 Dong Wang
Dong Wang Xinyue Ma
Xinyue Ma Linru Dong
Linru Dong Hairong Feng1
Hairong Feng1 Laurent Désaubry
Laurent Désaubry- 1Sino-French Joint Lab of Food Nutrition/Safety and Medicinal Chemistry, College of Biotechnology, Tianjin University of Science and Technology, Tianjin, China
- 2Laboratory of Medicinal Chemistry and Cardio-oncology, FRE2033, CNRS, Institut Le Bel, Strasbourg, France
We report a metal-free and stereoselective four-component reaction between α-ketoamides, amines, aromatic aldehydes and β-nitroalkenes or β-pivaloxy-nitroalkanes to obtain 2,3-dihydro-4-nitropyrroles functionalized in every position. The heterocycles accessible using this reaction may have utility in the synthesis of pharmacologically active compounds.
Introduction
The formation of novel chemical scaffold represents a critical step in drug discovery to generate original drug candidates (Kim et al., 2014). In this quest, multicomponent reactions are very attractive as they provide a wealth of complex products in only one step (Armstrong et al., 1996; Colombo and Peretto, 2008; Magedov et al., 2008; Domling et al., 2012; Bonne et al., 2013; Ruijter and Orru, 2013; Cores et al., 2014; Zarganes-Tzitzikas and Domling, 2014; Zhu et al., 2014; Herrera and Marques-López, 2015). Recently, our group reported a novel multicomponent condensation of amines, aromatic aldehydes and α-ketoamides to provide fully substituted 2,3-dihydropyrroles 5 in an excellent highly stereoselective, atom economic, and eco-friendly fashion (Scheme 1) (Wang et al., 2018).
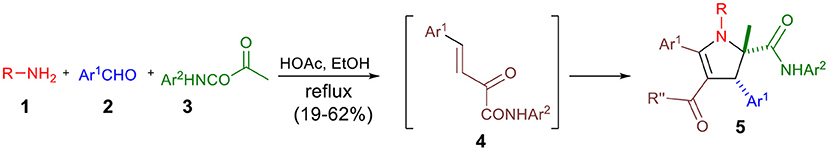
Scheme 1. Catalyst-free three-component synthesis of 2,3-dihydropyrroles 5.
The drug-like character of 2,3-dihydropyrroles prompted us to examine their pharmacological potential, which led to the discovery of potent α-glucosidase inhibitors, illustrating the utility of this novel methodology in medicinal chemistry (Wang et al., 2018).
To expand the scope of this reaction and generate 2,3-dihydropyrroles with new types of substituents, we considered using β-nitrostyrenes 6 susceptible to react similarly to the intermediate enone 4 (Scheme 2).
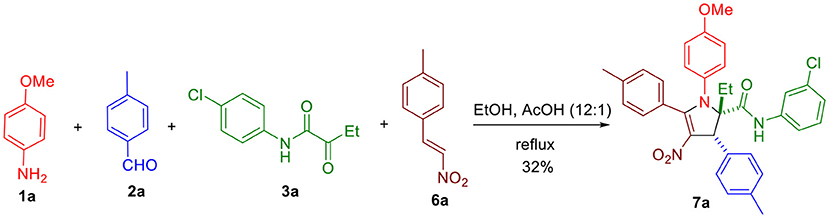
Scheme 2. Three-component synthesis of 2,3-dihydro-4-nitropyrrole 7a.
Results and Discussion
Using the optimized conditions identified in our previous study (10 h reflux in EtOH/AcOH 12:1, entry 1, Table 1), we were pleased to observe in our first attempt the generation of the expected 4-nitro-2,3-dihydropyrrole 7a in 32% yield, which was purified by flash chromatography. The singlet peak at 5.02 ppm in the proton NMR clearly reveals the existence of H1 (Figure 1, both at benzylic and allylic position), which is coherent with the one observed for compound 5a disclosed in our previous article (Wang et al., 2018). Moreover, two separate multiplets (at about 2.37 and 2.18 ppm) corresponding to CH2 group (Scheme 3) indicates they are diastereotopic protons, further confirming the structure (detailed experimental procedures and experimental data are disclosed in the Supplementary Material).
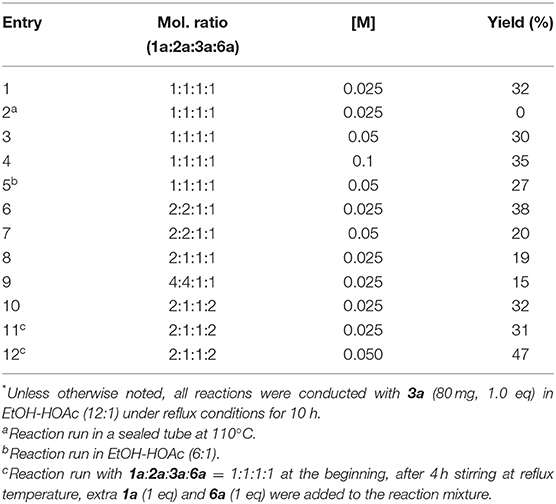
Table 1. Optimization of the synthesis of 2,3-dihydro-4-nitropyrrole 7a.*
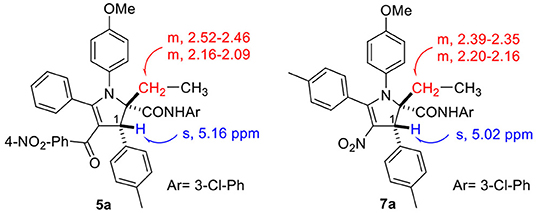
Figure 1. NMR features of 2,3-dihydropyrroles.
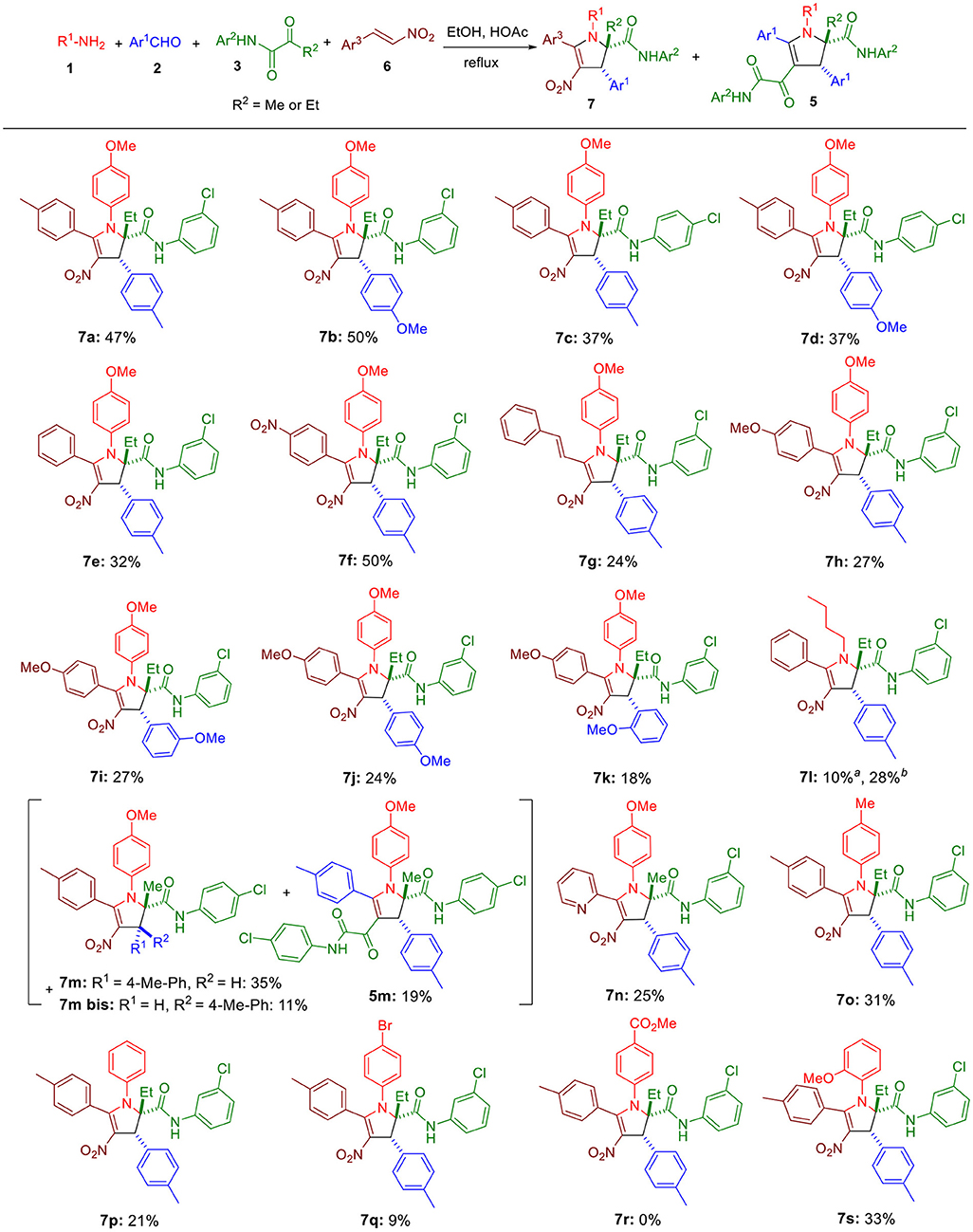
Scheme 3. Substrate scope for the four-component reaction. Unless otherwise noted, all reactions were conducted at 0.05 M concentration with an equimolar concentration of the four reagents in EtOH-AcOH (12:1) under reflux conditions, and an extra eq. of both 1 and 6 were added to the reaction medium after 4 h. All reactions were conducted for 10 h. aReaction was done without any acetic acid. bReaction was conducted with an equimolar concentration of the four reagents in EtOH (0.025 M) under reflux conditions.
Since homopyruvic amide 3a was not completely consumed, other reaction conditions were examined (Table 1). Heating the reaction to higher temperature did not yield the expected adduct (entry 2), probably due to thermal decomposition. More concentrated reaction media gave similar yields (entries 3 and 4). Similarly, adding more acetic acid to the reaction media did not offer improved yield either (entry 5). Doubling the amount of 1a and 2a slightly increased the yield to 38% (entry 6). However, changing the concentration or the molar ratio further did not lead to better result (entries 7–9). Since both amine 1a and β-nitrostyrene 6a were consumed after running the reaction for 4 h, we doubled their amount at the beginning of the reaction and also 4 h later, but it had little effect on the yield (entries 10 and 11). Eventually, performing this latter condition in a more concentrated condition increased the yield to 47% (entry 12).
Next, the substrate scope was explored. Replacing tolualdehyde by anisaldehyde did not significantly modify the yield (7b, Scheme 3). Moving the position of the chlorine in the ketoamide lowered the yield by about 10% (7c, 7d). Switching the methyl in the β-nitrostyrenes 6 to a nitro (7f) was well tolerated, but replacing it with a hydrogen (7e), extending the conjugation by adding an extra double bond (7g) or lowering the electrophilicity by introducing a 4-methoxy in the phenyl moiety (7h-7k) reduced the yields to 32% and 24–27%. We found that the replacement of anisidine by n-butylamine yielded the expected product 7l (28%) when acetic acid was absent in the medium, confirmining the suitability of aliphatic amines for this reaction. Using a pyruvic amide 3 (R2 = Me instead of Et), we observed the formation of the expected adduct 7m accompanied by its diastereomeric isomer 7m bis and the ketoamide 5m generated by the α,β-enones resulting from the condensation of aldehydes 2 and the pyruvic amide. Using a more electrophilic β-nitrostyrene such as 2-(2-nitrovinyl)pyridine yielded only the expected adduct 7n. Importantly, decreasing the basicity of the aniline 1 was highly detrimental (7o-7r), but shifting the methoxy of anisidine to the ortho position was well tolerated.
Next, we examined whether β-pivaloxy-nitroalkane could be used as a reactant (Scheme 4). Condensation with 8 afforded the expected adduct (9) unsubstituted at the C5-position, with yield similar to those observed with β-nitrostyrenes and aromatic aldehydes.
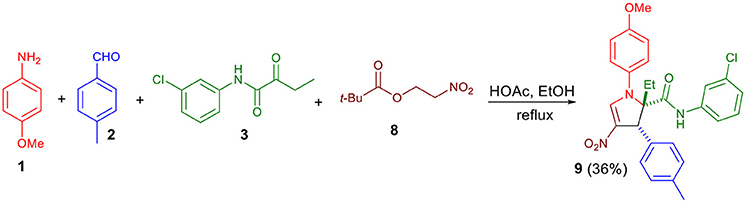
Scheme 4. Condensation reactions with β-pivaloxy-nitrsoalkane 8.
Furthermore, allylic alcohols 10a and 10b were also viable substrates that allowed the synthesis of 2,3-dihydro-4-nitropyrroles 11a-c unsubstituted at the C3-position (Scheme 5). Having the adduct 11c in hand, we examined whether this nitrostyrenic derivative could undergo a reductive cyclization using TiCl3 as a reductant (Tong et al., 2015). So far as we know, this approach to synthesize functionalized indoles from o-nitrostyrenes has never been reported with the nitro group appended to a vinyl moiety rather than an arene.
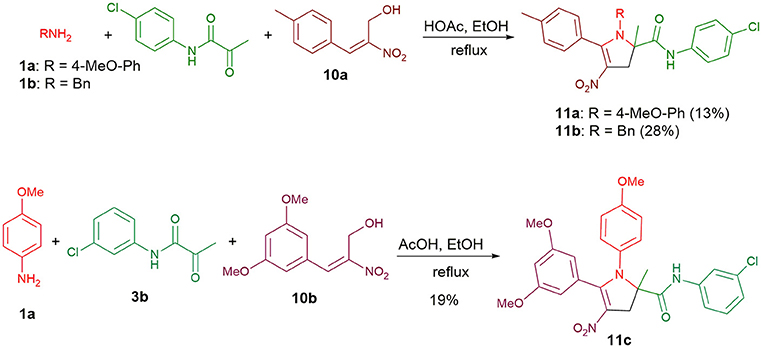
Scheme 5. Condensations with allylic alcohols 10a and 10b.
Our attempt to prepare the tetrahydropyrrolo[3,2-b]indole 12 from the adduct 11c by treatment with TiCl3 did not deliver this cyclized compound 12 (Scheme 6). Instead, we observed the formation of enamine 13a or ketone 14a depending on the reaction conditions (Table 2). Reduction of the nitro 11c by TiCl3 gave the enamine 13a as the major product in 14% yield, when the reaction was performed for 1.5 h at a 0.2 M concentration (entry 1, Table 2). Under more concentrated reaction conditions and with a reaction time extended to 14 h, only the pyrrolidinone 14a, resulting from the hydrolysis of 13a, was obtained (entry 2).
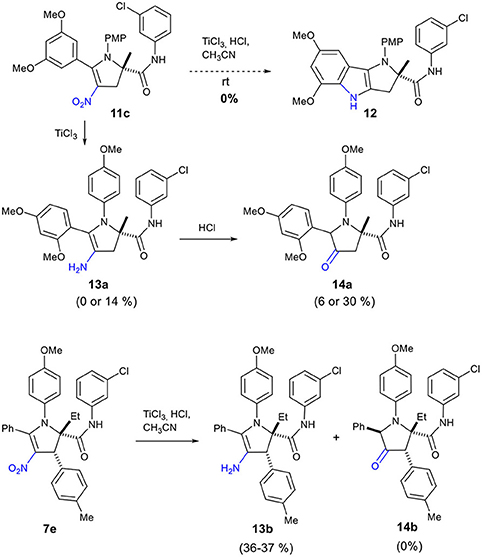
Scheme 6. Reductive transformation of 2,3-dihydro-4-nitropyrrole 11c to enamine 13a and ketone 14a and conversion of 7e to 13b.
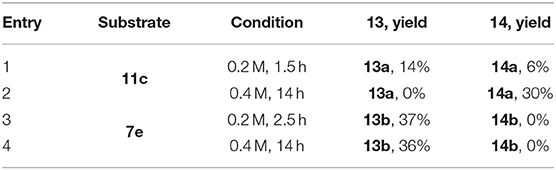
Table 2. Effect of reaction conditions on the reduction of 11c and 7e.
Surprisingly, application of this reaction to dihydro-4-nitropyrrole 7e provided dihydro-4-aminopyrrole 13b as the sole product in both conditions (entries 3 and 4), possibly due to the enhanced steric hindrance which prohibits the hydrolysis of the enamine. This sequence is noteworthy, as 1,2,4,5,5-pentasubstituted 3-amino-4,5-dihydro-1H-pyrroles have been scarcely reported, indicating a high potential to generate intellectual property in medicinal chemistry programs
On the basis of investigations previously reported by our group (Wang et al., 2018), we proposed that mechanism of this reaction involves the conjugate addition of imine 15 to nitrostyrene 6 to produce intermediate 16 that reacts with aldehyde 2, leading to 3-oxazine 18 (Scheme 7). Successive dehydration and deprotonation then generate the azomethine ylide 20 that undergoes an intramolecular 1,5-dipolar cycloaddition (Taylor, 1979) to afford the 2,3-dihydropyrrole 7. The stabilization of the intermediate 20 by π-stacking between the Ar1 and Ar2 probably account for the observed diastereospecificity of this reaction.
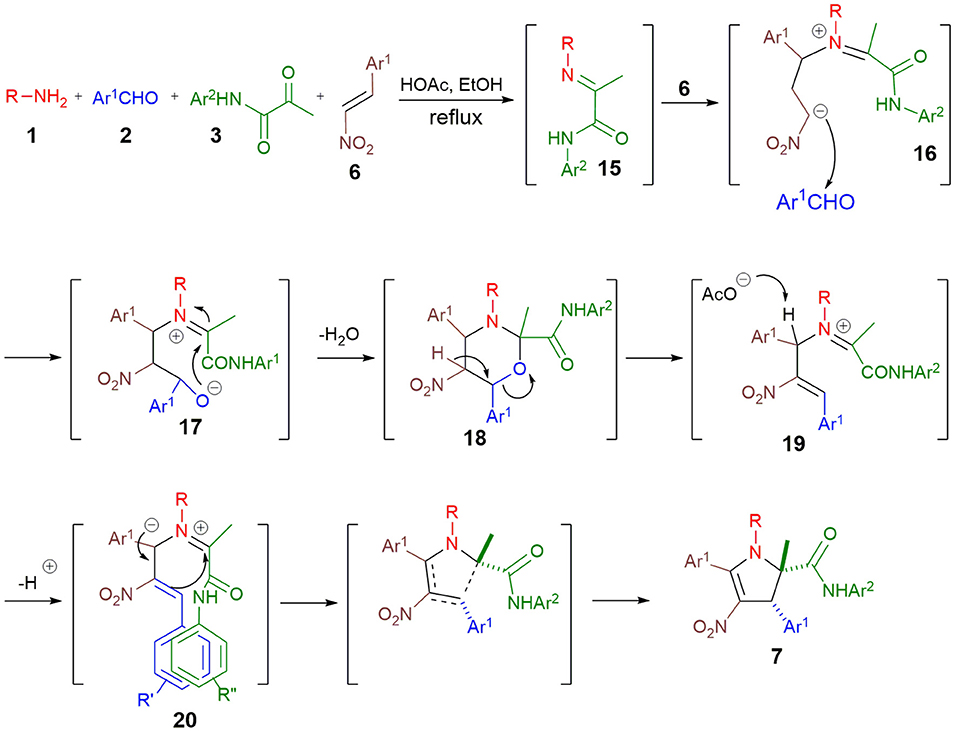
Scheme 7. Four-component synthesis of 2,3-dihydropyrroles 7 and proposed mechanism.
Conclusion
We have developed a multicomponent reaction that uses α-ketoamides, amines, aromatic aldehydes and β-nitrostyrenes or β-pivaloxy-nitroalkanes to deliver functionalized 2,3-dihydro-4-nitropyrroles in moderate yields (9–50%), however considering that this reaction generates four new bonds, it is still effective (55–84% average yield per bond formation). Combining our multicomponent reaction with a TiCl3-induced reduction gives access to novel polysubstituted dihydro-1H-pyrroles.
Data Availability Statement
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
LDé and DW were responsible for designing the experiments. XM, LDo, and HF performed the experimentations. LDé, DW, and PY analyzed the results and wrote the publication.
Funding
This work was supported in part by the National Natural Science Foundation of China (No. 81673296).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Miss Haijuan Qin (TUST) for assistance with 2D NMR experiments.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2019.00810/full#supplementary-material
References
Armstrong, R. W., Combs, A. P., Tempest, P. A., Brown, S. D., and Keating, T. A. (1996). Multiple-component condensation strategies for combinatorial library synthesis. Acc. Chem. Res. 29, 123–131. doi: 10.1021/ar9502083
Bonne, D., Constantieux, T., Coquerel, Y., and Rodriguez, J. (2013). Stereoselective multiple bond-forming transformations (MBFTs): the power of 1,2-and 1,3-dicarbonyl compounds. Chem. Eur. J. 19, 2218–2231. doi: 10.1002/chem.201204018
Colombo, M., and Peretto, I. (2008). Chemistry strategies in early drug discovery: an overview of recent trends. Drug Discov. Today 13, 677–684. doi: 10.1016/j.drudis.2008.03.007
Cores, A., Carbajales, C., and Coelho, A. (2014). Multicomponent reactions in antimitotic drug discovery. Curr. Top. Med. Chem. 14, 2209–2230. doi: 10.2174/1568026614666141127115130
Domling, A., Wang, W., and Wang, K. (2012). Chemistry and biology of multicomponent reactions. Chem. Rev. 112, 3083–3135. doi: 10.1021/cr100233r
Herrera, R. P., and Marques-López, E. (2015). Multicomponent Reactions: Concepts and Applications for Design and Synthesis. Wiley-VCH Verlag GmbH & Co. KGaA. Available online at: https://www.wiley.com/en-gb/Multicomponent+Reactions%3A+Concepts+and+Applications+for+Design+and+Synthesis-p-9781118016008
Kim, J., Kim, H., and Park (2014). Privileged structures: efficient chemical “navigators” toward unexplored biologically relevant chemical spaces. J. Am. Chem. Soc. 136, 14629–14638. doi: 10.1021/ja508343a
Magedov, I. V., Luchetti, G., Evdokimov, N. M., Manpadi, M., Steelant, W. F. A., Van slambrouck, S., et al. (2008). Novel three-component synthesis and antiproliferative properties of diversely functionalized pyrrolines. Bioorg. Med. Chem. Lett. 18, 1392–1396. doi: 10.1016/j.bmcl.2008.01.019
Ruijter, E., and Orru, R. V. A. (2013). Multicomponent reactions – opportunities for the pharmaceutical industry. Drug Discov. Today Technol. 10, e15–e20. doi: 10.1016/j.ddtec.2012.10.012
Tong, S., Xu, Z., Mamboury, M., Wang, Q., and Zhu, J. (2015). Aqueous titanium trichloride-promoted reductive cyclization of o-nitrostyrenes to indoles: development and application to the synthesis of rizatriptan and aspidospermidine. Angew. Chem. Int. Ed. 54, 11809–11812. doi: 10.1002/anie.201505713
Wang, D., Li, L., Feng, H., Sun, H., Almeida-Veloso, F., Charavin, M., et al. (2018). Catalyst-free three-component synthesis of highly functionalized 2,3-dihydropyrroles. Green Chem. 20, 2775–2780. doi: 10.1039/C8GC00987B
Zarganes-Tzitzikas, T., and Domling, A. (2014). Modern multicomponent reactions for better drug syntheses. Org. Chem. Front. 1, 834–837. doi: 10.1039/C4QO00088A
Zhu, J., Wang, Q., and Wang, M.-X. (2014). Multicomponent Reactions in Organic Synthesis. Wiley-VCH Verlag GmbH & Co. KGaA. Available online at: https://onlinelibrary.wiley.com/doi/book/10.1002/9783527678174
Keywords: multicomponent reaction, ketoamide, heterocycles, nitro, dihydropyrroles, cascade reaction, 1,5-dipolar cyclization
Citation: Wang D, Ma X, Dong L, Feng H, Yu P and Désaubry L (2019) Stereoselective Four-Component Synthesis of Functionalized 2,3-Dihydro-4-Nitropyrroles. Front. Chem. 7:810. doi: 10.3389/fchem.2019.00810
Received: 02 August 2019; Accepted: 11 November 2019;
Published: 26 November 2019.
Edited by:
Alexey Sukhorukov, N.D. Zelinsky Institute of Organic Chemistry (RAS), RussiaReviewed by:
Mohammad Bayat, Imam Khomeini International University, IranFedor Zubkov, Peoples' Friendship University of Russia, Russia
Copyright © 2019 Wang, Ma, Dong, Feng, Yu and Désaubry. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dong Wang, d2FuZ2RvbmdAdHVzdC5lZHUuY24=; Laurent Désaubry, ZGVzYXVicnlAdW5pc3RyYS5mcg==