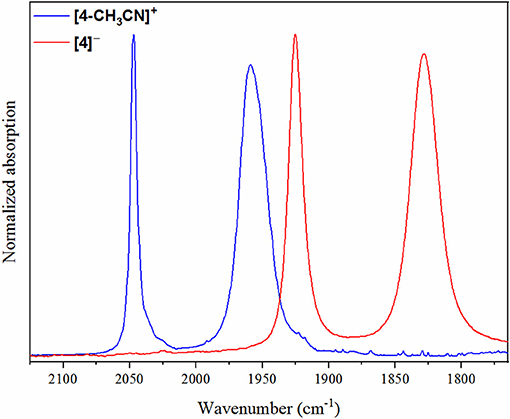
Figure 5. IR-SEC spectra recorded on [4-CH3CN]+ at the resting potential (blue) and upon two-electron reduction (red).
Voltammetry Under 1 Atm CO2 in the Presence of 0.3 % H2O
Prior studies of Mn(I) polypyridyl based electrocatalysts for CO2 reduction have only reported catalytic current upon addition of excess weak Brønsted acid with, for example, 2.71 M (5%) addition of H2O (Bourrez et al., 2011). In contrast to their Re counterparts, which have been established for some time to promote CO2 reduction in the absence of a proton donor, Mn(I) polypyridyl based electrocatalysts typically require the presence of a proton donor to promote intermediate metallocarboxylic acid formation due to the poorer nucleophilicity of Mn vs. Re catalysts (Riplinger and Carter, 2015). Cyclic voltammograms for pre-catalysts [1-CH3CN]+, [2-CH3CN]+, [3-CH3CN]+ and [4-CH3CN]+ recorded under 1 atm CO2 at 0.1 V s−1 in 0.1 M [Bu4N][PF6] acetonitrile electrolyte exhibit clear evidence of catalytic behavior for each complex (Figure 6). Although excess Brønsted acid was not added in these experiments, the reagent grade acetonitrile that was used still contains 0.17 M (0.3 %) residual H2O, which was recently demonstrated to be sufficient to facilitate proton-coupled CO2 reduction to form CO (McKinnon et al., 2019). This observation is logical due to the relatively larger overpotential applied, especially as the active catalyst under these experimental conditions is predicted by computation (vide infra) to be the more nucleophilic three-electron reduced dianion.
Figure 6. Cyclic voltammograms demonstrating catalytic activity of [1-CH3CN]+, [2-CH3CN]+, [3-CH3CN]+ and [4-CH3CN]+ under 1 atm CO2 (red) at υ = 0.1 V s−1 with 0.1 M [Bu4N][PF6] acetonitrile supporting electrolyte containing residual 0.17 M (0.3%) H2O as a Brønsted acid source. Cyclic voltammograms recorded under 1 atm of argon are also shown (blue). The current (y-axis) data are normalized with respect to the non-catalytic Faradaic response (ip).
Without knowledge of the in-situ pH of the electrolyte solution, the half-wave potential of the catalytic wave (Ecat/2) can be used to make a relative comparison of overpotentials under these experimental conditions. As expected, π-extension with the bqn ligand in pre-catalyst [4-CH3CN]+ gives rise to a significant positive shift of catalytic current with Ecat/2 = −2.42 V, whereas the bpy, phen and dmphen pre-catalysts exhibit Ecat/2 = −2.85, −2.64, and −2.74 V, respectively. One difficulty encountered under these experimental conditions was in attempting to establish steady-state catalytic conditions with respect to the rate-limiting consumption of CO2 within the electrochemical double-layer. As discussed later, this became less problematic once excess Brønsted acid was added. As such, TOFmax could not be satisfactorily determined under these experimental conditions. Thus, the TOF value is reported for each catalyst in Table 3 using Equation 3 and the icat/ip ratio determined at a scan rate of 0.1 V s−1, to aid their side-by-side comparison,
where F is the Faraday constant (96,485 s A mol−1), υ is the scan rate (V s−1), R is the universal gas constant (8.3145 V A s K−1 mol−1), T is the temperature (K), np is the number of electrons involved in the non-catalytic Faradaic current response (responsible for the non-catalytic Faradaic current, ip, as described by the Randles-Sevcik equation (Bard and Faulkner, 2001), and ncat is the number of electrons required for catalysis (two electrons for the reduction of CO2 to CO as shown in Equations 1 and 2). In our calculations of TOF, the first one-electron reduction wave (np = 1) was used for reference to determine the non-catalytic Faradaic current (ip) for [1-CH3CN]+, [2-CH3CN]+ and [3-CH3CN]+. In contrast, the first reduction wave of [4-CH3CN]+ exhibits a concerted two-electron event under non-catalytic conditions such that np = 2 at Epc = −1.30 V. The significance of this distinction is important when determining TOF from cyclic voltammetry analysis using Equation 3 as the ratio of can differ by a factor of 8 (1/4 vs. 8/4 for np = 1 or 2, respectively). However, this should, with ideal Randles-Sevcik behavior (ip ) of the two-electron reduction event, be completely offset by the 8 × smaller (icat/ip)2 ratio when np = 2. Thus, in theory, kinetic analysis of [4-CH3CN]+ can be conducted using either the concerted two-electron reduction event at Epc = −1.30 V (np = 2) or the subsequent one-electron reduction event at Epc = −2.51 V (np = 1) for reference in determining icat/ip. Using data recorded at υ=0.1 V s−1, Equation 3 results in different TOF values of 25 s−1 (icat/ip = 4.0, np = 2) or 14 s−1 (icat/ip =8.5, np = 1), indicating non-ideal behavior of the concerted two-electron reduction event, which is unsurprising considering this is an ECE mechanism and not a pure two-electron concerted EE mechanism. Scan rate analysis of this two-electron reduction event for [4-CH3CN]+ clearly demonstrates an increase in peak separation between its cathodic and anodic waves (Figure S9). More importantly, the 2.83-fold increase in current for a two-electron event, predicted by the Randles-Sevcik equation (ip ) assuming similar diffusion coefficients for each redox state, is in fact found to be significantly smaller (2.13-fold) when we compare the ip values of [4-CH3CN]+ at Epc = −1.30 (np = 2) and Epc = −2.51 V (np = 1). The TOF value of 25 s−1 (icat/ip = 4.0, np = 2) is therefore an overestimate of this catalyst's efficiency, and the value of TOF=14 s−1 is henceforth quoted for [4-CH3CN]+ (icat/ip = 8.5, np = 1) recorded at υ = 0.1 V s−1. In summary, the benchmark bpy-based pre-catalyst, [1-CH3CN]+ (icat/ip = 11.3) exhibits the highest TOF in this study of 25 s−1 at υ=0.1 V s−1, followed closely by [3-CH3CN]+ at 22 s−1 (icat/ip = 10.7), the bqn-based system [4-CH3CN]+ at 14 s−1 (icat/ip = 8.5) and finally [2-CH3CN]+ at just 11 s−1 (icat/ip = 7.4).
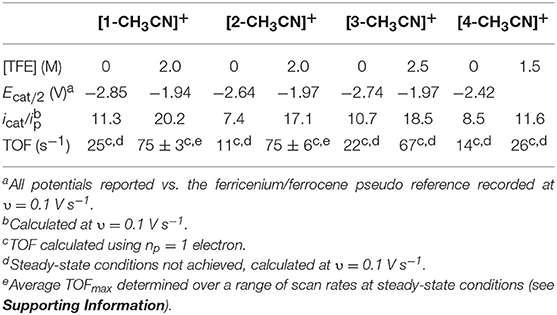
Table 3. Summary of electrocatalysis data derived from voltammetry experiments, in the absence of TFE and at optimum TFE concentrations.
Voltammetry Under 1 Atm CO2 in the Presence of Trifluoroethanol
To further probe the catalytic activity of [1-CH3CN]+, [2-CH3CN]+, [3-CH3CN]+ and [4-CH3CN]+, the voltammetry conditions were altered by adding incremental amounts of a non-aqueous proton source. For the purpose of this study, 2,2,2-trifluoroethanol (TFE, pKa(CH3CN) = 35.4(est.) Lam et al., 2015) was added as a Brønsted acid to observe and optimize proton-coupled catalytic CO2 reduction. The addition of at least 1.5 M TFE results in a plateau of catalytic current for [1-CH3CN]+, [2-CH3CN]+ and [3-CH3CN]+ when monitored at υ=0.1 V s−1 (Figure 7, Figures S16–S19). Notably, each of their catalytic waves exhibit a significant positive shift compared to experiments in the absence of excess Brønsted acid, with Ecat/2 = −1.94 V for [1-CH3CN]+ and Ecat/2 = −1.97 V for both [2-CH3CN]+ and [3-CH3CN]+. The predominant catalytic peak observed for [1-CH3CN]+, [2-CH3CN]+, and [3-CH3CN]+ is assigned, with the aid of computations, to the reduction-first pathway with the five-coordinate, two-electron reduced [fac-Mn0(N∧N•−)(CO)3]− monoanion being the active catalyst.
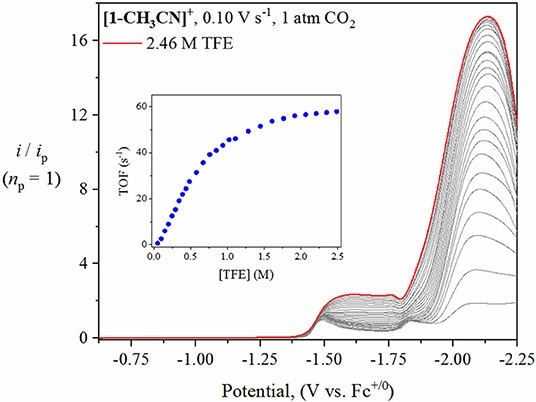
Figure 7. Linear sweep voltammetry of [1-CH3CN]+ recorded at υ=0.1 V s−1 under 1 atm CO2 with increasing TFE concentration (0–2.46 M). The current (y-axis) data are normalized with respect to the non-catalytic Faradaic response (ip). The inset plot of ‘TOF vs. TFE concentration’ demonstrates that zero-order conditions with respect to TFE concentration were achieved at 2.0 M TFE.
Interestingly, a weak grow-in of catalytic current can also be observed at Ecat/2 = −1.48 V for both [1-CH3CN]+ and [2-CH3CN]+ (a similar maximum is less discernable for [3-CH3CN]+). This lower overpotential weak catalytic current occurs directly from the one-electron reduced species and is thus attributed to catalytic activity of the Mn0-Mn0 dimer intermediate. Indirect support of this hypothesis is the fact that such weak catalytic current is not observed with the bqn pre-catalyst, [4-CH3CN]+ which we have already established, via IR-SEC studies, does not form a Mn0-Mn0 dimer intermediate. While appearance of this lower energy catalytic pathway is promising, the very weak current observed, and thus the minimal TOF (<1 s−1) prompted us to focus on the more efficient reduction-first pathway for these complexes. Furthermore, this mechanism has already been the focus of other independent studies (Bourrez et al., 2014; Neri et al., 2019).
In contrast to the above mentioned studies in the absence of excess Brønsted acid, the presence of optimal TFE concentrations benefitted the pursuit of steady-state catalytic conditions, thus allowing for an estimation of a maximum turnover frequency (TOFmax), at least in the case of [1-CH3CN]+ and [2-CH3CN]+. A collection of scan-rate dependent voltammetry data is provided in the Supporting Information for all pre-catalysts (Figures S20–S23) with kinetic analysis summarized in Table 3. Pre-catalysts [1-CH3CN]+ and [2-CH3CN]+ perform comparably with TOFmax values estimated at 75 s−1 under steady-state catalytic conditions. Pre-catalyst [3-CH3CN]+ still exhibits a scan-rate dependent current upon increasing scan rate >0.1 V s−1, thus preventing pure kinetic steady-state conditions, and hence TOFmax, to be obtained. Thus, an estimated TOF value of 67 s−1 is reported for [3-CH3CN]+ recorded at a scan rate of υ=0.1 V s−1 (Figure S22). Results from the addition of TFE to [4-CH3CN]+ were rather complicated due to the grow-in of an additional pre-wave at approx. −2.42 V which prevented accurate determination of Ecat/2 or TOFmax (Figure S23). At least from a qualitative perspective, it can be stated that TFE addition does give rise to a modest increase in the observed catalytic current maximum (Figure 8). Unfortunately, this phenomenon of multiple catalytic waves raises concerns about product selectivity and competitive side reactions, which are borne true following controlled potential electrolysis experiments, discussed below, which conclusively confirm hydrogen evolution as not just a competitive but a dominant process under these experimental conditions with [4-CH3CN]+. Mechanistic details of competitive hydrogen evolution vs. CO2 reduction are discussed in more detail below via computational analysis.
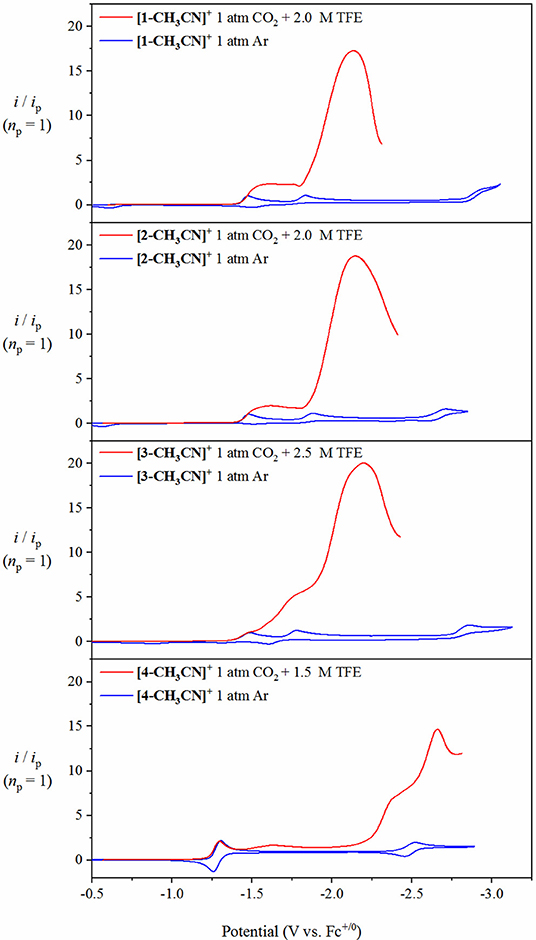
Figure 8. Overlay of cyclic voltammograms for [1-CH3CN]+, [2-CH3CN]+, [3-CH3CN]+ and [4-CH3CN]+ recorded under 1 atm of Ar (blue), with respective linear sweep voltammetry recorded under 1 atm CO2 in the presence of optimum TFE (red) at υ=0.1 V s−1 with 0.1 M [Bu4N][PF6] acetonitrile supporting electrolyte. The current (y-axis) data are normalized with respect to the non-catalytic Faradaic response (ip).
Controlled Potential Electrolysis
Product selectivity of all four pre-catalysts in the absence and presence of TFE under 1 atmosphere of CO2 was investigated by controlled potential electrolysis (CPE) with in-situ gas chromatography analysis used for CO and H2 quantification over a time period of 4 h. The potential bias applied in each case corresponded to Ecat/2 for specific experimental conditions as summarized in Table 3 for each catalyst. The tabulated Faradaic efficiency (FE) data in Table 4 represent the peak CO selectivity observed at a single time interval over the duration of the experiment, whereas the turnover number (TON) tabulated for both CO and H2 represents the total TON over the entire course of the experiment. Plots summarizing all electrolysis data are provided in Figures S28–S35. Ultimately, all pre-catalysts yielded CO product regardless of the conditions employed. Each of the pre-catalysts, [1-CH3CN]+, [2-CH3CN]+, and [3-CH3CN]+ exhibited a clear selectivity for CO production in the absence or presence of TFE with only [4-CH3CN]+ yielding significant H2 in the presence of 1.5 M TFE. Unfortunately, however, in most cases there is FE that is unaccounted for. This is especially true for pre-catalysts [2-CH3CN]+ and [3-CH3CN]+ which is likely due to rapid decomposition, consistent with their poor TONs for CO or H2 evolution. Pre-catalyst [1-CH3CN]+ performed significantly better, exhibiting percentage FE's of 62:2 CO:H2 in the absence of TFE, which increased to 84:2 in the presence of 2.0 M TFE. Efforts to quantify any formate () production to account for the full FE of all catalysts were in vain. This is likely due to the low turnover numbers of these catalysts, which as a whole performed quite poorly under the experimental conditions employed; a recognized problem for the [fac-Mn(N∧N)(CO)3]− class of electrocatalysts under homogeneous conditions (Grills et al., 2018). Indeed, two prior CPE studies of the [fac-MnBr(phen)(CO)3] pre-catalyst have each highlighted the poor performance of this system with the FECO ranging from 18 to 57% (Stanbury et al., 2017; Tignor et al., 2018). Interestingly, the bqn-derived pre-catalyst, [4-CH3CN]+ exhibited the greatest FECO of 98% in the absence of TFE, albeit with a very low TON of just 3. However, with such a low TON we cannot rule out catalyst decomposition as a contributing factor. As anticipated from its irregular voltammetry behavior, at least in comparison to [1-CH3CN]+, [2-CH3CN]+, and [3-CH3CN]+, upon the introduction of TFE, a dramatic shift in product selectivity was observed, with FE's of 14:69 CO:H2. As discussed below in the computational section, this shift in product selectivity is possibly a consequence of the reduced nucleophilicity of the two-electron reduced [4]− active catalyst, in large part due to the lower lying π* orbitals of the bqn ligand.
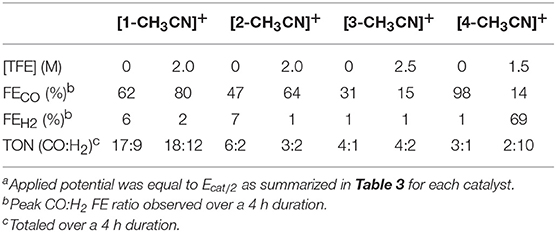
Table 4. Summary of controlled potential electrolysis data in the absence of TFE and at optimum TFE concentrationsa.
Computational Analysis
Density functional theory (DFT) calculations at the M06 level of theory (Zhao and Truhlar, 2008a,b, 2010) in conjunction with the SMD continuum solvation model (Marenich et al., 2009) for acetonitrile were performed to examine the catalyst activation and electrocatalytic CO2 reduction mechanisms of [1-CH3CN]+, [2-CH3CN]+, and [4-CH3CN]+. Mechanistic calculations on [3-CH3CN]+ were omitted for the sake of brevity due to its similarity in behavior to [2-CH3CN]+. The results are summarized in Schemes 1 and 2 along with tabulated energetics (Tables 5, 6). Further details on the electronic structures of selected reaction intermediates are provided in Supporting Information.
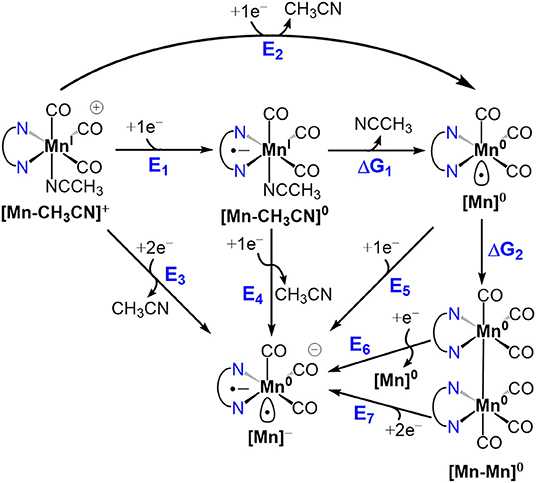
Scheme 1. Catalyst activation and dimerization pathways for MnI polypyridyl complexes [1-CH3CN]+, [2-CH3CN]+ and [4-CH3CN]+.
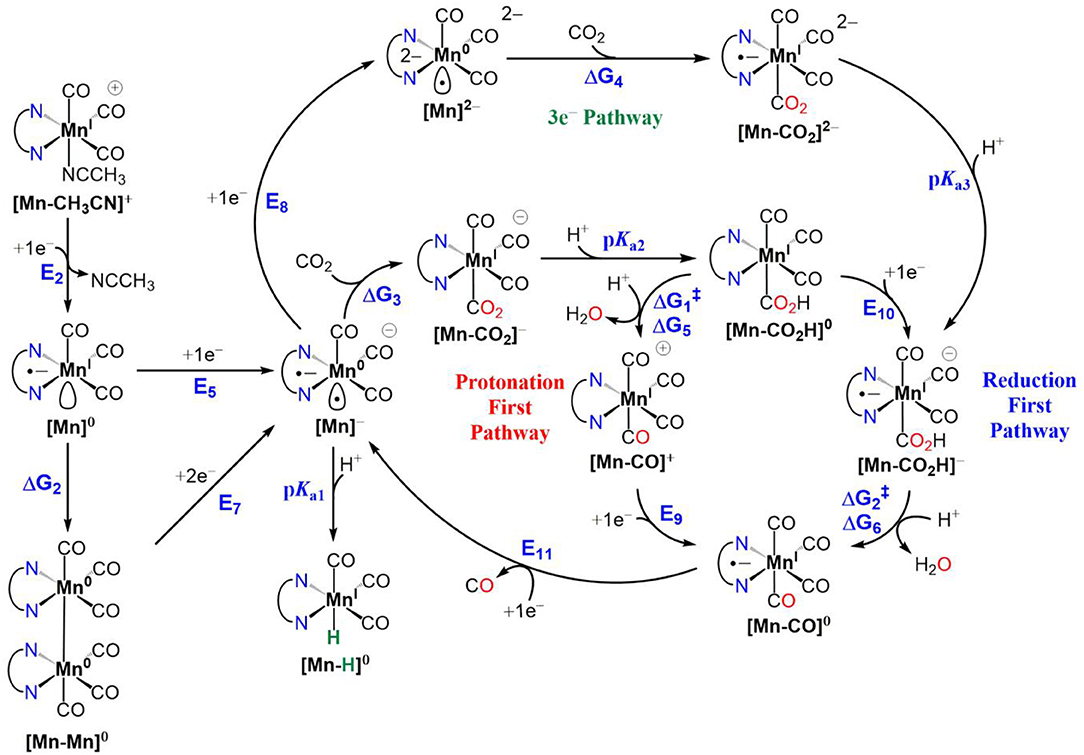
Scheme 2. Electrocatalytic reduction of CO2 to CO for MnI polypyridyl complexes of [1-CH3CN]+, [2-CH3CN]+ and [4-CH3CN]+, illustrating active catalyst generation, protonation-first and reduction-first pathways vs. the three-electron reduction pathway. Also included alongside are competitive Mn0-Mn0 dimer formation and MnI-H formation side reactions. For the C–OH bond cleavage steps, TFE is used as the Brønsted acid. The free energy changes (ΔG) and activation free energies (ΔG) are in units of kcal/mol and reduction potentials are in units of volts (V) vs. Fc+/0.
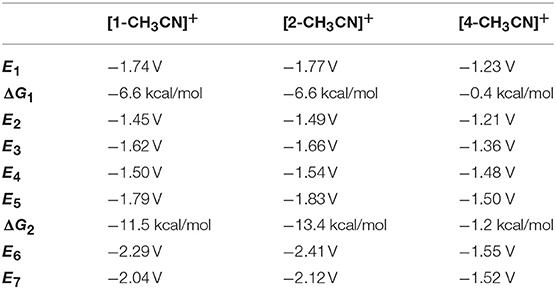
Table 5. Computed reduction potentials (V vs. Fc+/0) and free energy changes (ΔG, kcal/mol) of catalyst activation and dimerization pathways for MnI polypyridyl complexes [1-CH3CN]+, [2-CH3CN]+, and [4-CH3CN]+.
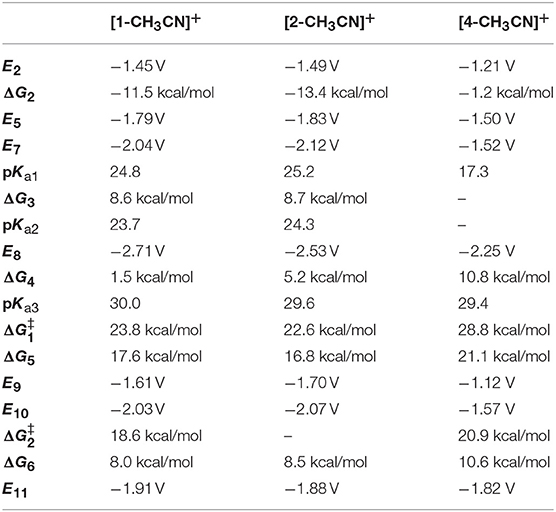
Table 6. Computed reduction potentials (V vs. Fc+/0), free energy changes (ΔG, kcal/mol) and activation free energies (ΔG‡, kcal/mol), and pKa's relevant for electrocatalytic reduction of CO2 to CO for MnI polypyridyl complexes [1-CH3CN]+, [2-CH3CN]+, and [4-CH3CN]+, shown in Scheme 2, comparing the protonation-first and reduction-first pathways vs. the three-electron reduction pathway.
Catalyst Activation and Dimer Formation
The activation of each catalyst starts with one-electron reduction of the solvent-coordinated [Mn-CH3CN]+ species. For all considered pathways in the present study, [1-CH3CN]+ and [2-CH3CN]+ exhibit similar energetics (Scheme 1), so henceforth we will focus on the activation of [1-CH3CN]+ to highlight the distinct behavior of [4-CH3CN]+. The one-electron reduction of [Mn-CH3CN]+ (E1) results in the formation of a ligand-based radical anion for all of the complexes investigated (Figure S36). Typically observed as an EC mechanism in voltammetry (E1ΔG1 in Scheme 1), subsequent dissociation of the acetonitrile ligand (ΔG1) leads to localization of the unpaired spin on the Mn center, generating a formally Mn0 species ([Mn]0) (Figure S36). The extensive π-conjugation of [4-CH3CN]+ introduces an ~+0.5 V anodic shift (E1 = −1.23 V) compared to that of [1-CH3CN]+ (E1 = −1.74 V), in line with the assumption of an initial ligand-based reduction. The computed energetics indicate that acetonitrile dissociation is very favorable upon reduction of [1-CH3CN]+ (ΔG1 = −6.6 kcal/mol) to generate pentacoordinate [1]0, further reduction of which (E5 = −1.79 V) will form [1]−. In contrast, theory predicts that [4-CH3CN]0 and [4]0 will coexist based on acetonitrile dissociation being nearly isoergic (ΔG1 = −0.4 kcal/mol). This distinct behavior is responsible for the experimentally observed concerted two-electron reduction of [4-CH3CN]+ to [4]− (E3 = −1.36 V), which requires a lower potential than the sequential one-electron reduction pathway, i.e., [4-CH3CN]0 to [4]− conversion (E4 = −1.48 V) in contrast to [1-CH3CN]+ (E3 = −1.62 V and E4 = −1.50 V).
Another intriguing difference is in the energetics of dimerization of [Mn]0 to [Mn-Mn]0, which is quite favorable for [1]0 (ΔG2 = −11.5 kcal/mol), in contrast to a nearly isoergic driving force in the case of [4]0 (ΔG2 = −1.2 kcal/mol). Closer inspection of the dimer geometries indicates that staggered conformations are more favorable and Mn–Mn distances are predicted as 2.96 Å and 3.16 Å for [1–1]0 and [4–4]0, respectively (Figure S37). This difference is partly attributed to ligand-induced steric effects that lead the Mn center to be out-of-plane with respect to the ligand in the case of [4]0 (for [4]0, Cbridge-N-Mn-N is 23.2°, compared to 1.1° for [1]0). It should be noted that dimerization of [4]0 is even further suppressed due to the predicted equilibrium between hexacoordinate [4-CH3CN]0 and pentacoordinate [4]0, even if the latter forms during electrocatalysis. As a result of the computational analysis described above, [1-CH3CN]+ and [2-CH3CN]+ are predicted to form [1]0 and [2]0, respectively, via an EC process followed by fast dimerization to form [1–1]0 and [2–2]0, respectively, which can then be reduced via sequential one-electron processes, or a concerted two-electron reduction process, to generate the catalytically active [1]− and [2]− species. We should note that two-electron reduction of the [Mn-Mn]0 dimer practically proceeds via two sequential one-electron reduction steps as the dimer is expected to decompose into [Mn]0 and [Mn]− upon the first reduction, and the computed reduction potential of [Mn]0 is more anodic than that of [Mn-Mn]0 (e.g., E5 = −1.79 V for [1]0 vs. E6 = −2.29 V for [1–1]0).
CO2 Binding and CO Evolution
Next, we turn our attention to CO2 binding to the two-electron reduced [Mn]− catalyst and subsequent steps of electrocatalytic CO2 reduction (Scheme 2). Similar to earlier reports (Riplinger et al., 2014), CO2 binding to [1]− is computed to be uphill (ΔG3 = 8.6 kcal/mol) and is driven by proton transfer from a Brønsted acid to generate [1-CO2H]0 (pKa1 = 24.8). Interestingly, we could not locate an optimized structure of CO2-bound [4-CO2]−, indicating that since the potential to generate [4]− is nearly +0.5 V more positive compared to that of [1]−, the pentacoordinate [4]− does not possess enough reducing power to activate CO2. On the other hand, further reduction to [4]2− (Figure S38), results in increased reactivity toward CO2 to generate [4-CO2]2− (ΔG4 = 10.8 kcal/mol), and subsequent protonation yields [4-CO2H]− (pKa3 = 29.4) (Scheme 2). It should be noted that the spin density (Figure S38) and total electron density difference (Figure S39) plots indicate that the third reduction is predominantly ligand centered in [4]2− and best characterized as [Mn0-bqn2−]2−, although the additional negative charge is shared between the metal center and the ligand in the case of [1]2− and [2]2− such that the electronic structure exhibits a resonance between [Mn−-(N∧N)•−]2− and [Mn0-(N∧N)2−]2−. [Mn-CO2H]− is predicted to be the common intermediate for both [1-CH3CN]+ and [4-CH3CN]+ electrocatalysts, as C–OH bond cleavage is expected to proceed predominantly via the reduction-first pathway (Scheme 2). Scission of the C–OH bond in [Mn-CO2H]− assisted by TFE leads to [Mn-CO]0. CO evolution may occur spontaneously from [Mn-CO]0 prior to one-electron reduction (Grice et al., 2013) and generation of [Mn]−. However, this mechanism is yet to be experimentally verified for this class of Mn catalyst. In contrast to [1-CH3CN]+, [4-CH3CN]+ produces H2 as the dominant product in the presence of TFE as a Brønsted acid, which is attributed to less favorable interaction of [4]− with CO2 compared to [1]−. However, we should also note that the pKa's of [1]− (pKa = 24.8) and [1]2− (pKa = 36.0) are significantly higher than their counterparts, [4]− (pKa = 17.3) and [4]2− (pKa = 29.0), indicating that hydride formation is not as favorable in the latter either. In the absence of a Brønsted acid as a proton source, all the catalysts are predicted to form the three-electron reduced [Mn]2− active catalyst before binding CO2 and subsequently producing CO and via interaction of a second CO2 molecule with [Mn-CO2]2−.
Conclusions
Through a systematic variation of the polypyridyl ligand from bpy to phen to dmphen and finally bqn, both steric- and electronic-based ligand influences on the activity of a [fac-Mn(N∧N)(CO)3(CH3CN)]+ class of CO2 reduction pre-catalysts has been established, providing critical insight into the manipulation of CO2 binding affinities and resulting product selectivity for these catalysts. Through a combination of IR-SEC and computational studies, the combined electronic and steric influences of the π-extended bqn ligand have been probed in [4-CH3CN]+, where formation of the [4–4]0 dimer is hindered and a concerted two-electron ECE mechanism for the generation of [4]− is favored. Computations have revealed how the lower lying π*-orbitals of the bqn ligand have rendered this pentacoordinate [4]− intermediate inactive with respect to CO2, even in the presence of a Brønsted acid. In contrast to the bpy, phen, and dmphen derived catalysts, this has resulted in a shift in product selectivity for the [4-CH3CN]+ pre-catalyst to favor H2 evolution in the presence of excess TFE. However, electrochemical and computational investigations have established successful CO2 activation following in-situ generation of a three-electron reduced [fac-Mn(−1)(N∧N•−)(CO)3]2− active catalyst, a first for any [fac-Mn(N∧N)(CO)3(CH3CN)]+ pre-catalyst. Although at the cost of additional overpotential, this three-electron pathway results in increased reactivity toward CO2 to generate the previously established [fac-Mn(I)(CO2H)(N∧N•−)(CO)3]− intermediate, which subsequently propagates the catalytic cycle via the standard reduction-first pathway involving rate-determining, proton-coupled C–OH bond cleavage.
Materials and Methods
Acetonitrile (ACS reagent grade, 99.5%), bromopentacarbonylmanganese(I) (98%), potassium carbonate (>99%), silver trifluoromethanesulfonate (>99%), tetrahydrofuran (anhydrous, 99.9%) and 2,2,2-trifluoroethanol (>99%) were purchased from Sigma Aldrich and used as received. Dichloromethane (ACS reagent grade, >99.9%) and diethyl ether were purchased from Pharmco-Aaper (ACS reagent grade, >99.9%) and used as received. The water content in ACS reagent grade acetonitrile was confirmed by Karl-Fisher titration to be 0.17 M (0.3%). Tetrabutylammonium hexafluorophosphate (99%, Sigma Aldrich) was recrystallized thrice from ethanol and dried under vacuum prior to electrolyte preparation. Steady-state FTIR spectra were recorded on a Thermo Nicolet 670 FTIR spectrophotometer using a liquid cell with CaF2 windows in spectrophotometric grade acetonitrile (99.5%, Sigma Aldrich) solvent. NMR spectra were recorded on an Agilent spectrometer operated at 399.80 MHz for 1H nuclei. CD3CN was used as received from Sigma Aldrich and its residual 1H solvent signal used as an internal reference for reporting the chemical shift (δ=1.96 ppm). Voltammetry and bulk electrolysis were carried out on a CH Instruments 620E potentiostat. A custom three-electrode cell was used for both voltammetry and bulk electrolysis experiments allowing airtight introduction of working, counter and reference electrodes as well as septa for gas purging. For cyclic voltammetry, glassy carbon (3 mm diameter) and Pt wire were used as working and counter electrodes, respectively, with 0.1 M [Bu4N][PF6] in ACS reagent grade acetonitrile as the supporting electrolyte. A non-aqueous reference electrode was used to minimize ohmic potential drop at the solvent interface. This consisted of a Ag wire in 0.10 M [Bu4N][PF6] acetonitrile supporting electrolyte isolated by a vycor frit and was calibrated in-situ using the ferricenium/ferrocene redox couple as an internal reference. Redox potentials (E) were determined from cyclic voltammetry as (Epa + Epc)/2, where Epa and Epc are the anodic and cathodic peak potentials, respectively. Where E could not be calculated due to irreversible behavior, Epc or Epa are reported accordingly. For CO2 concentration dependent studies, gas cylinders were ordered from Airgas containing pre-mixed ratios of Ar:CO2 (100:0, 80:20, 60:40, 50:50, 40:60, 20:80, 0:100). For controlled potential bulk electrolysis experiments a vitreous carbon (Duocell) working electrode soldered to a copper wire was used. A Pt gauze counter electrode was used, isolated from the main compartment by a fine porosity vycor tube+frit to minimize mass transfer resistance. Gas chromatography data were recorded on a custom Shimadzu GC-2014 instrument where a Ni “methanizer” catalyst was used to convert CO to CH4 prior to quantification of CH4 by the thermal conductivity detector. H2 was simultaneously monitored by a flame ionization detector during the same injection. The GC was pre-calibrated for CO and H2 quantification by mimicking bulk electrolysis conditions (i.e., 5 mL supporting electrolyte in the same cell, with electrodes, under 1 atm CO2). Standard curves for H2 and CO were generated using this cell where known volumes of the analyte gas (H2 or CO) were injected and the solution stirred for 30 min to allow equilibration of the analyte between the electrolyte and headspace prior to GC injection.
General Synthesis of [fac-Mn(N∧N)(CO)3(OTf)] Complexes 1-4
Bromopentacarbonyl manganese(I) (100 mg, 0.36 mmol) and silver trifluoromethanesulfonate (93.5 mg, 0.36 mmol) were charged to a 50-mL round bottomed flask with approximately 25 mL of degassed dichloromethane under 1 atmosphere of argon. The solution was allowed to mix in the dark for 1 h at room temperature after which the AgBr precipitate was removed by filtration through celite. The dichloromethane solvent was subsequently removed by a rotary evaporator. After confirming quantitative transformation to the [fac-Mn(OTf)(CO)5] intermediate by FTIR spectroscopy, the solid was transferred to a 10-mL microwave vial with 3 mL of tetrahydrofuran. To this solution was added a slight deficit of the appropriate ligand (0.33 mol). The vial was then sealed and reacted in a CEM Discovery microwave reactor at 70°C for 10 min. Each reaction afforded a bright yellow-orange solution. The tetrahydrofuran was reduced in volume to roughly 0.5 mL on a rotary evaporator at which point the pure product was immediately precipitated by addition of excess diethyl ether. The solid was isolated via vacuum filtration, rinsed with diethyl ether and dried under vacuum. No further purification was necessary.
[fac-Mn(OTf)(bpy)(CO)3] (1) FTIR (CH3CN) ν(CO): 2,050, 1,958 cm−1. 1H-NMR (CD3CN) δ: 7.70–7.73 (2H, m), 8.21–8.25 (2H, m), 8.40 (2H, d, J=8.0 Hz), 9.14 (2H, d, J=5.6 Hz) ppm. Anal. Calcd. for C14H8F3MnN2O6S: C, 37.85; H, 1.82; N, 6.31. Found: C, 38.27; H, 1.99; N, 6.02.
[fac-Mn(OTf)(phen)(CO)3] (2) FTIR (CH3CN) ν(CO): 2,050, 1,958 cm−1. 1H-NMR (CD3CN) δ: 8.04 (2H, dd, J1 = 4.0, J2 = 5.2 Hz), 8.20 (2H, s), 8.78 (2H, dd, J1 = 1.2, J2 = 8.0 Hz), 9.49 (2H, dd, J1 = 1.2, J2 = 4.8 Hz) ppm. Anal. Calcd. for C16H8F3MnN2O6S: C, 41.04; H, 1.72; N, 5.98. Found: C, 41.36; H, 1.88; N, 5.42.
[fac-Mn(OTf)(dmphen)(CO)3] (3) FTIR (CH3CN) ν(CO): 2,046, 1,959, 1,944(sh) cm−1. 1H-NMR (CD3CN) δ: 3.30 (6H, s), 7.90 (2H, d, J=8.0 Hz), 8.04 (2H, s), 8.57 (2H, d, J=8.0 Hz). Anal. Calcd. for C18H12F3MnN2O6S: C, 43.56; H, 2.44; N, 5.64. Found: C, 44.05; H, 2.69; N, 5.18.
[fac-Mn(OTf)(bqn)(CO)3] (4) FTIR (CH3CN) ν(CO): 2,047, 1,959 cm−1. 1H-NMR (CD3CN) δ: 7.90 (2H, dd, J1 = J2 = 8.0 Hz), 8.12 (2H, dd, J1 = J2 = 8.0 Hz), 8.21 (2H, d, J=8.0 Hz), 8.61 (2H, d, J=8.0 Hz), 8.83 (2H, d, J=8.0 Hz), 8.88 (2H, d, J=8.0 Hz) ppm. Anal. Calcd. for C22H12F3MnN2O6S: C, 48.54; H, 2.22; N, 5.15. Found: C, 49.10; H, 2.60; N, 4.91.
Computational Methods
Density Functional Theory
All geometries were fully optimized at the M06 level of density functional theory (Zhao and Truhlar, 2008a,b, 2010) with the SMD continuum solvation model (Marenich et al., 2009) for acetonitrile as solvent using the Stuttgart [8s7p6d2f | 6s5p3d1f] ECP10MDF contracted pseudopotential basis set (Dolg et al., 1987) on Mn and the 6-31G(d) basis set on all other atoms (Hehre et al., 1986). Non-analytical integrals were evaluated using the integral=grid=ultrafine option as implemented in the Gaussian 16 software package (Frisch et al., 2016). The nature of all stationary points was verified by analytic computation of vibrational frequencies, which were also used for the computation of zero-point vibrational energies, molecular partition functions, and for determining the reactants and products associated with each transition-state structure (by following the normal modes associated with imaginary frequencies). Partition functions were used in the computation of 298 K thermal contributions to the free energy employing the usual ideal-gas, rigid-rotator, harmonic oscillator approximation (Cramer, 2004). Free-energy contributions were added to single-point, SMD-solvated M06 electronic energies computed at the optimized geometries obtained with the initial basis with the SDD basis set on Mn and the larger 6-311+G(2df,p) basis set on all other atoms to arrive at final, composite free energies.
Solvation and Standard Reduction Potentials
As mentioned above, solvation effects for acetonitrile were accounted for by using the SMD continuum solvation model. A 1 M standard state was used for all species in solution (except for acetonitrile as solvent for which the standard state was assigned as 19.14 M). Thus, the free energy in solution is computed as the 1 atm gas-phase free energy, plus an adjustment for the 1 atm to 1 M standard-state concentration change of RT ln (24.5), or 1.9 kcal/mol, plus the 1 M to 1 M transfer (solvation) free energy computed from the SMD model. Standard reduction potentials were calculated for various possible redox couples to assess the energetic accessibility of different intermediates at various oxidation states. For a redox reaction of the form
where O and R denote the oxidized and reduced states of the redox couple, respectively, and n is the number of electrons involved in redox reaction, the reduction potential relative to SCE was computed as
where is the free energy change associated with Equation 1 (using Boltzmann statistics for the electron) and is taken as 0.141 V (Keith et al., 2013), which is required for the conversion of calculated vs. normal hydrogen electrode (NHE) in aqueous solution (ENHE = −4.281 V) (Kelly et al., 2006) to vs. the saturated calomel electrode (SCE) in acetonitrile (ESCE = −4.422 V) (Isse and Gennaro, 2010). We obtained reduction potentials referenced to the ferricenium/ferrocene couple by using a shift of −0.384 V from vs. SCE.
Data Availability Statement
All datasets generated for this study are included in the manuscript/Supplementary Files.
Author Contributions
MM, VB, and JR contributed to the synthesis, spectroscopic, and electrochemical characterization of all complexes studied. KN and DG contributed IR-SEC studies. ME contributed all computational studies. MM, ME, DG, and JR contributed equally to manuscript preparation.
Funding
JR thanks the National Science Foundation for support under Grant No. CHE-1800062. This work at BNL (DG and ME) was supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences & Biosciences, under Contract No. DE-SC0012704. KN was grateful to the DOE for an Office of Science Graduate Student Research (SCGSR) award.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00628/full#supplementary-material
Scan rate dependent voltammetry studies under inert and catalytic conditions; experimental and computational IR-SEC spectra; catalyst, acid and CO2 concentration dependence; controlled potential electrolysis plots; computational spin-density plots, total electron density difference plots, and cartesian coordinates. These materials are provided in the supporting information document.
References
Appel, A. M., and Helm, M. L. (2014). Determining the overpotential for a molecular electrocatalyst. ACS Catal. 4, 630–633. doi: 10.1021/cs401013v
Arakawa, H., Areasta, M., Armor, J. N., Barteau, M. A., Beckman, E. J., Bell, A. T., et al. (2001). Catalysis research of relevance to carbon management: progress, challenges, and opportunities. Chem. Rev. 101, 953–996. doi: 10.1021/cr000018s
Bard, A., and Faulkner, L. (2001). Electrochemical Methods: Fundamentals and Applications. Oxford: John Wiley & Sons, Inc.
Bourrez, M., Molton, F., Chardon-Noblat, S., and Deronzier, A. (2011). Mn(bipyridyl)(CO)3Br: an abundant metal carbonyl complex as efficient electrocatalyst for CO2 reduction. Angew. Chem. Int. Ed. 50, 9903–9906. doi: 10.1002/anie.201103616
Bourrez, M., Orio, M., Molton, F., Vezin, H., Duboc, C., Deronzier, A., et al. (2014). Pulsed-EPR evidence of a manganese(II) hydroxycarbonyl intermediate in the electrocatalytic reduction of carbon dioxide by a manganese bipyridyl derivative. Angew. Chem. Int. Ed. 53, 240–243. doi: 10.1002/anie.201306750
Cramer, C. J. (2004). Essentials of Computational Chemistry: Theories and Models. Chichester: John Wiley & Sons.
Dolg, M., Wedig, U., Stoll, H., and Preuss, H. (1987). Energy-adjusted Ab initio pseudopotentials for the first row transition elements. J. Chem. Phys. 86, 866–872. doi: 10.1063/1.452288
Francke, R., Schille, B., and Roemelt, M. (2018). Homogeneously catalyzed electroreduction of carbon dioxide—methods, mechanisms, and catalysts. Chem. Rev. 118, 4631–4701. doi: 10.1021/acs.chemrev.7b00459
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2016). Gaussian 16 Rev. A.03. Wallingford, CT: John Wiley & Sons.
Grice, K. A., Gu, N. X., Sampson, M. D., and Kubiak, C. P. (2013). Carbon monoxide release catalysed by electron transfer: electrochemical and spectroscopic investigations of [Re(bpy-R)(CO)4](OTf) complexes relevant to CO2 reduction. Dalton Trans. 42, 8498–8503. doi: 10.1039/c3dt50612f
Grills, D. C., Ertem, M. Z., McKinnon, M., Ngo, K. T., and Rochford, J. (2018). Mechanistic aspects of CO2 reduction catalysis with manganese-based molecular catalysts. Coord. Chem. Rev. 374, 173–217. doi: 10.1016/j.ccr.2018.05.022
Hartl, F., Rossenaar, B. D., Stor, G. J., and Stufkens, D. J. (1995). Role of an electron-transfer chain reaction in the unusual photochemical formation of five-coordinated anions [Mn(CO)3(α-diimine)]− from fac-[Mn(X)(CO)3(α-diimine)] (X = halide) at low temperatures. Recl. Trav. Chim. 114, 565–570. doi: 10.1002/recl.19951141123
Hehre, W. J., Radom, L., Schleyer, P. V. R., and Pople, J. A. (1986). Ab Initio Molecular Orbital Theory. New York, NY: Wiley.
Isse, A. A., and Gennaro, A. (2010). Absolute potential of the standard hydrogen electrode and the problem of interconversion of potentials in different solvents. J. Phys. Chem. B 114, 7894–7899. doi: 10.1021/jp100402x
Kaim, W., and Fiedler, J. (2009). Spectroelectrochemistry: the best of two worlds. Chem. Soc. Rev. 38, 3373–3382. doi: 10.1039/b504286k
Keith, J. A., Grice, K. A., Kubiak, C. P., and Carter, E. A. (2013). Elucidation of the selectivity of proton-dependent electrocatalytic CO2 reduction by fac-Re(bpy)(CO)3Cl. J. Am. Chem. Soc. 135, 15823–15829. doi: 10.1021/ja406456g
Kelly, C. P., Cramer, C. J., and Truhlar, D. G. (2006). Aqueous solvation free energies of ions and ion–water clusters based on an accurate value for the absolute aqueous solvation free energy of the proton. J. Phys. Chem. B 110, 16066–16081. doi: 10.1021/jp063552y
Kurtz, D. A., Dhakal, B., Hulme, R. J., Nichol, G. S., and Felton, G. A. N. (2015). Correlations between photophysical and electrochemical properties for a series of new Mn carbonyl complexes containing substituted phenanthroline ligands. Inorg. Chim. Acta 427, 22–26. doi: 10.1016/j.ica.2014.12.009
Lam, Y. C., Nielsen, R. J., Gray, H. B., and Goddard, W. A. (2015). A Mn bipyrimidine catalyst predicted to reduce CO2 at lower overpotential. ACS Catal. 5, 2521–2528. doi: 10.1021/cs501963v
Machan, C. W., Sampson, M. D., Chabolla, S. A., Dang, T., and Kubiak, C. P. (2014). Developing a mechanistic understanding of molecular electrocatalysts for CO2 reduction using infrared spectroelectrochemistry. Organometallics 33, 4550–4559. doi: 10.1021/om500044a
Marenich, A. V., Cramer, C. J., and Truhlar, D. G. (2009). Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 113, 6378–6396. doi: 10.1021/jp810292n
Matsubara, Y., Grills, D. C., and Kuwahara, Y. (2015). Thermodynamic aspects of electrocatalytic CO2 reduction in acetonitrile and with an ionic liquid as solvent or electrolyte. ACS Catal., 6440–6452. doi: 10.1021/acscatal.5b00656
McKinnon, M., Ngo, K. T., Sobottka, S., Sarkar, B., Ertem, M. Z., Grills, D. C., et al. (2019). Synergistic metal–ligand redox cooperativity for electrocatalytic CO2 reduction promoted by a ligand-based redox couple in Mn and Re tricarbonyl complexes. Organometallics 38, 1317–1329. doi: 10.1021/acs.organomet.8b00584
Neri, G., Donaldson, P. M., and Cowan, A. J. (2019). In situ study of the low overpotential “dimer pathway” for electrocatalytic carbon dioxide reduction by manganese carbonyl complexes. Phys. Chem. Chem. Phys. 21, 7389–7397. doi: 10.1039/c9cp00504h
Ngo, K. T., McKinnon, M., Mahanti, B., Narayanan, R., Grills, D. C., Ertem, M. Z., et al. (2017). Turning on the protonation-first pathway for electrocatalytic CO2 reduction by manganese bipyridyl tricarbonyl complexes. J. Am. Chem. Soc. 139, 2604–2618. doi: 10.1021/jacs.6b08776
Pegis, M. L., Roberts, J. A. S., Wasylenko, D. J., Mader, E. A., Appel, A. M., and Mayer, J. M. (2015). Standard reduction potentials for oxygen and carbon dioxide couples in acetonitrile and N,N-dimethylformamide. Inorg. Chem. 54, 11883–11888. doi: 10.1021/acs.inorgchem.5b02136
Riplinger, C., and Carter, E. A. (2015). Influence of weak brønsted acids on electrocatalytic CO2 reduction by manganese and rhenium bipyridine catalysts. ACS Catal. 5, 900–908. doi: 10.1021/cs501687n
Riplinger, C., Sampson, M. D., Ritzmann, A. M., Kubiak, C. P., and Carter, E. A. (2014). Mechanistic contrasts between manganese and rhenium bipyridine electrocatalysts for the reduction of carbon dioxide. J. Am. Chem. Soc. 136, 16285–16298. doi: 10.1021/ja508192y
Sampson, M. D., Nguyen, A. D., Grice, K. A., Moore, C. E., Rheingold, A. L., and Kubiak, C. P. (2014). Manganese catalysts with bulky bipyridine ligands for the electrocatalytic reduction of carbon dioxide: eliminating dimerization and altering catalysis. J. Am. Chem. Soc. 136, 5460–5471. doi: 10.1021/ja501252f
Scheiring, T., Kaim, W., and Fiedler, J. (2000). Geometrical and electronic structures of the acetyl complex Re(bpy)(CO)3(COCH3) and of [M(bpy)(CO)4](OTf), M = Mn,Re. J. Organomet. Chem. 598, 136–141. doi: 10.1016/S0022-328X(99)00691-9
Sinopoli, A., La Porte, N. T., Martinez, J. F., Wasielewski, M. R., and Sohail, M. (2018). Manganese carbonyl complexes for CO2 reduction. Coord. Chem. Rev. 365, 60–74. doi: 10.1016/j.ccr.2018.03.011
Smieja, J. M., Sampson, M. D., Grice, K. A., Benson, E. E., Froehlich, J. D., and Kubiak, C. P. (2013). Manganese as a substitute for rhenium in CO2 reduction catalysts: the importance of acids. Inorg. Chem. 52, 2484–2491. doi: 10.1021/ic302391u
Stanbury, M., Compain, J.-D., Trejo, M., Smith, P., Gour,é, E., and Chardon-Noblat, S. (2017). Mn-carbonyl molecular catalysts containing a redox-active phenanthroline-5,6-dione for selective electro- and photoreduction of CO2 to CO or HCOOH. Electrochim. Acta 240, 288–299. doi: 10.1016/j.electacta.2017.04.080
Stanbury, M., Compain, J. D., and Chardon-Noblat, S. (2018). Electro and photoreduction of CO2 driven by manganese-carbonyl molecular catalysts. Coord. Chem. Rev. 361, 120–137. doi: 10.1016/j.ccr.2018.01.014
Tignor, S. E., Kuo, H.-Y., Lee, T. S., Scholes, G. D., and Bocarsly, A. B. (2018). Manganese-based catalysts with varying ligand substituents for the electrochemical reduction of CO2 to CO. Organometallics 38, 1292–1299. doi: 10.1021/acs.organomet.8b00554
Zhao, Y., and Truhlar, D. G. (2008a). Density functionals with broad applicability in chemistry. Acc. Chem. Res. 41, 157–167. doi: 10.1021/ar700111a
Zhao, Y., and Truhlar, D. G. (2008b). The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 120, 215–241. doi: 10.1007/s00214-007-0310-x
Keywords: carbon dioxide reduction, electrocatalysis, manganese, carbon monoxide, hydrogen evolution, computational modeling
Citation: McKinnon M, Belkina V, Ngo KT, Ertem MZ, Grills DC and Rochford J (2019) An Investigation of Electrocatalytic CO2 Reduction Using a Manganese Tricarbonyl Biquinoline Complex. Front. Chem. 7:628. doi: 10.3389/fchem.2019.00628
Received: 01 March 2019; Accepted: 02 September 2019;
Published: 24 September 2019.
Edited by:
Federico Cesano, University of Turin, ItalyReviewed by:
Julio Lloret-Fillol, Institut Català d'Investigació Química, SpainCharles Machan, University of Virginia, United States
Copyright © 2019 McKinnon, Belkina, Ngo, Ertem, Grills and Rochford. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mehmed Z. Ertem, bXplcnRlbUBibmwuZ292; David C. Grills, ZGNncmlsbHNAYm5sLmdvdg==; Jonathan Rochford, am9uYXRoYW4ucm9jaGZvcmRAdW1iLmVkdQ==
†Present address: Ken T. Ngo, National Renewable Energy Laboratory (DOE), Golden, CO, United States