 Meng Zhao
Meng Zhao Yi Xiao1
Yi Xiao1 Satoshi Otsuka
Satoshi Otsuka Yoichi Nakao
Yoichi Nakao Tao Ye
Tao Ye- 1State Key Laboratory of Chemical Oncogenomics, Peking University Shenzhen Graduate School, Shenzhen, China
- 2Research Institute for Science and Engineering, Waseda University, Tokyo, Japan
- 3Department of Chemistry and Biochemistry, Graduate School of Advanced Science and Engineering, Waseda University, Tokyo, Japan
- 4Tsinghua Shenzhen International Graduate School, Xili, Shenzhen, China
Kakeromamide A (1), the first marine cyclopeptide inducing neural stem cells differentiation into astrocytes, was synthesized in 12 longest linear steps and 14% overall yield. Using this synthetic approach, four analogs of kakeromamide A were prepared and evaluated for neural differentiation- modulating activity.
Introduction
Marine cyanobacteria have been playing a pivotal role in producing structurally diversified and biologically intriguing peptides (Luesch et al., 2001; Guzman-Martinez et al., 2007; Zainuddin et al., 2007). In 2018, we reported the isolation of a novel cyclopeptide, kakeromamide A (Figure 1), from the marine cyanobacterium Moorea bouillonii collected near Japan (Nakamura et al., 2018). Preliminary biological tests revealed that kakeromamide A exhibits moderate cytotoxicity (IC50 = 10 μM) against Hela cells. To the best of our knowledge, this is the first example of marine cyclopeptide that can induce neural stem cells differentiation into astrocytes (2.5–10 μM). The constitutional information of kakeromamide A, featuring a peptidal macrocycle embedded with a thiazole ring and a β-amino acid, was revealed on the basis of in-depth spectroscopic analysis. The absolute configurations of kakeromamide A (1) were established by the advanced Marfey's method. Our laboratory's long-standing interest in the synthesis of bioactive marine natural products (Yang et al., 2000; Lei et al., 2014; Liao et al., 2016; Zhou et al., 2016; Guo et al., 2017; Cheng et al., 2018; Yu et al., 2019) prompted us to launch a synthetic program targeting kakeromamide A and its structural variants.
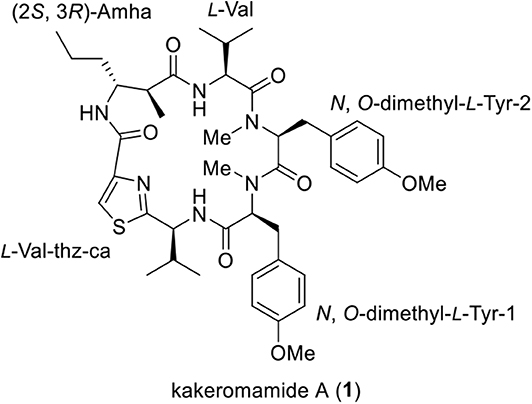
Figure 1. Structure of Kakeromamide A (1).
Materials and Methods
Experimental procedure and compound characterization data are furnished in the Supplementary Material (Data Sheet 1).
Results and Discussion
The retrosynthetic plan is depicted below (Figure 2). Strategic disconnection of the macrocycle at the least sterically demanding amide bond results in the linear pentapeptide 2. Further dissection of 2 reveals that the pentapeptide may be most conveniently constructed by the assembly of tripeptide 3 and dipeptide 4, derived from the disconnection of the bonds between two tyrosines. Tripeptide 3 and dipeptide 4 could be constructed from amino acid derivatives 5–8. The 3+2 strategy has the advantage in terms of convergence and efficiency. However, the amidation between polypeptidyl acids and N-methyl amines is sometimes unattainable because of notoriously latent epimerization (Humphrey and Chamberlin, 1997; Teixidó et al., 2005; Nabika et al., 2014). Therefore, as an alternative plan, we envisioned that the pentapeptide 2 could also be assembled in a linear fashion with corresponding amino acid synthons 5–8, which could circumvent the potential loss of stereo-integrity via oxazolone mechanism.
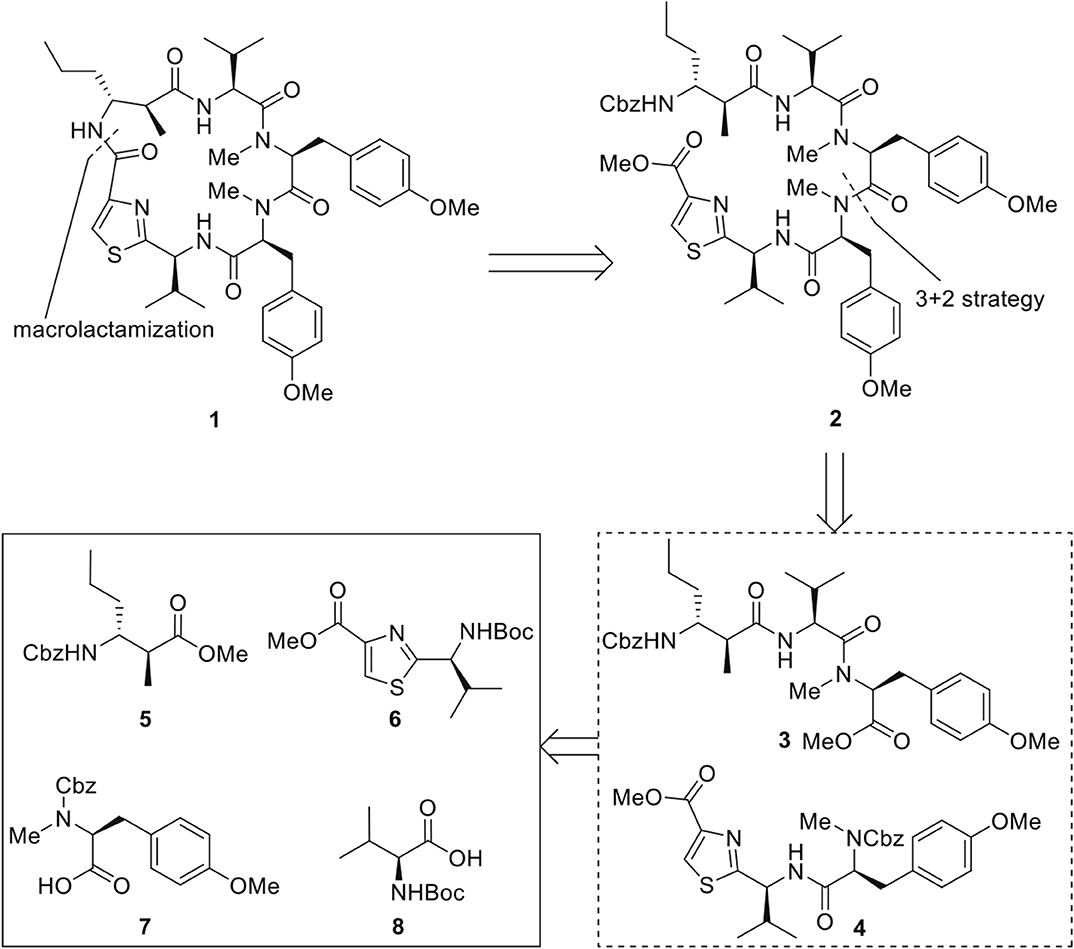
Figure 2. Retrosynthetic Plan of Kakeromamide A (1).
As shown in Figure 3, the synthesis of the nonconventional β-amino acid (11) commenced with the known N-tert-butanesulfinimine 9 (Staas et al., 2002). Following Ellman's protocol (Tang and Ellman, 1999, 2002), methyl propionate was treated with lithium diisopropylamide (LDA) and the following transmetalation with chlorotriisopropoxytitanium provided the titanium enolate, which added to sulfinimine 9 in THF at −78°C to furnish the α,β-disubstituted β-amino ester 10 in 82% yield. The high (10:1) diastereoselectivity observed in the addition process may be explained by analysis of Zimmerman–Traxler transition state Zimmerman and Traxler, 1957 during the nucleophilic addition step. Acidic cleavage of the chiral auxiliary was achieved by exposure of 10 to methanolic HCl solution and the resulting amine was protected by Cbz group to afford 5 in 83% yield over two steps. Saponification of methyl ester 5 with lithium hydroxide underwent smoothly to afford the desired acid 11, which was readily used in the following fragment coupling process without further purification.
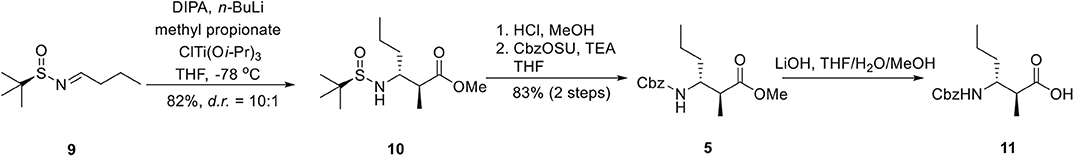
Figure 3. Synthesis of β-amino acid 11.
We next embarked on the assembly of the pentapeptide 2 (Figure 4). The synthesis began with the preparation of thiazole amino acid 6 from the known thioamide 12 (Aguilar and Meyers, 1994) following the modified Hantzsch conditions reported by Aguilar and Meyers (1994). Thus, thioamide 12 was treated with sodium bicarbonate and methyl bromopyruvate in dimethoxyethane and generated the intermediary hydroxythiazoline, which underwent dehydrative aromatization under the mediation of trifluoroacetic anhydride and 2,6-lutidine to afford thiazole amino acid 6 in 88% yield. Removal of the Boc group was accomplished by the treatment of thiazole amino acid 6 with trifluoroacetic acid to provide the corresponding ammonium salt, which was then subjected to 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) promoted amidation conditions and successfully incorporated the known L-tyrosine derivative 7 (Ramanjulu et al., 1997) to afford the dipeptide 4 in 77% yield over three steps. In parallel, the known L-tyrosine derivative 13 (Chen et al., 2002) was converted into tripeptide 3 via a three-step sequence of transformations including HATU/HOAt promoted coupling of amino ester 13 with N-Boc-L-valine (8) to provide dipeptide 20 in 96% yield; Cleavage of the N-terminal Boc protective group of 20 (HCl, MeOH) to give rise to the corresponding amine followed by coupling to Cbz protected beta amino acid 11 to provided 3 in 63% yield. With both dipeptide 4 and tripeptide 3 in hand, the stage was now set for their assembly to afford the corresponding macrocyclization precursor 2. In the event, methyl ester of 3 was saponified, in quantitative yield, to its corresponding carboxylic acid by the mild action of Me3SnOH. Reaction of this carboxylic acid with the amine derived from PdCl2 deprotection of Cbz-protected 4 under a variety of peptide coupling procedures including EDCI–HOAt, HATU, PyAOP, BOPCl, and Mukaiyama reagent, afforded at low yields of pentapeptide 2 as a diastereomeric mixtures. Apparently, somewhere along the line, most likely at the fragment coupling step, the conditions employed caused epimerization. This outcome, a consequence of the sensitivity of these molecules toward epimerization, forced upon us a change in strategy and tactics toward pentapeptide 2, as will be described below.
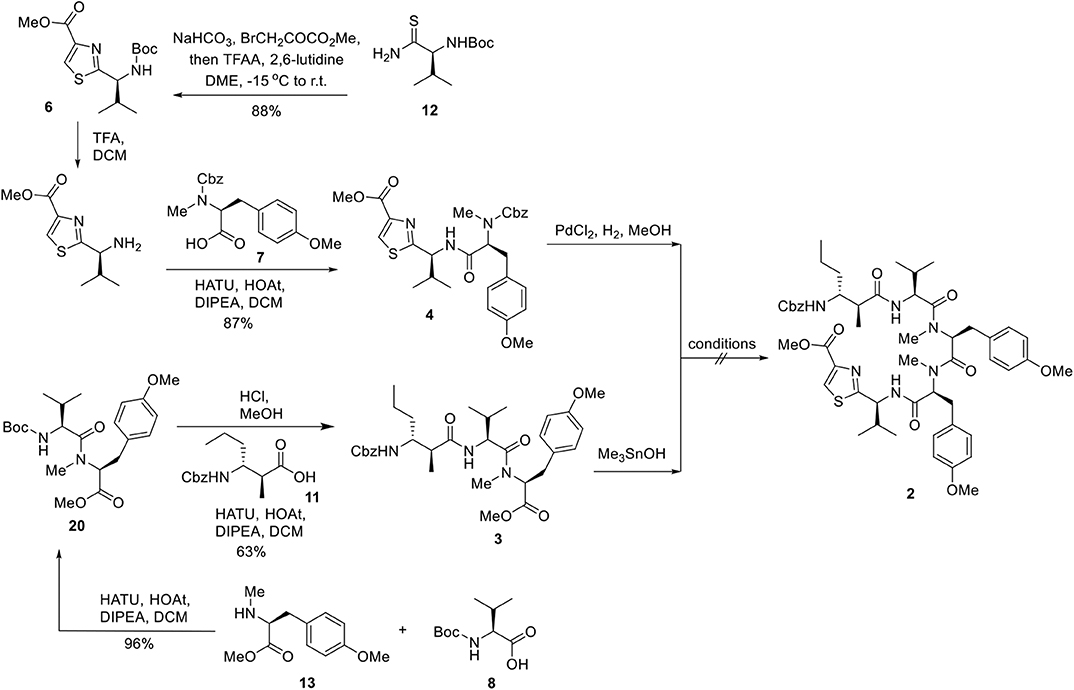
Figure 4. 3+2 Strategy for the Synthesis of Pentapeptide 2.
In order to circumvent the problems encountered in the 3+2 route (Figure 4), a more stepwise approach starting from dipeptide 4 was pursued. Thus, hydrogenolysis of the Cbz group in 4 using palladium chloride smoothly afforded the corresponding amine as its HCl salt, which was then condensed with the known L-tyrosine derivative 7 to provide tripeptide 14 in 77% yield over two steps. Removal of the Cbz group of 14 under identical conditions as for 4, and the resulting amine salt was coupled with N-Boc-L-valine in the presence of HATU/HOAt and DIPEA tetrapeptide 15 in 83% yield. To continue the linear chain elongation, Boc group was selectively removed with TFA/DCM, further coupling with the β-amino acid 11 promoted by HATU/HOAt and DIPEA to give rise to the desired linear pentapeptide 2 in 71% yield. To set the stage for the final macrocyclization (Figure 5), the C-terminal methyl ester was saponified with trimethyltin hydroxide (Furlán et al., 1996; Nicolaou et al., 2005) in toluene at 80°C. Cleavage of the N-terminal Cbz protective group under identical conditions as for 4 provided the corresponding amino acid cyclization precursor, which was immediately subjected to macrolactamization as promoted by pentafluorophenyl diphenylphosphinate (FDPP) (Chen and Xu, 1991; Pradhan et al., 2013) and DIPEA in DMF to provide kakeromamide A (1) in 40% yield over three steps. The spectroscopic data of the synthetic material were consistent with those for the natural kakeromamide A, as evident from the 1H and 13C NMR spectra and optical rotation = +6.15 (c 0.065, MeOH); versus the reported = +6.2 (c 0.065, MeOH); (Tables 1, 2). The synthesis therefore confirmed the relative and absolute configuration of the natural product and also sets the stage for a study of the structure–activity relationship of kakeromamide A, with analogs containing “point mutations” at every site within the cyclic compounds.
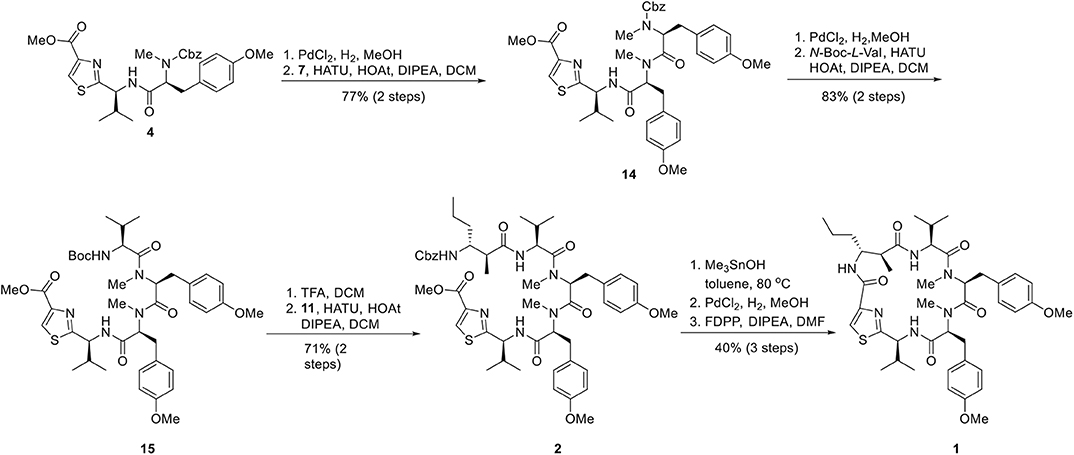
Figure 5. Total Synthesis of Kakeromamide A (1).
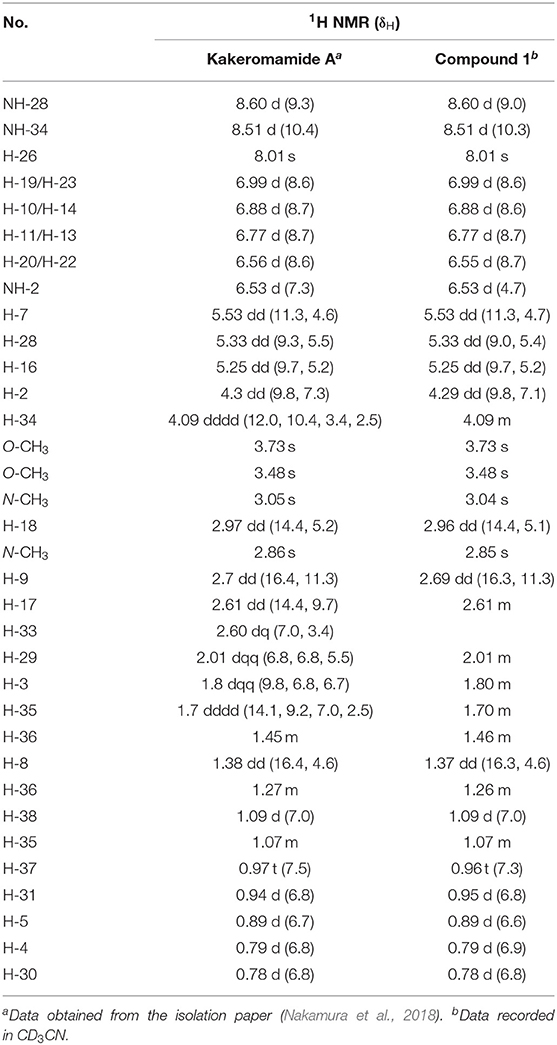
Table 1. Comparison of the 1H NMR Data of Synthesized 1 with Literature Data.
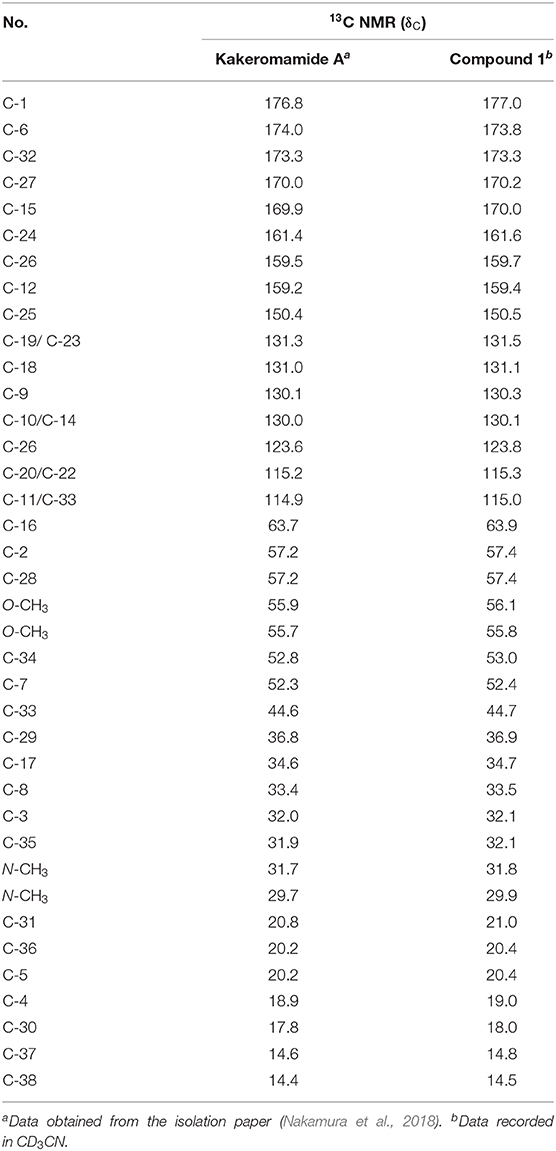
Table 2. Comparison of the 13C NMR Data of Synthesized 1 with Literature Data.
With completion of the total synthesis of kakeromamide A, we decided to employ the same approach in our analog synthesis. An alanine scan (Morrison and Weiss, 2001; Shaheen et al., 2012) of each residue of the cyclopeptide may identify key sites responsible for or contributing to the biological properties. The preparation of a complete set of individual residue analogs should allow an assessment of each structural feature of kakeromamide A and provide a detailed account of the structure function relationships. In the event, four analogs 16–19 (Figure 6) of kakeromamide A, constituting the alanine scan, were constructed from thioamide 12 (Figure 5). Attempts to replace the thiazole amino acid with alanine failed to deliver the desired cyclopeptide from the corresponding linear precursor, presumably due to the added conformational constraints of the linear precursor during macrolactamization.
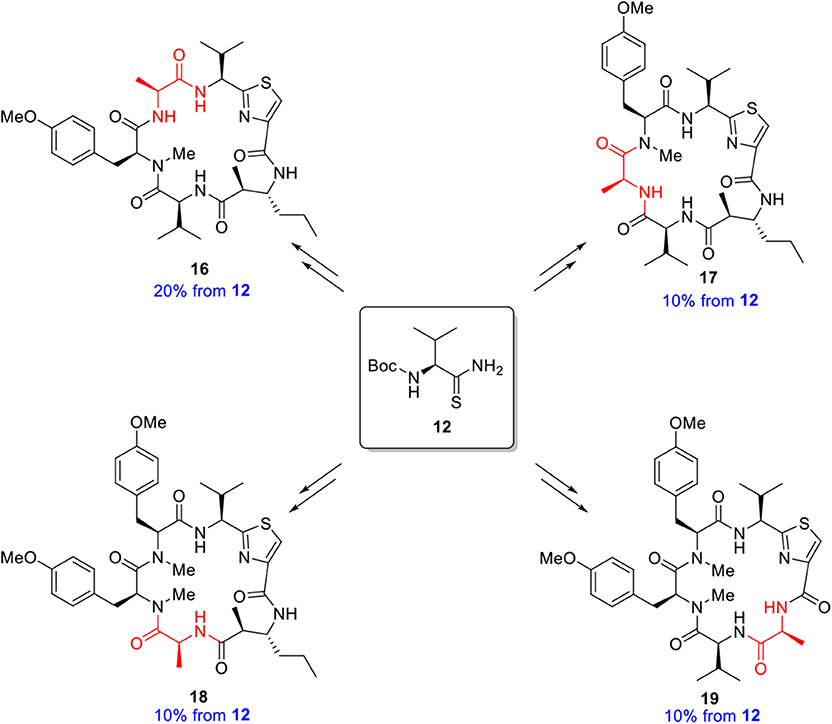
Figure 6. Synthesis of Structural Variants of Kakeromamide A.
Bioactive compounds modulating proliferation and differentiation of neural stem cells are required as chemical probes to study about mechanism of neural development or lead compounds for regenerative medicine. In order to evaluate neural differentiation-modulating activities of kakeromamide A and its analogs, we elected to perform the biological tests using the same procedure that we reported in the isolation paper (Nakamura et al., 2018). Neural stem cells were obtained following the procedure (Nakayama and Inoue, 2006; Iwata et al., 2016) with modification. 2.5, 5, and 10 μM of kakeromamide A or its respective derivative was added to astrocytes or neurons. The cell cytotoxicity of kakeromamide A and its analogs toward astrocytes or neurons was carried out using Hoechst 33342 staining (Figure 7). No significant cytotoxicity was observed upon the treatment with kakeromamide A and its analogs.
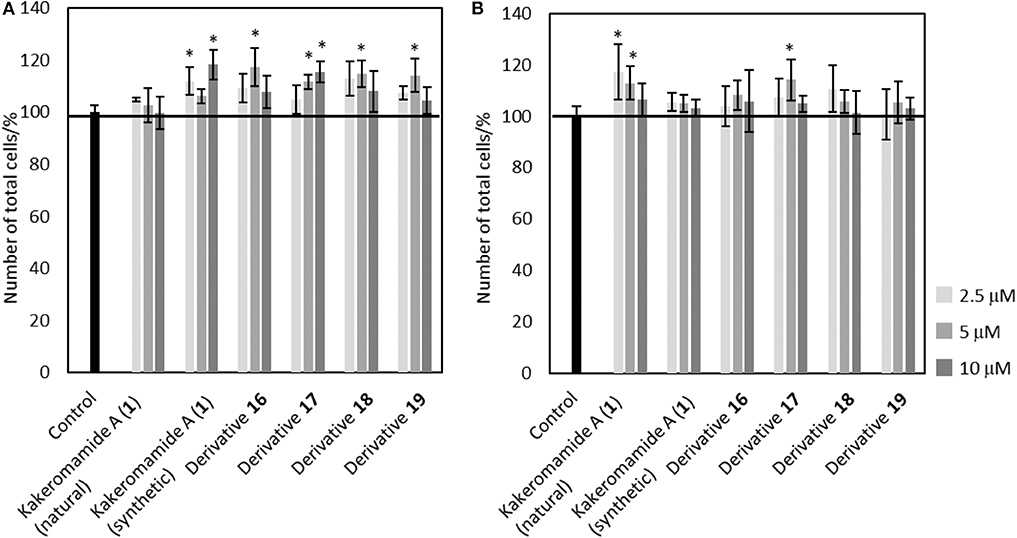
Figure 7. Cytotoxicity of Kakeromamide A and Its Analogs. (A) Percentages of cell numbers calculated by Hoechst 33342 positive cells, compared to that of control, in evaluating effects of kakeromamide A and its analogs for astrocytes (n = 3, means ± S.D., *p < 0.05). (B) Percentages of cell numbers calculated by Hoechst 33342 positive cells, compared to that of control, in evaluating effects of kakeromamide A and its analogs for neurons (n = 3, means ± S.D., *p < 0.05).
Next, kakeromamide A or its respective derivative was exposed at three concentrations (2.5, 5, and 10 μM) during differentiation of neural stem cells (NSCs). The effects of kakeromamide A and its analogs on the neural differentiation into neurons or astrocytes were evaluated by using NeuO (Er et al., 2015) a fluorescent probe which could selectively stain neurons, or immunostaining of an astrocyte marker GFAP (glial fibrillary acidic protein), respectively. The nuclei were stained by using Hoechst 33342 followed by the microscopic analysis. Their activities were determined by calculating a ratio NeuO or GFAP positive cells to whole cells and comparing with that of the control. As a result, the synthetic product of kakeromamide A (1) showed the same activity as the sample isolated from natural source, promoting differentiation of NSCs into astrocytes while inhibiting neuronal differentiation of NSCs (Figure 8). Furthermore, the astrocytic or neuronal differentiation were promoted or inhibited, respectively, by treatment with four analogs of compound 1. It was found that derivative 17 modulated the differentiation most strongly among derivatives. These results suggested that kakeromamide A and its analogs modulated neural differentiation of NSCs via common target (s) and especially derivative 17 had the highest affinity to the target (s).
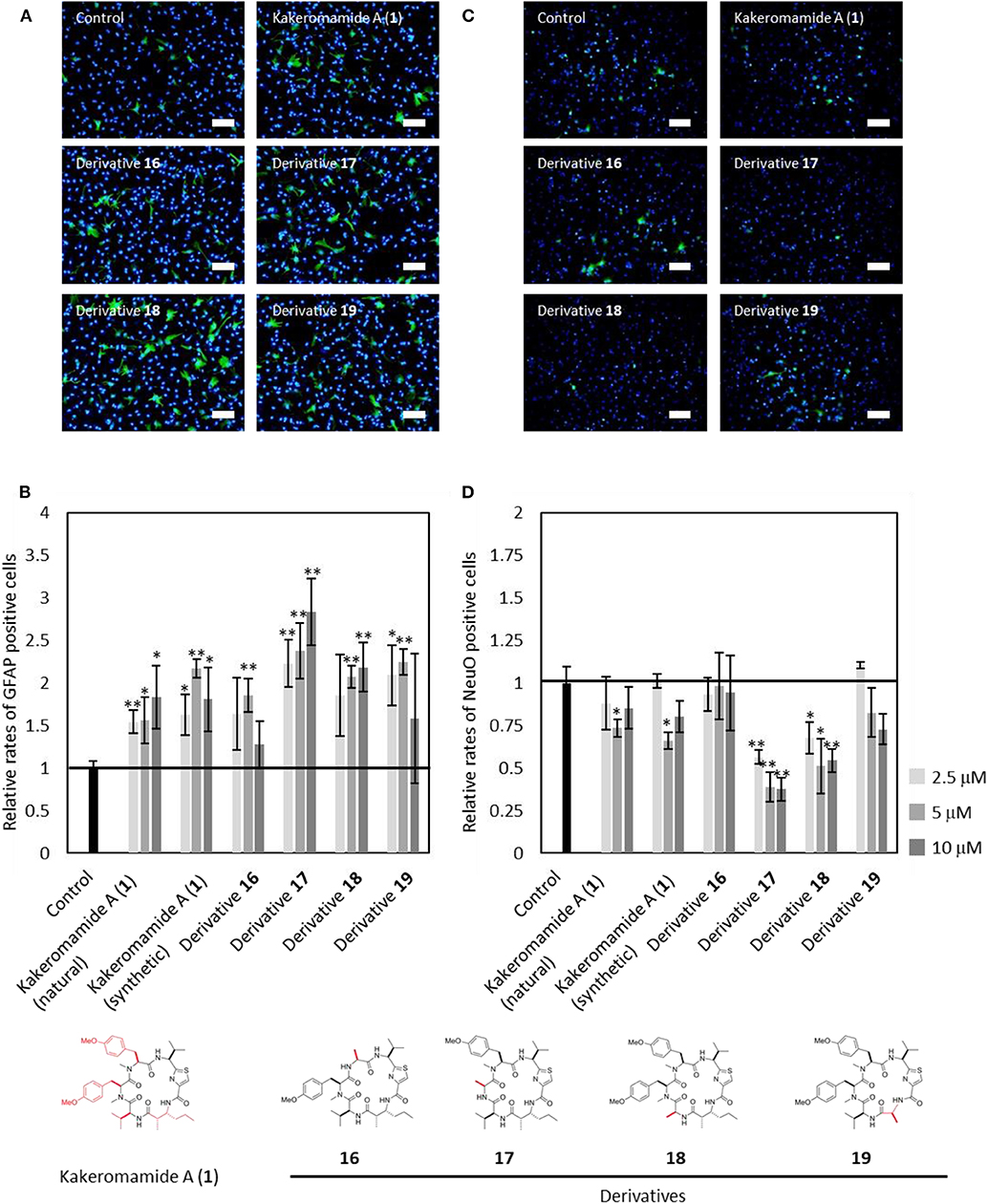
Figure 8. Neural Differentiation-Modulating Activity of Kakeromamide A and Its Analogs. (A) Fluorescent microscopic images of differentiation of neural stem cells (NSCs) into astrocyte. Astrocytes and nuclei were colored by anti-GFAP (glial fibrillary acidic protein, green) and Hoechst 33342 (blue), respectively (5 μM, Scale bar; 100 μm). (B) Rates for astrocytic differentiation of NSCs compared to that of control (n = 3, means ± S.D., *p < 0.05, **p < 0.01). (C) Fluorescent microscopic images of differentiation of NSCs into neuron. Neurons and nuclei were stained by NeuO (green) and Hoechst 33342 (blue), respectively (5 μM, Scale bar; 100 μm). (D) Rates for neuronal differentiation of NSCs compared to that of control (n = 3, means ± S.D., *p < 0.05, **p < 0.01).
Conclusions
In summary, we have achieved the first synthesis of a new cyclopeptide kakeromamide A (1) from the known thioamide 12 in 14% overall yield with the longest linear sequence being 12 steps. The present work confirms the originally assigned structure of kakeromamide A (1) and also allows access to four analogs. Neural differentiation-modulating activities of kakeromamide A and four analogs were performed, which revealed that kakeromamide A and its analogs promoted astrocytic differentiation while inhibited neuronal differentiation of NSCs and derivative 17 showed the strongest activity. Further analyses of the detailed mechanism for their biological activity are currently under investigation in our laboratories and will be described in due course.
Data Availability Statement
All datasets presented in this study are included in the article/Supplementary Material.
Author Contributions
MZ: designed and conducted chemical synthesis experiments, analyzed data. YX: contributed to analyzed data. SO: designed and conducted biological activity experiments, analyzed data. YG: contributed to chemical data analysis and writing of the paper. YN: contributed to biological data analysis and writing of the paper. TY: designed experiments, analyzed data, and wrote the paper.
Funding
We acknowledge financial support from the National Natural Science Foundation of China (21772009, 21901013), the Shenzhen Peacock Plan (KQTD2015071714043444), the Shenzhen Science and Technology Innovation Commission (JCYJ20170818090238288), the Shenzhen-Hong Kong Institute of Brain Science-Shenzhen Fundamental Research Institutions (2019SHIBS0004).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was inspired by the international and interdisciplinary environments of the JSPS Asian CORE Program, Asian Chemical Biology Initiative.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2020.00410/full#supplementary-material
References
Aguilar, E., and Meyers, A. I. (1994). Reinvestigation of a modified Hantzsch thiazole synthesis. Tetrahedron Lett. 35, 2473–2476. doi: 10.1016/S0040-4039(00)77147-4
Chen, S., and Xu, J. (1991). Pentafluorophenyl diphenylphosphinate a new efficient coupling reagent in peptide chemistry. Tetrahedron Lett. 32, 6711–6714. doi: 10.1016/S0040-4039(00)93583-4
Chen, Y., Bilban, M., Foster, C. A., and Boger, D. L. (2002). Solution-phase parallel synthesis of a pharmacophore library of HUN-7293 analogues: a general chemical mutagenesis approach to defining structure-function properties of naturally occurring cyclic (depsi)peptides. J. Am. Chem. Soc. 124, 5431–5440. doi: 10.1021/ja020166v
Cheng, Y., Tang, S., Guo, Y., and Ye, T. (2018). Total synthesis of anti-tuberculosis natural products ilamycins E1 and F. Org. Lett. 20, 6166–6169. doi: 10.1021/acs.orglett.8b02643
Er, J. C., Leong, C., Teoh, C. L., Yuan, Q., Merchant, P., Dunn, M., et al. (2015). NeuO: a fluorescent chemical probe for live neuron labeling. Angew. Chem. Int. Ed. 54, 2442–2446. doi: 10.1002/anie.201408614
Furlán, R. L. E., Mata, E. G., and Mascaretti, O. A. (1996). Cleavage of carboxylic esters effected by organotin oxides and hydroxides under classical heating and microwave irradiation. A comparative study. Tetrahedron Lett. 37, 5229–5232. doi: 10.1016/0040-4039(96)01071-4
Guo, Y., Zhao, M., Xu, Z., and Ye, T. (2017). Total synthesis and stereochemical assignment of actinoranone. Chem. Eur. J. 23, 3572–3576. doi: 10.1002/chem.201700476
Guzman-Martinez, A., Lamer, R., and VanNieuwenhze, M. S. (2007). Total synthesis of lysobactin. J. Am. Chem. Soc. 129, 6017–6021. doi: 10.1021/ja067648h
Humphrey, J. M., and Chamberlin, A. R. (1997). Chemical synthesis of natural product peptides: coupling methods for the incorporation of noncoded amino acids into peptides. Chem. Rev. 97, 2243–2266. doi: 10.1021/cr950005s
Iwata, T., Otsuka, S., Tsubokura, K., Kurbangalieva, A., Arai, D., Fukase, K., et al. (2016). One-pot evolution of ageladine a through a bio-inspired cascade towards selective modulators of neuronal differentiation. Chem. Eur. J. 22, 14707–14716. doi: 10.1002/chem.201602651
Lei, H., Yan, J., Yu, J., Liu, Y., Wang, Z., Xu, Z., et al. (2014). Total synthesis and stereochemical reassignment of mandelalide A. Angew. Chem. Int. Ed. 53, 6533–6537. doi: 10.1002/anie.201403542
Liao, L., Zhou, J., Xu, Z., and Ye, T. (2016). Concise total synthesis of nannocystin A. Angew. Chem. Int. Ed. 55, 13263–13266. doi: 10.1002/anie.201606679
Luesch, H., Yoshida, W. Y., Moore, R. E., Paul, V. J., and Corbett, T. H. (2001). Total structure determination of apratoxin A, a potent novel cytotoxin from the marine cyanobacterium Lyngbya majuscule. J. Am. Chem. Soc. 123, 5418–5423. doi: 10.1021/ja010453j
Morrison, K. L., and Weiss, G. A. (2001). Combinatorial alanine-scanning. Curr. Opin. Chem. Biol. 5, 302–307. doi: 10.1016/S1367-5931(00)00206-4
Nabika, R., Oishi, S., Misu, R., Ohno, H., and Fujii, N. (2014). Synthesis of IB-01212 by multiple N-methylations of peptide bonds. Bioorg. Med. Chem. Lett. 22, 6156–6162. doi: 10.1016/j.bmc.2014.08.036
Nakamura, F., Maejima, H., Kawamura, M., Arai, D., Okino, T., Zhao, M., et al. (2018). Kakeromamide A, a new cyclic pentapeptide inducing astrocyte differentiation isolated from the marine cyanobacterium Moorea bouillonii. Bioorg. Med. Chem. Lett. 28, 2206–2209. doi: 10.1016/j.bmcl.2018.04.067
Nakayama, T., and Inoue, N. (2006). Neural stem sphere method: induction of neural stem cells and neurons by astrocyte-derived factors in embryonic stem cells in vitro. Methods Mol. Biol. 330, 1–13. doi: 10.1385/1-59745-036-7:001
Nicolaou, K. C., Estrada, A. A., Zak, M., Lee, S. H., and Safina, B. S. (2005). A mild and selective method for the hydrolysis of esters with trimethyltin hydroxide. Angew. Chem. Int. Ed. 44, 1378–1382. doi: 10.1002/anie.200462207
Pradhan, T. K., Reddy, K. M., and Ghosh, S. (2013). Total synthesis of emericellamides A and B. Tetrahedron Asymmetry 24, 1042–1051. doi: 10.1016/j.tetasy.2013.07.012
Ramanjulu, J. M., Joullie, M. M., and Li, W. R. (1997). Syntheses of acyclic analogs of didemnin B. Synth. Commun. 27, 3259–3272. doi: 10.1080/00397919708004186
Shaheen, F., Rizvi, T. S., Musharraf, S. G., Ganesan, A., Xiao, K., Townsend, J. B., et al. (2012). Solid-phase total synthesis of cherimolacyclopeptide E and discovery of more potent analogues by alanine screening. J. Nat. Prod. 75, 1882–1887. doi: 10.1021/np300266e
Staas, D. D., Savage, K. L., Homnick, C. F., Tsou, N. N., and Ball, R. G. (2002). Asymmetric synthesis of α,α-difluoro-β-amino acid derivatives from enantiomerically pure N-tert-butylsulfinimines. J. Org. Chem. 67, 8276–8279. doi: 10.1021/jo0259313
Tang, T. P., and Ellman, J. A. (1999). The tert-butanesulfinyl group: an ideal chiral directing group and Boc-surrogate for the asymmetric synthesis and applications of β-amino acids. J. Org. Chem. 64, 12–13. doi: 10.1021/jo9820824
Tang, T. P., and Ellman, J. A. (2002). Asymmetric synthesis of beta-amino acid derivatives incorporating a broad range of substitution patterns by enolate additions to tert-butanesulfinyl imines. J. Org. Chem. 67, 7819–7832. doi: 10.1021/jo025957u
Teixidó, M., Albericio, F., and Giralt, E. (2005). Solid-phase synthesis and characterization of N-methyl-rich peptides. J. Pept. Res. 65, 153–166. doi: 10.1111/j.1399-3011.2004.00213.x
Yang, M., Peng, W., Guo, Y., and Ye, T. (2000). Total synthesis of dysoxylactam A. Org. Lett. 22, 1776–1779. doi: 10.1021/acs.orglett.0c00074
Yu, J., Yang, M., Guo, Y., and Ye, T. (2019). Total synthesis of psymberin (irciniastatin A). Org. Lett. 21, 3670–3673. doi: 10.1021/acs.orglett.9b01113
Zainuddin, E. N., Mentel, R., Wray, V., Jansen, R., Nimtz, M., Lalk, M., et al. (2007). Cyclic depsipeptides, ichthyopeptins A and B, from Microcystis ichthyoblabe. J. Nat. Prod. 70, 1084–1088. doi: 10.1021/np060303s
Zhou, J., Gao, B., Xu, Z., and Ye, T. (2016). Total synthesis and stereochemical assignment of callyspongiolide. J. Am. Chem. Soc. 138, 6948–6951. doi: 10.1021/jacs.6b03533
Keywords: kakeromamide A, total synthesis, biological evaluations, marine cyclopeptide, structural analogs
Citation: Zhao M, Xiao Y, Otsuka S, Nakao Y, Guo Y and Ye T (2020) Total Synthesis and Biological Evaluation of Kakeromamide A and Its Analogues. Front. Chem. 8:410. doi: 10.3389/fchem.2020.00410
Received: 20 March 2020; Accepted: 20 April 2020;
Published: 13 May 2020.
Edited by:
Zhendong Jin, University of Iowa, United StatesReviewed by:
Yuguo Du, Chinese Academy of Sciences (CAS), ChinaChristopher Pigge, University of Iowa, United States
Copyright © 2020 Zhao, Xiao, Otsuka, Nakao, Guo and Ye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yian Guo, YnJ5YW5fcGt1QHFxLmNvbQ==; Tao Ye, eWV0QHBrdXN6LmVkdS5jbg==