 Stefania Gambaro
Stefania Gambaro Pellegrino La Manna
Pellegrino La Manna Margherita De Rosa*
Margherita De Rosa* Annunziata Soriente
Annunziata Soriente Carmen Talotta*
Carmen Talotta* Carmine Gaeta
Carmine Gaeta Placido Neri
Placido Neri- Laboratory of Supramolecular Chemistry, Dipartimento di Chimica e Biologia “A. Zambelli”, Università degli Studi di Salerno, Salerno, Italy
Herein, we show that the hexameric resorcinarene capsule C is able to catalyze the formation of bis(heteroaryl)methanes by reaction between pyrroles or indoles and carbonyl compounds (α-ketoesters or aldehydes) in excellent yields and selectivity. Our results suggest that the capsule can play a double catalytic role as a H-bond catalyst, for the initial activation of the carbonyl substrate, and as a Brønsted acid catalyst, for the dehydration of the intermediate alcohol.
Introduction
Supramolecular organocatalysis is an emerging area in supramolecular chemistry whose principal aim is the design of novel systems able to perform catalytic functions mimicking the chemo-, regio-, and stereoselectivity of the natural enzymes (Conn and Rebek, 1997). At this regard, much attention has been focused on designing self-assembled molecular capsules (MCs) able to catalyze organic reaction by confinement of the reactants in their internal cavity (Borsato and Scarso, 2016; Catti et al., 2016; Gaeta et al., 2019). MCs are self-assembled structures sealed by weak non-covalent interactions between the single complementary units. Resembling to an enzyme pocket, the nanoconfined space inside a self-assembled molecular capsule allows the formation of a microenvironment with different physical and chemical features with respect to the external medium. In fact, the nanoconfinement of the reactants inside a MC slows down their molecular mobility determining a different stereo- and regiochemical outcome of the reaction with respect to the bulk conditions. Analogously to the natural systems, when the reactants are hosted inside a MC, the proximity effect between them and the stabilization of the intermediates and transition states induces a reaction acceleration.
Interestingly, Atwood and MacGillivray reported an interesting example of self-assembled capsule C (1)6·(H2O)8 (Figure 1; MacGillivray and Atwood, 1997), which is constituted by six resorcin[4]arene units 1 sealed by eight water molecules, and shows an hydrophobic cavity with an internal volume of 1,375 Å3. The six resorcinarene units and the eight water molecules are located, respectively, on the sides and on the corners of a cube, and the aggregate is sealed by 60 (O-H….O) hydrogen bonding interactions. The 8 bridged-water molecules establish H-bonds with the adjacent resorcinol OH groups and, in particular, four of them act as double H-bonds donor (Figure 1, H2O drawing in red) and single H-bond acceptor, saturating in this way their H-bonding valence. The other four bridged-water molecules act as single H-bond acceptor and single H-bond donor (Figure 1, blu), remaining with one H-bond donating free valence. Cohen et al. (Avram and Cohen, 2002b) demonstrated by NMR diffusion experiments, that the capsule C is self–assembled also in solution when water-saturated chloroform or benzene is used as a solvent.
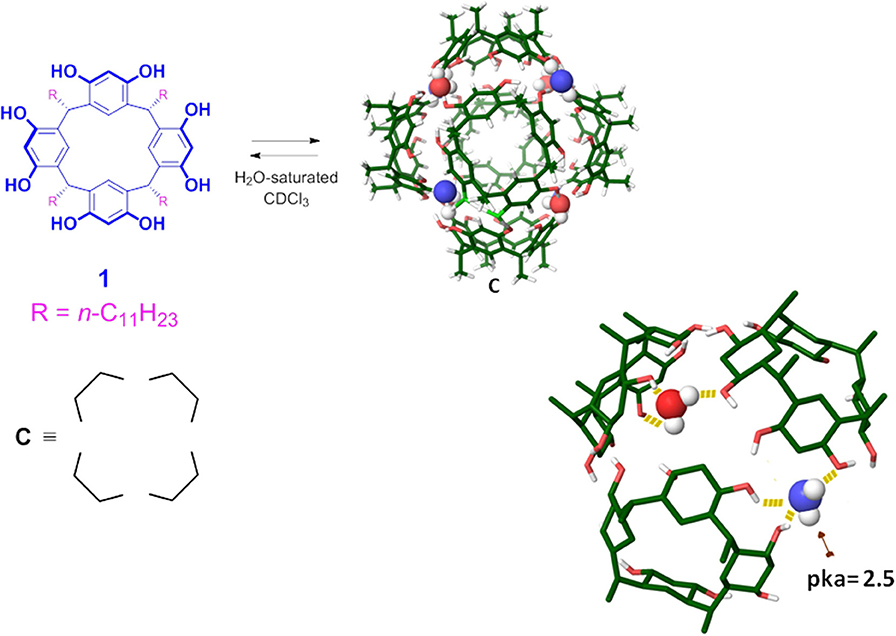
Figure 1. Chemical drawing of the C-undecyl-resorcin[4]arene 1. Tube model of the hexameric capsule C, the undecyl chains have been omitted for clarity. Chemical drawing of the model representing the hydrogen bond belt between the eight bridged water molecules and the six resorcinarene molecules, in blue the bridging water molecule with one H-bond donating free valence.
The capsule C is able to accommodate eight benzene (or chloroform) molecules inside its cavity (Avram and Cohen, 2002a,b, 2004; Shivanyuk and Rebek, 2003). Numerous studies showed that C is also able to host in its π-electron rich cavity, complementary guests by H-bonding and/or cation–π interactions (Shivanyuk and Rebek, 2001; Avram and Cohen, 2002a; Yamanaka et al., 2004; Evan-Salem et al., 2006). Tiefenbacher et al. demostrated that C behaves as a Brønsted acid (Zhang and Tiefenbacher, 2013; Köster and Tiefenbacher, 2018). In particular, their studies revealed that the hexameric aggregate has an estimated pKa value of about 5.5–6.0, a value certainly not comparable with that of the single resorcinarene unit. The acidic behavior of C is explained by the stabilization of its conjugate-base due to the delocalization of its negative charge over the phenolic groups and water molecules of the assembly. QM calculations, recently reported by our group (La Manna et al., 2018b) estimated a local pKa of ≈2.5 for the bridged-water molecules with one H-bond donating free valence (in blue in Figure 1), while the mean pKa value of all OH groups of C is 6.1, in agreement with the experimental datum.
Several reports clearly show that the mild Brønsted acidity of C and its ability to stabilize cationic transition states, are crucial factors for the catalytic activity of the capsule (Borsato and Scarso, 2016; Catti et al., 2016; Gaeta et al., 2019). Thus, amazing results have been reported in the last decade regarding the catalysis of chemical reactions into the nanoconfined space of the self-assembled capsule C, including the cyclization of terpenes (Zhang and Tiefenbacher, 2015, 2019; Zhang et al., 2017, 2018, 2019; Pahima et al., 2019), the hydration of the alkynes (La Sorella et al., 2016a), the carbonyl-olefin metathesis (Catti and Tiefenbacher, 2018), the sulfoxidation of thioethers (La Sorella et al., 2016b), the synthesis of substituted 1-H-tetrazoles (Giust et al., 2015), the activation of C-F bonds (Köster et al., 2019), and the iminium catalysis (Bräuer et al., 2017; La Manna et al., 2018a). Recently, we showed that the capsule C acts as a nanoreactor for a Friedel-Crafts alkylation of arenes and heteroarenes with benzyl chloride (La Manna et al., 2018b) under mild metal-free conditions. We showed that the bridged-water molecules with one H-bond donating free valence exert a crucial role in the activation of the C-Cl bond of benzyl chloride by H-bonding interaction. Analogously, the H-bond donor abilities of the water molecules of C have been exploited in the activation of β-nitrostyrenes toward the Michael reaction using pyrroles and indoles as nucleophiles (Gambaro et al., 2019).
As a part of our research program focused on the extension of the catalytic opportunities offered by the hexameric capsule C, we turned our attention to the synthesis of bis(heteroaryl)methanes (BHM) (Palmieri et al., 2010; Shiri et al., 2010; Shiri, 2012). BHM are fundamental building blocks in the synthesis of natural and unnatural porphyrin derivatives (Cho and Lee, 1998; Burrell et al., 2001; Laha et al., 2003). Moreover, they find applications in several fields, ranging from medicine (Sivaprasad et al., 2006; Awuah and You, 2012; Josefsen and Boyle, 2012) to environment and industry (Kursunlu et al., 2012). In particular, bis(indol)methanes (BIM) and bis(pyrrole)methanes, containing two simple or two substituted heteroaryl moieties are molecules with interesting biological properties (Sakemi and Sun, 1991; Gunasekera et al., 1994; Fürstner, 2003; Bao et al., 2005). This class of products is generally obtained by means of strategies relying upon the use of Brønsted (Palmieri et al., 2010; Shiri et al., 2010; Shiri, 2012) and Lewis acids (Ji et al., 2004; Guo et al., 2009; Ling et al., 2019; Qiang et al., 2019; Wu et al., 2019), strong Brønsted acids (Biaggi et al., 2006; Singh et al., 2011; Lucarini et al., 2013; Norouzi et al., 2018; Tran et al., 2018), and electrochemical methods (Du and Huang, 2018).
Results and Discussion
Prompted by these considerations and considering our interest in the development of novel organocatalytic strategies, we attempted the synthesis of BHMs derivatives by reaction between aromatic heterocycles and aldehydes and pyruvates in the presence of capsule C as a Brønsted acid catalyst. At this regard, as a model reaction for investigating the catalytic performance of C, we chose the reaction between pyrrole 2a and ethyl pyruvate 3a in Table 1.
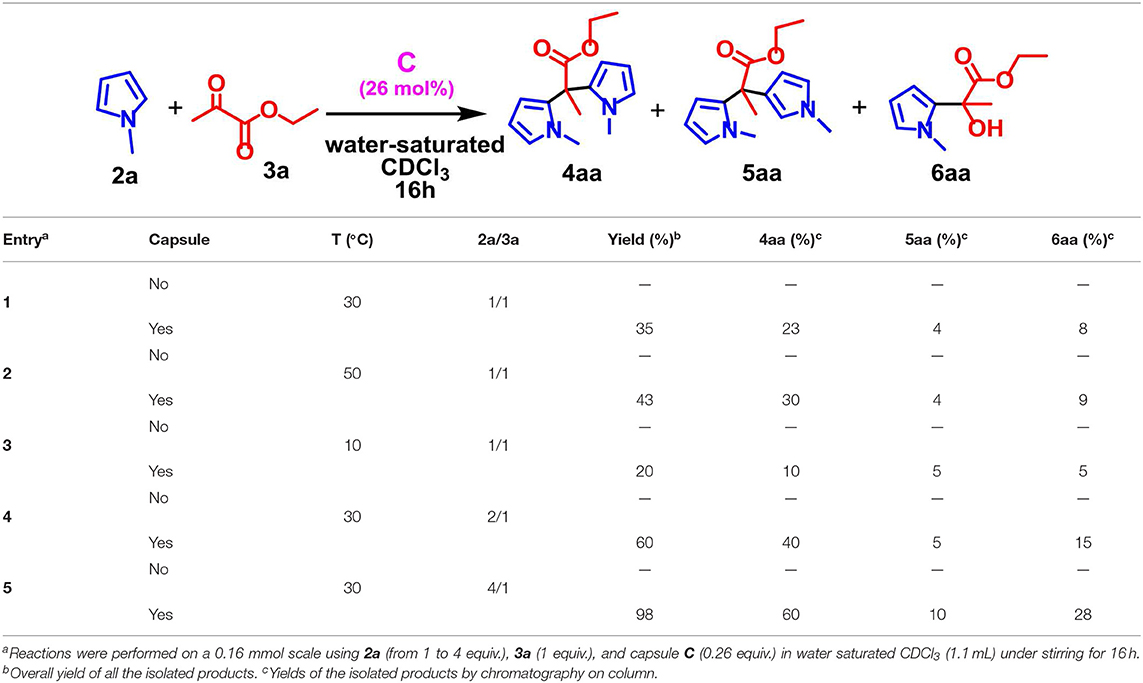
Table 1. Optimization of reaction conditions for the synthesis of BHMs catalyzed by C.
We started performing the reaction in Table 1 in the presence of capsule C in water-saturated CDCl3 at 30°C and with a 1/1 ratio of 2a/3a. It was found that the reaction proceeded smoothly to afford preferentially meso-α,α-substituted dipyrromethane 4aa in 23% yield, accompanied by a negligible amount of α,β-linked dipyrromethane 5aa and monoalkylated adduct 6aa (entry 1, Table 1). No evidence was detected of higher oligomers and other side products. In contrast, when the reaction in Table 1 was carried out under the same reaction conditions but in the absence of capsule C, no products could be evidenced (entry 1, Table 1). This result encouraged us to carry out a study for the optimisation of the reaction parameters in order to improve the reaction efficiency.
Initially, the influence of the reaction temperature was investigated (Table 1, entries 1–3). When the temperature was decreased to 10°C, both reaction efficiency and selectivity dropped (entry 3, Table 1), while an increase in the temperature had a little positive effect on the reaction outcome (entry 4, Table 1). Next, we moved to examine the molar ratio of 2a/3a on the yield of the reaction in Table 1. When an excess of 2a was used, an increase of the reaction efficiency in terms of yield was observed while keeping the selectivity for the adducts substantially unchanged, with the preferential formation of 4aa (entries 4–5, Table 1). These preliminary results indicated that capsule C was capable to promote the reaction in selective and efficient way and suggested that the reaction took place inside the cavity of C.
In order to confirm this conclusion, and in accord to a protocol previously reported by us and other groups (Bräuer et al., 2017; La Manna et al., 2018a), we performed a series of control experiments. In details, when the reaction between 2a and 3a was conducted under the conditions reported in Table 1 in the presence of C and of tetraethylammonium tetrafluoroborate, which is a known competitive guest, no hint of products were detected after 16 h at 50°C. Under these conditions, the ammonium guest occupying the cavity of capsule C acts as an inhibitor. In addition, the 1H NMR spectrum of the reaction mixture in the presence of tetraethylammonium tetrafluoroborate in Figure S3 featured shielded signals at negative chemical shifts values attributable to the cation inside the cavity of C. Finally, no hint of products was observed when the reaction reported in Table 1 was performed in the presence of DMSO (Figure S4), a hydrogen-bonding competitor solvent able to disaggregate the capsule C.
With these results in hand, we next studied the generality of the reaction with regard to both reactants (Table 2). Initially, we evaluated the influence of the α-ketoester structure on the reaction outcome. When α-ketoester 3c, bearing an isopropyl group, was reacted with 2a in the presence of C (26 mol%), the formation of the mono-alkylated adduct 6ac was observed with a yield of 55% (entry 3, Table 2), while no hint of other products was detected. Interestingly, under analogous conditions the α-ketoester 3b (R = Me) reacted with 2a giving the meso-dipyrromethane product 4ab (entry 2, Table 2) in 90% yield. Probably, by increasing the steric encumbrance of the R group of 3 from methyl (3b) to isopropyl (3c) the formation of the di-pyrromethane was hindered. When 3d (entry 4, Table 2), bearing a benzyloxy group, was used as substrate alongside 2a, then the formation of the double alkylated adducts α,α and α,β 4ad and 5ad was observed in a 1/1 ratio and with a complete loss of selectivity. Differently, using 3b (entry 2, Table 2) only the α,α adduct 4ab was obtained. Interestingly, when 3f bearing an electron-withdrawing trifluoromethyl group was used, the reaction in Table 2 was almost quantitative displaying a complete selectivity for the mono-alkylated adduct 6af and no evidence of bis-adduct or other side products (entry 6, Table 2). Finally, with α-ketoacid 3e no reaction took place and a decarboxylate product was recovered.
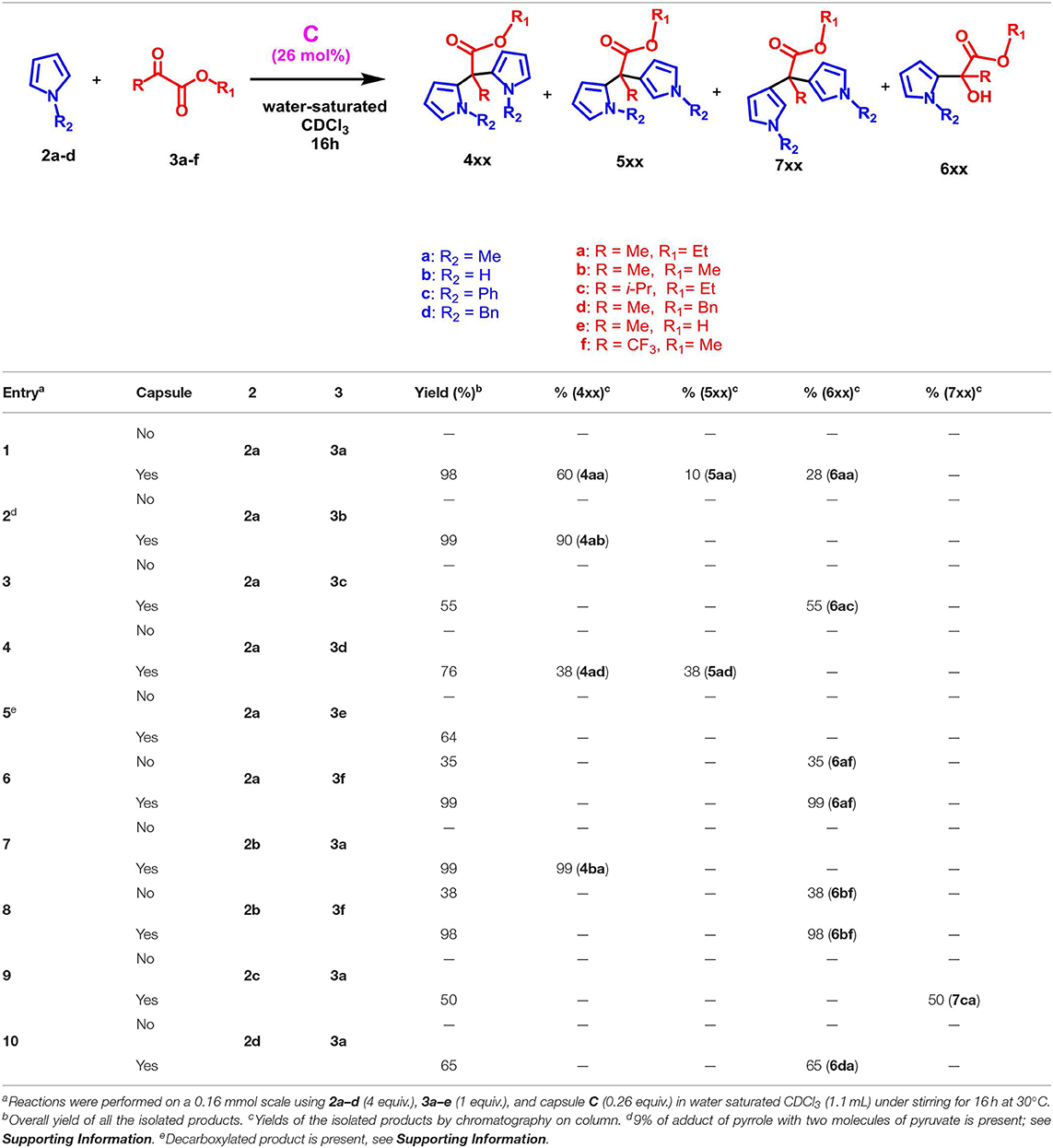
Table 2. Scope of the reaction between different pyrroles 2a–d and α-ketoesters 3a–f.
At this point, we examined effect of the substitution at the pyrrole nitrogen atom on the reaction outcome. The reaction between pyrrole 2b and 3a selectively delivered the meso bis-adduct 4ba in high yield (entry 7, Table 2). Even with pyrrole 2b, the reaction with 3f afforded to mono-adduct 6bf as the only reaction product (entry 8, Table 2), indicating that the choice of the ketoester influenced the outcome of the reaction.
When a more sterically demanding group was introduced on the nitrogen atom of pyrrole, the yield of the reaction in Table 2 decreased and the selectivity of the products was influenced. In fact, when pyrrole 2c, bearing a N-benzyl group, was used with 3a under the conditions reported in Table 2, then the mono-adduct 6da was obtained selectively and in good yield (entry 10, Table 2), whereas with N-phenyl pyrrole 2d we observed for the first time the selective formation of a β, β-di-adduct (7ca) (entry 9, Table 2). When the reaction was performed using indole derivatives (Table 3), only the formation of di-pyrromethane β, β-9 was observed in high yield independently of the substituents present on the benzene and pyrrole rings.
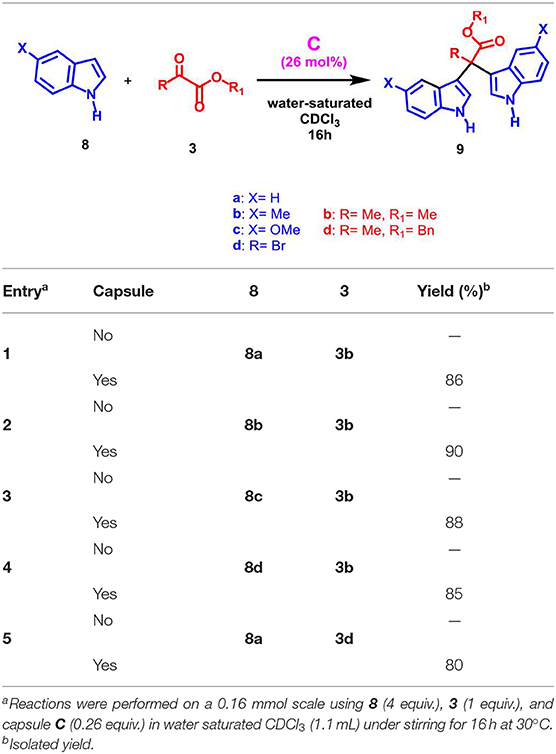
Table 3. Scope of the reaction with different indoles.
The mechanism proposed for the formation of α,α-substituted dipyrromethane 4xx and monoalkylated adduct 6xx in the nanoconfined space inside the capsule C, is outlined in Scheme 1. In detail, α-ketoester 3 is probably stabilized inside the capsule C through the formation of a H-bonding interaction with a bridged water molecule (Scheme 1).
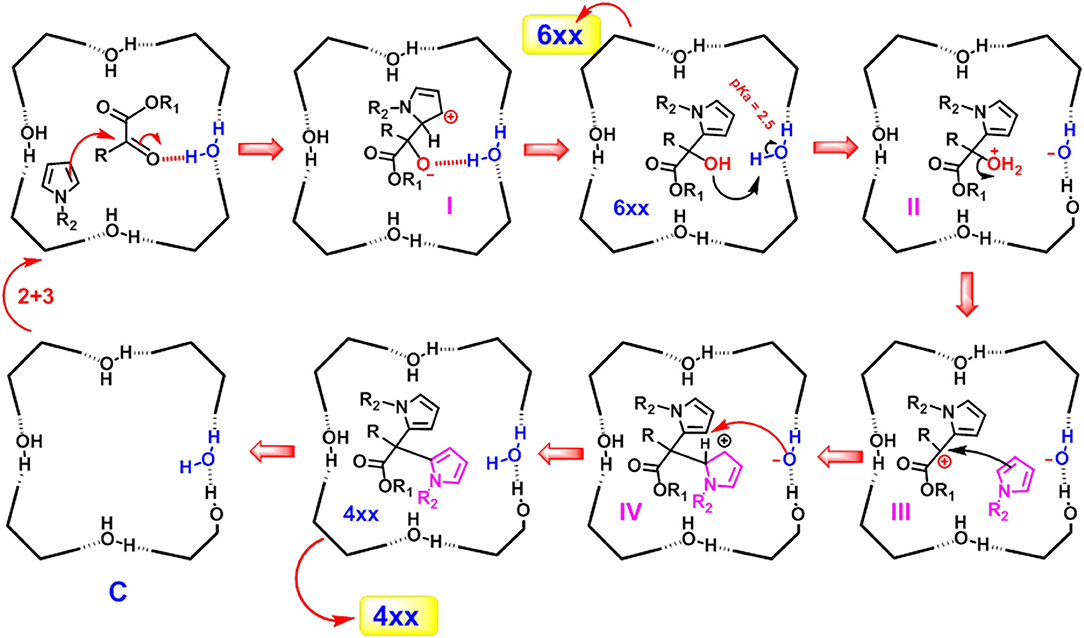
Scheme 1. Mechanism proposed for the formation of the products 4xx and 6xx in the nano-confined space inside the cavity of C.
Previously, we have already shown that pyrrole derivatives are hosted inside the cavity of C (La Manna et al., 2018b). At this point, an α-attack of pyrrole to the activated ketone group of 3 occurs inside the capsule, leading to intermediate I (Scheme 1) stabilized through H-bonding and cation···π interactions, which is re-aromatizated to 6xx. On the basis of the local acidity (pKa of ≈ 2.5) of the bridged water molecules with H-bond donating free valence, the product 6xx can be protonated inside the capsule C (II in Scheme 1) and converted to carbocation III by losing a water molecule. III undergoes an α-attack of a new pyrrole molecule to give the carbocation IV which is stabilized by cation···π interactions. This latter is rearomatizated to 4xx, by losing the β-proton and recovering the electroneutrality of the capsule C. The mechanism proposed in Scheme 1 is corroborated by the finding that α-ketoester 3f, bearing an electron-withdrawing trifluoromethyl moiety in α-position to ketone group, in the presence of C and 2a or 2b gives the mono-alkylated adduct 6af and 6bf in almost quantitative yields, while no evidence of di-adduct was detected. Probably, under these conditions, the presence of the electron-withdrawing trifluoromethyl group disfavours the formation of carbocation IV, which would have a positive charge on the carbon atom directly bonded to the electron-withdrawing trifluoromethyl group.
On the basis of these results and in order to extend the scope of the reaction between 2 and carbonyl compounds in the presence of C, we studied the procedure with a different carbonyl compound such as benzaldehyde 10a (Table 4). When the substrates 2a and 10a were mixed in 1/1 ratio in the presence of C in water-saturated CDCl3 then α,α-dipyrromethane 11a was obtained in 34% yield with a regioselectivity ratio of 8.5/1 (entry 1, Table 4) with respect to the α,β-isomer 12a. Interestingly, when the 2a/10a molar ratio was progressively increased to 2/1 and to 4/1 then the efficiency of the reaction was improved with a 54 and 87% yield of 11a, respectively (entries 2 and 3, Table 4). Interestingly, no hint of product 11a and 12a were detected in the reaction mixture in the absence of capsule C. The lowering of the reaction temperature from 50 to 25°C (entry 4 in Table 4) gives rise to a drop in the yield of 11a. Once the reaction conditions were optimized (Table 4), the substrate scope was then evaluated in order to determine the generality of the reaction.
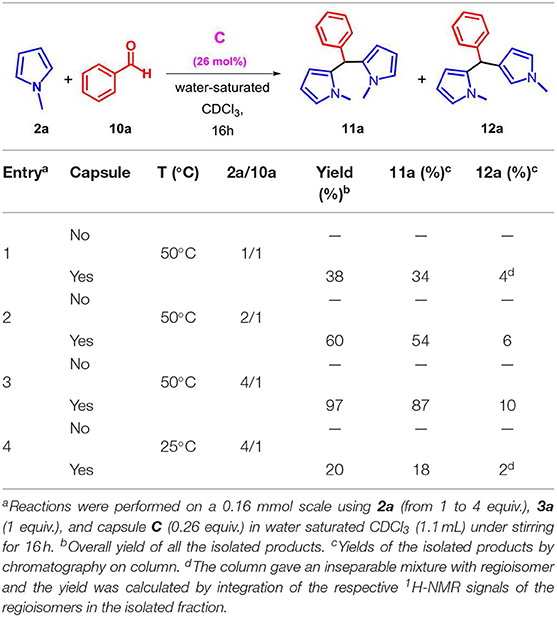
Table 4. Optimization of reaction conditions for the reaction between 2a and 10a.
As regards the effect of the substitution at the pyrrole nitrogen atom, we found that the introduction of a more hindering group, such as a phenyl or benzyl group, caused a complete loss of reactivity (entries 2–3, Table 5). Instead, the reaction with unsubstituted pyrrole 2b proceeded with a small decrease in yield but preserving the selectivity for adduct 11a (entry 1, Table 5). Interestingly, under the conditions reported in Table 5 no hint of mono-adduct heteroaryl methane was observed. Successively, we investigated the generality of the reaction between 2a and several aromatic aldehydes bearing electron-donating or -withdrawing groups (Table 5).
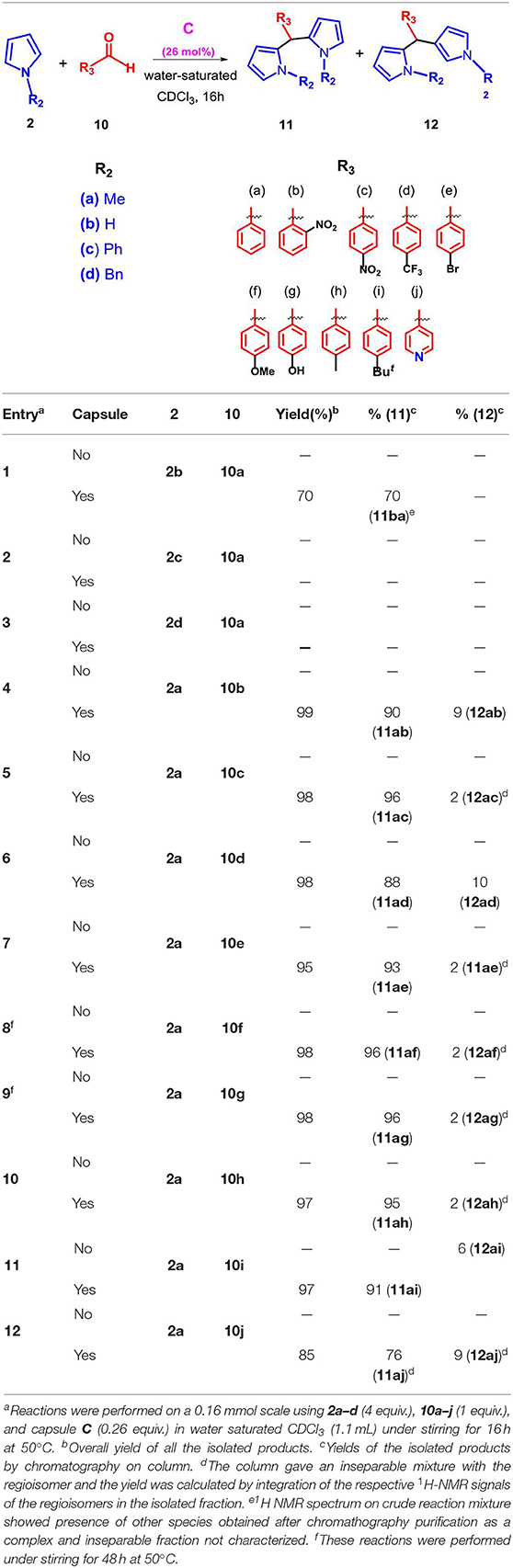
Table 5. Scope of the reaction with different pyrroles 2a–d and aldehydes 10a–j.
The protocol was found to be tolerant to a variety of aromatic aldehydes 10a–j, independently by the electronic nature and position of the substituents on the aryl group, affording α,α-adducts 11 in high yields and excellent regioselectivities. In fact, the double attack took place in a completely regioselective way to give 11 as almost the only product with a negligible amount of the corresponding isomer 12. No evidence of monoalkylated adduct was observed. Additionally, when the protocol was extended to the N-methyl indole 8e, the reaction proceeded smoothly and the adduct 13 was obtained as the only product in high yield (Table 6).
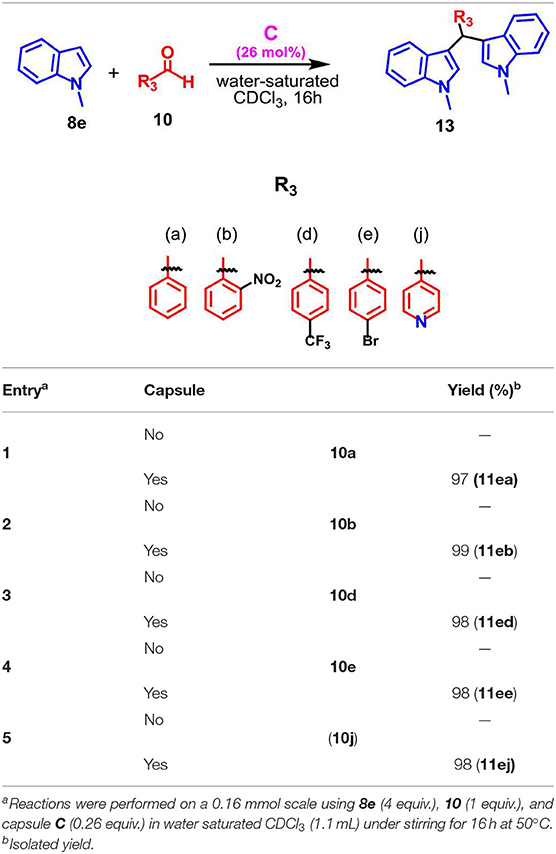
Table 6. Scope of the reaction between indole 8e and various aldehydes 10a, b, d, e, j.
Conclusions
The resorcinarene hexameric capsule C is able to catalyze the reaction between pyrroles or indoles and α-ketoesters or aldehydes for the formation of bis(heteroaryl)methanes. The reactions take place in the nanoconfined space inside the capsule C. The observed results suggested its double catalytic function: C can act as H-bond catalyst for the initial activation of the carbonyl functions and as a Brønsted acid catalyst for the dehydration of the intermediate alcohol. Generally, in the presence of C the formation of the α,α-bis(heteroaryl)methanes occurs with excellent yields and regioselectivity with respect to the α,β- or β,β-regioisomers.
Data Availability Statement
All experimental data are reported in the Supplementary Material.
Author Contributions
SG and PL performed the experiments. CT performed NMR studies. CG, AS, and PN participated in manuscript preparation. CT and MD prepared the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank the Regione Campania (POR CAMPANIAFESR 2007/2013 O.O.2.1, CUP B46D14002660009) for the FT-ICR mass spectrometer facilities, the Farma-BioNet (CUPB25C13000230007), and the Centro di Tecnologie Integrate per la Salute(CITIS) (project PONa3_00138) for the 600 MHz NMR facilities. Financial support is acknowledged from the Università di Salerno (FARB 2017 Composti Macrociclici come Organocatalizzatori Supramolecolari in Catalisi Biomimetica).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00687/full#supplementary-material
References
Avram, L., and Cohen, Y. (2002a). The role of water molecules in a resorcinarene capsule as probed by NMR diffusion measurements. Org. Lett. 4, 4365–4368. doi: 10.1021/ol0271077
Avram, L., and Cohen, Y. (2002b). Spontaneous formation of hexameric resorcinarene capsule in chloroform solution as detected by diffusion NMR. J. Am. Chem. Soc. 124, 15148–15149. doi: 10.1021/ja0272686
Avram, L., and Cohen, Y. (2004). Self-recognition, structure, stability, and guest affinity of pyrogallol[4]arene and resorcin[4]arene capsules in solution. J. Am. Chem. Soc. 126, 11556–11563. doi: 10.1021/ja047698r
Awuah, S. G., and You, Y. (2012). Boron dipyrromethene (BODIPY)-based photosensitizers for photodynamic therapy. RSC Adv. 2, 11169–11183. doi: 10.1039/c2ra21404k
Bao, B., Sun, Q., Yao, X., Hong, J., Lee, C. O., Sim, C., et al. (2005). Cytotoxic bisindole alkaloids from a marine sponge Spongosorites sp. J. Nat. Prod. 68, 711–715. doi: 10.1021/np049577a
Biaggi, C., Benaglia, M., Raimondi, L., and Cozzi, F. (2006). Organocatalytic synthesis of dipyrromethanes by the addition of N-methylpyrrole to aldehydes. Tetrahedron 62, 12375–12379. doi: 10.1016/j.tet.2006.09.104
Borsato, G., and Scarso, A. (2016). “Catalysis within the Self-Assembled Resorcin[4]arene Hexamer,” in Organic Nanoreactors, ed S. Sadjadi (London: Academic Press), 203–234. doi: 10.1016/B978-0-12-801713-5.00007-0
Bräuer, T. M., Zhang, Q., and Tiefenbacher, K. (2017). Iminium catalysis inside a self-assembled supramolecular capsule: scope and mechanistic studies. J. Am. Chem. Soc. 139, 17500–17507. doi: 10.1021/jacs.7b08976
Burrell, K. A., Officer, D. L., Plieger, P. G., and Reid, D. C. W. (2001). Synthetic routes to multiporphyrin arrays. Chem. Rev. 101, 2751–2796. doi: 10.1021/cr0000426
Catti, L., and Tiefenbacher, K. (2018). Brønsted acid-catalyzed carbonyl-olefin metathesis inside a self-assembled supramolecular host. Angew. Chem. Int. Ed. 57, 14589–14592. doi: 10.1002/anie.201712141
Catti, L., Zhang, Q., and Tiefenbacher, K. (2016). Advantages of catalysis in self-assembled molecular capsules. Chem. Eur. J. 22, 9060–9066. doi: 10.1002/chem.201600726
Cho, W.-S., and Lee, C.-H. (1998). Convenient synthesis of difurylmethanes and dithienylmethanes and their application to the syntheses of core-modified porphyrins. Bull. Korean Chem. Soc. 19, 314–319. doi: 10.1002/chin.199838113
Conn, M. M., and Rebek, J. Jr. (1997). Self-assembling capsules. Chem. Rev. 97, 1647–1668. doi: 10.1021/cr9603800
Du, K.-S., and Huang, J.-M. (2018). Electrochemical synthesis of bisindolylmethanes from indoles and ethers. Org. Lett. 20, 2911–2915. doi: 10.1021/acs.orglett.8b00968
Evan-Salem, T., Baruch, I., Avram, L., Cohen, Y., Palmer, L. C., and Rebek, J. Jr. (2006). Resorcinarenes are hexameric capsules in solution. Proc. Natl. Acad. Sci. U.S.A. 103, 12296–12300. doi: 10.1073/pnas.0604757103
Fürstner, A. (2003). Chemistry and biology of roseophilin and the prodigiosin alkaloids: a survey of the last 2500 years. Angew. Chem. Int. Ed. 42, 3582–3603. doi: 10.1002/anie.200300582
Gaeta, C., Talotta, C., De Rosa, M., La Manna, P., Soriente, A., and Neri, P. (2019). The hexameric resorcinarene capsule at work: supramolecular catalysis in confined spaces. Chem. Eur. J. 25, 4899–4913. doi: 10.1002/chem.201805206
Gambaro, S., De Rosa, M., Soriente, A., Talotta, C., Floresta, G., Rescifina, A., et al. (2019). A hexameric resorcinarene capsule as a hydrogen bonding catalyst in the conjugate addition of pyrroles and indoles to nitroalkenes. Org. Chem. Front. 6, 2339–2347. doi: 10.1039/C9QO00224C
Giust, S., La Sorella, G., Sperni, L., Fabris, F., Strukul, G., and Scarso, A. (2015), Supramolecular catalysis in the synthesis of substituted 1 H-tetrazoles from isonitriles by a self-assembled hexameric capsule. Asian J. Org. Chem. 4, 217–220. doi: 10.1002/ajoc.201402229
Gunasekera, S. P., McCarthy, P. J., and Kelly-Borges, M. (1994). Hamacanthins A and B, new antifungal bis indole alkaloids from the deepwater marine sponge. J. Nat. Prod. 57, 1437–1441. doi: 10.1021/np50112a014
Guo, X., Pan, S., Liu, J., and Li, Z. (2009). One-pot synthesis of symmetric and unsymmetric 1,1-bis-indolylmethanes via tandem iron-catalyzed C–H bond oxidation and C–O bond cleavage. J. Org. Chem. 74, 8848–8851. doi: 10.1021/jo902093p
Ji, S.-J., Zhou, M.-F., Gu, D.-G., Jiang, Z.-Q., and Loh, T.-P. (2004). Efficient FeIII-catalyzed synthesis of bis(indolyl)methanes in ionic liquids. Eur. J. Org. Chem. 2004, 1584–1587. doi: 10.1002/ejoc.200300719
Josefsen, L. B., and Boyle, R. W. (2012). Unique diagnostic and therapeutic roles of porphyrins and phthalocyanines in photodynamic therapy, imaging and theranostics. Theranostics 2, 916–966. doi: 10.7150/thno.4571
Köster, J. M., Häussinger, D., and Tiefenbacher, K. (2019). Activation of primary and secondary benzylic and tertiary alkyl(sp3)C-F bonds inside a self-assembled molecular container. Front. Chem. 6:639. doi: 10.3389/fchem.2018.00639
Köster, J. M., and Tiefenbacher, K. (2018). Elucidating the importance of hydrochloric acid as a cocatalyst for resorcinarene-capsule-catalyzed reactions. Chem. Cat. Chem. 10, 2941–2944. doi: 10.1002/cctc.201800326
Kursunlu, A. N., Guler, E., Ucan, H. I., and Boyle, R. W. (2012). A novel bodipy-dipyrrin fluorescent probe: synthesis and recognition behaviour towards Fe (II) and Zn (II). Dyes Pigm. 94, 496–502. doi: 10.1016/j.dyepig.2012.02.006
La Manna, P., De Rosa, M., Talotta, C., Gaeta, C., Soriente, A., Floresta, G., et al. (2018a). The hexameric resorcinarene capsule as an artificial enzyme: ruling the regio and stereochemistry of a 1,3-dipolar cycloaddition between nitrones and unsaturated aldehydes. Org. Chem. Front. 5, 827–837. doi: 10.1039/C7QO00942A
La Manna, P., Talotta, C., Floresta, G., De Rosa, M., Soriente, A., Rescifina, A., et al. (2018b). Mild friedel–crafts reactions inside a hexameric resorcinarene capsule: C–Cl bond activation through hydrogen bonding to bridging water molecules. Angew. Chem. Int. Ed. 57, 5423–5428. doi: 10.1002/anie.201801642
La Sorella, G., Sperni, L., Ballester, P., Strukul, G., and Scarso, A. (2016a). Hydration of aromatic alkynes catalyzed by a self-assembled hexameric organic capsule. Catal. Sci. Techol. 6, 6031–6036. doi: 10.1039/C6CY00307A
La Sorella, G., Sperni, L., Strukul, G., and Scarso, A. (2016b). Supramolecular activation of hydrogen peroxide in the selective sulfoxidation of thioethers by a self-assembled hexameric capsule. Adv. Synth. Catal. 358, 3443–3449. doi: 10.1002/adsc.201600430
Laha, J. K., Dhanalekshmi, S., Taniguchi, M., Ambroise, A., and Lindsey, J. S. (2003). A scalable synthesis of meso-substituted dipyrromethanes. Org. Process. Rev. Dev. 7, 799–812. doi: 10.1021/op034083q
Ling, Y., An, D., Zhou, Y., and Rao, W. (2019). Ga(OTf)3-catalyzed temperature-controlled regioselective Friedel–Crafts alkylation of trifluoromethylated 3-indolylmethanols with 2-substituted indoles: divergent synthesis of trifluoromethylated unsymmetrical 3,3′-and 3,6′-bis(indolyl)methanes. Org. Lett. 21, 3396–3401. doi: 10.1021/acs.orglett.9b01135
Lucarini, S., Mari, M., Piersanti, G., and Spadoni, G. (2013). Organocatalyzed coupling of indoles with dehydroalanine esters: synthesis of bis(indolyl)propanoates and indolacrylates. RSC Adv. 3, 19135–19143. doi: 10.1039/c3ra42922a
MacGillivray, L. R., and Atwood, J. L. (1997). A chiral spherical molecular assembly held together by 60 hydrogen bonds. Nature 389, 469–472. doi: 10.1038/38985
Norouzi, M., Elhamifar, D., and Mirbagheri, R. (2018). Self-assembled alkyl imidazolium based organosilica as efficient support for sulfonic acid catalyst in the synthesis of bis(indolyl)methanes. Polyhedron 154, 229–235. doi: 10.1016/j.poly.2018.07.047
Pahima, E., Zhang, Q., Tiefenbacher, K., and Major, D. T. (2019). Discovering monoterpene catalysis inside nanocapsules with multiscale modelling and experiments. J. Am. Chem. Soc. 141, 6234–6246. doi: 10.1021/jacs.8b13411
Palmieri, A., Petrini, M., and Shaikh, R. R. (2010). Synthesis of 3-substituted indolesviareactive alkylideneindolenine intermediates. Org. Biomol. Chem. 8, 1259–1127. doi: 10.1039/B919891A
Qiang, W., Liu, X., and Loh, T.-P. (2019). Supported iridium catalyst for the green synthesis of 3,3′-bis(indolyl)methanes using methanol as the bridging methylene source. ACS Sustain. Chem. Eng. 7, 8429–8439. doi: 10.1021/acssuschemeng.9b00094
Sakemi, S., and Sun, H. H. (1991). Nortopsentins A, B, and C. Cytotoxic and antifungal imidazolediylbis[indoles] from the sponge Spongosorites ruetzleri. J. Org. Chem. 56, 4304–4307. doi: 10.1021/jo00013a044
Shiri, M. (2012). Indoles in multicomponent processes (MCPs). Chem. Rev. 112, 3508–3549. doi: 10.1021/cr2003954
Shiri, M., Zolfigol, M. A., Kruger, H. G., and Tanbakouchian, Z. (2010). Bis- and trisindolylmethanes (BIMs and TIMs). Chem. Rev. 110, 2250–2293. doi: 10.1021/cr900195a
Shivanyuk, A., and Rebek, J. Jr. (2001). Reversible encapsulation by self-assembling resorcinarene subunits. Proc. Natl. Acad. Sci. U.S.A. 98, 9662–9665. doi: 10.1073/pnas.141226898
Shivanyuk, A., and Rebek, J. Jr. (2003). Assembly of resorcinarene capsules in wet solvents. J. Am. Chem. Soc. 125, 3432–3433. doi: 10.1021/ja027982n
Singh, K., Sharma, S., and Sharma, A. (2011). Unique versatility of Amberlyst 15. An acid and solvent-free paradigm towards synthesis of bis(heterocyclyl)methane derivatives. J. Mol. Catal. A Chem. 347, 34–37. doi: 10.1016/j.molcata.2011.07.007
Sivaprasad, G., Perumal, P. T., Prabavathy, V. R., and Mathivanan, N. (2006). Synthesis and anti-microbial activity of pyrazolylbisindoles–promising anti-fungal compounds. Bioorg. Med. Chem. Lett. 16, 6302–6305. doi: 10.1016/j.bmcl.2006.09.019
Tran, P. H., Nguyen, X.-T. T., and Chau, D.-K. N. (2018). A brønsted-acidic ionic liquid gel as an efficient and recyclable heterogeneous catalyst for the synthesis of bis(indolyl)methanes under solvent-free sonication. Asian J. Org. Chem. 7, 232–239. doi: 10.1002/ajoc.201700596
Wu, Z., Wang, G., Yuan, S., Wu, D., Liu, W., Ma, B., et al. (2019). Synthesis of bis(indolyl)methanes under dry grinding conditions, promoted by a Lewis acid surfactant–SiO2-combined nanocatalyst. Green Chem. 21:3542. doi: 10.1039/C9GC01073D
Yamanaka, M., Shivanyuk, A., and Rebek, J. (2004). Kinetics and thermodynamics of hexameric capsule formation. J. Am. Chem. Soc. 126, 2939–2943. doi: 10.1021/ja037035u
Zhang, Q., Catti, L., Pleiss, J., and Tiefenbacher, K. (2017). Terpene cyclizations inside a supramolecular catalyst: leaving-group-controlled product selectivity and mechanistic studies. J. Am. Chem. Soc. 139, 11482–11492. doi: 10.1021/jacs.7b04480
Zhang, Q., Catti, L., Syntrivanis, L.-D., and Tiefenbacher, K. (2019). En route to terpene natural products utilizing supramolecular cyclase mimetics. Nat. Prod. Rep. doi: 10.1039/C9NP00003H. [Epub ahead of print].
Zhang, Q., Rinkel, J., Goldfuss, B. M., Dickschat, J. S., and Tiefenbacher, K. (2018). Sesquiterpene cyclizations catalysed inside the resorcinarene capsule and application in the short synthesis of isolongifolene and isolongifolenone. Nat. Catal. 1, 609–615. doi: 10.3390/catal8120609
Zhang, Q., and Tiefenbacher, K. (2013). The hexameric resorcinarene capsule is a brønsted acid: investigation and application to synthesis and catalysis. J. Am. Chem. Soc. 135, 16213–16219. doi: 10.1021/ja4080375
Zhang, Q., and Tiefenbacher, K. (2015). Terpene cyclization catalysed inside a self-assembled cavity. Nat. Chem. 7, 197–202. doi: 10.1038/nchem.2181
Keywords: supramolecular organocatalysis, resorcinarene hexameric capsule, bis(heteroaryl)methanes, self-assembly, H-bond catalyst, Brønsted acid catalyst
Citation: Gambaro S, La Manna P, De Rosa M, Soriente A, Talotta C, Gaeta C and Neri P (2019) The Hexameric Resorcinarene Capsule as a Brønsted Acid Catalyst for the Synthesis of Bis(heteroaryl)methanes in a Nanoconfined Space. Front. Chem. 7:687. doi: 10.3389/fchem.2019.00687
Received: 30 August 2019; Accepted: 04 October 2019;
Published: 22 October 2019.
Edited by:
Ronald K. Castellano, University of Florida, United StatesReviewed by:
Richard J. Hooley, University of California, Riverside, United StatesAdam Urbach, Trinity University, United States
Konrad Tiefenbacher, ETH Zürich, Switzerland
Copyright © 2019 Gambaro, La Manna, De Rosa, Soriente, Talotta, Gaeta and Neri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Margherita De Rosa, bWFkZXJvc2FAdW5pc2EuaXQ=; Carmen Talotta, Y3RhbG90dGFAdW5pc2EuaXQ=