 Jiaocen Guo1,2†
Jiaocen Guo1,2† Fandong Kong1†Qingyun Ma1Qingyi Xie1Renshuai Zhang3
Fandong Kong1†Qingyun Ma1Qingyi Xie1Renshuai Zhang3 Haofu Dai1Yougen Wu2*
Haofu Dai1Yougen Wu2* Youxing Zhao1*
Youxing Zhao1*- 1Hainan Key Laboratory for Research and Development of Natural Product From Li Folk Medicine, Institute of Tropical Bioscience and Biotechnology, Chinese Academy of Tropical Agriculture Sciences, Haikou, China
- 2College of Horticulture, Hainan University, Haikou, China
- 3Qingdao Cancer Institute, The Affiliated Hospital of Qingdao University, Qingdao, China
Ganoderma fungi have long been used as functional foods and traditional medicines in Asian countries. Ganoderma ahmadii is one of the main species of Ganoderma fungi distributed in Hainan province of China, the fruiting bodies of which have been used in folk to lower blood sugar for a long time. A chemical investigation of the fruiting bodies of Ganoderma ahmadii led to the isolation of seven new meroterpenoids, named ganoduriporols F-L (1–7). The chemical structures of the compounds were elucidated by spectroscopic data including HRESIMS and 2D NMR. Compounds 5–7 represent the first examples of ganoduriporol-type meroterpenoids bearing oxepane rings in their skeletons. Compounds 1–4 showed inhibitory activity against protein tyrosine phosphatase 1B (PTP1B) comparable to the positive control Na3VO4, with IC50 values of 17, 20, 19, and 23 μM, respectively.
Introduction
Ganoderma fungi have been widely used as functional foods and traditional medicines, which have provided more efficient means for human health care, nutrition, medical care in China (Ma et al., 2019). It has been regarded as one of the most important medicinal fungi for preventing and treating various human diseases in Asian countries (Paterson, 2006). Previous studies have showed that the bioactive constituents of these fungi are mainly triterpenoids (Baby et al., 2015), polysaccharides (Wang et al., 2014), alkaloids (Zhao et al., 2015), and meroterpenoids (Yan et al., 2013) etc. These compounds with diverse structures displayed various biological effects, such as anti-tumor (Fu et al., 2019), anti-inflammatory (Lu et al., 2019), anti-diabetes (Wang et al., 2017), immunomodulation (Ji et al., 2007), and anti-oxidation activities (Qiu et al., 2016). Recently, a great deal of work on Ganoderma fungi have found that some constituents extracted from Ganoderma can promote the release of serum insulin and decrease the plasma glucose concentration in vivo (Huang et al., 2010; Li et al., 2017; Zhao and He, 2018).
Recent studies on the pathological mechanism revealed that type 2 diabetes has a close relationship with the protein tyrosine phosphatase family, which plays an important role in the negative regulator of insulin signaling by dephosphorylating the tyrosine residues of proteins (Tamrakar et al., 2014). PTP1B is an important member of the protein tyrosine phosphatase family and is responsible for insulin signaling pathway (Wang et al., 2015). Insulin resistance caused by expression of PTP1B as well as dephosphorylation of its target is one of the main causes of type 2 diabetes (Cai et al., 2015). Thus, PTP1B has been identified as a target for research and development of new drugs for the treatment of type 2 diabetes, and PTP1B inhibitors are potential lead compounds for such new drugs (Teng et al., 2011).
Ganoderma ahmadii is mainly distributed in Hainan, Yunnan, and Guizhou provinces in China (Wu and Dai, 2005), which have been used in folk medicine to lower blood sugar for a long time. As our ongoing search for bioactive constituents from the genus Ganoderma (Zhang et al., 2015; Huang et al., 2016, 2017), the bioactive constituents from G. ahmadii was studied, which led to the isolation of three new meroterpenoids with PTP1B inhibitory activity (Guo et al., 2019). A continuous research resulted in the isolation of another seven new meroterpenoids, named ganoduriporols F-L (1–7). Herein, the isolation, structural characterization, and PTP1B inhibitory activities of these compounds are reported.
Materials and Methods
General Experimental Procedures
The NMR spectra were recorded on a Bruker AV-500 spectrometer (Bruker, Bremen, Germany), and using tetramethylsilane (TMS) as an internal standard. Chemical shifts (δ) were expressed in ppm with reference to TMS. High Resolution Electrospray Ionization Mass Spectroscopy (HRESIMS) data were acquired using a mass spectrometer API QSTAR Pulsar (Bruker, Bremen, Germany). Optical rotations were measured using a JASCO P-1020 digital polarimeter. UV spectra were obtained with a Beckman DU 640 spectrophotometer. IR spectra were recorded on with a Shimadzu UV2550 spectrophotometer (Japan). Semipreparative high-performance liquid chromatography (HPLC) equipped with octadecyl silane (ODS) column (COSMOSIL-pack ODS-A, 10 × 250 nm, 5 μm, 4 ml/min) and phenyl (PH) column (COSMOSIL-pack ph, 10 × 250 nm, 5 μm, 4 ml/min) were used to isolate compounds. Silica gel (200-300 mesh; Qingdao Marine Chemical Inc., Qingdao, China) and Sephadex LH-20 (40–70 μm; Merck, Darmstadt, Germany) were used for column chromatography. Thin-layer chromatography (TLC) was carried out with precoated Si gel plates.
Plant Material
The Ganoderma ahmadii were collected in June 2017 Qiongzhong County, Hainan Province, China. The fungal material was identified by Prof. Zeng Nian-Kai (Hainan Medical University, China). The certified specimen (No.011-ZLZ) was deposited in the Institute of Tropical Bioscience and Biotechnology, Chinese Academy of Tropical Agricultural Sciences.
Extraction and Isolation
The dried and powdered fruiting bodies of G. ahmadii (5.0 kg) were extracted with 95% ethanol three times at room temperature. After filtration and evaporation, a gummy residue was obtained, which was taken up in H2O and with petroleum ether, ethyl acetate (EtOAc), and n-butanol. The EtOAc extract (55.0 g) was subjected to column chromatography (CC) on silica gel with gradient elution (petroleum ether-EtOAc, 8:1–1:2), which yielded seven fractions (Fr.1–Fr.7). Fr.6 (5.0 g) was further separated using an octadecyl silane silica gel column and eluted with gradient solvent of MeOH-H2O (30–100%) to give seven fractions (Fr.6.1–Fr.6.7), Fr.6.5 (116.0 mg) was purified by semipreparative HPLC [42% MeCN/H2O, containing 0.1% trifluoroacetic acid (TFA)] to afford compound 2 (tR 15.0 min; 3.5 mg). Fr.6.6 (140.0 mg) was further purified using semipreparative HPLC (70% MeOH/H2O, containing 0.1% TFA) to give compound 4 (tR 12.2 min; 3.0 mg). Fr.7 (6.0 g) was subjected to CC on an ODS elution with MeOH-H2O (20–100%) to give nine fractions (Fr.7.1–Fr.7.9). Fr.7.1 (2.2 g) was subjected to Sephadex LH-20 eluting with CHCI3-MeOH-petroleum ether (1:1:1) yielded three sub-fractions (Fr.7.1.1–7.1.3) based on TLC. Fr.7.1.2 (87.0 mg) was further purified by semipreparative HPLC (45% MeCN/H2O; containing 0.1% TFA) to afford compound 3 (tR 7.8 min; 3.0 mg). Fr.7.1.3 (70.0 mg) was subjected to semipreparative HPLC (65% MeOH/H2O; containing 0.1% TFA) to afford compound 6 (tR 32.0 min; 3.0 mg). Fr.7.2 (150.0 mg) was further purified by semipreparative HPLC (45% MeCN/H2O; containing 0.1% TFA) to afford compounds 7 (tR 20.7 min; 3.3 mg) and 5 (tR 22.2 min; 3.0 mg). Fr.7.7 (1.5 g) was subjected to Sephadex LH-20 (MeOH) to afford five fractions (Fr.7.7.1–Fr.7.7.5), in which Fr.7.7.4 (80.0 mg) was further prepared by semipreparative HPLC (72% MeOH/H2O, containing 0.1% TFA) to produce compound 1 (tR 14.2 min; 3.5 mg).
Characterization of Compounds 1–7
Ganoduriporol F (1): yellow oil; UV (MeOH) λ max (log ε) 307 (4.4), 224 (4.0) nm; IR (KBr) νmax (cm−1): 3436, 2925, 2851, 1630, 1388, 1168 (Figure S9); 1H and 13C NMR data, see Tables 1, 2; HRESIMS m/z 559.1938 [M + Na]+ (calcd for C30H32NaO9, 559.1939).
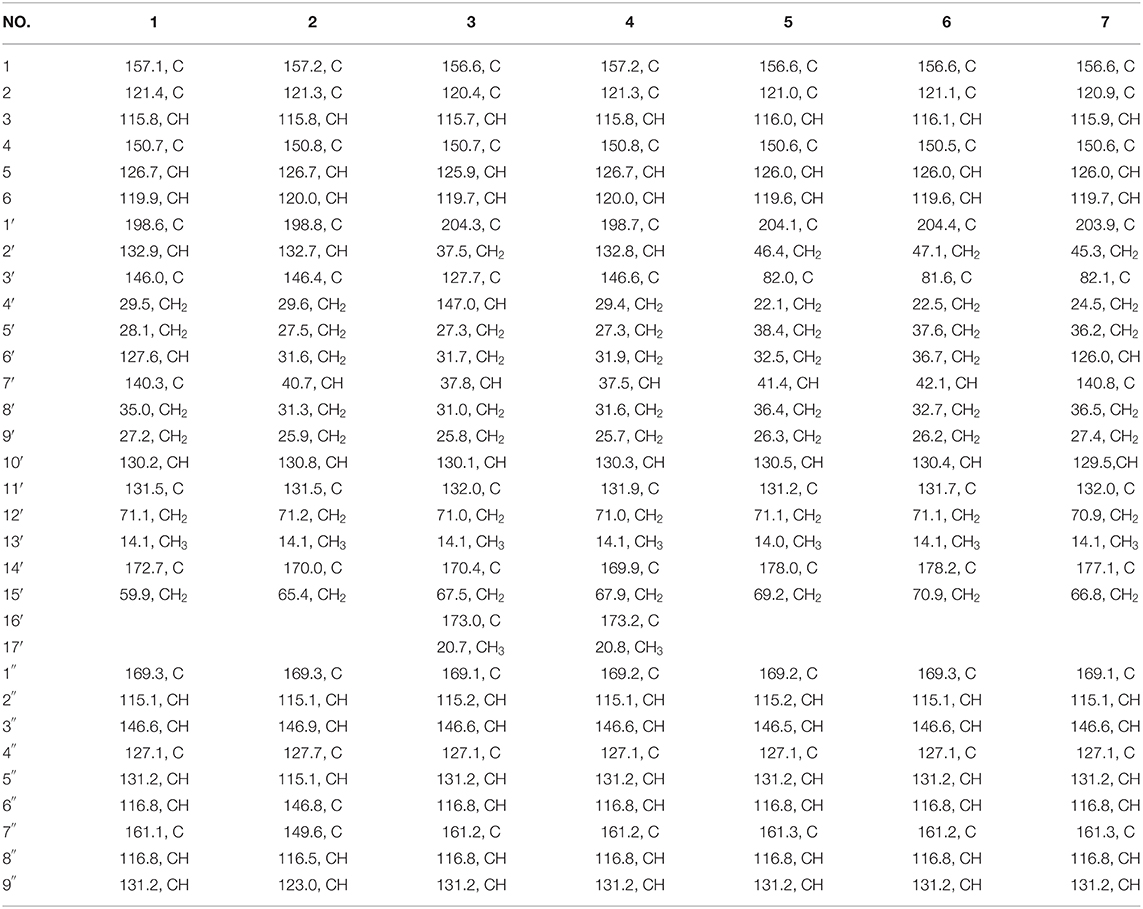
Table 1. 13C NMR (125 MHz) Data of Compounds 1–7 in CD3OD (δ in ppm).
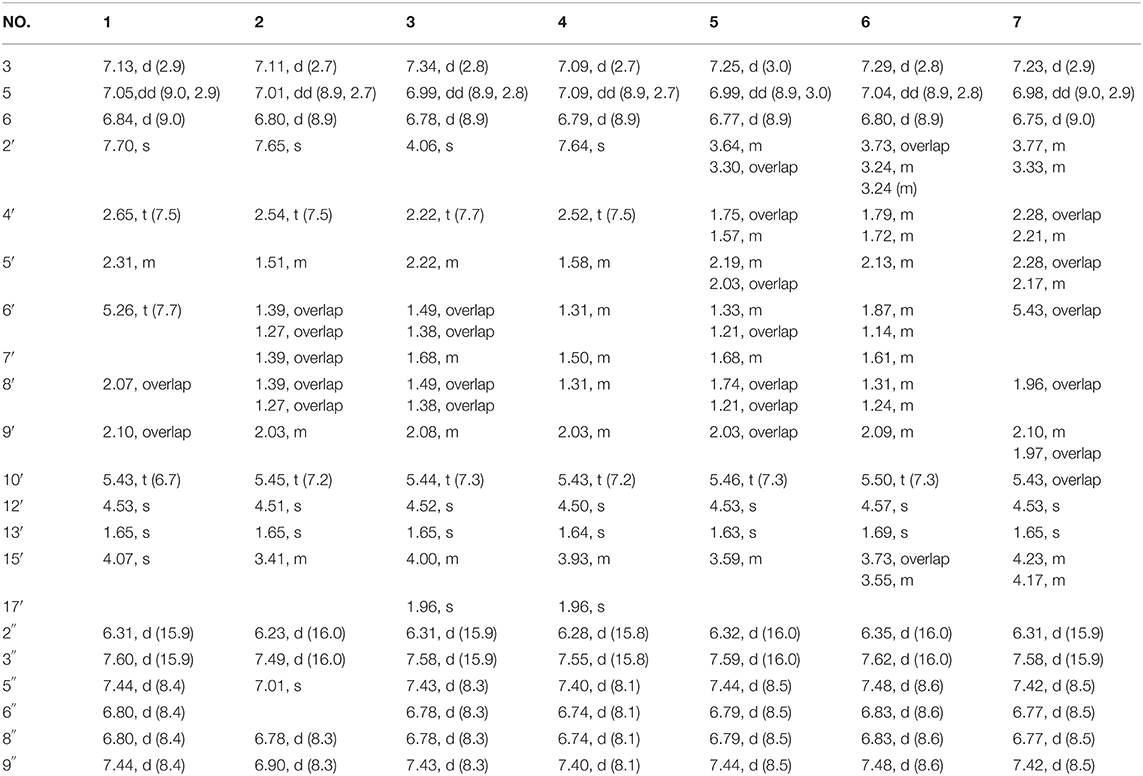
Table 2. 1H NMR (500 MHz) Data of Compounds 1-7 in CD3OD (δ in ppm, J in Hz).
Ganoduriporol G (2): yellow oil; +12 (c 0.1, MeOH); UV (MeOH) λ max (log ε) 328 (4.1), 288 (3.8) nm; IR (KBr) νmax (cm−1): 3414, 2927, 2859, 1610, 1474, 1263, 1195 (Figure S18); 1H and 13C NMR data, see Tables 1, 2; HRESIMS m/z 577.2046 [M + Na]+ (calcd for C30H34NaO10, 577.2044).
Ganoduriporol H (3): yellow oil; +10 (c 0.1, MeOH); UV (MeOH) λ max (log ε) 311 (4.3), 259 (4.0), 206 (4.8) nm; IR (KBr) νmax (cm−1): 3418, 2927, 2851, 1688, 1606, 1511, 1389, 1170 (Figure S27); 1H and 13C NMR data, see Tables 1, 2; HRESIMS m/z 603.2212 [M + Na]+ (calcd for C32H36NaO10, 603.2201).
Ganoduriporol I (4): yellow oil; +17 (c 0.1, MeOH); UV (MeOH) λ max (log ε) 292 (4.1), 247 (3.8) nm; IR (KBr) νmax (cm−1): 3435, 2926, 2851, 1685, 1603, 1449, 1268, 1169 (Figure S36); 1H and 13C NMR data, see Tables 1, 2; HRESIMS m/z 603.2169 [M + Na]+ (calcd for C32H36NaO10, 603.2201).
Ganoduriporol J (5): yellow oil; +8 (c 0.1, MeOH); UV (MeOH) λ max (log ε) 299 (4.2), 264 (4.0), 225 (4.5) nm; IR (KBr) νmax (cm−1): 3418, 2927, 2855, 1691, 1604, 1512, 1271, 1170 (Figure S45); 1H and 13C NMR data, see Tables 1, 2; HRESIMS m/z 561.2065 [M + Na]+ (calcd for C30H34NaO9, 561.2095).
Ganoduriporol K (6): yellow oil; +10 (c 0.1, MeOH); UV (MeOH) λ max (log ε) 312 (4.0), 224 (4.3) nm; IR (KBr) νmax (cm−1): 3422, 2928, 2847, 1682, 1602, 1474, 1280, 1198 (Figure S54); 1H and 13C NMR data, see Tables 1, 2; HRESIMS m/z 561.2147 [M + Na]+ (calcd for C30H34NaO9, 561.2095).
Ganoduriporol L (7): yellow oil; +14 (c 0.1, MeOH); UV (MeOH) λ max (log ε) 299 (4.4), 263 (4.1) nm; IR (KBr) νmax (cm−1): 3418, 2928, 1693, 1602, 1512, 1391, 1170 (Figure S63); 1H and 13C NMR data, see Tables 1, 2; HRESIMS m/z 559.1918 [M + Na]+ (calcd for C30H32NaO9, 559.1939).
PTP1B Inhibition Assay
The expressed and purified methods of recombinant PTP1B catalytic domain were the same as references (Liu et al., 2018). The dephosphorylation of p-nitrophenyl phosphate (pNPP) generated the product pNP, which can be monitored at an absorbance for 405 nm. The compounds were pre-incubated with the enzyme at 37 °C for 5 min. Assay was performed in the final volume of 100 μL in the active system containing 50 μL reaction buffer, 25.5 μL double distilled water, 2 μL test compounds and 10 μL enzymes. After incubation at 37°C for 15 min, the reaction was terminated using 0.5 M pNPP (12.5 μL). After initializing the enzymatic reaction, the plate was then read every 20 s for 15 min in the microplate reader at 405 nm. Sodium orthovanadate (Na3VO4) was used as a positive control. The equation used was: [(Ablank-Asample)/Ablank] × 100%. The IC50 values were determined by linear or non-linear regression analysis of the concentration-response data curve.
Cytotoxicity Assay
All isolated compounds 1-7 were evaluated for their cytotoxicity against BEL, K562, SGC7901, A549 and Hela cell lines. The cytotoxic activities were assayed by using the MTT method in 96 well plates according to the previous report (Shi et al., 2008).
Molecular Docking
Docking simulation referred to the published literature (Zhang et al., 2017). It was carried out by means of the SYBYL-X 2.0 software. All the ligand molecular were drawn using the standard parameters of SYBYL-X, then their geometric conformations were energy minimized employing the Tripos force field for 1,000 steps and Gasteiger-Huckel charges were calculated. Protein receptor (PDB: 1QXK) was prepared using the standard way. The H-bonds were shown using dotted line. Pymol was used as a viewer for interaction between ligands and protein receptor.
Results and Discussion
Identification of Compounds 1-7
Compound 1 was obtained as yellow oil, with a molecular formula of C30H32O9 from the molecular ion peak [M + Na]+ at m/z 559.1938 (calcd 559.1939) in the HRESIMS (Figure S8). The 1H NMR revealed diagnostic signals of a 2-substituted-1,4-dihydroxylbenzene moiety (δH 7.13, 7.05, and 6.84), a p-substituted hydroxybenzene substructure (δH 7.44 and 6.80), three olefinic singlets (δH 7.70, 5.26, and 5.43), two conjugated olefinic doublets (δH 6.31 and 7.60), and one methyl (δH 1.65). The 13C NMR and the DEPT spectra (Table 1, Figures S2, S3) showed a total of 30 carbon signals including one methyl, six methylenes with two oxygenated, twelve sp2 methines, and eleven sp2 quaternary carbons including three carboxylic or carbonyl carbons. Comparison of its 1H and 13C NMR spectral data (Tables 1, 2, Figures S1–S6) with those of ganoduriporol A (Chen et al., 2017) suggested that 1 had a similar structure to ganoduriporol A. Their main structural difference was that the /CH-3′ substructure in ganoduriporol A was dehydrogenated to form an tri-substituted double bond in 1, as suggested by heteronuclear multiple bond coherence spectroscopy (HMBC) correlations from the olefinic proton H-2′ (δH 7.70) to C-3′ (δC 146.0), C-4′ (δC 29.5), and C-1′ (δC 198.6). The configurations of the double bonds as 2′Z, 6′Z, 10′E, and 2″E were supported by rotating frame overhauser effect spectroscopy (ROESY) correlations (Figures 1, 2, Figure S7) of H-2′/H2-4′ (δH 2.65), H-6′ (δH 5.26)/H2-8′ (δH 2.07), H-10′ (δH 5.43)/H2-12′ (δH 4.53), and H-2″ (δH 6.31)/H-9″ (δH 7.44), respectively.
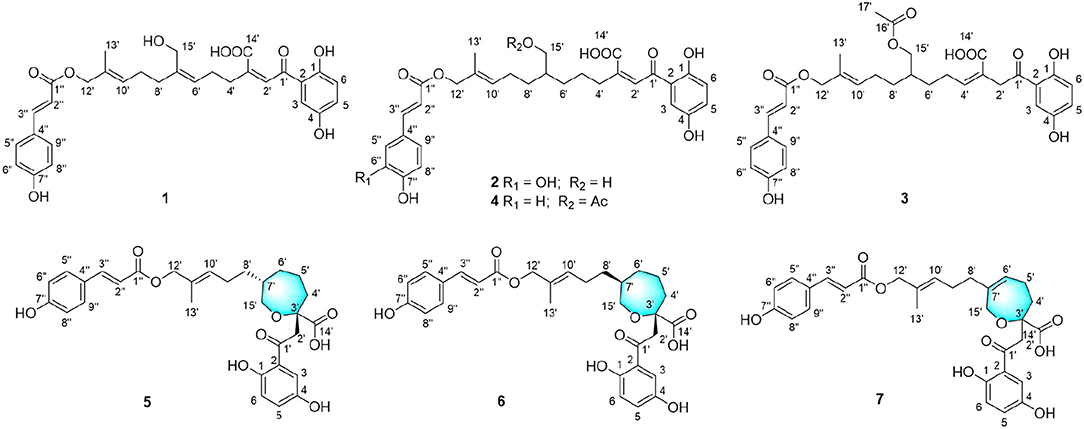
Figure 1. Structures of compounds 1–7.
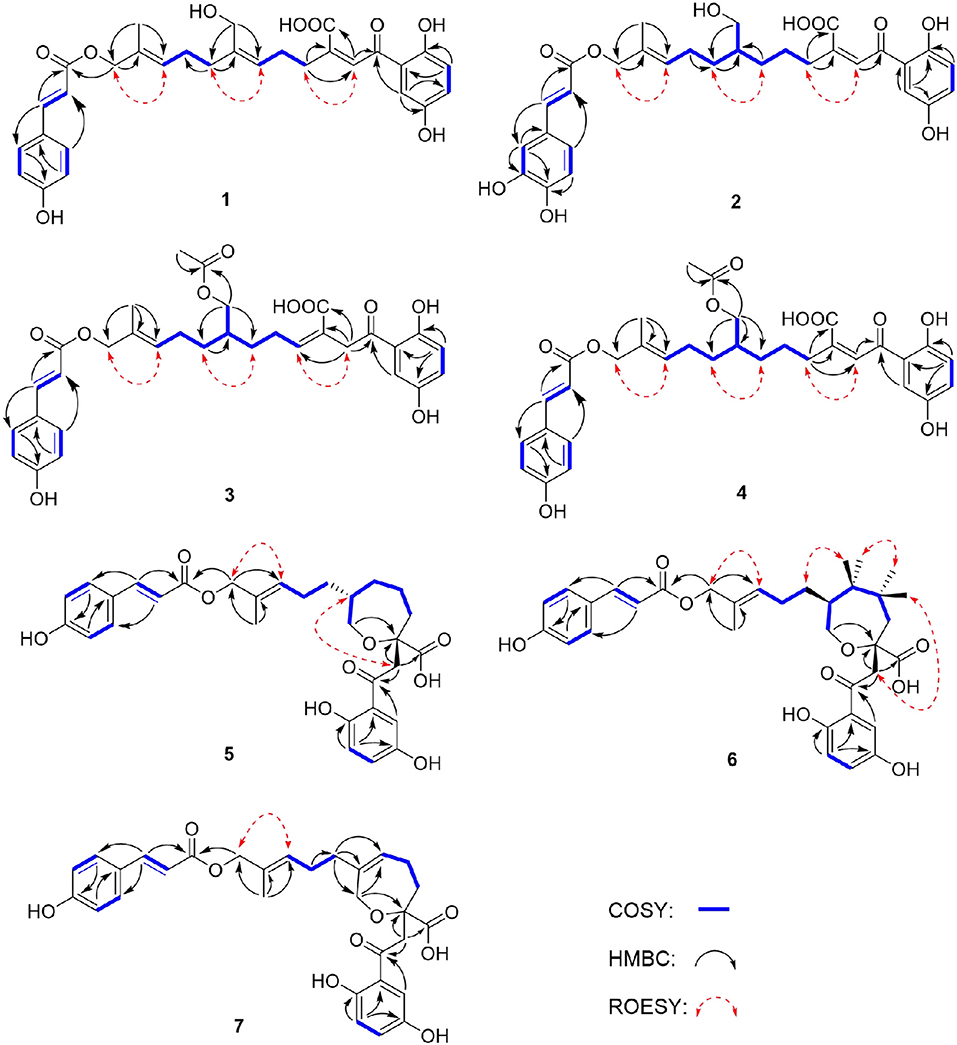
Figure 2. Key COSY, HMBC, and ROESY correlations of compounds 1–7.
Compound 2 was obtained as yellow oil with a molecular formula of C30H34O10 according to the HRESIMS data (Figure S17). Comparison of the NMR data (Tables 1, 2, Figures S10–S15) of 2 with those of 1 revealed the presence of an additional hydroxyl at C-6″ (δC 146.8) in 2, which was further confirmed by the HMBC correlations of H-5″ (δH 7.01) and H-8″ (δH 6.78) to C-6″ (δC 146.8). Besides, the presence of CH2-6′/CH-7′ fragment in 2 instead of olefinic double bond CH-6′/C-7′ as in 1 was revealed by HMBC correlations from H2-15′ (δH 3.41) to C-6′ (δC 31.6), C-7′ (δC 40.7), and C-8′ (δC 31.3) (Figures 1, 2). The configurations of 2′Z, 10′E and 2″E in compound 2 were revealed by ROESY correlations (Figure 2, Figure S16) of H-2′ (δH 7.65)/H2-4′ (δH 2.54), H-10′ (δH 5.45)/H2-12′ (δH 4.51), and H-2″ (δH 6.23)/H-5″.
Compound 3 was obtained as yellow oil, and its molecular formula C32H36O10 was concluded from the HRESIMS (Figure S26) m/z 603.2212 ([M + Na]+, calcd 603.2201), implying 15 degrees of unsaturation. Its 1H NMR and 13C NMR data (Tables 1, 2, Figures S19–S24) were very similar to those of ganoduriporol B (Chen et al., 2017) with the main difference being the presence of signals for an acetyl group, which located at C-15′ (δC 67.5) as suggested by HMBC correlations (Figures 1, 2) from H2-15′ (δH 4.00) and a singlet methyl proton signal at δH 4.00 to the ester carbonyl at δC 173.0. Besides, the CH-3′/CH2-4′ substructure in ganoduriporol B was dehydrogenated to afford an olefinic C-3′/CH-4′ double bond in 3, as revealed by HMBC correlations from (δH 4.06) to C-1′ (δC 204.3), C-3′ (δC 127.7), C-4′ (δC 147.0), and C-14′ (δC 170.4). The configurations of 3′Z, 10′E and 2″E in compound 3 were revealed by the ROESY correlations (Figure 2, Figure S25) of /H-4′ (δH 2.22), H-10′ (δH 5.44)/H2-12′ (δH 4.52), and H-2″ (δH 6.31)/H-5″ (δH 7.43).
Compound 4 was isolated as yellow oil. Its molecular formula was determined as C32H36O10 by HRESIMS (Figure S35), the same as that of 3. The NMR data of 4 were quite similar to those of 3 (Tables 1, 2). Analysis of the 2D NMR data (Figure 2, Figures S28–S33) of 4 suggested their only structural difference was the position of one double bond, which was located C-2′ (δC 132.8) and C-3′ (δC 146.6) in 4 instead of at C-3′ (δC 127.7) and C-4′ (δC 147.0) as in 3, as confirmed by HMBC correlations from the olefinic proton H-2′ (δH 7.64) to C-3′ (δC 146.6), C-4′ (δC 29.4), and C-1′ (δC 198.7). The configurations of 2′Z, 10′E, and 2″E in compound 4 were elucidated by ROESY correlations (Figures 1, 2, Figure S34) of H-2′/H2-4′ (δH 2.52), H-10′ (δH 5.43)/H2-12′ (δH 4.50), and H-2″ (δH 6.28)/H-9″ (δH 7.40).
Compound 5 was assigned the molecular formula as C30H34O9 on the basis of HRESIMS data (Figure S44), indicating 14 degrees of unsaturation. The 1H NMR and 13C NMR, together with 1H-detected heteronuclear single quantum coherence spectrum (HSQC), revealed the presence of one methyl, eight methylenes (two oxygenated), eleven methines, and ten quaternary carbons. Comparison of NMR data (Figures S37–S42) between compounds 5 and 1 found that the Δ2′ double bond in 1 was replaced by a methylene (δC/H 69.2/3.59) and an oxygenated quaternary carbon (δC 82.0), the latter of which was linked to C-2′ (δC 46.4) via an oxygen atom to form an oxepane rings, as suggested by HMBC correlations from (δH 3.64) to C-3′ (δC 82.0), C-14′ (δC 178.0), and C-1′ (δC 204.1) and from H2-15′ (δH 3.59) to C-3′. The other difference was that the double bond at C-6′ (δC 127.6) and C-7′ (δC 140.3) in 1 was replaced by a sp3 methine and methylene in 5, as confirmed by COSY correlations (Figures 1, 2) of H-7′ (δH 1.68) with H2-6′ (δH 1.21), H2-8′ (δH 1.74) and H2-15′. The E configurations of Δ10′ and Δ2″ double bonds were established by the ROESY correlations (Figure 2, Figure S43) from H-10′ (δH 5.46) to H2-12′ (δH 4.53) and the coupling constant (J = 16.0 Hz) between H-2″ (δH 6.32) and H-3″ (δH 7.59), respectively. ROESY correlations from (δH 3.64)/H-7′ (δH 1.68) suggested that and CH2-8′ were on the face opposite to each other.
Compound 6 was determined to have a molecular formula C30H34O9 based on HRESIMS (Figure S53) analysis, the same as that of 5. The 1D NMR data of 6 were almost identical to those of 5. Analysis of the 2D NMR data (Figures S46–S51) of 6 found that compounds 5 and 6 shared the same planar structure, indicating that they are a pair of stereoisomers. The E configuration of Δ10′ and Δ2″ double bonds were established by the ROESY correlations (Figures 1, 2, Figure S52) from H-10′ (δH 5.50) to (δH 4.57) and the coupling constant (J = 16.0 Hz) between H-2″ (δH 6.35) and H-3″ (δH 7.62), respectively. ROESY correlations from (δH 3.24)/H2-4′α (δH 1.72), and H2-4′β (δH 1.79)/β (δH 1.14), and H2-6′α (δH 1.87)/H2-8′α (δH 1.31) suggested that and were on the same face of the ring system, which is different with that of 5.
Compound 7 was obtained as a yellow oil with a molecular ion peak at m/z 559.1938 [M + Na]+ in HRESIMS (Figure S62), coinciding with the molecular formula C30H32O9. Comparison of the NMR data (Figures S55–S60) revealed that the structure of 7 was very similar to that of 5, with the difference being the CH2 (6′)-CH (7′) substructure in 5 was replaced by a tri-substituted double bond CH (6′)-C (7′) in 7, which was deduced from the HMBC correlations from H2-8′ (δH 1.96) to C-6′ (δC 126.0) and C-7′ (δC 140.8). The configurations of 6′Z, 10′E, and 2″E in compound 7 were elucidated by ROESY correlations (Figure S61) of H2-8′ (δH 1.96)/H-6′ (δH 5.43), H-10′ (δH 5.43)/H2-12′ (δH 4.53), and H-2″ (δH 6.31)/H-9″ (δH 7.42).
All compounds isolated were evaluated for their inhibitory activities against PTP1B using pNPP as a substrate and cytotoxicities against BEL, K562, SGC7901, A549, and Hela human cell lines using the MTT method. All of the compounds were non-cytotoxic against the tested tumor cell lines. Compounds 1-4 showed obvious inhibitory activity against PTP1B with IC50 values of 17, 20, 19, and 23 μM (Figure S64), respectively, comparable to the positive control Na3VO4 (IC50 = 12 μM). A positive effort was made to explain the activity of compound 1 against PTP1B by performing molecular docking (Figure 3). Docking results implied that 1 binds deep in the active site pocket and form H-bonds with ALA-217 and GLN-266, and the 7″-OH also formed H-bond with ARG-24 which was located in the so-called secondary binding site of PTP1B. Thus, it is a potent active molecule against PTP1B, with the ability to interact with both bonding sites. Based on the above research, we believed that it was feasible and reasonable to obtain PTP1B inhibitors with medicinal potential through appropriate structural modifications of these compounds. The cytotoxicity assessment also further provided evidence for this idea, for that all the compounds were inactive against the tested tumor cell lines.
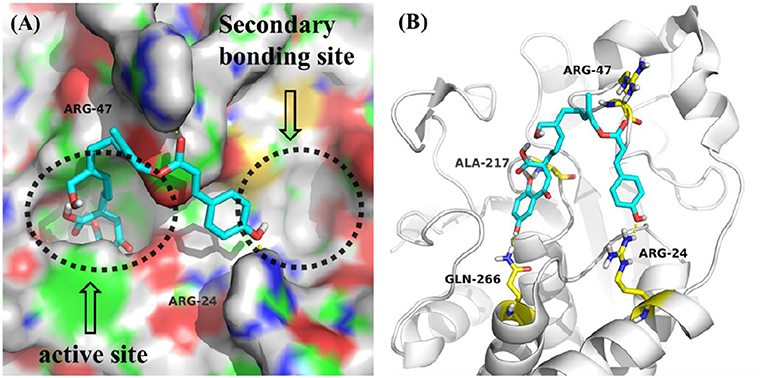
Figure 3. (A) Model of compound 1 (cyans) bound to PTP1B (PDB: 1QXK). (B) Interactions between compound 1 (cyans) and PTP1B. The key amino acids were shown in yellow.
Conclusions
In summary, seven new meroterpenoids were isolated and identified from the fruiting bodies of G. ahmadii. Among them, compounds 1–4 exhibited inhibitory activity against PTP1B but no cytotoxicity against the tested five tumor cell lines, suggesting that it has great potential to obtain new PTP1B inhibitors with medicinal use through appropriate structural modifications of these compounds. The possible mechanisms of these compounds against PTP1B were also revealed by molecular docking experiment. These findings once again proved the great medicinal values of Ganoderma fungi.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
JG and FK performed the experiments. QM contributed to the bioassays. QX collected the fruiting bodies of G. ahmadii. RZ was responsible for edited pictures. YZ and YW designed the work and revised the paper. All authors have approved the final version of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by Natural Science Foundation of Hainan Province (219MS078), Natural Science Foundation of China (81973568), China Agriculture Research System (CARS-21), and Central Public-interest Scientific Institution Basal Research Fund for Chinese Academy of Tropical Agricultural Sciences (17CXTD-15, 1630052016008).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2020.00279/full#supplementary-material
References
Baby, S., Johnson, A. J., and Govindan, B. (2015). Secondary metabolites from Ganoderma. Phytochemistry 114, 66–101. doi: 10.1016/j.phytochem.2015.03.010
Cai, J. Y., Zhao, L., and Tao, W. (2015). Potent protein tyrosine phosphatase 1B (PTP1B) inhibiting constituents from Anoectochilus chapaensis and moleculardocking studies. Pharm. Biol. 53, 1030–1034. doi: 10.3109/13880209.2014.957781
Chen, C., Liang, F., Chen, B., Sun, Z. Y., Xue, T. D., Yang, R. L., et al. (2017). Identifification of demethylincisterol A3 as a selective inhibitor of protein tyrosine phosphatase Shp2. Eur. J. Pharmacol. 795, 124–133. doi: 10.1016/j.ejphar.2016.12.012
Fu, Y. L., Shi, L., and Ding, K. (2019). Structure elucidation and anti-tumor activity in vivo of a polysaccharide from spores of Ganoderma lucidum (Fr.) Karst. Int. J. Biol. Macromol. 141, 693–699. doi: 10.1016/j.ijbiomac.2019.09.046
Guo, J. C., Ma, Q. Y., Kong, F. D., Xie, J. Y., Zhou, L. M., Ding, Q., et al. (2019). Meroterpenoids from the fruiting bodies of Ganoderma ahmadii Steyaret and their PTP1B inhibitory activities. Chin. J. Org. Chem. 39, 3264–3268. doi: 10.6023/cjoc201905010
Huang, C. Y., Chen, J. Y. F., Wu, J. E., Pu, Y. E., Liu, G. Y., Pan, M. H., et al. (2010). Ling-Zhi polysaccharides potentiate cytotoxic effects of anticancer drugs against drug-resistant urothelial carcinoma cells. J. Agric. Food. Chem. 58, 8798–8805. doi: 10.1021/jf1020158
Huang, S. Z., Cheng, B. H., Ma, Q. Y., Wang, Q., Kong, F. D., Dai, H. F., et al. (2016). Anti-allergic prenylated hydroquinonesand alkaloids from the fruiting body of Ganoderma calidophilum. RSC. Adv. 6, 21139–21147. doi: 10.1039/C6RA01466F
Huang, S. Z., Ma, Q. Y., Kong, F. D., Guo, Z. K., Cai, C. H., Hu, L. L., et al. (2017). Lanostane-type triterpenoids from the fruiting body of Ganoderma calidophilum. Phytochemistry 143, 104–110. doi: 10.1016/j.phytochem.2017.07.015
Ji, Z., Tang, Q. J., Zhang, J. S., Yang, Y., Jia, W., and Pan, Y. J. (2007). Immunomodulation of RAW264.7 macrophages by GLIS, a proteopolysaccharide from Ganoderma lucidum. J. Ethnopharmacol. 112, 445–450. doi: 10.1016/j.jep.2007.03.035
Li, K., Na, K., Sang, T. T., Wu, K. K., Wang, Y., and Wang, X. Y. (2017). The ethanol extracts of sporoderm-broken spores of Ganoderma lucidum inhibit colorectal cancer in vitro and in vivo. Oncol. Rep. 38, 2803–2813. doi: 10.3892/or.2017.6010
Liu, J. Q., Lian, C. L., Hu, T. Y., Wang, C. F., Xu, Y., Xiao, L., et al. (2018). Two new farnesyl phenolic compounds with anti-inflammatory activities from Ganoderma duripora. Food. Chem. 263, 155–162. doi: 10.1016/j.foodchem.2018.04.097
Lu, S. Y., Peng, X. R., Dong, J. R., Yan, H., Kong, Q. H., Shi, Q. Q., et al. (2019). Aromatic constituents from Ganoderma lucidum and their neuroprotective and anti-inflammatory activities. Fitoterapia 134, 58–64. doi: 10.1016/j.fitote.2019.01.013
Ma, L. F., Yan, J. J., Lang, H. Y., Jin, L. C., Qiu, F. J., Wang, Y. J., et al. (2019). Bioassay-guided isolation of lanostane-type triterpenoids as α-glucosidase inhibitors from Ganoderma hainanense. Phytochem. Lett. 29, 154–159. doi: 10.1016/j.phytol.2018.12.007
Paterson, R. R. M. (2006). Ganoderma-a therapeutic fungal biofactory. Phytochemistry 67, 1985–2001. doi: 10.1016/j.phytochem.2006.07.004
Qiu, J., Wang, X., and Song, C. (2016). Neuroprotective and antioxidant lanostanoid triterpenes from the fruiting bodies of Ganoderma atrum. Fitoterapia 109, 75–79. doi: 10.1016/j.fitote.2015.12.008
Shi, L., Yu, H. P., Zhou, Y. Y., Du, J. Q., Shen, Q., Li, J. Y., et al. (2008). Discovery of a novel competitive inhibitor of PTP1B by high-throughput screening. Acta. Pharmacol. Sin. 29, 278–284. doi: 10.1111/j.1745-7254.2008.00737.x
Tamrakar, A. K., Maurya, C. K., and Rai, A. K. (2014). PTP1B inhibitors for type 2 diabetes treatment: a patent review (2011-2014). Expert. Opin. Ther. Pat. 24, 1101–1115. doi: 10.1517/13543776.2014.947268
Teng, B. S., Wang, C. D., Yang, H. J., Wu, J. S., Zhang, D., Zhang, D., et al. (2011). A protein tyrosine phosphatase 1B activity inhibitor from the fruiting bodies of Ganoderma lucidum (Fr.) karst and its hypoglycemic potency on streptozotocin-induced type 2 diabetic mice. J. Agric. Food. Chem. 59, 6492–6500. doi: 10.1021/jf200527y
Wang, C. H., Hsieh, S. C., Wang, H. J., Chen, M. L., Lin, B. F., Chiang, B. H., et al. (2014). Concentration variation and molecular characteristics of soluble (1,3;1,6)-β-D-glucans in submerged cultivation products of Ganoderma lucidum mycelium. J. Agric. Food. Chem. 62, 634–641. doi: 10.1021/jf404533b
Wang, K., Bao, L., Ma, K., Zhang, J., Chen, B., Han, J., et al. (2017). A novel class of α-glucosidase and HMG-CoA reductase inhibitors from Ganoderma leucocontextum and the anti-diabetic properties of ganomycin I in KK-Ay mice. Eur. J. Med. Chem. 127, 1035–1046. doi: 10.1016/j.ejmech.2016.11.015
Wang, L. J., Jiang, B., Wu, N., Wang, S. Y., and Shi, D. Y. (2015). Small molecules as potent protein tyrosine phosphatase 1B (PTP1B) inhibitors documented in patents from 2009 to 2013. Mini. Rev. Med. Chem. 15, 104–122. doi: 10.2174/1389557515666150203144339
Wu, X. L., and Dai, Y. C. (2005). Coloured Illustartions of Ganodermatacete of China. Beijing: Science Press.
Yan, Y. M., Ai, J., Zhou, L. L., Chung, A. C. K., Li, R., Nie, J., et al. (2013). Lingzhiols, unprecedented rotary door-shaped meroterpenoids as potent and selective inhibitors of p-Smad3 from Ganoderma lucidum. Org. Lett. 15, 5488–5491. doi: 10.1021/ol4026364
Zhang, R., Yu, R., Xu, Q., Li, X., Luo, J., Jiang, B., et al. (2017). Discovery and evaluation of the hybrid of bromophenol and saccharide as potent and selective protein tyrosine phosphatase 1B inhibitors. Eur. J. Med. Chem. 134, 24–33. doi: 10.1016/j.ejmech.2017.04.004
Zhang, S. S., Wang, Y. G., Huang, S. Z., Hu, L. L., Dai, H. F., Yu, Z. F., et al. (2015). Three new lanostanoids from the mushroom Ganoderma tropicum. Molecules 20, 3281–3289. doi: 10.3390/molecules20023281
Zhao, R. L., and He, Y. M. (2018). Network pharmacology analysis of the anti-cancer pharmacological mechanisms of Ganoderma lucidum extract with experimental support using Hepa1-6-bearing C57 BL/6 mice. J. Ethnopharmacol. 210, 287–295. doi: 10.1016/j.jep.2017.08.041
Keywords: Ganoderma ahmadii, meroterpenoid, spectroscopic, PTP1B, cytotoxicity
Citation: Guo J, Kong F, Ma Q, Xie Q, Zhang R, Dai H, Wu Y and Zhao Y (2020) Meroterpenoids With Protein Tyrosine Phosphatase 1B Inhibitory Activities From the Fruiting Bodies of Ganoderma ahmadii. Front. Chem. 8:279. doi: 10.3389/fchem.2020.00279
Received: 31 December 2019; Accepted: 23 March 2020;
Published: 16 April 2020.
Edited by:
Prasat Kittakoop, Chulabhorn Graduate Institute, ThailandReviewed by:
Christophe Salome, SpiroChem AG, SwitzerlandFlorenci Vicent González, University of Jaume I, Spain
Copyright © 2020 Guo, Kong, Ma, Xie, Zhang, Dai, Wu and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yougen Wu, d3lnZW5nMjAwM0AxNjMuY29t; Youxing Zhao, emhhb3lvdXhpbmdAaXRiYi5vcmcuY24=
†These authors have contributed equally to this work