 Lingshan Lyu1
Lingshan Lyu1 Huiling Liu
Huiling Liu- 1Laboratory of Theoretical and Computational Chemistry, Institute of Theoretical Chemistry, Jilin University, Changchun, China
- 2College of Material and Chemical Engineering, Tongren University, Tongren, China
Palladium-catalyzed asymmetric allylic substitution, due to its valuable reactive profile, has become a quite useful tool in organic synthesis fields. In the present study, density functional theory (DFT) calculations were applied to investigate the important factors for palladium-catalyzed 3-butene-2-ol and methylaniline amination reaction, with tetrahydrofuran (THF) as solvent. We find that this catalytic protocol results in high regio- and stereoselectivity, which is in line with the experimental result. According to our calculations, the high regio- and stereoselectivity is caused by the steric hindrance between the substrate and the catalyst ligand. To verify this point, we further explore the reactive process with different axial chirality on the catalyst ligand (altering the steric hindrance), and the results suggest that the preponderant R chiral configuration product has reversed. These results could lead to a better understanding of the mechanism for 3-butene-2-ol amination reaction and are helpful for the design of the corresponding catalyst ligand in the industry.
Introduction
The N-alkylation of amines is an important kind of organic synthetic reaction. The products of such reactions relate to many important fields such as defense, chemical, and pharmaceutical. As the development of chemical and catalytic synthesis technologies, the C–N bond formation reaction via N-alkylation has become one of the focused researches in the organic chemistry. The early studies about the C–N bond formation reaction includes the famous Ullmann reaction (Ullmann and Bielecki, 1901) and Goldberg reaction (Goldberg, 1906). Subjected to the experimental condition, these approaches generally form much halogen hydride and acid wastewater, which is harmful for the environment. The reactive conditions are also tough due to the absence of a suitable catalyst, and the atom utilization and the product selectivity are also unsatisfactory. The discovery of the Buchwald–Hartwig N-alkylation method (Hartwig et al., 1996, 1999; Fors and Buchwald, 2010) boosts the development of the C–N bond's establishment and introduces palladium catalyzed into the aryl halide N-alkylation reactions. However, this method still suffers environment-unfriendly issues.
With the development of the green chemistry, exploring the reactions with higher efficiency and less toxicity has been the goal for scientists. Alcohol has drawn more and more attention because it has equal characters with the aryl halide in N-alkylation reactions (Trost and Crawley, 2003; Lu and Ma, 2008; Zhang et al., 2014). This reactive profile has many advantages: the raw reactants are cheap to obtain, the yield and the selectivity are considerable, and the only by-product is water. In 2002, Ozawa and coworkers found that allylic alcohols can be used as coupling factors of allylic substitution reactions catalyzed by Pd (Ozawa et al., 2002). Then, they found that the C–O bond dissociation is the rate-determining step in these reactions (Ozawa et al., 2004), which was also demonstrated in le Floch's work using density functional theory (DFT) calculation approaches (Piechaczyk et al., 2006). Further research revealed a more detailed reaction process of the oxidative addition step (Kinoshita et al., 2004; Raducan et al., 2012; Sundararaju et al., 2012). In 2014, Banerjee and co-workers chose Pd(dba)2 as catalyst to catalyze the 2-cyclohexene-1-ol and aniline amination reaction; they tested and optimized a series of reactive conditions (including ligands, additive, solvent, and temperature). Finally, they found that the yield was nearly 100% and the e.r. was 96:4–85:15 with L8 as the ligand, and 298K and tetrahydrofuran (THF) as the solvent (Banerjee et al., 2014). They also determined that the chiral phosphoric acid in the catalytic ligand could promote the formation of the active π-(allyl)-Pd intermediate, even changing the 2-cyclohexene-1-ol to a low-active chain alkyl allyl alcohol; the results of the reaction were also ideal. More broadly effective palladium catalysts for nucleophiles have also been determined over the years (Yang et al., 2015, 2016; Jiang et al., 2017). Recently, it has been reported that the chirality of catalytic ligand could significantly influence the stereoselective coordination (Major et al., 2019). In consideration of the enormous raw reactants and catalysts mentioned above, the mechanism of the high enantioselectivity for the N-alkylation reaction still needs further elaboration. Exploring the reactive process in silico thus becomes helpful and necessary.
Up to now, many corresponding issues have been successfully solved by the DFT approach (Herz et al., 2019; Ramakrishnan et al., 2019; Ying et al., 2019). Thus, in the present study, we explored the N-alkylation reactive process between 3-butene-2-ol and the methylaniline detailedly using the DFT approach. With THF as solvent and different axial-chirality ligands (L and L′) on the catalyst, the different attack directions of the nucleophile were considered, and the energetic profiles of the reaction were obtained. At an atomic view, the results will demonstrate the preferred formation of the chiral product and reveal the cause of the high regio- and stereoselectivity.
Methods
All calculations based on density functional theory were performed using the Gaussian 09 (Frisch et al., 2009) (Rev D. 01) software package. The structures were fully optimized by BP86 functional (Perdew, 1986; Becke, 1988), with def2-TZVP basis set on the Pd atom (Schäfer et al., 1992) and the 6-31G (d) basis set on the C, H, O, N, and P atoms (Hehre et al., 1972; Hariharan and Pople, 1973). The polarizable continuum model (PCM) method was used and THF was chosen as the solvent, and Grimme's version 3 dispersion corrections (DFT-D3, disp3 version) (Grimme et al., 2010) were introduced. Besides, the Gibbs free energy with thermal correction was obtained from the frequency calculations, and the single-point energies were also performed on BP86 level with an all-atom def2-TZVP basis set (Weigend et al., 2003), empirical dispersion corrections (DFT-D3), and PCM method for THF solvent. Intrinsic reaction coordinate (IRC) calculations (Fukui, 1970, 1981) were performed to verify if the transition states coordinated with the intermediate correctly. The atomic dipole-corrected Hirshfeld (ADCH) (Lu and Chen, 2012b) was calculated by the Multiwfn 3.7 (Lu and Chen, 2012a) program. The bond length of the key step is shown in Schemes S1–S6 and Tables S1–S6, respectively (detailed in the supporting information, they are not shown in the manuscript for clarity). In addition, the configuration of the phosphoramidite ligand (L) is shown in Figure 1A. The ligand was simplified by removing the bulky backbone atoms of naphthalene and just remained the chiral character. The whole catalytic process is illustrated in Figure 1B, and the accurate configuration of each state are shown in the corresponding figures in each later section.
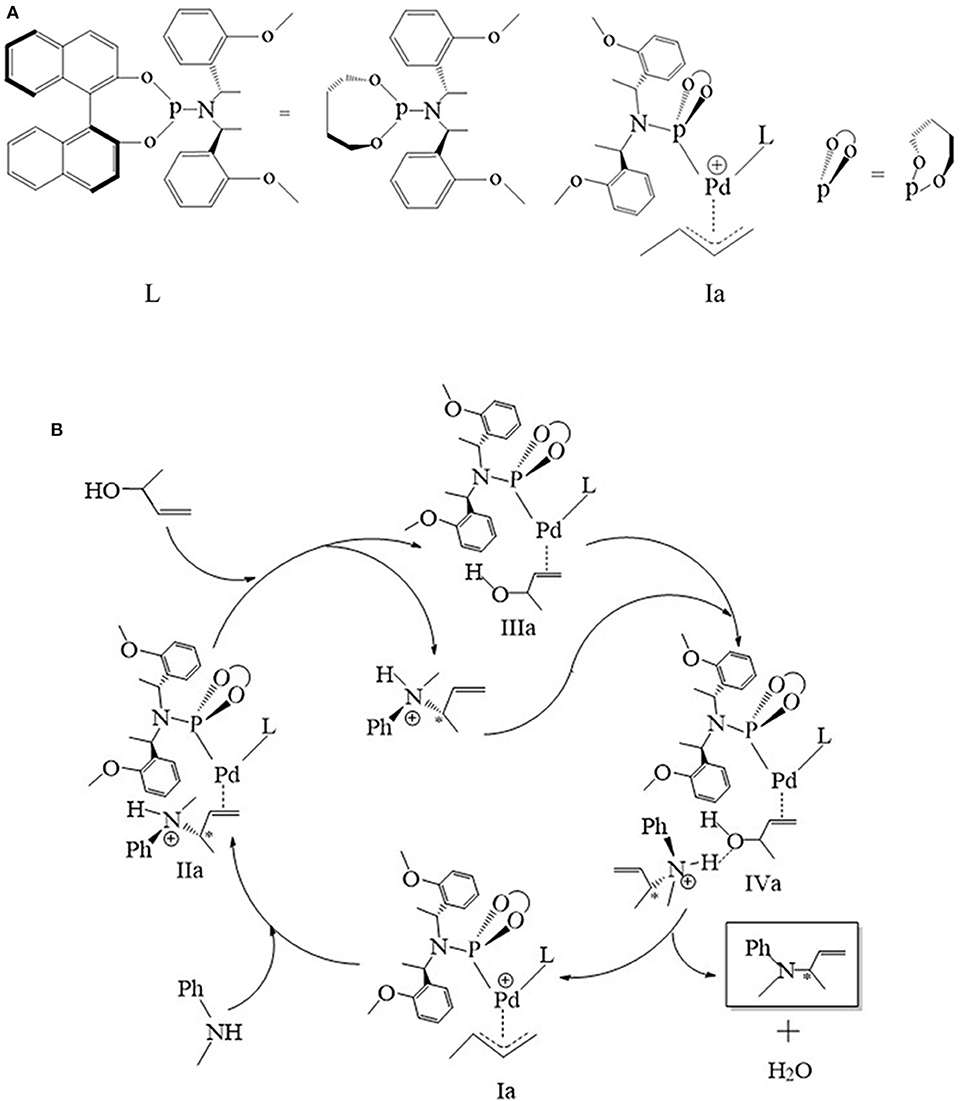
Figure 1. (A) The structure of phosphoramidite ligand (L). (B) The schematic representation of the mechanism of palladium-catalyzed the 3-butene-2-ol amination reaction.
Result and Discussion
The 3-Butene-2-ol Amination Reaction Process in Gas
The overall free energetic profile for the 3-butene-2-ol amination reaction process in gas is outlined in Figure 2. The total energy of palladium-butenyl intermediate Ia, 3-butene-2-ol and methylaniline, was set to the energy reference. Butenyl, which coordinates with Pd of Ia, was identified as having a three-center–four-electron π bond, which causes it to lack electron, while the N of methylaniline has lone pair electrons and could act as the nucleophile. Thus, the methylaniline could nucleophilic attack the butenyl moiety of complex Ia in two directions (named as up-direction and down-direction, as shown in Figure 3), and form two transition states (see Figure 2). For the up-attack direction of the nucleophile, it could form the S chiral configuration transition state, TSI-IIa-S (18.7 kcal/mol); for the down-attack direction, it forms the R chiral configuration transition state, TSI-IIa-R (18.4 kcal/mol). The energy barrier of forming these two configuration transition states are very similar. The distance between the N of methylaniline and the C of the butenyl is 1.962 Å in TSI-IIa-R and 2.215 Å in TSI-IIa-S, while it decreases to 1.582 Å in IIa-R and 1.565 Å in IIa-S (Table S1), which suggests that the methylaniline forms the covalent bond with the butenyl. In IIa-R/S, the conjugative effect between the butenyl and Pd decreases as the covalent binding between the butenyl and the N of methylaniline increases, which causes the total electron transfers from N to the Pd. Then, the 3-butene-2-ol participates in the reaction, and its butenyl moiety coordinates with the palladium and replaces the N-butenyl-N-methylaniline, forming IIIa-R (36.2 kcal/mol) and IIIa-S (36.1 kcal/mol). It seems that the energy value of these two states are a little high. The abnormal energy values indicate that the reactive process is not so satisfactory in gas condition; therefore, the influence of the solvation effect on the reaction is considered for further exploration.
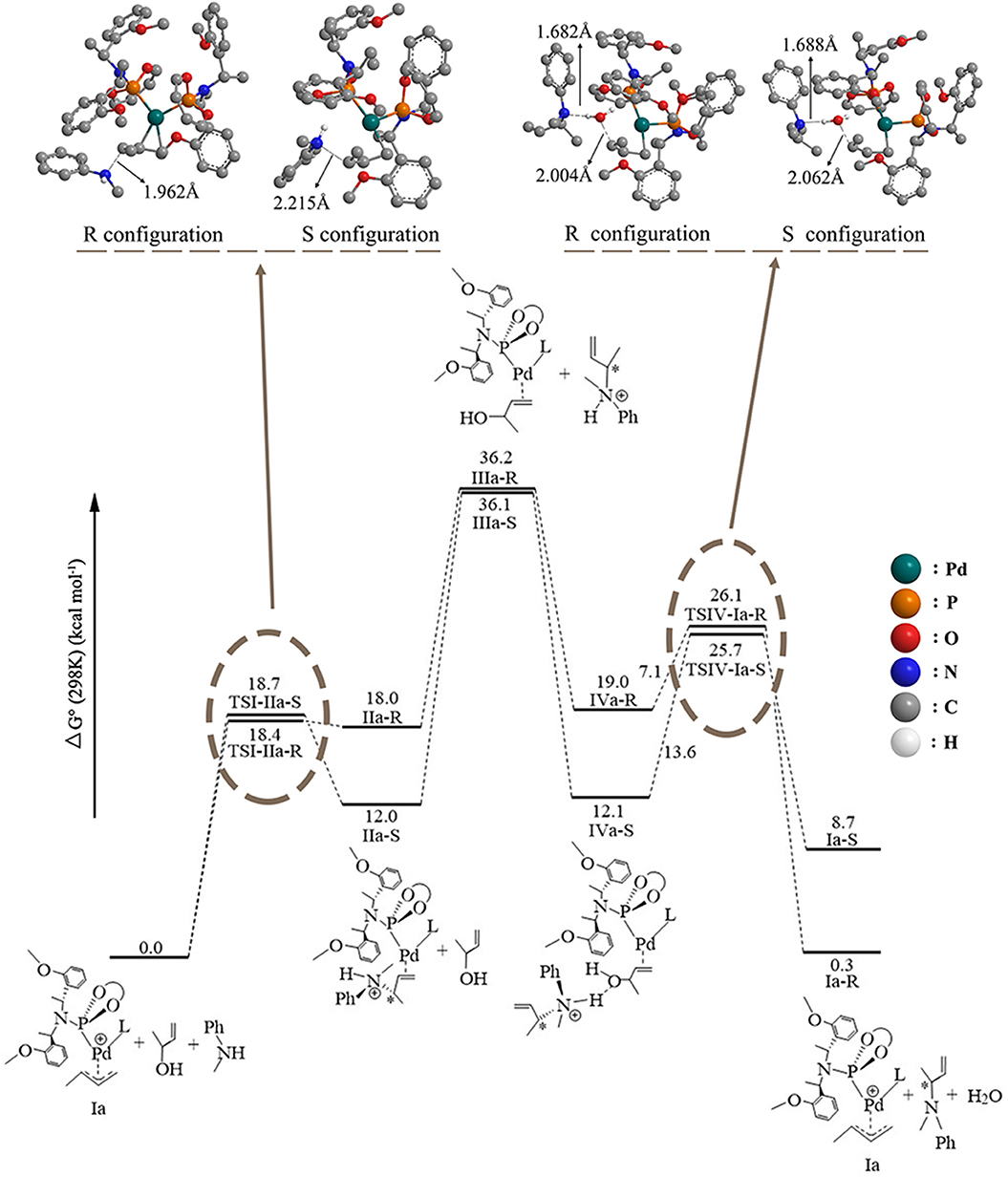
Figure 2. The overall free energetic profiles for the 3-butene-2-ol amination reaction process in gas catalyzed by phosphoramidite palladium.
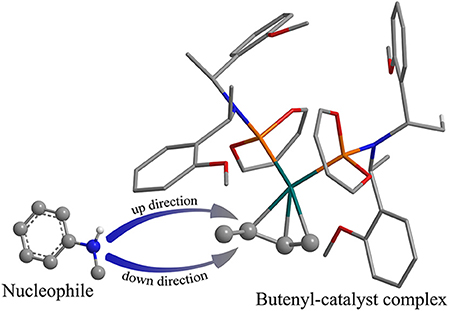
Figure 3. The two attack directions of the nucleophile to approach the substrate complex.
The 3-Butene-2-ol Amination Reaction Process in THF
To consider the effect of the solvent for the reaction, THF was chosen as the representative solvent in our work because it was proved to be the best solvent for the reaction (Banerjee et al., 2014). The overall free energetic profile is shown in Figure 4. The butenyl that coordinates with Pd of Ib was found having a three-center–four-electron π bond, and the electron density is unsaturated at this portion, while the N of methylaniline has lone pair electrons and could act as the nucleophilic reactive site. There are two nucleophilic attack directions for N, which could form the R and S configuration transition states, respectively. The distance between the N of methylaniline and the C of butenyl is 2.086 Å in TSI-IIb-R and 2.254 Å in TSI-IIb-S, while it decreases to 1.575 Å in IIb-R and 1.559 Å in IIb-S (Table S3), which suggests that the methylaniline forms the covalent bond with the butenyl. For the up-attack direction of the nucleophile, it could form the S chiral configuration transition state, TSI-IIb-S (19.6 kcal/mol); for the down-attack direction, it forms the R chiral configuration transition state, TSI-IIb-R (14.3 kcal/mol). Compared with the corresponding energy values in gas, TSI-IIa-S (18.7 kcal/mol) and TSI-IIa-R (18.4 kcal/mol), the influence of solvent environment to the reaction has been demonstrated clearly. The energy barrier of forming the R configuration is lower than the S configuration, which means that it is easier for the nucleophile to attack the butenyl form the down direction. To figure out the difference in the energy barrier, we visualized the structure of the TSI-IIb-R state and the TSI-IIb-S state. As shown in Figure 5, it could be seen that, for the down-attack direction, the methylaniline locates just below the butenyl. Thus, it can coordinate with the butenyl directly. However, for the up-attack direction, the methyl of the methylaniline forms steric hindrance with the phosphoramidite ligand (L), which makes methylaniline combining difficult and enlarges the energy barrier. In the next step, the 3-butene-2-ol participates in the reaction, and its butenyl moiety coordinates with the palladium, replacing the N-butenyl-N-methylaniline, and forms IIIb-R (21.0 kcal/mol) and IIIb-S (21.1 kcal/mol). The energy value of the IIIb-R state and the IIIb-S state becomes distinctly lower than that in gas (36.2 and 36.1 kcal/mol), which may be related to the existence of the THF. Thus, we can suggest that the existence of the THF increases the strength of the coordination bond, which would be described at the charge analysis part. The dissociative cationic N-butenyl-N-methylaniline could further form a hydrogen bond with the substrate hydroxy of IIIb-R and IIIb-S, producing a relatively stable state IVb-R (18.2 kcal/mol) and IVb-S (7.7 kcal/mol).
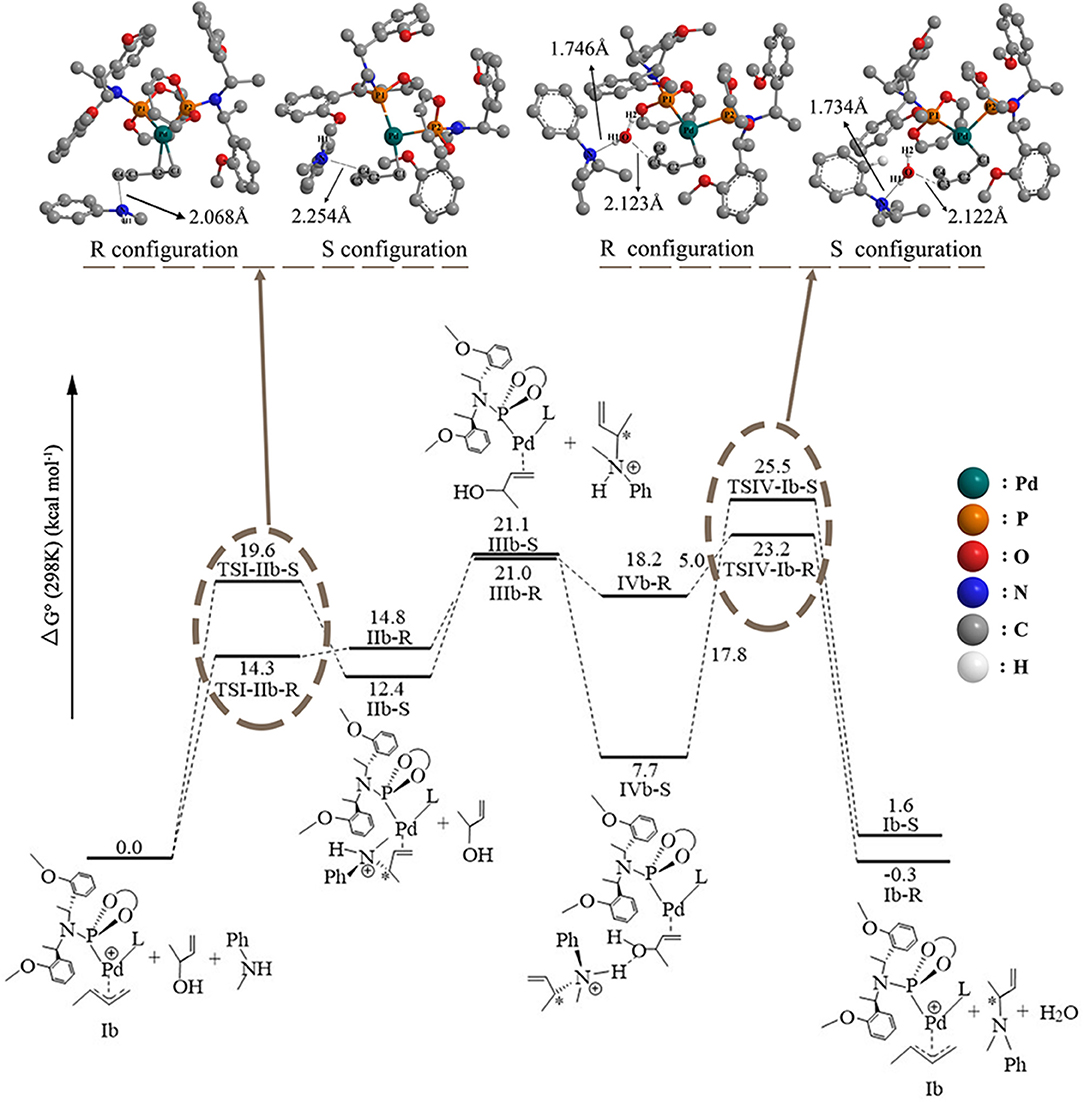
Figure 4. The overall free energetic profiles for the 3-butene-2-ol amination reaction process in THF catalyzed by phosphoramidite Pd-L. The structure of the TSI-IIb-R/S and TSIV-Ib-R/S has been shown in ball-and-stick style; the important distance between atoms has also been shown in the corresponding place.
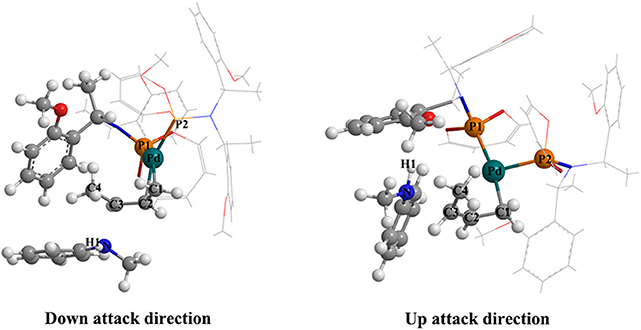
Figure 5. Visualizing the structure of the TSI-IIb-R state (left picture) and the TSI-IIb-S state (right picture).
The last step of the catalytic reaction contains the oxidative addition of the C–O bond in 3-butene-2-ol substrate, which produces water and deprotonated N-butenyl-N-methylaniline and regenerates the palladium-butenyl intermediate Ib. In the IVb-R/S state, the distance between the C and O of the 3-butene-2-ol is 1.467 Å/1.459 Å, and the distance between the N and H of the N-butenyl-N-methylaniline moiety is 1.073 Å/1.075 Å (Table S4). While in the TSIV-Ib-R/S state, the distance increases to 2.123 Å/2.122 Å and 1.746 Å/1.734 Å, which shows the elimination trend of the water molecule. The energy value of the IVb-S state (7.7 kcal/mol) is lower than the IVb-R state (18.2 kcal/mol), which seems that the IVb-S state is more stable. The energy barrier of forming the TSIV-Ib-R state is 5.0 kcal/mol, much lower than that of forming the TSIV-Ib-S state (17.8 kcal/mol). Besides, the energy value of the TSIV-Ib-S state is kind of high (25.5 kcal/mol), which causes it hard to be formed. To find the reason why the energetic barrier of the two states is so different, we visualized the structure of the TSIV-Ib-R and TSIV-Ib-S states (shown in Figure 6); it could be seen that, for the down-attack direction, the benzene ring of the N-butenyl-N-methylaniline locates just between the two benzene of the ligand, which is favorable for the reaction to proceed. However, for the up-attack direction, the benzene ring of the N-butenyl-N-methylaniline is close to the benzene ring of the ligand, which would form a strong steric hindrance and significantly enlarge the energy barrier. What is more, the final energy value of the Ib-R state is −0.3 kcal/mol, suggesting it is a slight exothermic reactive process, while the final energy value of the Ib-S state is 1.6 kcal/mol, suggesting it is a slight endothermic reactive process. These results identify that the down-attack direction of the nucleophile is the preferred reactive route. Combining the energy barrier values of the TSI-IIb-R/S, we can draw the conclusion: it is much easier to form R rather than S configuration product, under the Pd-L catalytic system condition with THF solvent. These results are in line with the experimental results (Banerjee et al., 2014) and could well-explain the highly regio- and stereoselective of the present catalytic protocol.
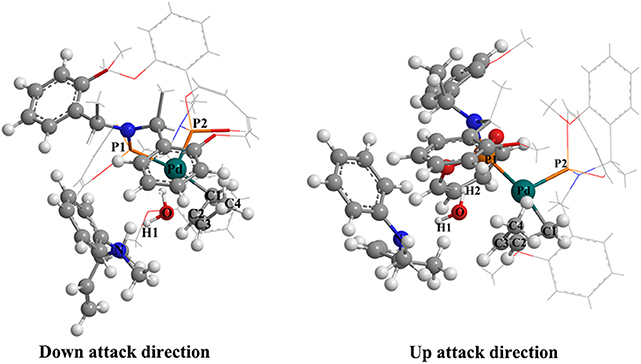
Figure 6. Visualizing the structure of the TSIV-Ib-R state (left picture) and the TSIV-Ib-S state (right picture).
To better describe the reactive process of each step, we also calculate the ADCH population charge (Lu and Chen, 2012b) for the N (nucleophile) and the Pd (Table 1), owing to the main charge transformation taking place in these two atoms. In the beginning, due to the existence of the lone pair electrons, the ADCH charge for N is −0.406, an obvious negative value. The ADCH charge value for the Pd is 0.276, a positive value, because of its conjugation with the butenyl. After the nucleophilic attack of the nucleophile, in the IIb-R/S state, the methylaniline forms covalent bond with the butenyl, and the N shares electrons with the butenyl, which causes the ADCH charge of N increasing to 0.050/0.081 (IIb-R/S). The ADCH charge for the Pd decreases to 0.174/0.157 (IIb-R/S) as butenyl tends to form the conjugation with the nucleophile, which means that the electrons flow to the Pd. In the IIIb-R/S state, the ADCH charge for N and Pd basically maintains their value. However, due to the obvious energy difference existing in the IIIa-R/S and IIIb-R/S states, we also calculate the ADCH charge for the 3-butene-2-ol of the IIIa-R/S and IIIb-R/S states, and the value is −0.177 and −0.197, respectively. The more negative the ADCH charge is, the more stable the bond strength would be, since the total charge of the complex is electrically neutral. Therefore, the coordination bond between the 3-butene-2-ol and the Pd of the IIIb-R/S state is more stable than that of the IIIa-R/S state, which is in agreement with the existence of the THF increasing the strength of the coordination bond. In the IVb-R/S state, the ADCH charge for N is 0.072/0.050, and for Pd, ADCH charge is 0.108/0.147, respectively. The whole flow direction of the charge indicates that the down-attack direction of the methylaniline is the preferred reactive route in the Pd-L catalytic system, which enriches the reactive details.
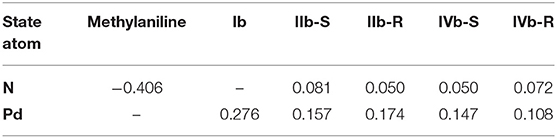
Table 1. The ADCH charge of the N and the Pd in different states for Pd-L catalytic system.
The Influence of the Ligand Axial Chirality on the Reaction
To explore the influence of the ligand axial chirality on the reaction, the phosphoramidite ligand with different axial chirality of L, L′ (Figure 7A, L′ was also treated by the same way as L) was introduced. The whole free energetic profile has been shown in Figure 7B. The overall reactive mode is similar to that of the Pd-L catalytic system: For the up-attack direction of nucleophile, it could form the S chiral configuration, TSI-IIb′-S (17.4 kcal/mol); for the down-attack direction, it forms the R chiral configuration, TSI-IIb′-R (11.9 kcal/mol). Compared to the corresponding energy values of the Pd-L catalytic system, the TSI-IIb-S state (19.6 kcal/mol) and the TSI-IIb-R state (14.3 kcal/mol), the energy barrier of forming these two states becomes lower. The energy value of the IIb′-R/S state is 10.6 and 9.3 kcal/mol, respectively, which is also lower than the corresponding values of the IIb-R/S state (14.8 and 12.4 kcal/mol). That is to say, the Pd-L′ catalytic system shows a better reactive ability to some extent. The distance between the N of methylaniline and the C of butenyl is 2.098 Å in the TSI-IIb′-R state and 2.252 Å in the TSI-IIb′-S state, while it decreases to 1.627 Å in the IIb′-R state and 1.556 Å in the IIb′-S state (Table S5), which is consistent with the covalent bonding trend between the methylaniline and the butenyl. Next, 3-butene-2-ol participates in the reaction, and its butenyl moiety coordinates with palladium, replacing the N-butenyl-N-methylaniline, forming IIIb′-R (15.0 kcal/mol) and IIIb′-S (15.2 kcal/mol). The dissociative cationic N-butenyl-N-methylaniline could further form a hydrogen bond with the substrate hydroxy of IIIb′-R and IIIb′-S, producing a relative stable-state IVb′-R (11.2 kcal/mol) and IVb′-S (8.7 kcal/mol). From Figure 7, we can see that the energy barriers of forming the TSIV-Ib′-R and TSIV-Ib′-S states are 15.3 and 10.4 kcal/mol, respectively. Based on the above results, the C–O bond dissociation is the rate-determining step for the reaction, which has also been proven by the experiment (Ozawa et al., 2004). Therefore, for the Pd-L′ catalytic system, it is the S configuration product that seems easier to be formed, and this result is in contrast with the Pd-L catalytic system.
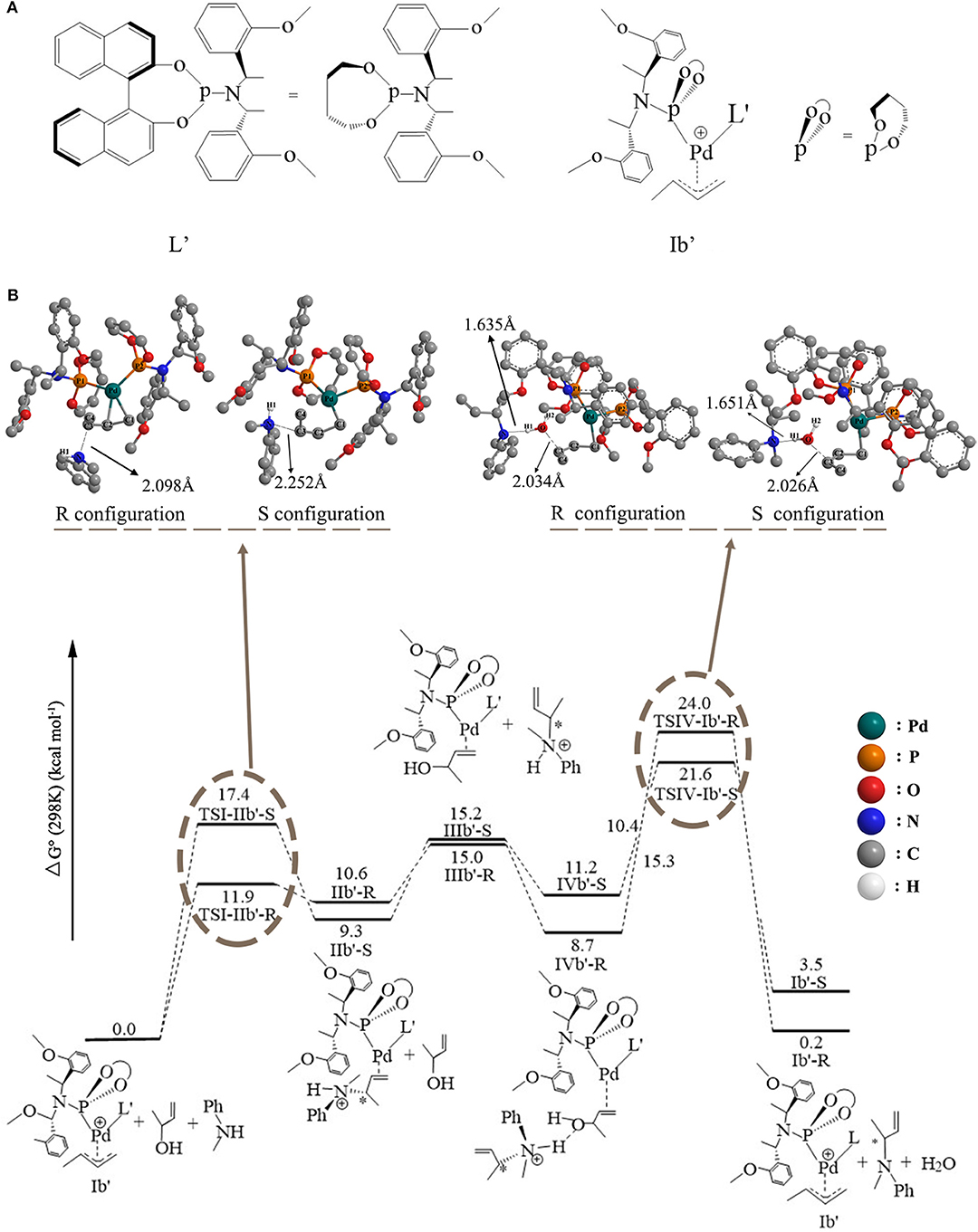
Figure 7. (A) The structure of phosphoramidite ligand (L′). (B) The overall free energetic profiles and structures for the 3-butene-2-ol amination reaction process in THF catalyzed by phosphoramidite Pd-L′.
To figure out the reason why the ligand axial chirality could significantly influence the preponderant reactive route, we visualized the corresponding structure of the TSIV-Ib′-R and the TSIV-Ib′-S state. As shown in Figure 8, for the TSIV-Ib′-R state, the space around the butenyl of the N-butenyl-N-methylaniline is relative smaller than that of the TSIV-Ib′-S state. The relative small space could lead to a stronger steric hindrance in the R chiral configuration structure, which corresponds to the energy barriers of TSIV-Ib′-S (10.4 kcal/mol) and TSIV-Ib′-R (15.3 kcal/mol). Therefore, the axial chiral ligand greatly affected the preponderant reactive route by the steric hindrance. What is more, most energy values of the Pd-L′ catalytic system are lower than that of the Pd-L catalytic system, suggesting that the Pd-L′ catalytic system might be superior to the Pd-L catalytic system.
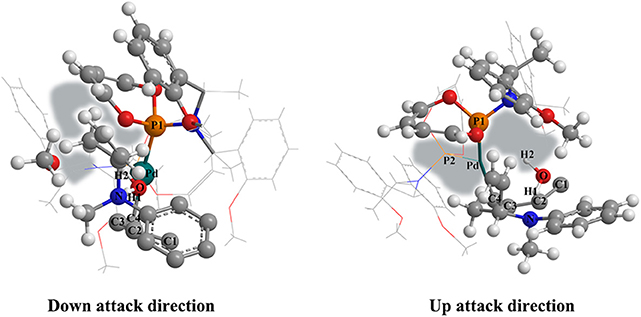
Figure 8. Visualizing the structure of the TSIV-Ib′-R state (left picture) and the TSIV-Ib′-S state (right picture). The space around the butenyl of the N-butenyl-N-methylaniline has been painted in light gray.
We also calculate the ADCH charge for the N (methylaniline) and the Pd (Table 2), which is similar to that of the Pd-L catalytic system process; the ADCH charge for N is −0.406, and the ADCH charge value for the Pd is 0.275. After the nucleophilic attack of the methylaniline, there is a covalent bond formed between the N and the butenyl. In the IIb′-R/S state, the ADCH charge of N increases to 0.016/0.070. In addition, because the butenyl tends to form the conjugation with the methylaniline, the electrons flow back to the Pd from the butenyl, which leads to the ADCH charge for the Pd decreasing to 0.085/0.086 (IIb′-R/S). In the IIIb′-R/S state, the ADCH charge for N and Pd basically maintains their value. The ADCH charge for the N is 0.070/0.082 in the IVb′-R/S state. The ADCH charge for the Pd is 0.159/0.110 in the IVb′-R/S state. These changes of the ADCH charge values also indicate that the up-attack direction of the methylaniline is the preferred reactive route in the Pd-L′ catalytic system and could help us to better understand the reactive mechanism.
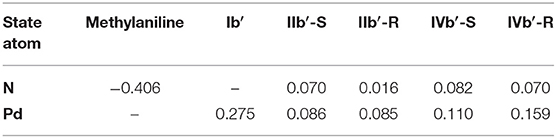
Table 2. The ADCH charge of the N and the Pd in different states for Pd-L′ catalytic system.
Conclusion
As the palladium-catalyzed asymmetry unsaturated olefin substitution reactions become more and more popular, understanding the catalytic mechanism has been an essential mission. In the present study, we utilized the DFT approach to explore the mechanism of palladium-catalyzed 3-butene-2-ol and methylaniline amination reaction. According to our computational results, the high regio- and stereoselectivity of the catalyst could influence the reactive route of the reaction, which induces different preponderant chiral product. This view could be proved by the high ratio of the chiral products in the experiment (Kimura et al., 2003; Piechaczyk et al., 2006; Banerjee et al., 2014). The high regio- and stereoselectivity is caused by the steric hindrance between the substrate and the ligand. Furthermore, this point is verified by introducing the axial chirality on the catalyst ligand. In the Pd-L catalytic system, down-attack direction of the nucleophile acts as the preponderant reaction route, during which process it tends to form R configuration product; in contrast, the up-attack direction of the nucleophile becomes the preponderant reaction route, and it tends to form S configuration product in the Pd-L′ catalytic system. These results shed light on the formation process of the 3-butene-2-ol and methylaniline amination reaction, which might lead a deeper understanding between the catalyst ligand and the substrate and provide theoretical instructions for the catalyst ligand design and optimization in industry.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
LL performed research and wrote the article. WF and SY analyzed the data. HL and XH designed the research and wrote the article.
Funding
This work was supported by the National Natural Science Foundation of China (No. 21573090) and the Scientific Research Fund of Jilin Provincial Education Department (2015437).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2020.00048/full#supplementary-material
References
Banerjee, D., Junge, K., and Beller, M. (2014). Cooperative catalysis by palladium and a chiral phosphoric acid: enantioselective amination of racemic allylic alcohols. Angew. Chem. Int. Ed. 53, 13049–13053. doi: 10.1002/anie.201405511
Becke, A. D. (1988). Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A Gen. Phys. 38, 3098–3100. doi: 10.1103/PhysRevA.38.3098
Fors, B. P., and Buchwald, A. C. S. (2010). A multiligand based Pd catalyst for C– N cross-coupling reactions. J. Am. Chem. Soc. 132, 15914–15917. doi: 10.1021/ja108074t
Frisch, M., Trucks, G., Schlegel, H., Scuseria, G., Robb, M., Cheeseman, J., et al. (2009). Gaussian 09 Revision D. Wallingford: Gaussian Inc.
Fukui, K. (1970). A formulation of the reaction coordinate. J. Phys. Chem. 74:4161. doi: 10.1021/j100717a029
Fukui, K. (1981). The path of chemical reactions - the IRC approach. Accoun. Chem. Res. 14, 363–368. doi: 10.1021/ar00072a001
Goldberg, I. (1906). Ueber phenylirungen bei gegenwart von kupfer als katalysator. Berichte der Deutschen Chemischen Gesellschaft 39, 1691–1692. doi: 10.1002/cber.19060390298
Grimme, S., Antony, J., Ehrlich, S., and Krieg, H. (2010). A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132:154104. doi: 10.1063/1.3382344
Hariharan, P. C., and Pople, J. A. (1973). The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 28, 213–222. doi: 10.1007/BF00533485
Hartwig, J. F., Kawatsura, M., Hauck, S. I., Shaughnessy, K. H., and Alcazar-Roman, L. M. (1999). Room-temperature palladium-catalyzed amination of aryl bromides and chlorides and extended scope of aromatic C– N bond formation with a commercial ligand. J. Org. Chem. 64, 5575–5580. doi: 10.1021/jo990408i
Hartwig, J. F., Richards, S., Barañano, D., and Paul, A. C. S. (1996). Influences on the relative rates for C– N bond-forming reductive elimination and β-hydrogen elimination of amides. A case study on the origins of competing reduction in the palladium-catalyzed amination of aryl halides. J. Am. Chem. Soc. 118, 3626–3633. doi: 10.1021/ja954121o
Hehre, W. J., Ditchfield, K., and Pople, J. A. (1972). Self-consistent molecular orbital methods. XII. Further extensions of gaussian-type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 56, 2257–2261. doi: 10.1063/1.1677527
Herz, K., Podewitz, M., Stohr, L., Wang, D. R., Frey, W., Liedl, K. R., et al. (2019). Mechanism of olefin metathesis with neutral and cationic molybdenum imido alkylidene N-heterocyclic carbene complexes. J. Am. Chem. Soc. 141, 8264–8276. doi: 10.1021/jacs.9b02092
Jiang, G. B., Li, J. X., Zhu, C. L., Wu, W. Q., and Jiang, H. F. (2017). Palladium-catalyzed sequential nucleophilic addition/oxidative annulation of bromoalkynes with benzoic acids to construct functionalized isocoumarins. Org. Lett. 19, 4440–4443. doi: 10.1021/acs.orglett.7b01919
Kimura, M., Futamata, M., Shibata, K., and Tamaru, Y. (2003). Pd center dot Et3B-catalyzed alkylation of amines with allylic alcohols. Chem. Commun. 234–235. doi: 10.1039/b210920d
Kinoshita, H., Shinokubo, H., and Oshima, K. (2004). Water enables direct use of allyl alcohol for Tsuji-Trost reaction without activators. Org. Lett. 6, 4085–4088. doi: 10.1021/ol048207a
Lu, T., and Chen, F. (2012a). Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592. doi: 10.1002/jcc.22885
Lu, T., and Chen, F. W. (2012b). Atomic dipole moment corrected hirshfeld population method. J. Theor. Comput. Chem. 11, 163–183. doi: 10.1142/S0219633612500113
Lu, Z., and Ma, S. M. (2008). Metal-catalyzed enantioselective allylation in asymmetric synthesis. Angew. Chem. Int. Ed. 47, 258–297. doi: 10.1002/anie.200605113
Major, M. M., Rovid, G., Balogh, S., Benyei, A., Lendvay, G., Frigyes, D., et al. (2019). Double stereoselective coordination of chiral N,S ligands: synthesis, coordination chemistry and utilization in Pd-catalyzed allylic alkylation. Appl. Organomet. Chem. 33:e4726. doi: 10.1002/aoc.4726
Ozawa, F., Ishiyama, T., Yamamoto, S., Kawagishi, S., and Murakami, H. (2004). Catalytic C-O bond cleavage of allylic alcohols using diphosphinidenecyclobutene-coordinated palladium complexes. A mechanistic study. Organometallics 23, 1698–1707. doi: 10.1021/om030682+
Ozawa, F., Okamoto, H., Kawagishi, S., Yamamoto, S., Minami, T., and Yoshifuji, M. (2002). (pi-allyl)palladium complexes bearing diphosphinidenecyclobutene ligands (DPCB): Highly active catalysts for direct conversion of allylic alcohols. J. Am. Chem. Soc. 124, 10968–10969. doi: 10.1021/ja0274406
Perdew, J. P. (1986). Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 33, 8822–8824. doi: 10.1103/PhysRevB.33.8822
Piechaczyk, O., Thoumazet, C., Jean, Y., and le Floch, P. (2006). DFT study on the palladium-catalyzed allylation of primary amines by allylic alcohol. J. Am. Chem. Soc. 128, 14306–14317. doi: 10.1021/ja0621997
Raducan, M., Alam, R., and Szabo, K. J. (2012). Palladium-catalyzed synthesis and isolation of functionalized allylboronic acids: selective, direct allylboration of ketones. Angew. Chem. Int. Ed. 51, 13050–13053. doi: 10.1002/anie.201207951
Ramakrishnan, S., Moretti, R. A., and Chidsey, C. E. D. (2019). Mapping free energy regimes in electrocatalytic reductions to screen transition metal-based catalysts. Chem. Sci. 10, 7649–7658. doi: 10.1039/c9sc01766f
Schäfer, A., Horn, H., and Ahlrichs, R. (1992). Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 97, 2571–2577. doi: 10.1063/1.463096
Sundararaju, B., Achard, M., and Bruneau, C. (2012). Transition metal catalyzed nucleophilic allylic substitution: activation of allylic alcohols via pi-allylic species. Chem. Soc. Rev. 41, 4467–4483. doi: 10.1039/c2cs35024f
Trost, B. M., and Crawley, M. L. (2003). Asymmetric transition-metal-catalyzed allylic alkylations: Applications in total synthesis. Chem. Rev. 103, 2921–2943. doi: 10.1021/cr020027w
Ullmann, F., and Bielecki, G. (1901). Ueber synthesen in der biphenylreihe. Berichte der Deutschen Chemischen Gesellschaft 34, 2174–2185. doi: 10.1002/cber.190103402141
Weigend, F., Furche, F., and Ahlrichs, R. (2003). Gaussian basis sets of quadruple zeta valence quality for atoms H-Kr. J. Chem. Phys. 119, 12753–12762. doi: 10.1063/1.1627293
Yang, K. S., Gurak, J. A., Liu, Z., and Engle, K. M. (2016). Catalytic, regioselective hydrocarbofunctionalization of unactivated alkenes with diverse C-H nucleophiles. J. Am. Chem. Soc. 138, 14705–14712. doi: 10.1021/jacs.6b08850
Yang, Q., Wang, Q. F., and Yu, Z. K. (2015). Substitution of alcohols by N-nucleophiles via transition metal-catalyzed dehydrogenation. Chem. Soc. Rev. 44, 2305–2329. doi: 10.1039/c4cs00496e
Ying, F., Zhang, Y. T., Xiang, C. Y., Song, Z. J., Xie, H. J., and Bao, W. L. (2019). Key mechanistic features in palladium-catalyzed methylcyclopropanation of norbornenes with vinyl bromides: insights from DFT calculations. Front. Chem. 7:169. doi: 10.3389/fchem.2019.00169
Keywords: amination, alkene alcohol, palladium catalyzed, axial chirality, DFT calculations
Citation: Lyu L, Feng W, Yang S, Liu H and Huang X (2020) Exploring the Mechanism of the Palladium-Catalyzed 3-Butene-2-ol Amination Reaction: A DFT Study. Front. Chem. 8:48. doi: 10.3389/fchem.2020.00048
Received: 26 November 2019; Accepted: 15 January 2020;
Published: 21 February 2020.
Edited by:
Yong Wang, Ningbo University, ChinaCopyright © 2020 Lyu, Feng, Yang, Liu and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huiling Liu, aHVpbGluZ0BqbHUuZWR1LmNu; Xuri Huang, aHVhbmd4ckBqbHUuZWR1LmNu