 Qiaoling Li1
Qiaoling Li1 Tianran Zhang2Xiaofei Yu1Xiaoyu Wu1Xinghua Zhang1
Tianran Zhang2Xiaofei Yu1Xiaoyu Wu1Xinghua Zhang1 Zunming Lu1
Zunming Lu1 Xiaojing Yang1*
Xiaojing Yang1* Yang Huang1*Lanlan Li1*
Yang Huang1*Lanlan Li1*- 1Key Lab for Micro- and Nano-Scale Boron Nitride Materials in Hebei Province, School of Materials Science and Engineering, Hebei University of Technology, Tianjin, China
- 2Department of Chemical and Biomolecular Engineering, National University of Singapore, Singapore, Singapore
The development of efficient, stable, and low-cost catalytic material for the oxygen reduction reaction (ORR) is currently highly desirable but challenging. In this work, based on first-principles calculation, the stabilities, catalytic activities and catalytic mechanisms of isolated Au atom supported on defective porous BN (p-BN) have been studied in detail. The results reveal that the defective p-BN anchor Au atom strongly to ensure the stability of Au/p-BN. Based on frontier molecular orbital and charge-density analysis, isolated Au atom supported on porous BN with VN defect (Au/p-BN-VN) is an effective ORR catalyst. Especially, the low barriers of the formation (0.38 eV) and dissociation (0.31 eV) of *OOH and the instability of H2O2 on Au/p-BN-VN catalyst suggest that ORR proceeds via 4-electron pathway. Along the favorable pathway, the reduction of O2 to *OOH is the rate-limiting step with the largest activation barrier of 0.38 eV and the maximum free energy change is 1.88 eV. Our results provide a useful guidance for the design and fabrication of new Au-base catalyst with high-efficiency and are beneficial for the developing of novel isolated metal atom catalysts for ORR.
Introduction
The large-scale commercial applications of electrochemical devices such as polymer electrolyte membrane fuel cells (PEMFCs) are significantly obstructed by sluggish kinetic cathode oxygen reduction reaction (ORR) (Debe, 2012; Sharma and Pollet, 2012; Chen et al., 2019b). Pt-based materials are the best choice for current fuel cell cathodic catalysts. Nevertheless, Pt-based catalysts also suffer from many problems such as high cost, less abundance, the poor tolerance to CO poison and poor stability in an electrochemical environment (Nie et al., 2015). Therefore, great efforts have been devoted to developing novel electrocatalysts for ORR with high efficiency and long-term stability based on low cost and abundant materials.
Au has attracted attention as a promising Pt-alternative catalyst due to its extremely similar crystal structure, low cost, and more abundant than Pt. However, the ORR activity of traditional Au-based catalysts is much lower than that of commercial Pt/C. To date, there are several strategies, such as controlling the shape or morphology of Au nanocrystals (Deng et al., 2015; Alba-Molina et al., 2019), alloying Au with foreign metals (Kodama et al., 2016; Xue et al., 2018), or depositing Au nanoparticles on functional substrate supports (Uosaki et al., 2014; Nie et al., 2015; Kobayashi et al., 2016; Ostojic et al., 2018; Peng et al., 2019). Unfortunately, the catalytic activity of these Au nanoparticles catalysts generally has strongly particle size/shape–dependented and the overall efficiency has been rather low because only the local interface atoms are active for catalysis. Deeply, the fully filled d shell of Au (5 d10) is energetically stable, which limits the adsorption and activation of O2 and ORR intermediates (Nørskov et al., 2004; Bhatt et al., 2017). Therefore, on the Au-based catalyst, ORR mainly produces H2O2 via 2-electron process. A rationally design to decrease Au usage and alter Au intrinsic chemical inertness is crucial in enhancing its ORR catalytic activity. Considering the noble nature of Au, downsizing the nanometer-scale to single-atoms could effectively reduce the use of precious metal and maximize atom utilization efficiency. However, up to now, it is not clear whether single-atom Au catalyst can promote ORR process.
As a kind of high-efficiency catalyst, single-atom catalysts (SACs) exhibit extraordinary catalytic activity and selectivity toward various reactions (Bayatsarmadi et al., 2017; Peng et al., 2018; Xiao et al., 2018). Generally, an appropriate support plays a crucial role in realizing isolated metal atom dispersion and immobilize metal atom (Du et al., 2019). Moreover, the strong interaction between the metal and support can modulate the electronic structure of metal catalyst and further improve the catalytic performance. As a catalytic support, BN nanosheet, a structural analog of grapheme, featured with a local polarity B-N bond, has attracted considerable attention due to its excellent acid-base resistance, extraordinary oxidation resistance, and thermal stability (Lin, 2002; Pakdel et al., 2014; Sun et al., 2016; Tran-Thuy et al., 2017). Uosaki et al. demonstrated the possibility to functionalize inert Au substrate to become ORR catalysts by BN modified, reducing the overpotential by ca. 0.27 V (Uosaki et al., 2014). Later, Elumalai and coworkers further reduced the overpotential ca. 50 mV by boron nitride nanosheet (BNNS) supported gold nanoparticles (Elumalai et al., 2016; Lyalin et al., 2017). In addition, BNNS with vacancy defect has been proved to be excellent support for SACs. Feng et al. theoretically predicted that Fe-embedded BNNS with N vacancy defect exhibits a good catalytic activity for ORR as its strong hybridization between the Fe atom and the sp2 dangling bonds of nitrogen atoms (Feng et al., 2015). In another study, the catalytic activity of Cu, Co, Au, Pt et al. transition metal atoms doped h-BN nanosheets were also proposed as viable catalysts for CO oxidation (Lin et al., 2013; Deng et al., 2018, 2019).
In this work, a systematic density functional theory (DFT) is performed to explore the potential application of atomic Au anchored on defective porous BN (Au/p-BN) for ORR activity and the ORR mechanism were investigated in detail. Our calculation results revealed that p-BN with vacancy defect (both VB and VN) can efficiently anchor Au atom and modify the electronic structure of Au. Moreover, Au/p-BN-VN is a promising catalyst for ORR, because O2 is activated effectively on Au/p-BN-VN and reduced to H2O via desirable 4-electron process. These results are beneficial to develop new Au-base catalyst with high-efficiency and design novel single-atom catalysts for ORR.
Computational Models
All the calculations were performed at a spin-polarized density functional theory (DFT) level as implemented in the DMol3 code (Delley, 1990, 2000). Functional of generalized gradient approximation (GGA) with the Perdew, Burke and Ernzerhof (PBE) was used to describe exchange-correlation potential (Perdew et al., 1996). All Electron Relativistic was used for core treatment and the double numerical plus polarization (DNP) was chosen as the atomic orbital basis set (Koelling and Harmon, 1977). The real-space global orbital cutoff radius of 4.6 Å was adopted to ensure high-quality results. The self-consistent field (SCF) procedure was used with a convergence threshold of 10−6 au in the energy and electron density (Pulay, 1982). The Brillouin zone was sampled with a 5 × 5 × 1 k-points grid generated automatically by the Monkhorst–Pack method for geometric optimization, while a 9 × 9 × 1 k-points grid was used for electronic structures computations. A conductor-like screening model (COSMO) was used to simulate a H2O solvent environment. The dielectric constant was set as 78.54 for H2O solvent (Feng et al., 2015; Wang et al., 2015).
As shown in Figure S1, a 3 × 3 × 2 dimension supercell of h-BN was used to construct potential p-BN by introducing vacancies as our previously reported (Li et al., 2016, 2018). The periodic images were separated by a vacuum space of 18 Å in the z direction. The geometry optimizations with the convergence tolerances of energy, maximum force, and displacement on each atom were set to 1.0 × 10−6 Ha, 0.001 Ha/Å, and 0.005 Å, respectively. The full optimized unit cell of p-BN contains a central porous ring of 12 atoms. A 2 × 2 × 1 p-BN supercell with 48 B and 48 N atoms was adopted to prevent interactions in periodic images in our study and the lattice parameter was calculated to be 13.65 Å. The interlayer spacing of p-BN was calculated to be 3.821 Å. In this theoretical level, results are in good consistent with those of theorical (Tang et al., 2014) and experimental (Weng et al., 2013; Ye et al., 2016; Tian et al., 2018) studies.
The binding energy (Ebind) of isolated metal atom on p-BN was calculated as:
where Emetal, Ep−BN, and Emetal/p−BN is the total energy of the isolated metal atom, the p-BN substrate and metal atom/p-BN, respectively.
For the adsorption energy (Eads), it was defined by:
where Eadsorbate, Esubstrate, and Eadsorbate/substrate, denote the total energy of the free ORR species, substrate and adsorbed species on substrates, respectively. By this definition, positive Eads indicates an energetically favorable exothermic process.
The overall binding strength of Au atom in unsupported Aun clusters is characterized by the cohesive energy (Ecov), which is defined by:
where E(Au) and E(Aun) represent the calculated energies of an Au atom and the unsupported Aun cluster, respectively and n is the number of atom in the cluster.
For Aun clusters supported on p-BN-VN, Ecov can be computed as the following:
where E(p-BN-VN) and E(Aun/p-BN-VN) represent the calculated energies of p-BN-VN support and the Aun/p-BN-VN, respectively.
The transition states for ORR elemental steps were obtained by complete LST/QST tools in DMol3 code (Govind et al., 2003), and frequency calculations were performed to confirm the obtained transition states. Free energies of the ORR intermediates in electrochemical reaction pathways were calculated based on the computational hydrogen electrode (CHE) model proposed by Peterson et al. (2010). The Gibbs free energy (ΔG) change of each elementary step in the ORR was determined by:
The reaction energy (ΔE) is the total energy change directly obtained by DFT calculation. ΔEZPE and ΔS are the zero–point energy difference and the entropy change between the products and reactants, respectively. T is the system temperature in this work (T = 298.15 K). For each system, its EZPE can be implemented by summing vibrational frequencies over all normal modes. The entropies of the free molecules (O2, H2, H2O, H2O2) were taken from the NIST database [Computational Chemistry Comparison and Benchmark Database.http://cccbdb.nist.gov/].
Results and Discussion
Geometry and Stability of Au/p-BN
We firstly examined all the possible anchored sites for single Au atom on both perfect p-BN and defective p-BN including single nitrogen (VN) and boron (VB) vacancies and the most stable configurations for Au atom anchored on p-BN are shown in Figure 1. As shown in Figure 1A, the Au atom locates on the bridge site over boron-nitride bond for perfect p-BN and the bond length for Au-B and Au-N are calculated to be 2.929 and 2.387 Å respectively, which are much shorter than the van der Waals distance of 3.790 and 3.210 Å. With the introduction of VN (VB) defects (Au/p-BN-VN, Au/p-BN-VB), Au atom locates at the center of the defect, forming three non-equivalent Au-B (Au-N) bonds with the distance of 2.126, 1.988, and 2.020 Å (1.906, 2.471, and 1.880 Å), respectively (Figures 1B,C). Compared with Au/p-BN, the shorter bond lengths reveal the stronger interaction between the Au atom and the support. As radius of the Au atom is larger than that of the missing N/B atom, the anchored Au atom protrude from the basal plane of p-BN surface by about 0.631 and 0.544 Å for Au/p-BN-VN and Au/p-BN-VB respectively.
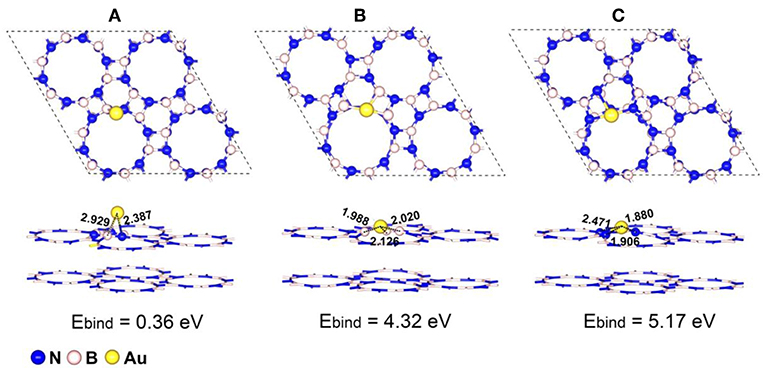
Figure 1. Optimized geometric configurations with the corresponding binding energy (Ebind/eV) of single atom Au anchored on a perfect p-BN (A), p-BN with VN defect (B), and p-BN with VB defect (C), respectively. The bond lengths are given in Å.
For an effective SACs, the strong binding strength between metal atoms and support is an ultimate prerequisite to protect metal atom from its aggregation and thus keep its good stability for long-term uses (Yang et al., 2013). In order to evaluate the stability of isolated Au atom on p-BN, the binding energies (Ebind /eV) of Au atom on both perfect and defective p-BN were calculated. For perfect p-BN, the Ebind was calculated to be 0.36 eV. With the introduction of vacancies, the Ebind increased to be 4.32 and 5.17 eV for VN and VB, respectively, which is larger than corresponding cohesive energy (Ecov = 3.76 eV, Figure S2) evidently, indicating that the defect structures are crucial in producing stable Au/p-BN catalyst. It is noteworthy that the Ebind for Au atom on defective p-BN is much larger than that for Au on other substrates reported such as on defective h-BN (3.48 eV for VN and 3.72 eV for VB, PBE) (Gao et al., 2012), on CeO2 (2.11 eV, PBE+U) (Song and Hensen, 2013), and on TiO2 with oxygen vacancy (1.28 eV, PBE) (Wan et al., 2018) etc. The larger Ebind of Au atoms on defective p-BN also makes the diffusion of Au atom to its neighboring site is considerably difficult in terms of the large endothermicity and high energy barrier, thus vigorously excluding the clustering of anchored Au atom on this support. We further considered the migration of Au atom on VN by the diffusion energy diagrams (Figure S3) (Chen et al., 2019a), which is relevant for particle aggregation. Two different pathways for the diffusion of the most stable Au on the VN were considered and the activation energies are calculated to be 3.46 and 2.36 eV, respectively. These large migration activation energies suggest that the Au atom is dynamically stable on the adjacent 3B site on VN. Therefore, these results clearly show that the defective p-BN anchor Au atom strongly to ensure the stability of Au/p-BN.
To gain a better understanding of the interaction between the Au atom and defect sites of p-BN, we performed electronic structure analyses by calculating the partial density of states (PDOS), frontier molecular orbital and total charge density and charge-density difference. As shown in Figures 2A,B, the PDOS for Au/p-BN-VB and Au/p-BN-VN demonstrates that the impurity states mainly consist of the 5d orbital of Au atom and 2p orbital of its adjacent N and B atoms, respectively. Meanwhile, the evident resonance between the 5d orbital of the Au atom and the 2p orbital of N/B atoms are observed, revealing a strong interaction between the Au atom and B/N-vacancy. The strongly interaction indicates that Au atom can be favorably supported on the defect of p-BN in thermodynamics. This is due to so-generated defect states in defective p-BN that affected the system's adsorption ability and catalytic activity (Figure S4) (Li et al., 2016, 2018). In particular, it is found that there is one evident half-occupied d state cross the Fermi level for Au/p-BN-VN (Figure 2B). This forceful d state around the Fermi level is generally considered to be an indicator of the high activity, which play a crucial role in catalysis (Deng et al., 2019).
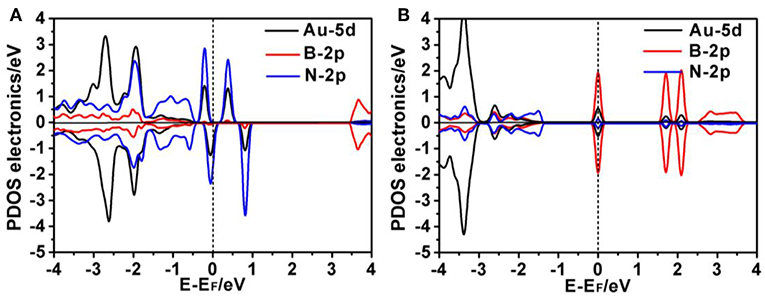
Figure 2. Projected density of states (PDOS) for the Au/p-BN-VB (A) and Au/p-BN-VN (B), respectively. The position of the Fermi level was marked in dashed line.
Figure 3A displays the highest occupied molecular orbital (HOMO) energies of the Au/p-BN, Au/p-BN-VN, Au/p-BN-VB catalyst and the lowest unoccupied molecular orbital (LUMO) energy of O2 molecule. For comparison, pure Au13 cluster and Pt13 cluster were also calculated and displayed. In the case of the ORR occurring on catalyst, the O2 molecule obtains electrons from the catalyst and then is reduced to H2O, that is, electrons on the HOMO of catalyst transfer to the LUMO of O2 during ORR. From the viewpoint of frontal molecular orbit theory (Zhou, 1993), the smaller the energy difference between the HOMO and the LUMO, the more easily an electron transfer takes place. As shown in Figure 3A, on pure Pt13, the energy difference between the HOMO of catalyst and the LUMO of O2 is less than that on pure Au13, indicating that Pt is a better catalyst than Au for ORR. The HOMO shifts to lower energy after Au has been loaded onto both p-BN and VB support. Consequently, the energy difference between the HOMO of catalyst and the LUMO of O2 increased and the electron transfer between Au/p-BN (Au/p-BN-VB) and O2 is more difficult even than Au13 cluster. Hence, neither Au/p-BN nor Au/p-BN-VB is suitable as the catalyst for ORR. Whereas, among all of the modeled catalyst, Au/p-BN-VN catalyst shares the minimized energy difference between the HOMO of catalyst and the LUMO of O2 and this energy difference is even less than that of pure Pt13. In other words, electron transfer can more smoothly proceed from the HOMO of Au/p-BN-VN to the LUMO of O2. These results indicate that the VN modifies the Au/p-BN-VN electron structure such that Au/p-BN-VN may be more beneficial than Pt for ORR. Thus, we would only investigate the ORR activity and mechanism of isolated atomic Au loaded on p-BN with VN defect (Au/p-BN-VB) in the following discussion.
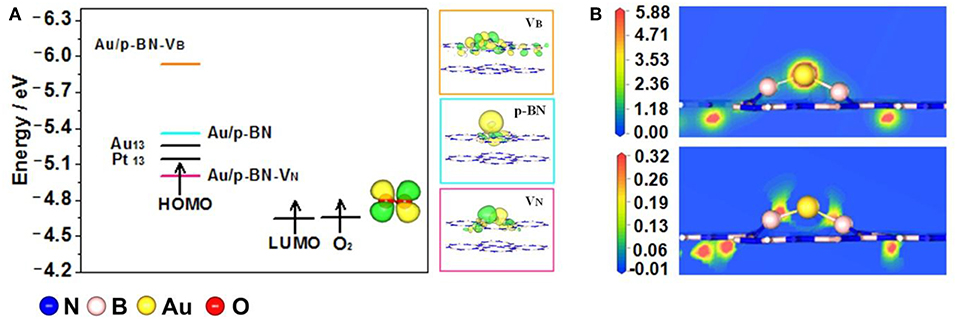
Figure 3. Frontier molecular orbital diagram shows the interaction of acceptor O2 with donor catalysts Au/p-BN, Au/p-BN with VN, VB, Au13, and Pt13. HOMO shapes of Au supported on Au/p-BN, Au/p-BN with VN, VB, and LUMO shape of O2 (A). Total charge density and Charge-density difference plots of Au anchored on VN (B).
To further understand the regulation of Au electronic structure by VN defect, we analyze the total charge density and charge-density difference as shown in Figure 3B. From the contours of charge-density difference, there is obvious charge enrichment in B atoms together with charge depletion in Au atom, indicating electrons are transferred from the adsorbed Au to the defective p-BN. Indeed, the bader charge analysis confirms that 0.595 e were transferred from Au atom to the B atoms around VN. This is due to the B atoms around VN have less electrons than the N atoms, indicating the B p orbitals accepting electrons from the Au d orbital (Figure S5).
The Adsorption of the ORR Intermediates
In this section, we considered the adsorption of ORR key intermediates (*O2, *OOH, *O, *OH, and H2O) on Au/p-BN-VN to investigate its catalytic activity, where, the * denotes the adsorption state on the Au/p-BN-VN surface. For each species, different adsorption sites and various adsorption configurations were considered, and the favored configurations are shown in Figure 4.
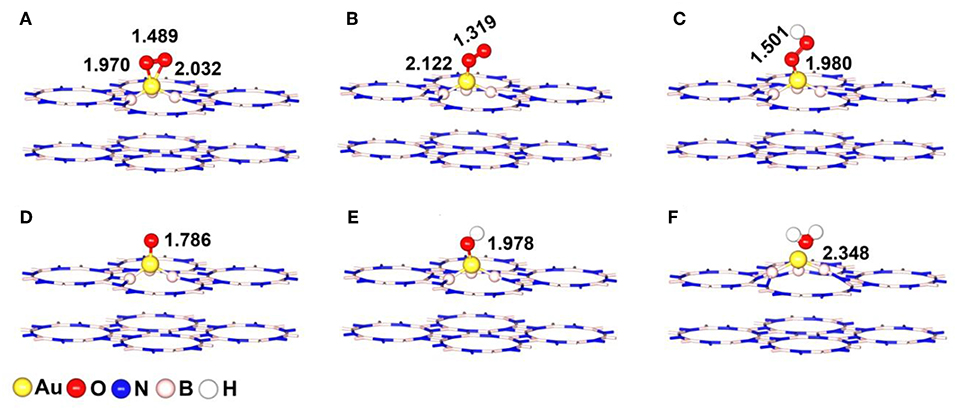
Figure 4. The most stable adsorption configurations of ORR intermediates on Au/p-BN-VN catalyst (A) *O2-Bridge, (B) *O2-Pauling, (C) *OOH, (D) *O, (E) *OH, and (F) H2O. The key bond lengths are given in Å.
The chemisorption of molecular O2 on catalyst surface is a crucial step for effective ORR as its initial adsorption manner plays an important role in the subsequent reaction pathways. Hence, we discuss the adsorption of O2 firstly by considering two initial configurations including a Bridge (side-on) and a Pauling (end-on) with the minimum energy configurations (Figures 4A,B). For Bridge-adsorption, two nonequivalent Au-O bonds are formed with the distances of 1.970 Å and 2.032 Å, respectively. Both are shorter than that of Au-O (2.122 Å) for pauling-adsorption. After adsorption, the O-O bond length of *O2 elongates from 1.225 Å to 1.489 Å and 1.319 Å for the Bridge and Pauling configurations, respectively. It is worth noting that the O-O bond length not only longer than that of *O2 adsorbed on Au13 cluster but also longer that of *O2 adsorbed Pt13 cluster, as the depiction in Figure 5A. The elongated O-O bond suggests that O2 can be more easily activated and dissociated, that is, Au/p-BN-VN catalyst benefits the activation and dissociation of molecular *O2 easier. To get more insight into the activation of *O2 by Au/p-BN-VN, Au13 and Pt13 catalysts, the Hirshfeld charge (Hirshfeld, 1977) were calculated. The results reveal that there are 0.513 and 0.259 e were transformed from the Au/p-BN-VN to O2 molecule for the Bridge and Pauling configurations, respectively, which larger than those from pure Au13 (0.257 e and 0.151 e) and Pt13 (0.260 e and 0.152 e). Moreover, from contours of the charge-density difference plots (Figure S6), it can be seen that certain amount of charge is transferred from Au atoms around VN vacancy to the O2 molecule, concomitant with the formation of the O-Au bond between O2 and Au/p-BN-VN. The increases in charge transfer lead to the elongation of the O–O bond of *O2, subsequently the activation of *O2 on Au/p-BN-VN. In addition, we further calculated the O2 adsorption on isolated Pt and Pd supported on defective p-BN to compare the ORR catalytic activity. The Au/p-BN-VN possess more superior ORR catalytic performance than Pt and Pd atom loaded on p-BN due to the large Eads value, O-O bond elongation, and more electron transfer between O2 and catalyst (Table S1).
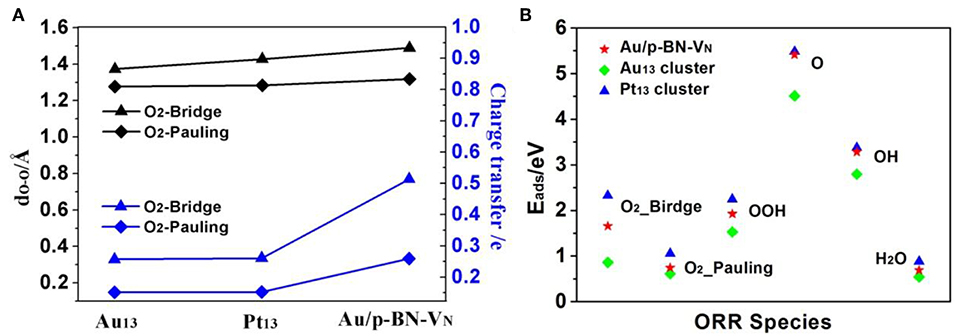
Figure 5. The O-O bond lengths and charge transfer amount of the adsorbed *O2 species on Au/p-BN-VN, Pt13, and Au13 catalyst (A). The adsorption energies (Eads) of the ORR intermediates adsorption on Au/p-BN-VN, Pt13 and Au13 (B).
For the OOH, O, OH and H2O species, the most favorable adsorption configurations were displayed in Figures 4C–F. After adsorbed on Au/p-BN-VN, the O-Au (dO−Au) bond is formed and the bond length is calculated to be 1.980, 1.786, and 1.978 Å for *OOH, *O, and *OH species, respectively. In the case of *OOH species, the O-OH is elongated from 1.341 Å of the isolated OOH to 1.501 Å, indicating the activation of the O-OH bond. Furthermore, the H2O is 2.348 Å far away from the catalyst surface, suggesting weak interaction between H2O and Au/p-BN-VN catalyst.
Further, the adsorption energies (Eads) of *O2 and other ORR intermediates including *OOH, *O, *OH and H2O on Au/p-BN-VN, Au13, and Pt13 cluster are summarized in Figure 5B. The Eads of these species on pure Au13 cluster are all the least, revealing the Au cluster itself is very inert and the adsorption on Au13 cluster is unfavorable. The Eads of *O2 on Au/p-BN-VN was computed to be 1.66 eV and 0.72 eV for the Bridge and Pauling models, respectively, which is larger than that of *O2 on Au13 cluster (0.86 and 0.62 eV) but smaller than that of *O2 on Pt13 cluster (2.33 and 1.05 eV). In particular, the Eads of *O and *OH species on Au/p-BN-VN is large up to 5.41 and 3.28 eV respectively, comparable to the Eads of *O and *OH species on Pt13 (5.47 and 3.37 eV). Generally, the adsorption strength of *O and *OH species on transition metal catalyst depends upon the electronic configuration of d-electrons of metal, which is primarily affected by whether the d shell is fully occupied or not. The fully filled d shell of Au is energetically stable, which is likely to have a lower the d-band center (Bhatt et al., 2017). Therefore, pure Au cluster is energetically stable as its fully filled d shell. But for Au/p-BN-VN catalyst, the Au atom with a partially filled d shell has a stronger Eads for oxygenated species. The smaller Eads illustrate the weak interaction between H2O and catalysts, suggesting that the final product H2O can speedy drift away from the catalyst surface, avoiding the flooding of the catalyst. In addition, we consider the effects of water environment on the adsorption by using a conductor-like screening model (COSMO) to simulate a H2O solvent environment, which is shown to be an effective method to describe the solvation and may exhibit excellent consistency with experiment (Wang et al., 2015). As shown in Table S2, the adsorption energies of *O2 species on Au/p-BN-VN are increased slightly by solvation, while few impact on other species. Thus, water slimly stabilizes the adsorbed O2 species that exhibits positive effects on the ORR process.
The deterioration of the cathode catalyst can largely be attributed to H2O2 species, an intermediate by a 2e− transfer reaction of ORR. Here, we examined the adsorption of *H2O2 molecules on Au/p-BN-VN, Au13, and Pt13 catalysts as shown in Figure S7. The adsorption of *H2O2 on Au13 cluster is stable with a small bond length change on bond length of H2O2 molecule (dO−O = 1.525 Å). When adsorbed on Pt13 cluster, the O-O bond length of *H2O2 elongate from 1.469 Å of isolated H2O2 molecule to 1.796 Å, suggesting the activation of O-O bond. Notably, the O-O bond length is further elongated to 2.832 Å on the Au/p-BN-VN catalyst, accompanying with the generation of a H2O molecule and retention of O species, revealing that *H2O2 is unstable on the Au/p-BN-VN catalyst and is likely to decompose into H2O and O species immediately. Accordingly, ORR process occurs on the Au/p-BN-VN catalyst surface via a direct 4e− pathway. Combined the frontal molecular orbit analysis, the activation of O-O bond and the adsorption of ORR intermediates, we can ascertain that Au/p-BN-VN is beneficial to the catalysis of ORR.
Mechanism of ORR on Au/p-BN-VN Catalyst
The elementary reactions mechanism for the ORR on Au/p-BN-VN were further investigated, including: *O2 dissociation, *OOH formation or dissociation, *H2O2 formation, *OH formation, and H2O formation, as the follows:
As shown in Figure 6, we confirmed the initial states (IS) and final states (FS) of various elementary reactions of the ORR on Au/p-BN-VN according to the most favorable adsorption sites, corresponding transition states (TS) are determined by the complete LST/QST method subsequently. The atomic configurations at various states along ORR reaction paths and corresponding heats of reaction and activation energies are also summarized in Figure 6.
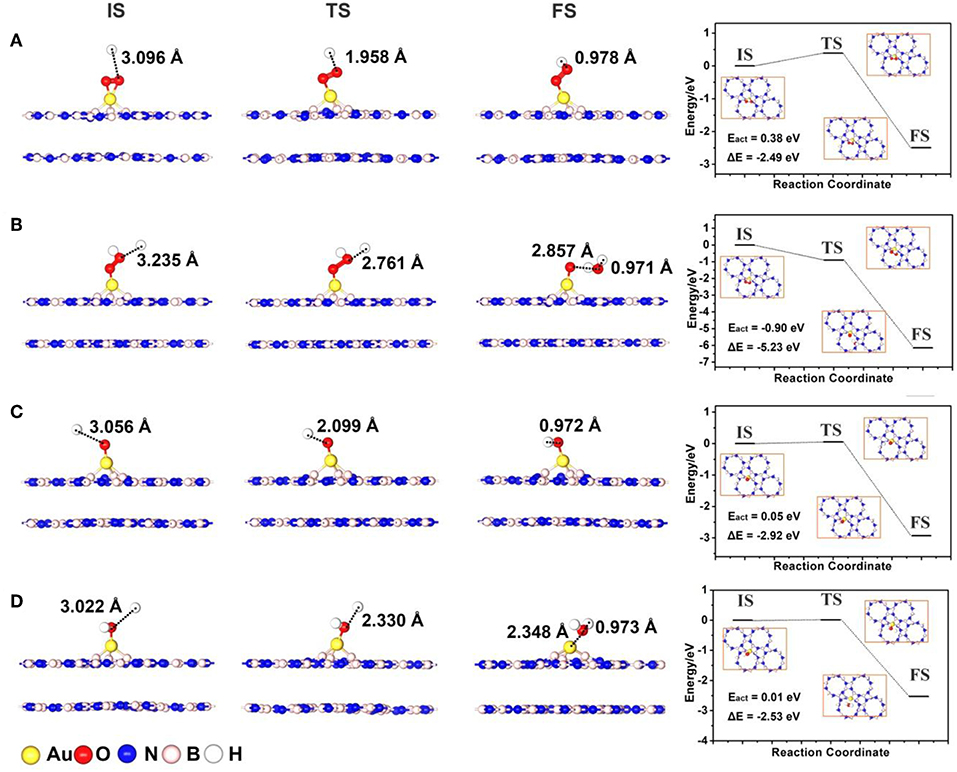
Figure 6. The structures for initial state (IS), transition state (TS), final state (FS), and potential energy profile for *O2 + H+ + e− → *OOH (A), *OOH + H+ + e− → H2O + *O (B), *O + H+ + e− → *OH (C) and *OH + H+ + e− → H2O (D) on a Au atom supported p-BN with VN.
After the *O2 adsorption, it can either undertake decomposition to form two adsorbed *O (1-a) or generate *OOH with the capture of one H+ and an additional electron (1-b). In pathway (1-a) (Figure S8A), the direct *O2 dissociation begins with the minimum energy state of adsorbed *O2 with Bridge configuration. The dissociation proceeds with the stretching of O-O bond above Au site, such that each O atom is near a Au-B bridge bond at the transition state. The calculated activation barrier (Eact = 1.32 eV) for *O2 dissociation step on Au/p-BN-VN is similar with that on Au13 cluster (Eact = 1.01 eV, Figure S9A), as well as the reaction energy is less favorable (ΔE = 1.14 eV). Obviously, the direct dissociation of *O2 on Au/p-BN-VN via pathway (1-a) is not the most favorable way. Contrarily, the activated *O2 on Au/p-BN-VN can easily capture one H+ and an additional e− to form an *OOH species via the pathway (1-b). Remarkably, this step is exothermic with 2.49 eV and the reaction energy barrier is only 0.38 eV (Figure 6A), indicating the feasibility of *O2 protonation process on the Au/p-BN-VN layers.
Next, three possible reaction pathways (2a-c) were also identified, corresponding to the protonation and dissociation process of *OOH on catalyst. Generally, the next reaction may proceed in the following ways: (1) the further hydrogenation of *OOH may result in the formation of H2 (by the pathway 2-a) leading to unfavorable 2-electron process, (2) the *OOH undergoes further protonation reduction to form a H2O molecule and one *O species on catalyst surface (by the pathway 2-b), and (3) O-OH dissociate into*O and *OH (by the pathway 2-c). However, the 2e− reduction pathway 2-a is impossible as the adsorption of the H2O2 on Au/p-BN-VN is unstable discussed in the previous section 3.2. Therefore, we only consider the pathway 2-b and 2-c. Amazingly, the protonation process of OOH* on the Au/p-BN-VN catalyst (*OOH + H+→*O + H2O) is exothermic by −5.23 eV with no energy barrier at all (Figure 6B). Therefore, once the *OOH species is formed, it can be further hydrogenated by reacting with another H+. In contrast, the *OOH are easy to form H2O2 on the Au13 with a low activation energy barrier (Eact = 0.08 eV) as shown in Figure S9B, which is consist with the reported results (Eact = 0.07 eV) of Au(111) catalyst (Yang et al., 2017). In addition, the *OOH group also can directly dissociate into *O + *OH via pathway (2-c) on the Au/p-BN-VN catalyst with a small activation energy barrier (Eact = 0.31 eV) and benefited exothermic reaction energy (−1.95 eV) as shown in Figure S8B. Finally, we also considered the kinetics for the subsequent reductions of *O to *OH and *OH to H2O, the activation energy barriers were 0.05 eV and 0.01 eV, respectively (Figures 6C,D). Overall, the ORR on the Au/p-BN-VN is a 4e− transformation pathway, which breaks the convention of a 2e− reduction from O2 to H2O2 on other Au-based catalysts (Uosaki et al., 2014; Yang et al., 2017). Along the favorable pathway, the reduction of O2 to *OOH is the rate-limiting step with the largest activation barrier of 0.38 eV on Au/p-BN-VN catalyst. This value of Eact is smaller than that of the reported single Pt on TiC(111) catalysts (Eact = 0.51 eV) and pure Pt(111) surface (Eact = 0.79 eV) (Duan and Wang, 2011; Tak et al., 2018).
Finally, the free energy change of each ORR step of the most favorable reaction route (as shown in Figure S10) on Au/p-BN-VN was listed in Figure 7. Here, the energy state of the first step is the total energy of both Au/p-BN-VN and O2 molecule. Then, the reference energy state of the next reduction steps is the total energy of the products of previous reaction step and H+ + e−. The ORR proceeds mainly through three forms are characterized as the O2 activation, hydrogenation reduction, and O-OH breaking processes. For the first step of the reaction, the adsorption of O2, is exothermic and downhill in the free energy profile by 0.89 eV. Then, the adsorbed O2 molecule prefers to be hydrogenated to form an OOH molecule and the Gibbs free energy decreases by of 1.88 eV. Furthermore, the formed OOH species is hydrogenated to form a H2O and leave one *O on the Au/p-BN-VN site with the ΔG of 5.15 eV. Subsequently, the *O atom is converted into OH with the help of H and this process is downhill in the free energy profile by 2.32 eV. Finally, the *OH is further reduced to form another H2O molecule with a downhill the free energy profile by 2.51 eV.
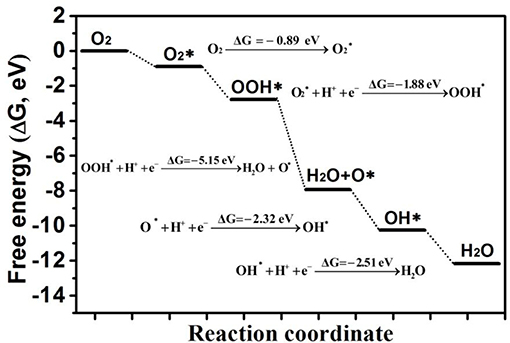
Figure 7. The free energies profile of whole ORR pathways on Au/p-BN-VN.
Conclusions
In summary, by density functional theory computations, the stability as well as its catalytic activities for ORR of the single atom Au supported on p-BN layers was systematically investigated. The study demonstrated that Au/p-BN-VN exhibits both high stability and excellent catalytic activities for ORR. Specifically, Au/p-BN-VN has a large Ebind value (4.32 eV) which is even higher than the Ecov of bulk Au (3.76 eV) and higher migration barrier (Eact = 2.36~3.46 eV), which can prevent the aggregation and ensure the stability of Au/p-BN-VN catalyst. It was found that the positive charge density of B3 site among VN defect is beneficial to the electrons transfer from the d shell filled Au atom to B atoms, which is beneficial to break the inert state of Au-d10 and contribute to the adsorption and activation of ORR species. Notably, the *H2O2 is found to be quite unstable on the Au/p-BN-VN catalyst and tend to decompose into H2O and *O species immediately. Moreover, the activation energy of each reaction pathway and the free energies profile of whole ORR pathways on Au/p-BN-VN were estimated and analyzed. By comparison of activation energy of each elementary reaction, we find that the reduction of the O2 to OOH is the rate-limited step with the largest activation barrier of 0.38 eV. Overall, these results would shed light on the understanding of Au-based catalyst for ORR and motivate the investigations on ORR kinetic behavior of isolated metal atom supported on p-BN catalysts.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Author Contributions
All authors have made a substantial contribution to the work and approved its publication. QL and LL designed the protocol, made calculations, and wrote the paper. TZ made calculations. XYu, XW, XZ, and ZL entered the discussion. XYa, YH, and LL revised the paper.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Nos. 21603052, 51772075, 51871088, 51771068, 51771067) and the Natural Science Foundation of Hebei Province (No. B2018202167, E2019202048).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00674/full#supplementary-material
References
Alba-Molina, D., Puente Santiago, A. R., Giner-Casares, J. J., Martín-Romero, M. T., Camacho, L., Luque, R., et al. (2019). Citrate-stabilized gold nanoparticles as high-performance electrocatalysts: the role of size in the electroreduction of oxygen. J. Phys. Chem. C 123, 9807–9812. doi: 10.1021/acs.jpcc.9b00249
Bayatsarmadi, B., Zheng, Y., Vasileff, A., and Qiao, S. Z. (2017). Recent advances in atomic metal doping of carbon-based nanomaterials for energy conversion. Small 13:1700191. doi: 10.1002/smll.201700191
Bhatt, M. D., Lee, G., and Lee, J. S. (2017). Screening of oxygen-reduction-reaction-efficient electrocatalysts based on Ag-M (M = 3d, 4d, and 5d transition metals) nanoalloys: a density functional theory study. Energy Fuels 31, 1874–1881. doi: 10.1021/acs.energyfuels.6b02991
Chen, D., Rui, X., Zhang, Q., Geng, H., Gan, L., Zhang, W., et al. (2019a). Persistent zinc-ion storage in mass-produced V2O5 architectures. Nano Energy 60, 171–178. doi: 10.1016/j.nanoen.2019.03.034
Chen, D., Tan, H., Rui, X., Zhang, Q., Feng, Y., Geng, H., et al. (2019b). Oxyvanite V3O5: a new intercalation-type anode for lithium-ion battery. InfoMat. 1, 251–259. doi: 10.1002/inf2.12011
Debe, M. K. (2012). Electrocatalyst approaches and challenges for automotive fuel cells. Nature 486, 43–51. doi: 10.1038/nature11115
Delley, B. (1990). An all-electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 92, 508–517. doi: 10.1063/1.458452
Delley, B. (2000). From molecules to solids with the DMol3 approach. J. Chem. Phys. 113, 7756–7764. doi: 10.1063/1.1316015
Deng, C., He, R., Shen, W., Li, M., and Zhang, T. (2019). Single-atom catalyst of cobalt supported on defective two-dimensional boron nitride material as a promising electrocatalyst for oxygen reduction reaction: a DFT study Phys. Chem. Chem. Phys. 21, 6900–6907. doi: 10.1039/C9CP00452A
Deng, C., He, R., Wen, D., Shen, W., and Li, M. (2018). Theoretical study on the origin of activity for oxygen reduction reaction of metal doped two-dimensional boron nitride materials. Phys. Chem. Chem. Phys. 20, 10240–10246. doi: 10.1039/C8CP00838H
Deng, Y. J., Tripkovic, V., Rossmeisl, J., and Arenz, M. (2015). Oxygen reduction reaction on Pt overlayers deposited onto a gold film: ligand, strain, and ensemble effect. ACS Catal. 6, 671–676. doi: 10.1021/acscatal.5b02409
Du, W., Geng, H., Yang, Y., Zhang, Y., Rui, X., and Li, C. C. (2019). Pristine graphene for advanced electrochemical energy applications. J. Power Sources 437:226899. doi: 10.1016/j.jpowsour.2019.226899
Duan, Z., and Wang, G. (2011). A first principles study of oxygen reduction reaction on a Pt (111) surface modified by a subsurface transition metal M (M = Ni, Co, or Fe). Phys. Chem. Chem. Phys. 13, 20178–20187. doi: 10.1039/c1cp21687b
Elumalai, G., Noguchi, H., Lyalin, A., Taketsugu, T., and Uosaki, K. (2016). Gold nanoparticle decoration of insulating boron nitride nanosheet on inert gold electrode toward an efficient electrocatalyst for the reduction of oxygen to water. Electrochem. Commun. 66, 53–57. doi: 10.1016/j.elecom.2016.02.021
Feng, L., Liu, Y., and Zhao, J. (2015). Iron-embedded boron nitride nanosheet as a promising electrocatalyst for the oxygen reduction reaction (ORR): a density functional theory (DFT) study. J. Power Sources 287, 431–438. doi: 10.1016/j.jpowsour.2015.04.094
Gao, M., Lyalin, A., and Taketsugu, T. (2012). Catalytic activity of Au and Au2 on the h-BN Surface: adsorption and activation of O2. J. Phys. Chem. C 116, 9054–9062. doi: 10.1021/jp300684v
Govind, N., Petersen, M., Fitzgerald, G., King-Smith, D., and Andzelm, J. (2003). A generalized synchronous transit method for transition state location. Comp. Mater. Sci. 28, 250–258. doi: 10.1016/S0927-0256(03)00111-3
Hirshfeld, F. L. (1977). Bonded-atom fragments for describing molecular charge densities. Theoret. Chim. Acta 44, 129–138. doi: 10.1007/BF00549096
Kobayashi, H., Teranishi, M., Negishi, R., Naya, S. I., and Tada, H. (2016). Reaction mechanism of the multiple-electron oxygen reduction reaction on the surfaces of gold and platinum nanoparticles loaded on titanium(IV) oxide. J. Phys. Chem. Lett. 7, 5002–5007. doi: 10.1021/acs.jpclett.6b02026
Kodama, K., Jinnouchi, R., Takahashi, N., Murata, H., and Morimoto, Y. (2016). Activities and stabilities of Au-modified stepped-Pt single-crystal electrodes as model cathode catalysts in polymer electrolyte fuel cells. J. Am. Chem. Soc. 138, 4194–4200. doi: 10.1021/jacs.6b00359
Koelling, D. D., and Harmon, B. N. (1977). A technique for relativistic spin-polarised calculations. J. Phys. C 10, 3107–3114. doi: 10.1088/0022-3719/10/16/019
Li, L., Yu, X., Yang, X., Fang, Y., Zhang, X., Xu, X., et al. (2016). Porous BN with vacancy defects for selective removal of CO from H2 feed gas in hydrogen fuel cells: a DFT study. J. Mate. Chem. A 4, 15631–15637. doi: 10.1039/C6TA03208G
Li, Q., Liu, Y., Yu, X., Li, L., Zhang, X., Lu, Z., et al. (2018). Removal of Cr(iii)/Cr(vi) from wastewater using defective porous boron nitride: a DFT study. Inorg.Chem. Front. 5, 1933–1940. doi: 10.1039/C8QI00416A
Lin, C. (2002). Characterization of boron-nitride-supported Pt catalysts for the deep oxidation of benzene. J. Catal. 210, 39–45. doi: 10.1006/jcat.2002.3638
Lin, S., Ye, X., Johnson, R. S., and Guo, H. (2013). First-principles investigations of metal (Cu, Ag, Au, Pt, Rh, Pd, Fe, Co, and Ir) doped hexagonal boron nitride nanosheets: stability and catalysis of CO oxidation. J. Phys. Chem. C 117, 17319–17326. doi: 10.1021/jp4055445
Lyalin, A., Uosaki, K., and Taketsugu, T. (2017). Oxygen reduction reaction catalyzed by small gold cluster on h-BN/Au(111) support. Electrocatalysis 9, 182–188. doi: 10.1007/s12678-017-0395-5
Nie, Y., Li, L., and Wei, Z. (2015). Recent advancements in Pt and Pt-free catalysts for oxygen reduction reaction. Chem. Soc. Rev. 44, 2168–2201. doi: 10.1039/C4CS00484A
Nørskov, J. K., Rossmeisl, J., Logadottir, A., and Lindqvist, L. (2004). Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 108, 17886–17892. doi: 10.1021/jp047349j
Ostojic, N., Duan, Z., Galyamova, A., Henkelman, G., and Crooks, R. M. (2018). Electrocatalytic study of the oxygen reduction reaction at gold nanoparticles in the absence and presence of interactions with SnO-x supports. J. Am. Chem. Soc. 140, 13775–13785. doi: 10.1021/jacs.8b08036
Pakdel, A., Bando, Y., and Golberg, D. (2014). Nano boron nitride flatland. Chem. Soc. Rev. 43, 934–959. doi: 10.1039/C3CS60260E
Peng, Y., Lu, B., and Chen, S. (2018). Carbon-supported single atom catalysts for electrochemical energy conversion and storage. Adv. Mater. 30:e1801995. doi: 10.1002/adma.201801995
Peng, Y., Lu, B., Wang, N., Lu, J. E., Lic, C., Ping, Y., et al. (2019). Oxygen reduction reaction catalyzed by black-phosphorus-supported metal nanoparticles: Impacts of interfacial charge transfer. ACS Appl. Mater. Interfaces 11, 24707–24714. doi: 10.1021/acsami.9b05471
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized gradient approximation made simple. Phy. Rev. Lett. 77, 3865–3868. doi: 10.1103/PhysRevLett.77.3865
Peterson, A. A., Abild-Pedersen, F., Studt, F., Rossmeisl, J., and Nørskov, J. K. (2010). How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 3, 1311–1315. doi: 10.1039/c0ee00071j
Pulay, P. (1982). Improved SCF convergence acceleration. J. Comput. Chem. 3, 556–560. doi: 10.1002/jcc.540030413
Sharma, S., and Pollet, B. G. (2012). Support materials for PEMFC and DMFC electrocatalysts—A review. J. Power Sources 208, 96–119. doi: 10.1016/j.jpowsour.2012.02.011
Song, W., and Hensen, E. J. M. (2013). Structure sensitivity in CO oxidation by a single Au atom supported on ceria. J. Phys. Chem. C 117, 7721–7726. doi: 10.1021/jp400977m
Sun, W., Meng, Y., Fu, Q., Wang, F., Wang, G., Gao, W., et al. (2016). High-yield production of boron nitride nanosheets and its uses as a catalyst support for hydrogenation of nitroaromatics. ACS appl. Mater. Interfaces 8, 9881–9888. doi: 10.1021/acsami.6b01008
Tak, Y. J., Yang, S., Lee, H., Lim, D. H., and Soon, A. (2018). Examining the rudimentary steps of the oxygen reduction reaction on single-atomic Pt using Ti-based non-oxide supports. J. Ind. Eng. Chem. 58, 208–215. doi: 10.1016/j.jiec.2017.09.027
Tang, Q., Bao, J., Li, Y., Zhou, Z., and Chen, Z. (2014). Tuning band gaps of BN nanosheets and nanoribbons via interfacial dihalogen bonding and external electric field. Nanoscale 6, 8624–8634. doi: 10.1039/C4NR00008K
Tian, Z., Sun, J., Wang, S., Zeng, X., Zhou, S., Bai, S., et al. (2018). A thermal interface material based on foam-templated three-dimensional hierarchical porous boron nitride. J. Mater. Chem. A, 6, 17540–17547. doi: 10.1039/C8TA05638B
Tran-Thuy, T.-M., Chen, C.-C., and Lin, S. D. (2017). Spectroscopic studies of how moisture enhances CO oxidation over Au/BN at ambient temperature. ACS Catal. 7, 4304–4312. doi: 10.1021/acscatal.7b01374
Uosaki, K., Elumalai, G., Noguchi, H., Masuda, T., Lyalin, A., Nakayama, A., et al. (2014). Boron nitride nanosheet on gold as an electrocatalyst for oxygen reduction reaction: theoretical suggestion and experimental proof. J. Am. Chem. Soc. 136, 6542–6545. doi: 10.1021/ja500393g
Wan, J., Chen, W., Jia, C., Zheng, L., Dong, J., Zheng, X., et al. (2018). Defect effects on TiO2 nanosheets: Stabilizing single atomic site Au and promoting catalytic properties. Adv. Mater. 30:1705369. doi: 10.1002/adma.201705369
Wang, Y., Yuan, H., Li, Y., and Chen, Z. (2015). Two-dimensional iron-phthalocyanine (Fe-Pc) monolayer as a promising single-atom-catalyst for oxygen reduction reaction: a computational study. Nanoscale 7, 11633–11641. doi: 10.1039/C5NR00302D
Weng, Q., Wang, X., Zhi, C., Bando, Y., and Golberg, D. (2013). Boron nitride porous microbelts for hydrogen storage. Acs Nano 7, 1558–1565. doi: 10.1021/nn305320v
Xiao, B., Zhu, H., Liu, H., Jiang, X., and Jiang, Q. (2018). The activity improvement of the TM3(hexaiminotriphenylene)2 monolayer for oxygen reduction electrocatalysis: A density functional theory study. Front. Chem. 6:351. doi: 10.3389/fchem.2018.00351
Xue, Q., Bai, J., Han, C., Chen, P., Jiang, J. X., and Chen, Y. (2018). Au nanowires@Pd-polyethylenimine nanohybrids as highly active and methanol-tolerant electrocatalysts toward oxygen reduction reaction in alkaline media. ACS Catal. 8, 11287–11295. doi: 10.1021/acscatal.8b03447
Yang, X. F., Wang, A., and Qiao, B. (2013). Single-atom catalysts: a new frontier in heterogeneous catalysis. Acc. Chem. Res. 46, 1740–1748. doi: 10.1021/ar300361m
Yang, Y., Dai, C., Fisher, A., Shen, Y., and Cheng, D. (2017). A full understanding of oxygen reduction reaction mechanism on Au(111) surface. J. Phys. 29:365201. doi: 10.1088/1361-648X/aa7db6
Ye, J., Zhu, X., Cheng, B., Yu, J., and Jiang, C. (2016). Few-layered graphene-like boron nitride: a highly efficient adsorbent for indoor formaldehyde removal. Environ. Sci. Technol. Lett. 4, 20–25. doi: 10.1021/acs.estlett.6b00426
Keywords: porous boron nitride (p-BN), isolated Au atom, catalysis, oxygen reduction reaction (ORR), density functional theory (DFT)
Citation: Li Q, Zhang T, Yu X, Wu X, Zhang X, Lu Z, Yang X, Huang Y and Li L (2019) Isolated Au Atom Anchored on Porous Boron Nitride as a Promising Electrocatalyst for Oxygen Reduction Reaction (ORR): A DFT Study. Front. Chem. 7:674. doi: 10.3389/fchem.2019.00674
Received: 30 August 2019; Accepted: 25 September 2019;
Published: 17 October 2019.
Edited by:
Xiaopeng Han, Tianjin University, ChinaReviewed by:
Tianyi Ma, University of Newcastle, AustraliaXianhong Rui, Guangdong University of Technology, China
Li Li, Chongqing University, China
Copyright © 2019 Li, Zhang, Yu, Wu, Zhang, Lu, Yang, Huang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaojing Yang, eWFuZ3hpYW9qaW5nQGhlYnV0LmVkdS5jbg==; Yang Huang, aHVhbmd5YW5nQGhlYnV0LmVkdS5jbg==; Lanlan Li, bGlhYmNAaGVidXQuZWR1LmNu